
Therapeutic Advances in Diabetes, Autoimmune, and Neurological Diseases

Abstract
1. Introduction
1.1. Small Molecule and Antibody-Based Therapeutics
1.2. Peptide-Based Therapeutics
1.3. Cell and Gene Therapies
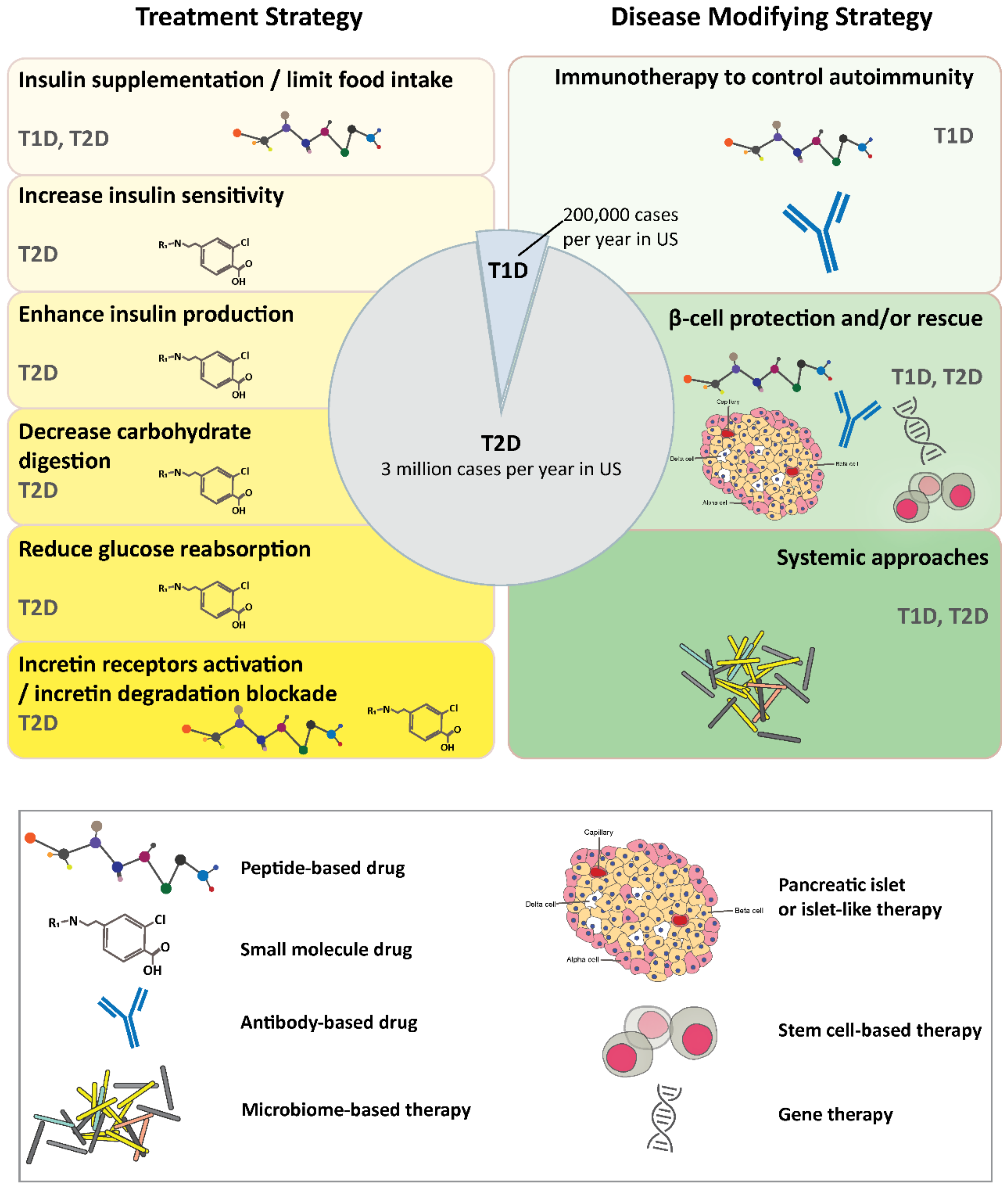
2. Diabetes
2.1. Treatment Strategies
2.2. Disease Modifying Strategies
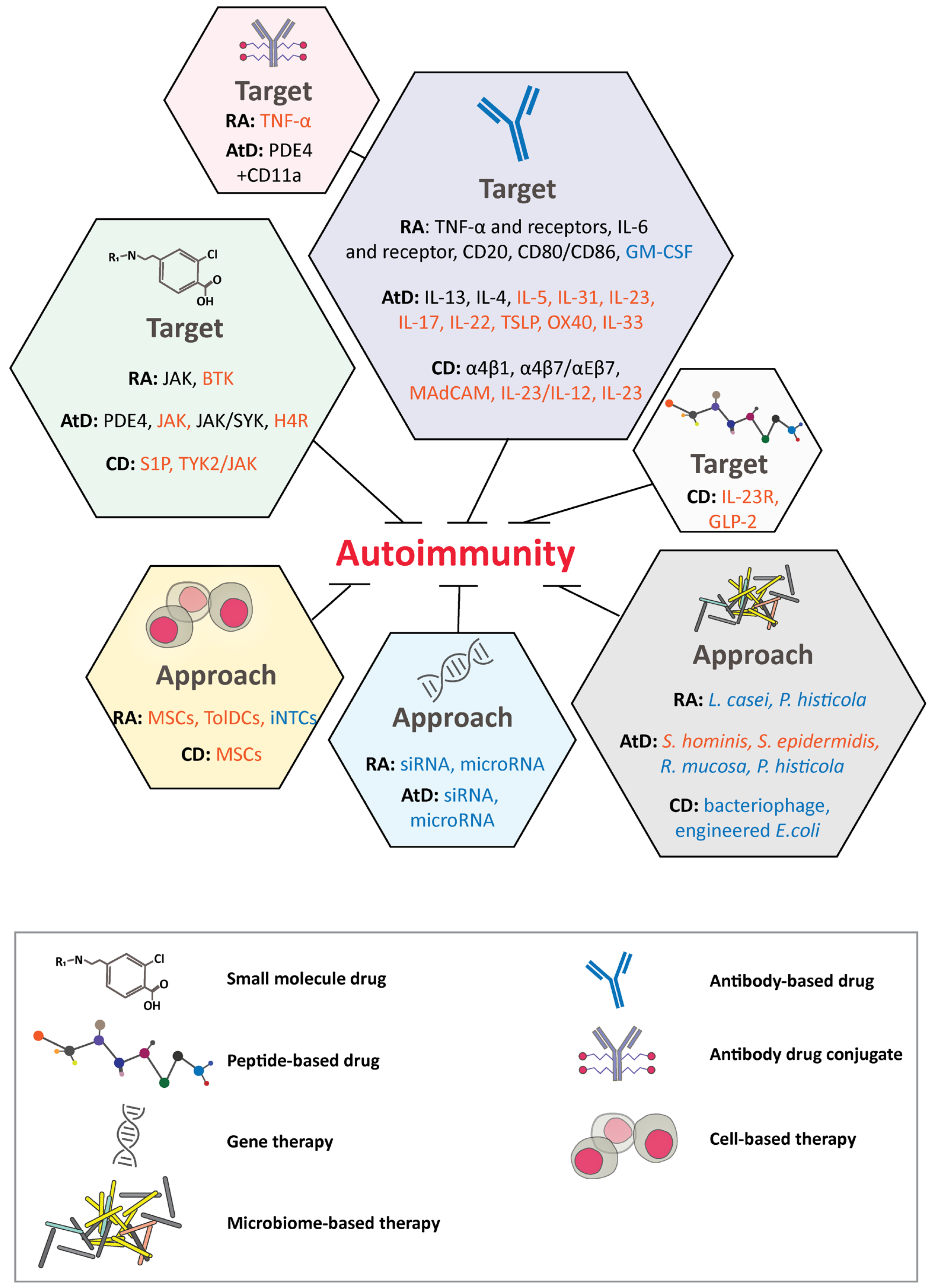
3. Autoimmune Diseases
3.1. Rheumatoid Arthritis
3.2. Atopic Dermatitis
3.3. Crohn’s Disease
4. Neurological Diseases
4.1. Chronic Pain
4.1.1. Osteoarthritis
4.1.2. Migraine
4.2. Neurodegenerative Disease
4.2.1. Alzheimer’s Disease

{kind=link}
{kind=link}
| Drug | Approval Date | Mechanism of Action | Indication | Status | Reference |
|---|---|---|---|---|---|
| Tacrine | 1995 | AChEI | mild to moderate AD | Discontinued | [478] |
| Donepezil | 1996 | AChEI | mild to moderate AD | Approved | [479] |
| Rivastigmine | 1997 | AChEI | mild to moderate AD | Approved | [480] |
| Galantamine | 2001 | AChEI | mild to moderate AD | Approved | [481] |
| Memantine | 2003 | NMDA receptor agonist | Moderate to severe AD | Approved | [482] |
4.2.2. Parkinson’s Disease
5. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| FDA | U.S. Food and Drug Administration |
| CDER | The Center for Drug Evaluation and Research |
| CBER | The Center for Biologics Evaluation and Research |
| WHO | World health organization |
| mAb | Monoclonal antibody |
| bsAb | Bispecific antibody |
| ADC | Antibody drug conjugate |
| CAR T | Chimeric antigen receptor T cell |
| TNF | Tumor necrosis factor |
| GPCR | G-protein coupled receptor |
| GLP-1 | Glucagon-like peptide-1 |
| GIP | Glucose-dependent insulinotropic polypeptide |
| AAV | Adeno-associated virus |
| GM-CSF | Granulocyte-macrophage colony stimulating factor |
| ON | Oligonucleotide |
| ASO | Antisense oligonucleotide |
| siRNA | Short interfering RNA |
| GalNac | N-acetylgalactosamine |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| T1D | Type 1 diabetes mellitus |
| T2D | Type 2 diabetes mellitus |
| HbA1c | Hemoglobin A1c |
| HSC | Hematopoietic stem cell |
| MSC | Mesenchymal stem cell |
| NF-κB | Nuclear factor -κB |
| JAK | Janus kinase |
| STAT | Transducer and activator of transcription |
| SYK | Spleen tyrosine kinase |
| TYK2 | Tyrosine kinase 2 |
| BTK | Bruton’s tyrosine kinase |
| AtD | Atopic dermatitis |
| Th | T helper |
| PDE4 | Phosphodiesterase 4 |
| EASI | Eczema area and severity index |
| H4R | Histamine H4 receptor |
| MAdCAM | Mucosal addressin cell adhesion molecule |
| S1P | Sphingosine 1-phosphate |
| CNS | Central nervous system |
| OA | Osteoarthritis |
| NSAID | Nonsteroidal anti-inflammatory drug |
| COX | Cyclooxygenase |
| NGF | Nerve growth factor |
| MMP | Matrix metalloproteinase |
| CGRP | Calcitonin gene-related peptide |
| ND | Neurodegenerative disease |
| AD | Alzheimer’s disease |
| PD | Parkinson’s disease |
| AChEIs | Acetylcholinesterase inhibitors |
| NMDA | N-methyl-D-aspartic acid |
| DMTs | Disease-modifying treatments |
References
- FDA. FDA-Regulated Products and Facilities. 2019. Available online: https://www.fda.gov/about-fda/fda-basics/fact-sheet-fda-glance (accessed on 1 August 2020).
- Meier, C.; Cairns-Smith, S.; Schulze, U. Can emerging drug classes improve R&D productivity? Drug Discov. Today 2013, 18, 607–609. [Google Scholar] [CrossRef] [PubMed]
- David, E.; Tramontin, T.; Zemmel, R. Pharmaceutical R&D: The road to positive returns. Nat. Rev. Drug Discov. 2009, 8, 609–610. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef]
- Yu, J.X.; Hubbard-Lucey, V.M.; Tang, J. Immuno-oncology drug development goes global. Nat. Rev. Drug Discov. 2019, 18, 899–900. [Google Scholar] [CrossRef]
- Czechowicz, A.; Palchaudhuri, R.; Scheck, A.; Hu, Y.; Hoggatt, J.; Saez, B.; Pang, W.W.; Mansour, M.K.; Tate, T.A.; Chan, Y.Y.; et al. Selective hematopoietic stem cell ablation using CD117-antibody-drug-conjugates enables safe and effective transplantation with immunity preservation. Nat. Commun. 2019, 10, 617. [Google Scholar] [CrossRef]
- Gillard, G.R.; Proctor, J.; Brooks, M.; Lamothe, T.; Hyzy, S.; McDonough, S.; Palchaudhuri, R.; Bhat, A.; Sarma, G.; Bhattarai, P.; et al. Administration of a CD45 Antibody Drug Conjugate as a Novel, Targeted Approach to Achieve Immune System Reset: A Single Dose of CD45-targeted ADC Safely Conditions for Autologous Transplant and Ameliorates Disease in Multiple Models of Autoimmune Disease [abstract]. Arthritis Rheumatol. 2019, 71 (Suppl. 10), 120. [Google Scholar]
- Jovcevska, I.; Muyldermans, S. The Therapeutic Potential of Nanobodies. BioDrugs 2020, 34, 11–26. [Google Scholar] [CrossRef]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Ting, J.P.; Liu, J.; Al-Azzam, S.; Pandya, P.; Afshar, S. Impact of non-proteinogenic amino acids in the discovery and development of peptide therapeutics. Amino Acids 2020, 52, 1207–1226. [Google Scholar] [CrossRef]
- Valeur, E.; Gueret, S.M.; Adihou, H.; Gopalakrishnan, R.; Lemurell, M.; Waldmann, H.; Grossmann, T.N.; Plowright, A.T. New Modalities for Challenging Targets in Drug Discovery. Angew. Chem. Int. Ed. Engl. 2017, 56, 10294–10323. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Afshar, S. In Vitro Assays: Friends or Foes of Cell-Penetrating Peptides. Int. J. Mol. Sci. 2020, 21, 4719. [Google Scholar] [CrossRef]
- Henninot, A.; Collins, J.C.; Nuss, J.M. The Current State of Peptide Drug Discovery: Back to the Future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef]
- Yang, W.; Gadgil, P.; Krishnamurthy, V.R.; Landis, M.; Mallick, P.; Patel, D.; Patel, P.J.; Reid, D.L.; Sanchez-Felix, M. The Evolving Druggability and Developability Space: Chemically Modified New Modalities and Emerging Small Molecules. AAPS J. 2020, 22, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Njardarson, J.T. Top 200 Brand Name Drugs by Retail Sales in 2019. 2019. Available online: https://njardarson.lab.arizona.edu/content/top-pharmaceuticals-poster (accessed on 2 August 2020).
- McGrath, N.A.; Brichacek, M.; Njardarson, J.T. A Graphical Journey of Innovative Organic Architectures That Have Improved Our Lives. J. Chem. Educ. 2010, 87, 1348–1349. [Google Scholar] [CrossRef]
- Drucker, D.J. Advances in oral peptide therapeutics. Nat. Rev. Drug Discov. 2020, 19, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Brayden, D.J.; Hill, T.A.; Fairlie, D.P.; Maher, S.; Mrsny, R.J. Systemic delivery of peptides by the oral route: Formulation and medicinal chemistry approaches. Adv. Drug Deliv. Rev. 2020, 157, 2–36. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, T.A.; Teijeiro-Osorio, D.; Rosa, M.; Coulter, I.S.; Alonso, M.J.; Brayden, D.J. Current status of selected oral peptide technologies in advanced preclinical development and in clinical trials. Adv. Drug Deliv. Rev. 2016, 106, 223–241. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, H.; Ghosh, D.; Williams, R.O., 3rd. Just how prevalent are peptide therapeutic products? A critical review. Int. J. Pharm. 2020, 587, 119491. [Google Scholar] [CrossRef] [PubMed]
- Ashmore-Harris, C.; Fruhwirth, G.O. The clinical potential of gene editing as a tool to engineer cell-based therapeutics. Clin. Transl. Med. 2020, 9, 15. [Google Scholar] [CrossRef]
- Kim, H.; Kim, J.S. A guide to genome engineering with programmable nucleases. Nat. Rev. Genet. 2014, 15, 321–334. [Google Scholar] [CrossRef]
- Katrekar, D.; Chen, G.; Meluzzi, D.; Ganesh, A.; Worlikar, A.; Shih, Y.R.; Varghese, S.; Mali, P. In vivo RNA editing of point mutations via RNA-guided adenosine deaminases. Nat. Methods 2019, 16, 239–242. [Google Scholar] [CrossRef]
- Nishikura, K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 2016, 17, 83–96. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Ma, W.; Mei, C.; Chen, X.; Yao, Y.; Liu, Y.; Qin, X.; Yuan, Y. Description of CRISPR/Cas9 development and its prospect in hepatocellular carcinoma treatment. J. Exp. Clin. Cancer Res. 2020, 39, 1–11. [Google Scholar] [CrossRef]
- Stein, R. First U.S. Patients Treated With CRISPR As Human Gene-Editing Trials Get Underway. 2019. Available online: https://www.npr.org/sections/health-shots/2019/04/16/712402435/first-u-s-patients-treated-with-crispr-as-gene-editing-human-trials-get-underway (accessed on 2 August 2020).
- Henderson, H. CRISPR Clinical Trials: A 2019 Update; Innovative Genomics Institute: Berkeley, CA, USA, 2019. [Google Scholar]
- Rosenbaum, L. New Data From First Human Crispr Trials Shows Promising Results. 2019. Available online: https://www.forbes.com/sites/leahrosenbaum/2019/11/19/human-crispr-trials-promising/?sh=7e470f132daa (accessed on 10 August 2020).
- Bacman, S.R.; Kauppila, J.H.K.; Pereira, C.V.; Nissanka, N.; Miranda, M.; Pinto, M.; Williams, S.L.; Larsson, N.G.; Stewart, J.B.; Moraes, C.T. MitoTALEN reduces mutant mtDNA load and restores tRNA(Ala) levels in a mouse model of heteroplasmic mtDNA mutation. Nat. Med. 2018, 24, 1696–1700. [Google Scholar] [CrossRef] [PubMed]
- Gammage, P.A.; Viscomi, C.; Simard, M.L.; Costa, A.S.H.; Gaude, E.; Powell, C.A.; Van Haute, L.; McCann, B.J.; Rebelo-Guiomar, P.; Cerutti, R.; et al. Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat. Med. 2018, 24, 1691–1695. [Google Scholar] [CrossRef] [PubMed]
- Mok, B.Y.; de Moraes, M.H.; Zeng, J.; Bosch, D.E.; Kotrys, A.V.; Raguram, A.; Hsu, F.; Radey, M.C.; Peterson, S.B.; Mootha, V.K.; et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 2020, 583, 631–637. [Google Scholar] [CrossRef]
- Neklesa, T.; Snyder, L.B.; Willard, R.R.; Vitale, N.; Pizzano, J.; Gordon, D.A.; Bookbinder, M.; Macaluso, J.; Dong, H.; Ferraro, C.; et al. ARV-110: An oral androgen receptor PROTAC degrader for prostate cancer. J. Clin. Oncol. 2019, 37, 259. [Google Scholar] [CrossRef]
- Flanagan, J.J.; Qian, Y.; Gough, S.M.; Andreoli, M.; Bookbinder, M.; Cadelina, G.; Bradley, J.; Rousseau, E.; Willard, R.; Pizzano, J.; et al. Abstract P5-04-18: ARV-471, an oral estrogen receptor PROTAC degrader for breast cancer. Cancer Res. 2019, 79. [Google Scholar] [CrossRef]
- Banik, S.M.; Pedram, K.; Wisnovsky, S.; Ahn, G.; Riley, N.M.; Bertozzi, C.R. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 2020, 584, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, D.; Moriyama, J.; Nakamura, T.; Miki, E.; Takahashi, E.; Sato, A.; Akaike, T.; Itto-Nakama, K.; Arimoto, H. AUTACs: Cargo-Specific Degraders Using Selective Autophagy. Mol. Cell 2019, 76, 797–810. [Google Scholar] [CrossRef]
- Li, Z.; Zhu, C.; Ding, Y.; Fei, Y.; Lu, B. ATTEC: A potential new approach to target proteinopathies. Autophagy 2020, 16, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Fei, Y.; Lu, B. Emerging New Concepts of Degrader Technologies. Trends Pharmacol. Sci. 2020, 41, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, M.; Langer, R.; Traverso, G. Microbial therapeutics: New opportunities for drug delivery. J. Exp. Med. 2019, 216, 1005–1009. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Das, P.; Buschmann, M.; Gilbert, J.A. The Future of Microbiome-Based Therapeutics in Clinical Applications. Clin. Pharmacol. Ther. 2020, 107, 123–128. [Google Scholar] [CrossRef]
- Garrett, W.S. Immune recognition of microbial metabolites. Nat. Rev. Immunol. 2020, 20, 91–92. [Google Scholar] [CrossRef]
- Skelly, A.N.; Sato, Y.; Kearney, S.; Honda, K. Mining the microbiota for microbial and metabolite-based immunotherapies. Nat. Rev. Immunol. 2019, 19, 305–323. [Google Scholar] [CrossRef]
- Kowalski, K.; Mulak, A. Brain-Gut-Microbiota Axis in Alzheimer’s Disease. J. Neurogastroenterol. Motil. 2019, 25, 48–60. [Google Scholar] [CrossRef]
- He, Y.; Li, B.; Sun, D.; Chen, S. Gut Microbiota: Implications in Alzheimer’s Disease. J. Clin. Med. 2020, 9, 2042. [Google Scholar] [CrossRef]
- Charbonneau, M.R.; Isabella, V.M.; Li, N.; Kurtz, C.B. Developing a new class of engineered live bacterial therapeutics to treat human diseases. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- International Diabetes Federation. IDF Diabetes Atlas 2019. 2019. Available online: https://www.diabetesatlas.org/en/ (accessed on 20 August 2020).
- American Diabetes Association. Economic Costs of Diabetes in the U.S. in 2017. Diabetes Care 2018, 41, 917–928. [Google Scholar] [CrossRef]
- Bluestone, J.A.; Herold, K.; Eisenbarth, G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature 2010, 464, 1293–1300. [Google Scholar] [CrossRef]
- Daneman, D. Type 1 diabetes. Lancet 2006, 367, 847–858. [Google Scholar] [CrossRef]
- Redondo, M.J.; Fain, P.R.; Eisenbarth, G.S. Genetics of type 1A diabetes. Recent Prog. Horm. Res. 2001, 56, 69–89. [Google Scholar] [CrossRef]
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef]
- Burrack, A.L.; Martinov, T.; Fife, B.T. T Cell-Mediated Beta Cell Destruction: Autoimmunity and Alloimmunity in the Context of Type 1 Diabetes. Front. Endocrinol. 2017, 8, 343. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.E.; Hull, R.L.; Utzschneider, K.M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006, 444, 840–846. [Google Scholar] [CrossRef]
- American Diabetes, A. Gestational diabetes mellitus. Diabetes Care 2003, 26 (Suppl. S1), S103–S105. [Google Scholar] [CrossRef]
- Melmer, A.; Laimer, M. Treatment Goals in Diabetes. Endocr. Dev. 2016, 31, 1–27. [Google Scholar] [CrossRef]
- Kahanovitz, L.; Sluss, P.M.; Russell, S.J. Type 1 Diabetes—A Clinical Perspective. Point Care 2017, 16, 37–40. [Google Scholar] [CrossRef]
- Pickup, J.C. Insulin-pump therapy for type 1 diabetes mellitus. N. Engl. J. Med. 2012, 366, 1616–1624. [Google Scholar] [CrossRef]
- Ryan, G.J.; Jobe, L.J.; Martin, R. Pramlintide in the treatment of type 1 and type 2 diabetes mellitus. Clin. Ther 2005, 27, 1500–1512. [Google Scholar] [CrossRef]
- Woods, S.C.; Lutz, T.A.; Geary, N.; Langhans, W. Pancreatic signals controlling food intake; insulin, glucagon and amylin. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2006, 361, 1219–1235. [Google Scholar] [CrossRef] [PubMed]
- Ratner, R.E.; Dickey, R.; Fineman, M.; Maggs, D.G.; Shen, L.; Strobel, S.A.; Weyer, C.; Kolterman, O.G. Amylin replacement with pramlintide as an adjunct to insulin therapy improves long-term glycaemic and weight control in Type 1 diabetes mellitus: A 1-year, randomized controlled trial. Diabet Med. 2004, 21, 1204–1212. [Google Scholar] [CrossRef]
- Inzucchi, S.E.; Lipska, K.J.; Mayo, H.; Bailey, C.J.; McGuire, D.K. Metformin in patients with type 2 diabetes and kidney disease: A systematic review. JAMA 2014, 312, 2668–2675. [Google Scholar] [CrossRef] [PubMed]
- Werner, A.L.; Travaglini, M.T. A review of rosiglitazone in type 2 diabetes mellitus. Pharmacotherapy 2001, 21, 1082–1099. [Google Scholar] [CrossRef] [PubMed]
- Beysen, C.; Murphy, E.J.; Nagaraja, H.; Decaris, M.; Riiff, T.; Fong, A.; Hellerstein, M.K.; Boyle, P.J. A pilot study of the effects of pioglitazone and rosiglitazone on de novo lipogenesis in type 2 diabetes. J. Lipid Res. 2008, 49, 2657–2663. [Google Scholar] [CrossRef]
- Tessier, D.; Dawson, K.; Tetrault, J.P.; Bravo, G.; Meneilly, G.S. Glibenclamide vs gliclazide in type 2 diabetes of the elderly. Diabet Med. 1994, 11, 974–980. [Google Scholar] [CrossRef]
- Foster, R.H.; Plosker, G.L. Glipizide. A review of the pharmacoeconomic implications of the extended-release formulation in type 2 diabetes mellitus. Pharmacoeconomics 2000, 18, 289–306. [Google Scholar] [CrossRef] [PubMed]
- Langtry, H.D.; Balfour, J.A. Glimepiride. A review of its use in the management of type 2 diabetes mellitus. Drugs 1998, 55, 563–584. [Google Scholar] [CrossRef]
- Crowley, M.F.; Wolff, F.W.; Bloom, A. Tolbutamide in diabetes; some clinical and biochemical studies. Br. Med. J. 1957, 2, 327–331. [Google Scholar] [CrossRef]
- Chiasson, J.L.; Josse, R.G.; Gomis, R.; Hanefeld, M.; Karasik, A.; Laakso, M.; Group, S.-N.T.R. Acarbose for prevention of type 2 diabetes mellitus: The STOP-NIDDM randomised trial. Lancet 2002, 359, 2072–2077. [Google Scholar] [CrossRef]
- Scott, L.J.; Spencer, C.M. Miglitol: A review of its therapeutic potential in type 2 diabetes mellitus. Drugs 2000, 59, 521–549. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Hisauchi, I.; Taguchi, I.; Sezai, A.; Toyoda, S.; Tomiyama, H.; Sata, M.; Ueda, S.; Oyama, J.I.; Kitakaze, M.; et al. Effects of canagliflozin in patients with type 2 diabetes and chronic heart failure: A randomized trial (CANDLE). ESC Heart Fail. 2020, 7, 1585–1594. [Google Scholar] [CrossRef]
- Deeks, E.D.; Scheen, A.J. Canagliflozin: A Review in Type 2 Diabetes. Drugs 2017, 77, 1577–1592. [Google Scholar] [CrossRef]
- Dhillon, S. Dapagliflozin: A Review in Type 2 Diabetes. Drugs 2019, 79, 1135–1146. [Google Scholar] [CrossRef]
- Frampton, J.E. Empagliflozin: A Review in Type 2 Diabetes. Drugs 2018, 78, 1037–1048. [Google Scholar] [CrossRef]
- Lorenz, M.; Evers, A.; Wagner, M. Recent progress and future options in the development of GLP-1 receptor agonists for the treatment of diabesity. Bioorg. Med. Chem. Lett. 2013, 23, 4011–4018. [Google Scholar] [CrossRef] [PubMed]
- Nauck, M. Incretin therapies: Highlighting common features and differences in the modes of action of glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors. Diabetes Obes. Metab. 2016, 18, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Aroda, V.R. A review of GLP-1 receptor agonists: Evolution and advancement, through the lens of randomised controlled trials. Diabetes Obes. Metab. 2018, 20 (Suppl. S1), 22–33. [Google Scholar] [CrossRef]
- Keating, G.M. Alogliptin: A review of its use in patients with type 2 diabetes mellitus. Drugs 2015, 75, 777–796. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J. Sitagliptin: A Review in Type 2 Diabetes. Drugs 2017, 77, 209–224. [Google Scholar] [CrossRef]
- Garnock-Jones, K.P. Saxagliptin/Dapagliflozin: A Review in Type 2 Diabetes Mellitus. Drugs 2017, 77, 319–330. [Google Scholar] [CrossRef]
- McGill, J.B. Linagliptin for type 2 diabetes mellitus: A review of the pivotal clinical trials. Ther. Adv. Endocrinol. Metab. 2012, 3, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Linagliptin: A review of its use in the management of type 2 diabetes mellitus. Drugs 2012, 72, 1793–1824. [Google Scholar] [CrossRef] [PubMed]
- Wallia, A.; Molitch, M.E. Insulin therapy for type 2 diabetes mellitus. JAMA 2014, 311, 2315–2325. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, E.; Brennan, P. The effectiveness of continuous subcutaneous insulin pumps with continuous glucose monitoring in outpatient adolescents with type 1 diabetes: A systematic review. JBI Libr. Syst. Rev. 2012, 10, 1–10. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Freeland, B.; Farber, M.S. A Review of Insulin for the Treatment of Diabetes Mellitus. Home Healthc. Now 2016, 34, 416–423. [Google Scholar] [CrossRef]
- Njardarson Group. Top Pharmaceuticals Poster. Available online: https://njardarson.lab.arizona.edu/content/top-pharmaceuticals-poster (accessed on 21 August 2020).
- Global Diabetes Drugs Market. Available online: https://www.fortunebusinessinsights.com/industry-reports/infographics/diabetes-drugs-market-100570 (accessed on 1 September 2020).
- Saboo, B. Key elements of successful intensive therapy in patients with type 1 diabetes. Indian J. Endocrinol. Metab. 2015, 19, S44–S46. [Google Scholar] [CrossRef]
- Fineberg, S.E.; Kawabata, T.T.; Finco-Kent, D.; Fountaine, R.J.; Finch, G.L.; Krasner, A.S. Immunological responses to exogenous insulin. Endocr. Rev. 2007, 28, 625–652. [Google Scholar] [CrossRef] [PubMed]
- PhRMA. Follow the Dollar Report. Available online: https://www.phrma.org/report/follow-the-dollar-report (accessed on 10 August 2020).
- Seino, Y.; Fukushima, M.; Yabe, D. GIP and GLP-1, the two incretin hormones: Similarities and differences. J. Diabetes Investig. 2010, 1, 8–23. [Google Scholar] [CrossRef]
- Hui, H.; Farilla, L.; Merkel, P.; Perfetti, R. The short half-life of glucagon-like peptide-1 in plasma does not reflect its long-lasting beneficial effects. Eur. J. Endocrinol. 2002, 146, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Cowart, K. Oral Semaglutide: First-in-Class Oral GLP-1 Receptor Agonist for the Treatment of Type 2 Diabetes Mellitus. Ann. Pharmacother. 2020, 54, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Hedrington, M.S.; Davis, S.N. Oral semaglutide for the treatment of type 2 diabetes. Expert Opin. Pharmacother. 2019, 20, 133–141. [Google Scholar] [CrossRef]
- Coskun, T.; Sloop, K.W.; Loghin, C.; Alsina-Fernandez, J.; Urva, S.; Bokvist, K.B.; Cui, X.; Briere, D.A.; Cabrera, O.; Roell, W.C.; et al. LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: From discovery to clinical proof of concept. Mol. Metab. 2018, 18, 3–14. [Google Scholar] [CrossRef]
- Frias, J.P.; Nauck, M.A.; Van, J.; Kutner, M.E.; Cui, X.; Benson, C.; Urva, S.; Gimeno, R.E.; Milicevic, Z.; Robins, D.; et al. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: A randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet 2018, 392, 2180–2193. [Google Scholar] [CrossRef]
- Hartman, M.L.; Sanyal, A.J.; Loomba, R.; Wilson, J.M.; Nikooienejad, A.; Bray, R.; Karanikas, C.A.; Duffin, K.L.; Robins, D.A.; Haupt, A. Effects of Novel Dual GIP and GLP-1 Receptor Agonist Tirzepatide on Biomarkers of Nonalcoholic Steatohepatitis in Patients With Type 2 Diabetes. Diabetes Care 2020, 43, 1352–1355. [Google Scholar] [CrossRef]
- Lilly Investors. Lilly’s Tirzepatide Significantly Reduced A1C and Body Weight in People with Type 2 Diabetesdiabetes. 2020. Available online: https://investor.lilly.com/news-releases/news-release-details/lillys-tirzepatide-significantly-reduced-a1c-and-body-weight (accessed on 9 December 2020).
- Willard, F.S.; Douros, J.D.; Gabe, M.B.; Showalter, A.D.; Wainscott, D.B.; Suter, T.M.; Capozzi, M.E.; van der Velden, W.J.; Stutsman, C.; Cardona, G.R.; et al. Tirzepatide is an imbalanced and biased dual GIP and GLP-1 receptor agonist. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Andersen, A.; Lund, A.; Knop, F.K.; Vilsboll, T. Glucagon-like peptide 1 in health and disease. Nat. Rev. Endocrinol. 2018, 14, 390–403. [Google Scholar] [CrossRef]
- Samms, R.J.; Coghlan, M.P.; Sloop, K.W. How May GIP Enhance the Therapeutic Efficacy of GLP-1? Trends Endocrinol. Metab. 2020, 31, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Sturchler, E.; Zhu, J.; Nieto, A.; Cistrone, P.A.; Xie, J.; He, L.; Yea, K.; Jones, T.; Turn, R.; et al. Autocrine selection of a GLP-1R G-protein biased agonist with potent antidiabetic effects. Nat. Commun. 2015, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Freedman, N.J.; Lefkowitz, R.J. Desensitization of G protein-coupled receptors. Recent Prog. Horm. Res. 1996, 51, 319–351, discussion 352–313. [Google Scholar]
- Kim, S.J.; Nian, C.; McIntosh, C.H. Adipocyte expression of the glucose-dependent insulinotropic polypeptide receptor involves gene regulation by PPARgamma and histone acetylation. J. Lipid Res. 2011, 52, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Ceperuelo-Mallafre, V.; Duran, X.; Pachon, G.; Roche, K.; Garrido-Sanchez, L.; Vilarrasa, N.; Tinahones, F.J.; Vicente, V.; Pujol, J.; Vendrell, J.; et al. Disruption of GIP/GIPR axis in human adipose tissue is linked to obesity and insulin resistance. J. Clin. Endocrinol. Metab. 2014, 99, E908–E919. [Google Scholar] [CrossRef] [PubMed]
- Kulina, G.R.; Rayfield, E.J. The Role of Glucagon in the Pathophysiology and Management of Diabetes. Endocr. Pract. 2016, 22, 612–621. [Google Scholar] [CrossRef]
- Haedersdal, S.; Lund, A.; Knop, F.K.; Vilsboll, T. The Role of Glucagon in the Pathophysiology and Treatment of Type 2 Diabetes. Mayo Clin. Proc. 2018, 93, 217–239. [Google Scholar] [CrossRef]
- Glucagon nasal powder (Baqsimi) for severe hypoglycemia. Med. Lett. Drugs Ther. 2019, 61, 148–149.
- Jall, S.; Sachs, S.; Clemmensen, C.; Finan, B.; Neff, F.; DiMarchi, R.D.; Tschop, M.H.; Muller, T.D.; Hofmann, S.M. Monomeric GLP-1/GIP/glucagon triagonism corrects obesity, hepatosteatosis, and dyslipidemia in female mice. Mol. Metab. 2017, 6, 440–446. [Google Scholar] [CrossRef]
- Finan, B.; Yang, B.; Ottaway, N.; Smiley, D.L.; Ma, T.; Clemmensen, C.; Chabenne, J.; Zhang, L.; Habegger, K.M.; Fischer, K.; et al. A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat. Med. 2015, 21, 27–36. [Google Scholar] [CrossRef]
- Tai, J.; Liu, W.; Li, Y.; Li, L.; Holscher, C. Neuroprotective effects of a triple GLP-1/GIP/glucagon receptor agonist in the APP/PS1 transgenic mouse model of Alzheimer’s disease. Brain Res. 2018, 1678, 64–74. [Google Scholar] [CrossRef]
- Stiller, C.R.; Dupre, J.; Gent, M.; Heinrichs, D.; Jenner, M.R.; Keown, P.A.; Laupacis, A.; Martell, R.; Rodger, N.W.; Wolfe, B.M.; et al. Effects of cyclosporine in recent-onset juvenile type 1 diabetes: Impact of age and duration of disease. J. Pediatr. 1987, 111, 1069–1072. [Google Scholar] [CrossRef]
- Feutren, G.; Papoz, L.; Assan, R.; Vialettes, B.; Karsenty, G.; Vexiau, P.; Du Rostu, H.; Rodier, M.; Sirmai, J.; Lallemand, A.; et al. Cyclosporin increases the rate and length of remissions in insulin-dependent diabetes of recent onset. Results of a multicentre double-blind trial. Lancet 1986, 2, 119–124. [Google Scholar] [CrossRef]
- Baekkeskov, S.; Nielsen, J.H.; Marner, B.; Bilde, T.; Ludvigsson, J.; Lernmark, A. Autoantibodies in newly diagnosed diabetic children immunoprecipitate human pancreatic islet cell proteins. Nature 1982, 298, 167–169. [Google Scholar] [CrossRef]
- Zimmet, P. Antibodies to glutamic acid decarboxylase in the prediction of insulin dependency. Diabetes Res. Clin. Pract. 1996, 34 (Suppl. S1), S125–S131. [Google Scholar] [CrossRef]
- Tian, J.; Clare-Salzler, M.; Herschenfeld, A.; Middleton, B.; Newman, D.; Mueller, R.; Arita, S.; Evans, C.; Atkinson, M.A.; Mullen, Y.; et al. Modulating autoimmune responses to GAD inhibits disease progression and prolongs islet graft survival in diabetes-prone mice. Nat. Med. 1996, 2, 1348–1353. [Google Scholar] [CrossRef] [PubMed]
- Tisch, R.; Liblau, R.S.; Yang, X.D.; Liblau, P.; McDevitt, H.O. Induction of GAD65-specific regulatory T-cells inhibits ongoing autoimmune diabetes in nonobese diabetic mice. Diabetes 1998, 47, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, M.J.; Jaramillo, A.; Zipris, D.; Lazarus, A.H.; Serreze, D.V.; Leiter, E.H.; Cyopick, P.; Danska, J.S.; Delovitch, T.L. Interleukin 4 reverses T cell proliferative unresponsiveness and prevents the onset of diabetes in nonobese diabetic mice. J. Exp. Med. 1993, 178, 87–99. [Google Scholar] [CrossRef]
- Wherrett, D.K.; Bundy, B.; Becker, D.J.; DiMeglio, L.A.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Greenbaum, C.J.; Herold, K.C.; Marks, J.B.; et al. Antigen-based therapy with glutamic acid decarboxylase (GAD) vaccine in patients with recent-onset type 1 diabetes: A randomised double-blind trial. Lancet 2011, 378, 319–327. [Google Scholar] [CrossRef]
- Ludvigsson, J.; Faresjo, M.; Hjorth, M.; Axelsson, S.; Cheramy, M.; Pihl, M.; Vaarala, O.; Forsander, G.; Ivarsson, S.; Johansson, C.; et al. GAD treatment and insulin secretion in recent-onset type 1 diabetes. N. Engl. J. Med. 2008, 359, 1909–1920. [Google Scholar] [CrossRef] [PubMed]
- Diabetes Prevention Trial--Type 1 Diabetes Study, G. Effects of insulin in relatives of patients with type 1 diabetes mellitus. N. Engl. J. Med. 2002, 346, 1685–1691. [Google Scholar] [CrossRef]
- Ludvigsson, J.; Cheramy, M.; Axelsson, S.; Pihl, M.; Akerman, L.; Casas, R.; Clinical GAD-Study Group in Sweden. GAD-treatment of children and adolescents with recent-onset type 1 diabetes preserves residual insulin secretion after 30 months. Diabetes Metab. Res. Rev. 2014, 30, 405–414. [Google Scholar] [CrossRef]
- Elias, D.; Avron, A.; Tamir, M.; Raz, I. DiaPep277 preserves endogenous insulin production by immunomodulation in type 1 diabetes. Ann. N. Y. Acad. Sci. 2006, 1079, 340–344. [Google Scholar] [CrossRef]
- Lazar, L.; Ofan, R.; Weintrob, N.; Avron, A.; Tamir, M.; Elias, D.; Phillip, M.; Josefsberg, Z. Heat-shock protein peptide DiaPep277 treatment in children with newly diagnosed type 1 diabetes: A randomised, double-blind phase II study. Diabetes Metab. Res. Rev. 2007, 23, 286–291. [Google Scholar] [CrossRef]
- Chatenoud, L. Immune therapy for type 1 diabetes mellitus-what is unique about anti-CD3 antibodies? Nat. Rev. Endocrinol. 2010, 6, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Herold, K.C.; Hagopian, W.; Auger, J.A.; Poumian-Ruiz, E.; Taylor, L.; Donaldson, D.; Gitelman, S.E.; Harlan, D.M.; Xu, D.; Zivin, R.A.; et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N. Engl. J. Med. 2002, 346, 1692–1698. [Google Scholar] [CrossRef]
- Herold, K.C.; Gitelman, S.; Greenbaum, C.; Puck, J.; Hagopian, W.; Gottlieb, P.; Sayre, P.; Bianchine, P.; Wong, E.; Seyfert-Margolis, V.; et al. Treatment of patients with new onset Type 1 diabetes with a single course of anti-CD3 mAb Teplizumab preserves insulin production for up to 5 years. Clin. Immunol. 2009, 132, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Hagopian, W.; Ferry, R.J., Jr.; Sherry, N.; Carlin, D.; Bonvini, E.; Johnson, S.; Stein, K.E.; Koenig, S.; Daifotis, A.G.; Herold, K.C.; et al. Teplizumab preserves C-peptide in recent-onset type 1 diabetes: Two-year results from the randomized, placebo-controlled Protege trial. Diabetes 2013, 62, 3901–3908. [Google Scholar] [CrossRef] [PubMed]
- Herold, K.C.; Gitelman, S.E.; Willi, S.M.; Gottlieb, P.A.; Waldron-Lynch, F.; Devine, L.; Sherr, J.; Rosenthal, S.M.; Adi, S.; Jalaludin, M.Y.; et al. Teplizumab treatment may improve C-peptide responses in participants with type 1 diabetes after the new-onset period: A randomised controlled trial. Diabetologia 2013, 56, 391–400. [Google Scholar] [CrossRef]
- Sherry, N.; Hagopian, W.; Ludvigsson, J.; Jain, S.M.; Wahlen, J.; Ferry, R.J., Jr.; Bode, B.; Aronoff, S.; Holland, C.; Carlin, D.; et al. Teplizumab for treatment of type 1 diabetes (Protege study): 1-year results from a randomised, placebo-controlled trial. Lancet 2011, 378, 487–497. [Google Scholar] [CrossRef]
- Washington Business Journal. MacroGenics, Lilly Abandon Diabetes Drug 2010: Washington Business Journal. Available online: https://www.bizjournals.com/washington/quick_news/2010/10/macrogenics-lilly-abandon-diabetes-drug.html (accessed on 5 September 2020).
- Herold, K.C.; Bundy, B.N.; Long, S.A.; Bluestone, J.A.; DiMeglio, L.A.; Dufort, M.J.; Gitelman, S.E.; Gottlieb, P.A.; Krischer, J.P.; Linsley, P.S.; et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N. Engl. J. Med. 2019, 381, 603–613. [Google Scholar] [CrossRef]
- Mulvey, A. FDA Breakthrough Therapy Designation for Teplizumab—Based on the First Study to Delay the Onset of T1D for 2+ Years. 2019. Available online: https://www.jdrf.org/blog/2019/08/05/fda-breakthrough-therapy-designation-teplizumab-based-first-study-delay-onset-t1d-2-years/ (accessed on 10 August 2020).
- Keymeulen, B.; Vandemeulebroucke, E.; Ziegler, A.G.; Mathieu, C.; Kaufman, L.; Hale, G.; Gorus, F.; Goldman, M.; Walter, M.; Candon, S.; et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N. Engl. J. Med. 2005, 352, 2598–2608. [Google Scholar] [CrossRef] [PubMed]
- Ambery, P.; Donner, T.W.; Biswas, N.; Donaldson, J.; Parkin, J.; Dayan, C.M. Efficacy and safety of low-dose otelixizumab anti-CD3 monoclonal antibody in preserving C-peptide secretion in adolescent type 1 diabetes: DEFEND-2, a randomized, placebo-controlled, double-blind, multi-centre study. Diabet Med. 2014, 31, 399–402. [Google Scholar] [CrossRef]
- Aronson, R.; Gottlieb, P.A.; Christiansen, J.S.; Donner, T.W.; Bosi, E.; Bode, B.W.; Pozzilli, P.; Group, D.I. Low-dose otelixizumab anti-CD3 monoclonal antibody DEFEND-1 study: Results of the randomized phase III study in recent-onset human type 1 diabetes. Diabetes Care 2014, 37, 2746–2754. [Google Scholar] [CrossRef]
- Buzzetti, R. Diabetes: Immunotherapy for T1DM--still not there yet. Nat. Rev. Endocrinol. 2013, 9, 697–698. [Google Scholar] [CrossRef]
- Orban, T.; Bundy, B.; Becker, D.J.; DiMeglio, L.A.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Greenbaum, C.J.; Marks, J.B.; Monzavi, R.; et al. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: A randomised, double-blind, placebo-controlled trial. Lancet 2011, 378, 412–419. [Google Scholar] [CrossRef]
- Greenbaum, C.J.; Schatz, D.A.; Haller, M.J.; Sanda, S. Through the fog: Recent clinical trials to preserve beta-cell function in type 1 diabetes. Diabetes 2012, 61, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Linsley, P.S.; Greenbaum, C.J.; Rosasco, M.; Presnell, S.; Herold, K.C.; Dufort, M.J. Elevated T cell levels in peripheral blood predict poor clinical response following rituximab treatment in new-onset type 1 diabetes. Genes Immun. 2019, 20, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Pescovitz, M.D.; Greenbaum, C.J.; Krause-Steinrauf, H.; Becker, D.J.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Marks, J.B.; McGee, P.F.; Moran, A.M.; et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N. Engl. J. Med. 2009, 361, 2143–2152. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, A.I.; Le Page, M.A.; Maxwell, M.J.; Stolp, J.; Guo, H.; Jayasimhan, A.; Hibbs, M.L.; Santamaria, P.; Miller, J.F.; Plebanski, M.; et al. The CD19 signalling molecule is elevated in NOD mice and controls type 1 diabetes development. Diabetologia 2013, 56, 2659–2668. [Google Scholar] [CrossRef]
- Lee, M.; Park, H.; Youn, J.; Oh, E.T.; Ko, K.; Kim, S.; Park, Y. Interleukin-10 plasmid construction and delivery for the prevention of type 1 diabetes. Ann. N. Y. Acad. Sci. 2006, 1079, 313–319. [Google Scholar] [CrossRef]
- ActoBio Therapeutics. ActoBio Therapeutics Greenlighted by FDA to Commence a Phase Ib/IIa Trial with AG019 for the Treatment of Early Onset Type 1 Diabetes. 2018. Available online: https://www.prnewswire.com/news-releases/actobio-therapeutics-greenlighted-by-fda-to-commence-a-phase-ibiia-trial-with-ag019-for-the-treatment-of-early-onset-type-1-diabetes-300621670.html (accessed on 21 August 2020).
- ActoBio Therapeutics. ActoBio Therapeutics™ Progresses AG019 to Next Stage of a Phase Ib/IIa Clinical Study for the Treatment of Type 1 Diabetes. 2019. Available online: https://www.prnewswire.com/news-releases/actobio-therapeutics-progresses-ag019-to-next-stage-of-a-phase-ibiia-clinical-study-for-the-treatment-of-type-1-diabetes-300878301.html (accessed on 21 August 2020).
- Thrower, S.L.; James, L.; Hall, W.; Green, K.M.; Arif, S.; Allen, J.S.; Van-Krinks, C.; Lozanoska-Ochser, B.; Marquesini, L.; Brown, S.; et al. Proinsulin peptide immunotherapy in type 1 diabetes: Report of a first-in-man Phase I safety study. Clin. Exp. Immunol. 2009, 155, 156–165. [Google Scholar] [CrossRef]
- Gremizzi, C.; Vergani, A.; Paloschi, V.; Secchi, A. Impact of pancreas transplantation on type 1 diabetes-related complications. Curr. Opin. Organ Transplant. 2010, 15, 119–123. [Google Scholar] [CrossRef]
- Gondolesi, G.E.; Aguirre, N.F.; Ramisch, D.A.; Mos, F.A.; Pedraza, N.F.; Fortunato, M.R.; Gutierrez, L.M.; Fraguas, H.; Marrugat, R.; Rabin, G.E.; et al. Pancreas Transplantation at a Single Latin-American Center; Overall Results with Type 1 and Type 2 Diabetes Mellitus. Transplant. Proc. 2018, 50, 1475–1481. [Google Scholar] [CrossRef]
- Al-Qaoud, T.M.; Odorico, J.S.; Redfield, R.R., 3rd. Pancreas transplantation in type 2 diabetes: Expanding the criteria. Curr. Opin. Organ Transplant. 2018, 23, 454–460. [Google Scholar] [CrossRef]
- Stratta, R.J.; Fridell, J.A.; Gruessner, A.C.; Odorico, J.S.; Gruessner, R.W. Pancreas transplantation: A decade of decline. Curr. Opin. Organ Transplant. 2016, 21, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Najarian, J.S.; Sutherland, D.E.; Matas, A.J.; Steffes, M.W.; Simmons, R.L.; Goetz, F.C. Human islet transplantation: A preliminary report. Transplant. Proc. 1977, 9, 233–236. [Google Scholar] [PubMed]
- Shapiro, A.M.; Lakey, J.R.; Ryan, E.A.; Korbutt, G.S.; Toth, E.; Warnock, G.L.; Kneteman, N.M.; Rajotte, R.V. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N. Engl. J. Med. 2000, 343, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Bruni, A.; Gala-Lopez, B.; Pepper, A.R.; Abualhassan, N.S.; Shapiro, A.J. Islet cell transplantation for the treatment of type 1 diabetes: Recent advances and future challenges. Diabetes Metab. Syndr. Obes. 2014, 7, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.M.; Ricordi, C.; Hering, B.J.; Auchincloss, H.; Lindblad, R.; Robertson, R.P.; Secchi, A.; Brendel, M.D.; Berney, T.; Brennan, D.C.; et al. International trial of the Edmonton protocol for islet transplantation. N. Engl. J. Med. 2006, 355, 1318–1330. [Google Scholar] [CrossRef] [PubMed]
- Hering, B.J.; Kandaswamy, R.; Ansite, J.D.; Eckman, P.M.; Nakano, M.; Sawada, T.; Matsumoto, I.; Ihm, S.H.; Zhang, H.J.; Parkey, J.; et al. Single-donor, marginal-dose islet transplantation in patients with type 1 diabetes. JAMA 2005, 293, 830–835. [Google Scholar] [CrossRef]
- Koh, A.; Senior, P.; Salam, A.; Kin, T.; Imes, S.; Dinyari, P.; Malcolm, A.; Toso, C.; Nilsson, B.; Korsgren, O.; et al. Insulin-heparin infusions peritransplant substantially improve single-donor clinical islet transplant success. Transplantation 2010, 89, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Takita, M.; Chaussabel, D.; Noguchi, H.; Shimoda, M.; Sugimoto, K.; Itoh, T.; Chujo, D.; SoRelle, J.; Onaca, N.; et al. Improving efficacy of clinical islet transplantation with iodixanol-based islet purification, thymoglobulin induction, and blockage of IL-1beta and TNF-alpha. Cell Transplant. 2011, 20, 1641–1647. [Google Scholar] [CrossRef]
- Faradji, R.N.; Tharavanij, T.; Messinger, S.; Froud, T.; Pileggi, A.; Monroy, K.; Mineo, D.; Baidal, D.A.; Cure, P.; Ponte, G.; et al. Long-term insulin independence and improvement in insulin secretion after supplemental islet infusion under exenatide and etanercept. Transplantation 2008, 86, 1658–1665. [Google Scholar] [CrossRef] [PubMed]
- Posselt, A.M.; Bellin, M.D.; Tavakol, M.; Szot, G.L.; Frassetto, L.A.; Masharani, U.; Kerlan, R.K.; Fong, L.; Vincenti, F.G.; Hering, B.J.; et al. Islet transplantation in type 1 diabetics using an immunosuppressive protocol based on the anti-LFA-1 antibody efalizumab. Am. J. Transplant. 2010, 10, 1870–1880. [Google Scholar] [CrossRef]
- Turgeon, N.A.; Avila, J.G.; Cano, J.A.; Hutchinson, J.J.; Badell, I.R.; Page, A.J.; Adams, A.B.; Sears, M.H.; Bowen, P.H.; Kirk, A.D.; et al. Experience with a novel efalizumab-based immunosuppressive regimen to facilitate single donor islet cell transplantation. Am. J. Transplant. 2010, 10, 2082–2091. [Google Scholar] [CrossRef] [PubMed]
- Balcazar, N.; Sathyamurthy, A.; Elghazi, L.; Gould, A.; Weiss, A.; Shiojima, I.; Walsh, K.; Bernal-Mizrachi, E. mTORC1 activation regulates beta-cell mass and proliferation by modulation of cyclin D2 synthesis and stability. J. Biol. Chem. 2009, 284, 7832–7842. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.; Fung, J.J.; Lu, L.; Qian, S. Tolerance-inducing strategies in islet transplantation. Int. J. Endocrinol. 2012, 2012. [Google Scholar] [CrossRef]
- Liu, C.; Noorchashm, H.; Sutter, J.A.; Naji, M.; Prak, E.L.; Boyer, J.; Green, T.; Rickels, M.R.; Tomaszewski, J.E.; Koeberlein, B.; et al. B lymphocyte-directed immunotherapy promotes long-term islet allograft survival in nonhuman primates. Nat. Med. 2007, 13, 1295–1298. [Google Scholar] [CrossRef]
- Cabello-Olmo, M.; Arana, M.; Radichev, I.; Smith, P.; Huarte, E.; Barajas, M. New Insights into Immunotherapy Strategies for Treating Autoimmune Diabetes. Int. J. Mol. Sci. 2019, 20, 4789. [Google Scholar] [CrossRef]
- Zhao, Y.; Jiang, Z.; Delgado, E.; Li, H.; Zhou, H.; Hu, W.; Perez-Basterrechea, M.; Janostakova, A.; Tan, Q.; Wang, J.; et al. Platelet-Derived Mitochondria Display Embryonic Stem Cell Markers and Improve Pancreatic Islet beta-cell Function in Humans. Stem Cells Transl. Med. 2017, 6, 1684–1697. [Google Scholar] [CrossRef]
- Kang, E.M.; Zickler, P.P.; Burns, S.; Langemeijer, S.M.; Brenner, S.; Phang, O.A.; Patterson, N.; Harlan, D.; Tisdale, J.F. Hematopoietic stem cell transplantation prevents diabetes in NOD mice but does not contribute to significant islet cell regeneration once disease is established. Exp. Hematol. 2005, 33, 699–705. [Google Scholar] [CrossRef]
- Voltarelli, J.C.; Couri, C.E.; Stracieri, A.B.; Oliveira, M.C.; Moraes, D.A.; Pieroni, F.; Coutinho, M.; Malmegrim, K.C.; Foss-Freitas, M.C.; Simoes, B.P.; et al. Autologous nonmyeloablative hematopoietic stem cell transplantation in newly diagnosed type 1 diabetes mellitus. JAMA 2007, 297, 1568–1576. [Google Scholar] [CrossRef]
- D’Addio, F.; Valderrama Vasquez, A.; Ben Nasr, M.; Franek, E.; Zhu, D.; Li, L.; Ning, G.; Snarski, E.; Fiorina, P. Autologous nonmyeloablative hematopoietic stem cell transplantation in new-onset type 1 diabetes: A multicenter analysis. Diabetes 2014, 63, 3041–3046. [Google Scholar] [CrossRef]
- Gu, B.; Miao, H.; Zhang, J.; Hu, J.; Zhou, W.; Gu, W.; Wang, W.; Ning, G. Clinical benefits of autologous haematopoietic stem cell transplantation in type 1 diabetes patients. Diabetes Metab. 2018, 44, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Snarski, E.; Milczarczyk, A.; Halaburda, K.; Torosian, T.; Paluszewska, M.; Urbanowska, E.; Krol, M.; Boguradzki, P.; Jedynasty, K.; Franek, E.; et al. Immunoablation and autologous hematopoietic stem cell transplantation in the treatment of new-onset type 1 diabetes mellitus: Long-term observations. Bone Marrow Transplant. 2016, 51, 398–402. [Google Scholar] [CrossRef]
- Moreira, A.; Kahlenberg, S.; Hornsby, P. Therapeutic potential of mesenchymal stem cells for diabetes. J. Mol. Endocrinol. 2017, 59, R109–R120. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, P.O.; Schwarcz, E.; Korsgren, O.; Le Blanc, K. Preserved beta-cell function in type 1 diabetes by mesenchymal stromal cells. Diabetes 2015, 64, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Yu, X.; Wang, Z.; Wang, F.; Wang, L.; Gao, H.; Chen, Y.; Zhao, W.; Jia, Z.; Yan, S.; et al. Long term effects of the implantation of Wharton’s jelly-derived mesenchymal stem cells from the umbilical cord for newly-onset type 1 diabetes mellitus. Endocr. J. 2013, 60, 347–357. [Google Scholar] [CrossRef]
- Thakkar, U.G.; Trivedi, H.L.; Vanikar, A.V.; Dave, S.D. Insulin-secreting adipose-derived mesenchymal stromal cells with bone marrow-derived hematopoietic stem cells from autologous and allogenic sources for type 1 diabetes mellitus. Cytotherapy 2015, 17, 940–947. [Google Scholar] [CrossRef]
- Vanikar, A.V.; Dave, S.D.; Thakkar, U.G.; Trivedi, H.L. Cotransplantation of adipose tissue-derived insulin-secreting mesenchymal stem cells and hematopoietic stem cells: A novel therapy for insulin-dependent diabetes mellitus. Stem Cells Int. 2010, 2010. [Google Scholar] [CrossRef]
- Dave, S.D.; Vanikar, A.V.; Trivedi, H.L.; Thakkar, U.G.; Gopal, S.C.; Chandra, T. Novel therapy for insulin-dependent diabetes mellitus: Infusion of in vitro-generated insulin-secreting cells. Clin. Exp. Med. 2015, 15, 41–45. [Google Scholar] [CrossRef]
- Liu, X.; Zheng, P.; Wang, X.; Dai, G.; Cheng, H.; Zhang, Z.; Hua, R.; Niu, X.; Shi, J.; An, Y. A preliminary evaluation of efficacy and safety of Wharton’s jelly mesenchymal stem cell transplantation in patients with type 2 diabetes mellitus. Stem Cell Res. Ther. 2014, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bhansali, S.; Dutta, P.; Kumar, V.; Yadav, M.K.; Jain, A.; Mudaliar, S.; Bhansali, S.; Sharma, R.R.; Jha, V.; Marwaha, N.; et al. Efficacy of Autologous Bone Marrow-Derived Mesenchymal Stem Cell and Mononuclear Cell Transplantation in Type 2 Diabetes Mellitus: A Randomized, Placebo-Controlled Comparative Study. Stem Cells Dev. 2017, 26, 471–481. [Google Scholar] [CrossRef]
- Jiang, R.; Han, Z.; Zhuo, G.; Qu, X.; Li, X.; Wang, X.; Shao, Y.; Yang, S.; Han, Z.C. Transplantation of placenta-derived mesenchymal stem cells in type 2 diabetes: A pilot study. Front. Med. 2011, 5, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Skyler, J.S.; Fonseca, V.A.; Segal, K.R.; Rosenstock, J.; Investigators, M.-D. Allogeneic Mesenchymal Precursor Cells in Type 2 Diabetes: A Randomized, Placebo-Controlled, Dose-Escalation Safety and Tolerability Pilot Study. Diabetes Care 2015, 38, 1742–1749. [Google Scholar] [CrossRef]
- Chen, P.; Huang, Q.; Xu, X.J.; Shao, Z.L.; Huang, L.H.; Yang, X.Z.; Guo, W.; Li, C.M.; Chen, C. The effect of liraglutide in combination with human umbilical cord mesenchymal stem cells treatment on glucose metabolism and beta cell function in type 2 diabetes mellitus. Zhonghua Nei Ke Za Zhi 2016, 55, 349–354. [Google Scholar] [CrossRef]
- Hildreth, C. Top Companies Developing Cell Therapy Treatments for Diabetes. 2019. Available online: https://bioinformant.com/stem-cells-for-diabetes/ (accessed on 20 August 2020).
- Zhao, Y.; Lin, B.; Darflinger, R.; Zhang, Y.; Holterman, M.J.; Skidgel, R.A. Human cord blood stem cell-modulated regulatory T lymphocytes reverse the autoimmune-caused type 1 diabetes in nonobese diabetic (NOD) mice. PLoS ONE 2009, 4, e4226. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y. Stem cell educator therapy and induction of immune balance. Curr. Diabetes Rep. 2012, 12, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Jiang, Z.; Zhao, T.; Ye, M.; Hu, C.; Zhou, H.; Yin, Z.; Chen, Y.; Zhang, Y.; Wang, S.; et al. Targeting insulin resistance in type 2 diabetes via immune modulation of cord blood-derived multipotent stem cells (CB-SCs) in stem cell educator therapy: Phase I/II clinical trial. BMC Med. 2013, 11, 160. [Google Scholar] [CrossRef]
- Zhao, Y.; Jiang, Z.; Zhao, T.; Ye, M.; Hu, C.; Yin, Z.; Li, H.; Zhang, Y.; Diao, Y.; Li, Y.; et al. Reversal of type 1 diabetes via islet beta cell regeneration following immune modulation by cord blood-derived multipotent stem cells. BMC Med. 2012, 10, 3. [Google Scholar] [CrossRef]
- Delgado, E.; Perez-Basterrechea, M.; Suarez-Alvarez, B.; Zhou, H.; Revuelta, E.M.; Garcia-Gala, J.M.; Perez, S.; Alvarez-Viejo, M.; Menendez, E.; Lopez-Larrea, C.; et al. Modulation of Autoimmune T-Cell Memory by Stem Cell Educator Therapy: Phase 1/2 Clinical Trial. EBioMedicine 2015, 2, 2024–2036. [Google Scholar] [CrossRef]
- Tenspolde, M.; Zimmermann, K.; Weber, L.C.; Hapke, M.; Lieber, M.; Dywicki, J.; Frenzel, A.; Hust, M.; Galla, M.; Buitrago-Molina, L.E.; et al. Regulatory T cells engineered with a novel insulin-specific chimeric antigen receptor as a candidate immunotherapy for type 1 diabetes. J. Autoimmun. 2019, 103, 102289. [Google Scholar] [CrossRef]
- Zhang, L.; Sosinowski, T.; Cox, A.R.; Cepeda, J.R.; Sekhar, N.S.; Hartig, S.M.; Miao, D.; Yu, L.; Pietropaolo, M.; Davidson, H.W. Chimeric antigen receptor (CAR) T cells targeting a pathogenic MHC class II:peptide complex modulate the progression of autoimmune diabetes. J. Autoimmun. 2019, 96, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Crawford, F.; Yu, L.; Michels, A.; Nakayama, M.; Davidson, H.W.; Kappler, J.W.; Eisenbarth, G.S. Monoclonal antibody blocking the recognition of an insulin peptide-MHC complex modulates type 1 diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 2656–2661. [Google Scholar] [CrossRef] [PubMed]
- Furuyama, K.; Chera, S.; van Gurp, L.; Oropeza, D.; Ghila, L.; Damond, N.; Vethe, H.; Paulo, J.A.; Joosten, A.M.; Berney, T.; et al. Diabetes relief in mice by glucose-sensing insulin-secreting human alpha-cells. Nature 2019, 567, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, H.; Ko, U.H.; Oh, Y.; Lim, A.; Sohn, J.W.; Shin, J.H.; Kim, H.; Han, Y.M. Islet-like organoids derived from human pluripotent stem cells efficiently function in the glucose responsiveness in vitro and in vivo. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ramli, R.; Reddy, M.; Oliver, N. Artificial Pancreas: Current Progress and Future Outlook in the Treatment of Type 1 Diabetes. Drugs 2019, 79, 1089–1101. [Google Scholar] [CrossRef] [PubMed]
- FDA. What Is the Pancreas? What Is an Artificial Pancreas Device System? 2018. Available online: https://www.fda.gov/medical-devices/artificial-pancreas-device-system/what-pancreas-what-artificial-pancreas-device-system (accessed on 10 August 2020).
- Saunders, A.; Messer, L.H.; Forlenza, G.P. MiniMed 670G hybrid closed loop artificial pancreas system for the treatment of type 1 diabetes mellitus: Overview of its safety and efficacy. Expert Rev. Med. Devices 2019, 16, 845–853. [Google Scholar] [CrossRef]
- Brown, S.A.; Kovatchev, B.P.; Raghinaru, D.; Lum, J.W.; Buckingham, B.A.; Kudva, Y.C.; Laffel, L.M.; Levy, C.J.; Pinsker, J.E.; Wadwa, R.P.; et al. Six-Month Randomized, Multicenter Trial of Closed-Loop Control in Type 1 Diabetes. N. Engl. J. Med. 2019, 381, 1707–1717. [Google Scholar] [CrossRef] [PubMed]
- NIH. Artificial Pancreas System Better Controls Blood Glucose Levels Than Current Technology. 2019. Available online: https://www.nih.gov/news-events/news-releases/artificial-pancreas-system-better-controls-blood-glucose-levels-current-technology (accessed on 9 September 2020).
- Abai, A.M.; Hobart, P.M.; Barnhart, K.M. Insulin delivery with plasmid DNA. Hum. Gene Ther. 1999, 10, 2637–2649. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; McLane, B.; Kim, E.H.; Yoon, J.W.; Jun, H.S. Remission of diabetes by insulin gene therapy using a hepatocyte-specific and glucose-responsive synthetic promoter. Mol. Ther. 2011, 19, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Candon, S.; Perez-Arroyo, A.; Marquet, C.; Valette, F.; Foray, A.P.; Pelletier, B.; Milani, C.; Ventura, M.; Bach, J.F.; Chatenoud, L. Correction: Antibiotics in Early Life Alter the Gut Microbiome and Increase Disease Incidence in a Spontaneous Mouse Model of Autoimmune Insulin-Dependent Diabetes. PLoS ONE 2016, 11, e0147888. [Google Scholar] [CrossRef]
- Candon, S.; Perez-Arroyo, A.; Marquet, C.; Valette, F.; Foray, A.P.; Pelletier, B.; Milani, C.; Ventura, M.; Bach, J.F.; Chatenoud, L. Antibiotics in early life alter the gut microbiome and increase disease incidence in a spontaneous mouse model of autoimmune insulin-dependent diabetes. PLoS ONE 2015, 10, e0125448. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Ley, R.E.; Volchkov, P.Y.; Stranges, P.B.; Avanesyan, L.; Stonebraker, A.C.; Hu, C.; Wong, F.S.; Szot, G.L.; Bluestone, J.A.; et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature 2008, 455, 1109–1113. [Google Scholar] [CrossRef]
- Davis-Richardson, A.G.; Ardissone, A.N.; Dias, R.; Simell, V.; Leonard, M.T.; Kemppainen, K.M.; Drew, J.C.; Schatz, D.; Atkinson, M.A.; Kolaczkowski, B.; et al. Bacteroides dorei dominates gut microbiome prior to autoimmunity in Finnish children at high risk for type 1 diabetes. Front. Microbiol. 2014, 5, 678. [Google Scholar] [CrossRef]
- De Goffau, M.C.; Fuentes, S.; van den Bogert, B.; Honkanen, H.; de Vos, W.M.; Welling, G.W.; Hyoty, H.; Harmsen, H.J. Aberrant gut microbiota composition at the onset of type 1 diabetes in young children. Diabetologia 2014, 57, 1569–1577. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Li, Z.; Zhou, Z. Gut microbiome in type 1 diabetes: A comprehensive review. Diabetes Metab. Res. Rev. 2018, 34, e3043. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; Cassader, M. Interactions between gut microbiota and host metabolism predisposing to obesity and diabetes. Annu. Rev. Med. 2011, 62, 361–380. [Google Scholar] [CrossRef]
- Beer, K. The Gut Microbiome in Type 2 Diabetes. Clin. Rev. 2018, 28, 13–14. [Google Scholar]
- Tai, N.; Wong, F.S.; Wen, L. The role of gut microbiota in the development of type 1, type 2 diabetes mellitus and obesity. Rev. Endocr. Metab. Disord. 2015, 16, 55–65. [Google Scholar] [CrossRef]
- Forslund, K.; Hildebrand, F.; Nielsen, T.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; Pedersen, H.K.; et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 2015, 528, 262–266. [Google Scholar] [CrossRef]
- Napolitano, A.; Miller, S.; Nicholls, A.W.; Baker, D.; Van Horn, S.; Thomas, E.; Rajpal, D.; Spivak, A.; Brown, J.R.; Nunez, D.J. Novel gut-based pharmacology of metformin in patients with type 2 diabetes mellitus. PLoS ONE 2014, 9, e100778. [Google Scholar] [CrossRef]
- Wu, H.; Esteve, E.; Tremaroli, V.; Khan, M.T.; Caesar, R.; Manneras-Holm, L.; Stahlman, M.; Olsson, L.M.; Serino, M.; Planas-Felix, M.; et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat. Med. 2017, 23, 850–858. [Google Scholar] [CrossRef]
- Wang, Z.; Saha, S.; Van Horn, S.; Thomas, E.; Traini, C.; Sathe, G.; Rajpal, D.K.; Brown, J.R. Gut microbiome differences between metformin- and liraglutide-treated T2DM subjects. Endocrinol. Diabetes Metab. 2018, 1, e00009. [Google Scholar] [CrossRef] [PubMed]
- Weickert, M.O.; Pfeiffer, A.F.H. Impact of Dietary Fiber Consumption on Insulin Resistance and the Prevention of Type 2 Diabetes. J. Nutr. 2018, 148, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Aydin, O.; Nieuwdorp, M.; Gerdes, V. The Gut Microbiome as a Target for the Treatment of Type 2 Diabetes. Curr. Diabates Rep. 2018, 18, 1–11. [Google Scholar] [CrossRef]
- Sharma, S.; Tripathi, P. Gut microbiome and type 2 diabetes: Where we are and where to go? J. Nutr. Biochem. 2019, 63, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Theofilopoulos, A.N.; Kono, D.H.; Baccala, R. The multiple pathways to autoimmunity. Nat. Immunol. 2017, 18, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Coronel-Restrepo, N.; Posso-Osorio, I.; Naranjo-Escobar, J.; Tobón, G.J. Autoimmune diseases and their relation with immunological, neurological and endocrinological axes. Autoimmun. Rev. 2017, 16, 684–692. [Google Scholar] [CrossRef]
- Cooper, G.S.; Stroehla, B.C. The epidemiology of autoimmune diseases. Autoimmun. Rev. 2003, 2, 119–125. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid arthritis. Lancet 2016, 388, 2023–2038. [Google Scholar] [CrossRef]
- WHO. Chronic Diseases and Health Promotion. Available online: https://www.who.int/chp/topics/rheumatic/en/ (accessed on 4 October 2020).
- Ezerioha, M. Let’s Dig into Everything about RA; Rheumatoid Arthritis Support Network. 2018. Available online: https://www.rheumatoidarthritis.org (accessed on 15 September 2020).
- McInnes, I.B.; Schett, G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet 2017, 389, 2328–2337. [Google Scholar] [CrossRef]
- Giannini, D.; Antonucci, M.; Petrelli, F.; Bilia, S.; Alunno, A.; Puxeddu, I. One year in review 2020: Pathogenesis of rheumatoid arthritis. Clin. Exp. Rheumatol. 2020, 38, 387–397. [Google Scholar]
- Ramos, P.S.; Shedlock, A.M.; Langefeld, C.D. Genetics of autoimmune diseases: Insights from population genetics. J. Hum. Genet. 2015, 60, 657–664. [Google Scholar] [CrossRef]
- Calabresi, E.; Petrelli, F.; Bonifacio, A.F.; Puxeddu, I.; Alunno, A. One year in review 2018: Pathogenesis of rheumatoid arthritis. Clin. Exp. Rheumatol. 2018, 36, 175–184. [Google Scholar] [PubMed]
- Croia, C.; Bursi, R.; Sutera, D.; Petrelli, F.; Alunno, A.; Puxeddu, I. One year in review 2019: Pathogenesis of rheumatoid arthritis. Clin. Exp. Rheumatol. 2019, 37, 347–357. [Google Scholar]
- Gregersen, P.K.; Silver, J.; Winchester, R.J. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987, 30, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Watad, A.; Amital, H. ACPAs Are Much More Than Diagnostic Autoantibodies. Rambam Maimonides Med. J. 2016, 7. [Google Scholar] [CrossRef]
- Kurowska, W.; Kuca-Warnawin, E.H.; Radzikowska, A.; Maśliński, W. The role of anti-citrullinated protein antibodies (ACPA) in the pathogenesis of rheumatoid arthritis. Cent. Eur. J. Immunol. 2017, 42, 390. [Google Scholar] [CrossRef]
- Harre, U.; Georgess, D.; Bang, H.; Bozec, A.; Axmann, R.; Ossipova, E.; Jakobsson, P.J.; Baum, W.; Nimmerjahn, F.; Szarka, E.; et al. Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J. Clin. Investig. 2012, 122, 1791–1802. [Google Scholar] [CrossRef]
- Nielen, M.M.; van Schaardenburg, D.; Reesink, H.W.; van de Stadt, R.J.; van der Horst-Bruinsma, I.E.; de Koning, M.H.; Habibuw, M.R.; Vandenbroucke, J.P.; Dijkmans, B.A. Specific autoantibodies precede the symptoms of rheumatoid arthritis: A study of serial measurements in blood donors. Arthritis Rheum. 2004, 50, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Li, S.; Yang, Y.; Zhang, K.; Dong, S.; Wang, X.; Liu, X.; Ren, Y.; Zhang, M.; Yan, X. T helper 17 and T helper 1 cells are increased but regulatory T cells are decreased in subchondral bone marrow microenvironment of patients with rheumatoid arthritis. Am. J. Transl. Res. 2016, 8, 2956. [Google Scholar]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef]
- Malemud, C.J. The role of the JAK/STAT signal pathway in rheumatoid arthritis. Ther. Adv. Musculoskelet. Dis. 2018, 10, 117–127. [Google Scholar] [CrossRef]
- Winthrop, K.L. The emerging safety profile of JAK inhibitors in rheumatic disease. Nat. Rev. Rheumatol. 2017, 13, 234–243. [Google Scholar] [CrossRef]
- Nakayamada, S.; Kubo, S.; Iwata, S.; Tanaka, Y. Recent Progress in JAK Inhibitors for the Treatment of Rheumatoid Arthritis. BioDrugs 2016, 30, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Vaz, A.; Lisse, J.; Rizzo, W.; Albani, S. Discussion: DMARDs and biologic therapies in the management of inflammatory joint diseases. Expert Rev. Clin. Immunol. 2009, 5, 291–299. [Google Scholar] [CrossRef]
- Gerriets, V.; Bansal, P.; Goyal, A.; Khaddour, K. Tumor Necrosis Factor (TNF) Inhibitors. In StatPearls; StatPearls Publishing Copyright © 2020; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2020. [Google Scholar]
- Kalden, J.R.; Schulze-Koops, H. Immunogenicity and loss of response to TNF inhibitors: Implications for rheumatoid arthritis treatment. Nat. Rev. Rheumatol. 2017, 13, 707–718. [Google Scholar] [CrossRef] [PubMed]
- AbbVie. Novel Antibody Drug Conjugate ABBV-3373 Shows Improvement in Disease Activity in Phase 2a Study of Patients with Rheumatoid Arthritis. 2020. Available online: https://news.abbvie.com/news/press-releases/novel-antibody-drug-conjugate-abbv-3373-shows-improvement-in-disease-activity-in-phase-2a-study-patients-with-rheumatoid-arthritis.htm (accessed on 10 September 2020).
- Xu, Z.; Bouman-Thio, E.; Comisar, C.; Frederick, B.; Van Hartingsveldt, B.; Marini, J.C.; Davis, H.M.; Zhou, H. Pharmacokinetics, pharmacodynamics and safety of a human anti-IL-6 monoclonal antibody (sirukumab) in healthy subjects in a first-in-human study. Br. J. Clin. Pharmacol. 2011, 72, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Ogata, A.; Kato, Y.; Higa, S.; Yoshizaki, K. IL-6 inhibitor for the treatment of rheumatoid arthritis: A comprehensive review. Mod. Rheumatol. 2019, 29, 258–267. [Google Scholar] [CrossRef]
- Genovese, M.; Fleischmann, R.; Fiore, S.; Radin, A.; Fan, C.; Huizinga, T. SAT0117 sarilumab, a subcutaneously-administered, fully-human monoclonal antibody inhibitor of the IL-6 receptor: Relationship between eular responses and change from baseline of selected clinical parameters. Ann. Rheum. Dis. 2013, 72, A620. [Google Scholar] [CrossRef]
- Rafique, A.; Martin, J.; Blome, M.; Huang, T.; Ouyang, A.; Papadopoulos, N. AB0037 Evaluation of the binding kinetics and functional bioassay activity of sarilumab and tocilizumab to the human il-6 receptor (il-6r) alpha. Ann. Rheum. Dis. 2013, 72, A797. [Google Scholar] [CrossRef]
- Bae, S.-C.; Lee, Y.H. Comparison of the efficacy and tolerability of tocilizumab, sarilumab, and sirukumab in patients with active rheumatoid arthritis: A Bayesian network meta-analysis of randomized controlled trials. Clin. Rheumatol. 2018, 37, 1471–1479. [Google Scholar] [CrossRef] [PubMed]
- Mok, C.C. Rituximab for the treatment of rheumatoid arthritis: An update. Drug Des. Dev. Ther. 2014, 8, 87. [Google Scholar] [CrossRef] [PubMed]
- Du, F.H.; Mills, E.A.; Mao-Draayer, Y. Next-generation anti-CD20 monoclonal antibodies in autoimmune disease treatment. Autoimmun. Highlights 2017, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A.; Deeks, E.D. Abatacept: A review in rheumatoid arthritis. Drugs 2017, 77, 1221–1233. [Google Scholar] [CrossRef]
- Avci, A.B.; Feist, E.; Burmester, G.R. Targeting GM-CSF in rheumatoid arthritis. Clin. Exp. Rheumatol. 2016, 34, 39–44. [Google Scholar]
- Crotti, C.; Raimondo, M.G.; Becciolini, A.; Biggioggero, M.; Favalli, E.G. Spotlight on mavrilimumab for the treatment of rheumatoid arthritis: Evidence to date. Drug Des. Dev. Ther. 2017, 11, 211–223. [Google Scholar] [CrossRef]
- Taylor, P.C.; Saurigny, D.; Vencovsky, J.; Takeuchi, T.; Nakamura, T.; Matsievskaia, G.; Hunt, B.; Wagner, T.; Souberbielle, B. Efficacy and safety of namilumab, a human monoclonal antibody against granulocyte-macrophage colony-stimulating factor (GM-CSF) ligand in patients with rheumatoid arthritis (RA) with either an inadequate response to background methotrexate therapy or an inadequate response or intolerance to an anti-TNF (tumour necrosis factor) biologic therapy: A randomized, controlled trial. Arthritis Res. Ther. 2019, 21, 1–13. [Google Scholar] [CrossRef]
- Genovese, M.C.; van Vollenhoven, R.F.; Pacheco-Tena, C.; Zhang, Y.; Kinnman, N. VX-509 (Decernotinib), an oral selective JAK-3 inhibitor, in combination with methotrexate in patients with rheumatoid arthritis. Arthritis Rheumatol. 2016, 68, 46–55. [Google Scholar] [CrossRef]
- Whang, J.A.; Chang, B.Y. Bruton’s tyrosine kinase inhibitors for the treatment of rheumatoid arthritis. Drug Discov. Today 2014, 19, 1200–1204. [Google Scholar] [CrossRef] [PubMed]
- Schafer, P.H.; Kivitz, A.J.; Ma, J.; Korish, S.; Sutherland, D.; Li, L.; Azaryan, A.; Kosek, J.; Adams, M.; Capone, L.; et al. Spebrutinib (CC-292) Affects Markers of B Cell Activation, Chemotaxis, and Osteoclasts in Patients with Rheumatoid Arthritis: Results from a Mechanistic Study. Rheumatol. Ther. 2020, 7, 101–119. [Google Scholar] [CrossRef] [PubMed]
- Gillooly, K.M.; Pulicicchio, C.; Pattoli, M.A.; Cheng, L.; Skala, S.; Heimrich, E.M.; McIntyre, K.W.; Taylor, T.L.; Kukral, D.W.; Dudhgaonkar, S.; et al. Bruton’s tyrosine kinase inhibitor BMS-986142 in experimental models of rheumatoid arthritis enhances efficacy of agents representing clinical standard-of-care. PLoS ONE 2017, 12, e0181782. [Google Scholar] [CrossRef]
- Watterson, S.H.; Liu, Q.; Beaudoin Bertrand, M.; Batt, D.G.; Li, L.; Pattoli, M.A.; Skala, S.; Cheng, L.; Obermeier, M.T.; Moore, R.; et al. Discovery of Branebrutinib (BMS-986195): A Strategy for Identifying a Highly Potent and Selective Covalent Inhibitor Providing Rapid in Vivo Inactivation of Bruton’s Tyrosine Kinase (BTK). J. Med. Chem. 2019, 62, 3228–3250. [Google Scholar] [CrossRef]
- Caldwell, R.D.; Qiu, H.; Askew, B.C.; Bender, A.T.; Brugger, N.; Camps, M.; Dhanabal, M.; Dutt, V.; Eichhorn, T.; Gardberg, A.S.; et al. Discovery of Evobrutinib: An Oral, Potent, and Highly Selective, Covalent Bruton’s Tyrosine Kinase (BTK) Inhibitor for the Treatment of Immunological Diseases. J. Med. Chem. 2019, 62, 7643–7655. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Tuckwell, K.; Katsumoto, T.R.; Zhao, R.; Galanter, J.; Lee, C.; Rae, J.; Toth, B.; Ramamoorthi, N.; Hackney, J.A. Fenebrutinib Versus Placebo or Adalimumab in Rheumatoid Arthritis: A Randomized, Double-Blind, Phase II Trial. Arthritis Rheumatol. 2020, 72, 1435–1446. [Google Scholar] [CrossRef]
- Norman, P. Investigational Bruton’s tyrosine kinase inhibitors for the treatment of rheumatoid arthritis. Expert Opin. Investig. Drugs 2016, 25, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Esfandiari, E.; Gevorkyan, H.; Faessel, H.; Zhao, Z.; Hanna, K.; Smithson, G.; Wagner, J.; Fedyk, E.R.; Mclean, L. SAT0226 A phase 1, randomized, double-blind, placebo-controlled, single- and multiple-rising dose study of the btk inhibitor tak-020 in healthy volunteers. Ann. Rheum. Dis. 2018, 77, 974. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Wang, L.; Huang, S.; Li, S.; Li, M.; Shi, J.; Bai, W.; Wang, Q.; Zheng, L.; Liu, Y. Efficacy and Safety of Umbilical Cord Mesenchymal Stem Cell Therapy for Rheumatoid Arthritis Patients: A Prospective Phase I/II Study. Drug Des. Dev. Ther. 2019, 13, 4331–4340. [Google Scholar] [CrossRef]
- Steinman, R.M.; Hawiger, D.; Nussenzweig, M.C. Tolerogenic dendritic cells. Ann. Rev. Immunol. 2003, 21, 685–711. [Google Scholar] [CrossRef]
- Ahmed, M.; Bae, Y.-S. Dendritic cell-based immunotherapy for rheumatoid arthritis: From bench to bedside. Immune Netw. 2016, 16, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Bell, G.M.; Anderson, A.E.; Diboll, J.; Reece, R.; Eltherington, O.; Harry, R.A.; Fouweather, T.; MacDonald, C.; Chadwick, T.; McColl, E.; et al. Autologous tolerogenic dendritic cells for rheumatoid and inflammatory arthritis. Ann. Rheum. Dis. 2017, 76, 227–234. [Google Scholar] [CrossRef]
- Álvaro-Gracia, J.M.; Jover, J.A.; García-Vicuña, R.; Carreño, L.; Alonso, A.; Marsal, S.; Blanco, F.; Martínez-Taboada, V.M.; Taylor, P.; Martín-Martín, C.; et al. Intravenous administration of expanded allogeneic adipose-derived mesenchymal stem cells in refractory rheumatoid arthritis (Cx611): Results of a multicentre, dose escalation, randomised, single-blind, placebo-controlled phase Ib/IIa clinical trial. Ann. Rheum. Dis. 2017, 76, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Liu, H.; Wang, Y.; Chen, S.; Liu, J.; Li, W.; Dou, H.; Hou, W.; Meng, M. Study of the adoptive immunotherapy on rheumatoid arthritis with Thymus-derived invariant natural killer T cells. Int. Immunopharmacol. 2019, 67, 427–440. [Google Scholar] [CrossRef]
- Wu, X.; He, B.; Liu, J.; Feng, H.; Ma, Y.; Li, D.; Guo, B.; Liang, C.; Dang, L.; Wang, L. Molecular insight into gut microbiota and rheumatoid arthritis. Int. J. Mol. Sci. 2016, 17, 431. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Pan, H.; Guo, R.; Ju, Y.; Wang, Q.; Zhu, J.; Xie, Y.; Zheng, Y.; Li, T.; Liu, Z.; Lu, L.; et al. A single bacterium restores the microbiome dysbiosis to protect bones from destruction in a rat model of rheumatoid arthritis. Microbiome 2019, 7, 1–11. [Google Scholar] [CrossRef]
- Marietta, E.V.; Murray, J.A.; Luckey, D.H.; Jeraldo, P.R.; Lamba, A.; Patel, R.; Luthra, H.S.; Mangalam, A.; Taneja, V. Suppression of inflammatory arthritis by human gut-derived Prevotella histicola in humanized mice. Arthritis Rheumatol. 2016, 68, 2878–2888. [Google Scholar] [CrossRef]
- Rai, M.F.; Pan, H.; Yan, H.; Sandell, L.J.; Pham, C.T.N.; Wickline, S.A. Applications of RNA interference in the treatment of arthritis. Transl. Res. 2019, 214, 1–16. [Google Scholar] [CrossRef]
- Cohen, J.L.; Shen, Y.; Aouadi, M.; Vangala, P.; Tencerova, M.; Amano, S.U.; Nicoloro, S.M.; Yawe, J.C.; Czech, M.P. Peptide- and Amine-Modified Glucan Particles for the Delivery of Therapeutic siRNA. Mol. Pharm. 2016, 13, 964–978. [Google Scholar] [CrossRef]
- Plavcová, Z.; Šalamúnová, P.; Saloň, I.; Štěpánek, F.; Hanuš, J.; Hošek, J. Curcumin encapsulation in yeast glucan particles promotes its anti-inflammatory potential in vitro. Int. J. Pharm. 2019, 568, 118532. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Zhang, Y.P.; Wu, J.; Jie, L.G.; Deng, J.X.; Zhao, D.B.; Yu, Q.H. Downregulated microRNA-135a ameliorates rheumatoid arthritis by inactivation of the phosphatidylinositol 3-kinase/AKT signaling pathway via phosphatidylinositol 3-kinase regulatory subunit 2. J. Cell. Physiol. 2019, 234, 17663–17676. [Google Scholar] [CrossRef] [PubMed]
- Nutten, S. Atopic dermatitis: Global epidemiology and risk factors. Ann. Nutr. Metab. 2015, 66 (Suppl. S1), 8–16. [Google Scholar] [CrossRef]
- Spergel, J.M. Epidemiology of atopic dermatitis and atopic march in children. Immunol. Allergy Clin. 2010, 30, 269–280. [Google Scholar] [CrossRef]
- Badloe, F.M.S.; De Vriese, S.; Coolens, K.; Schmidt-Weber, C.B.; Ring, J.; Gutermuth, J.; Krohn, I.K. IgE autoantibodies and autoreactive T cells and their role in children and adults with atopic dermatitis. Clin. Transl. Allergy 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Sullivan, M.; Silverberg, N.B. Current and emerging concepts in atopic dermatitis pathogenesis. Clin. Dermatol. 2017, 35, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Nedoszytko, B.; Reszka, E.; Gutowska-Owsiak, D.; Trzeciak, M.; Lange, M.; Jarczak, J.; Niedoszytko, M.; Jablonska, E.; Romantowski, J.; Strapagiel, D. Genetic and Epigenetic Aspects of Atopic Dermatitis. Int. J. Mol. Sci. 2020, 21, 6484. [Google Scholar] [CrossRef] [PubMed]
- Brunner, P.M.; Guttman-Yassky, E.; Leung, D.Y. The immunology of atopic dermatitis and its reversibility with broad-spectrum and targeted therapies. J. Allergy Clin. Immunol. 2017, 139, S65–S76. [Google Scholar] [CrossRef] [PubMed]
- Gittler, J.K.; Shemer, A.; Suárez-Fariñas, M.; Fuentes-Duculan, J.; Gulewicz, K.J.; Wang, C.Q.; Mitsui, H.; Cardinale, I.; de Guzman Strong, C.; Krueger, J.G. Progressive activation of TH2/TH22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J. Allergy Clin. Immunol. 2012, 130, 1344–1354. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Kim, J.S.; Cho, D.H.; Park, H.J. Molecular Mechanisms of Cutaneous Inflammatory Disorder: Atopic Dermatitis. Int. J. Mol. Sci. 2016, 17, 1234. [Google Scholar] [CrossRef]
- Nakahara, T.; Kido-Nakahara, M.; Tsuji, G.; Furue, M. Basics and recent advances in the pathophysiology of atopic dermatitis. J. Dermatol. 2020, 48, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Moreno, A.S.; McPhee, R.; Arruda, L.K.; Howell, M.D. Targeting the T helper 2 inflammatory axis in atopic dermatitis. Int. Arch. Allergy Immunol. 2016, 171, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Sonkoly, E.; Muller, A.; Lauerma, A.I.; Pivarcsi, A.; Soto, H.; Kemeny, L.; Alenius, H.; Dieu-Nosjean, M.-C.; Meller, S.; Rieker, J. IL-31: A new link between T cells and pruritus in atopic skin inflammation. J. Allergy Clin. Immunol. 2006, 117, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Wollina, U. Microbiome in atopic dermatitis. Clin. Cosmet. Investig. Dermatol. 2017, 10, 51. [Google Scholar] [CrossRef] [PubMed]
- Renert-Yuval, Y.; Guttman-Yassky, E. New treatments for atopic dermatitis targeting beyond IL-4/IL-13 cytokines. Ann. Allergy Asthma Immunol. 2020, 124, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Moyle, M.; Cevikbas, F.; Harden, J.L.; Guttman-Yassky, E. Understanding the immune landscape in atopic dermatitis: The era of biologics and emerging therapeutic approaches. Exp. Dermatol. 2019, 28, 756–768. [Google Scholar] [CrossRef]
- Guttman-Yassky, E.; Hanifin, J.M.; Boguniewicz, M.; Wollenberg, A.; Bissonnette, R.; Purohit, V.; Kilty, I.; Tallman, A.M.; Zielinski, M.A. The role of phosphodiesterase 4 in the pathophysiology of atopic dermatitis and the perspective for its inhibition. Exp. Dermatol. 2019, 28, 3–10. [Google Scholar] [CrossRef]
- Yang, H.; Wang, J.; Zhang, X.; Zhang, Y.; Qin, Z.-l.; Wang, H.; Luo, X.-y. Application of topical phosphodiesterase 4 inhibitors in mild to moderate atopic dermatitis: A systematic review and meta-analysis. JAMA Dermatol. 2019, 155, 585–593. [Google Scholar] [CrossRef]
- Simpson, E.L.; Imafuku, S.; Poulin, Y.; Ungar, B.; Zhou, L.; Malik, K.; Wen, H.C.; Xu, H.; Estrada, Y.D.; Peng, X.; et al. A Phase 2 Randomized Trial of Apremilast in Patients with Atopic Dermatitis. J. Invest. Dermatol. 2019, 139, 1063–1072. [Google Scholar] [CrossRef]
- Yu, S.; Pearson, A.D.; Lim, R.K.; Rodgers, D.T.; Li, S.; Parker, H.B.; Weglarz, M.; Hampton, E.N.; Bollong, M.J.; Shen, J.; et al. Targeted Delivery of an Anti-inflammatory PDE4 Inhibitor to Immune Cells via an Antibody-drug Conjugate. Mol. Ther. 2016, 24, 2078–2089. [Google Scholar] [CrossRef] [PubMed]
- Furue, M.; Ulzii, D.; Nakahara, T.; Tsuji, G.; Furue, K.; Hashimoto-Hachiya, A.; Kido-Nakahara, M. Implications of IL-13Rα2 in atopic skin inflammation. Allergol. Int. 2020, 69, 412–416. [Google Scholar] [CrossRef]
- D’Ippolito, D.; Pisano, M. Dupilumab (Dupixent): An Interleukin-4 Receptor Antagonist for Atopic Dermatitis. Pharm. Ther. 2018, 43, 532–535. [Google Scholar]
- Wollenberg, A.; Howell, M.D.; Guttman-Yassky, E.; Silverberg, J.I.; Kell, C.; Ranade, K.; Moate, R.; van der Merwe, R. Treatment of atopic dermatitis with tralokinumab, an anti-IL-13 mAb. J. Allergy Clin. Immunol. 2019, 143, 135–141. [Google Scholar] [CrossRef]
- Guttman-Yassky, E.; Blauvelt, A.; Eichenfield, L.F.; Paller, A.S.; Armstrong, A.W.; Drew, J.; Gopalan, R.; Simpson, E.L. Efficacy and Safety of Lebrikizumab, a High-Affinity Interleukin 13 Inhibitor, in Adults With Moderate to Severe Atopic Dermatitis: A Phase 2b Randomized Clinical Trial. JAMA Dermatol. 2020, 156, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.G.; Narayana, P.K.; Pouliquen, I.J.; Lopez, M.C.; Ferreira-Cornwell, M.C.; Getsy, J.A. Efficacy and safety of mepolizumab administered subcutaneously for moderate to severe atopic dermatitis. Allergy 2020, 75, 950–953. [Google Scholar] [CrossRef] [PubMed]
- Silverberg, J.I.; Pinter, A.; Pulka, G.; Poulin, Y.; Bouaziz, J.-D.; Wollenberg, A.; Murrell, D.F.; Alexis, A.; Lindsey, L.; Ahmad, F. Phase 2B randomized study of nemolizumab in adults with moderate-to-severe atopic dermatitis and severe pruritus. J. Allergy Clin. Immunol. 2020, 145, 173–182. [Google Scholar] [CrossRef]
- McKeage, K.; Duggan, S. Risankizumab: First Global Approval. Drugs 2019, 79, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Guttman-Yassky, E.; Brunner, P.M.; Neumann, A.U.; Khattri, S.; Pavel, A.B.; Malik, K.; Singer, G.K.; Baum, D.; Gilleaudeau, P.; Sullivan-Whalen, M.; et al. Efficacy and safety of fezakinumab (an IL-22 monoclonal antibody) in adults with moderate-to-severe atopic dermatitis inadequately controlled by conventional treatments: A randomized, double-blind, phase 2a trial. J. Am. Acad. Dermatol. 2018, 78, 872–881. [Google Scholar] [CrossRef]
- Simpson, E.L.; Lacour, J.P.; Spelman, L.; Galimberti, R.; Eichenfield, L.F.; Bissonnette, R.; King, B.A.; Thyssen, J.P.; Silverberg, J.I.; Bieber, T.; et al. Baricitinib in patients with moderate-to-severe atopic dermatitis and inadequate response to topical corticosteroids: Results from two randomized monotherapy phase III trials. Br. J. Dermatol. 2020. [Google Scholar] [CrossRef]
- Kim, B.S.; Howell, M.D.; Sun, K.; Papp, K.; Nasir, A.; Kuligowski, M.E. Treatment of atopic dermatitis with ruxolitinib cream (JAK1/JAK2 inhibitor) or triamcinolone cream. J. Allergy Clin. Immunol. 2020, 145, 572–582. [Google Scholar] [CrossRef]
- AbbVie. RINVOQ™(Upadacitinib) Monotherapy Shows Improvement in Skin Clearance and Itch in First Phase 3 Study for Atopic Dermatitis. 2020. Available online: https://news.abbvie.com/news/press-releases/rinvoq-upadacitinib-monotherapy-shows-improvement-in-skin-clearance-and-itch-in-first-phase-3-study-for-atopic-dermatitis.htm (accessed on 4 September 2020).
- Silverberg, J.I.; Simpson, E.L.; Thyssen, J.P.; Gooderham, M.; Chan, G.; Feeney, C.; Biswas, P.; Valdez, H.; DiBonaventura, M.; Nduaka, C.; et al. Efficacy and Safety of Abrocitinib in Patients With Moderate-to-Severe Atopic Dermatitis: A Randomized Clinical Trial. JAMA Dermatol. 2020, 156, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Bissonnette, R.; Maari, C.; Forman, S.; Bhatia, N.; Lee, M.; Fowler, J.; Tyring, S.; Pariser, D.; Sofen, H.; Dhawan, S.; et al. The oral Janus kinase/spleen tyrosine kinase inhibitor ASN002 demonstrates efficacy and improves associated systemic inflammation in patients with moderate-to-severe atopic dermatitis: Results from a randomized double-blind placebo-controlled study. Br. J. Dermatol. 2019, 181, 733–742. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Guttman-Yassky, E. JAK Inhibitors for Atopic Dermatitis: An Update. Am. J. Clin. Dermatol. 2019, 20, 181–192. [Google Scholar] [CrossRef]
- Ohsawa, Y.; Hirasawa, N. The role of histamine H1 and H4 receptors in atopic dermatitis: From basic research to clinical study. Allergol. Int. 2014, 63, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Werfel, T.; Layton, G.; Yeadon, M.; Whitlock, L.; Osterloh, I.; Jimenez, P.; Liu, W.; Lynch, V.; Asher, A.; Tsianakas, A.; et al. Efficacy and safety of the histamine H(4) receptor antagonist ZPL-3893787 in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2019, 143, 1830–1837. [Google Scholar] [CrossRef]
- Simpson, E.L.; Parnes, J.R.; She, D.; Crouch, S.; Rees, W.; Mo, M.; van der Merwe, R. Tezepelumab, an anti–thymic stromal lymphopoietin monoclonal antibody, in the treatment of moderate to severe atopic dermatitis: A randomized phase 2a clinical trial. J. Am. Acad. Dermatol. 2019, 80, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J.; Soumelis, V.; Watanabe, N.; Ito, T.; Wang, Y.H.; Malefyt Rde, W.; Omori, M.; Zhou, B.; Ziegler, S.F. TSLP: An epithelial cell cytokine that regulates T cell differentiation by conditioning dendritic cell maturation. Annu. Rev. Immunol. 2007, 25, 193–219. [Google Scholar] [CrossRef]
- Guttman-Yassky, E.; Pavel, A.B.; Zhou, L.; Estrada, Y.D.; Zhang, N.; Xu, H.; Peng, X.; Wen, H.C.; Govas, P.; Gudi, G.; et al. GBR 830, an anti-OX40, improves skin gene signatures and clinical scores in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2019, 144, 482–493. [Google Scholar] [CrossRef]
- Nakagawa, H.; Iizuka, H.; Nemoto, O.; Shimabe, M.; Furukawa, Y.; Kikuta, N.; Ootaki, K. Safety, tolerability and efficacy of repeated intravenous infusions of KHK4083, a fully human anti-OX40 monoclonal antibody, in Japanese patients with moderate to severe atopic dermatitis. J. Dermatol. Sci. 2020, 99, 82–89. [Google Scholar] [CrossRef]
- MorphoSys. MorphoSys AG: MOR106 Clinical Development in Atopic Dermatitis Stopped. 2019. Available online: https://www.morphosys.com/media-investors/media-center/morphosys-ag-mor106-clinical-development-in-atopic-dermatitis-stopped (accessed on 28 September 2020).
- Ungar, B.; Pavel, A.B.; Li, R.; Kimmel, G.; Nia, J.; Hashim, P.; Kim, H.J.; Chima, M.; Vekaria, A.S.; Estrada, Y. Phase 2 randomized, double-blind study of IL-17 targeting with secukinumab in atopic dermatitis. J. Allergy Clin. Immunol. 2020, 147, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y. Interleukin-33 in atopic dermatitis. J. Dermatol. Sci. 2019, 96, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Gutowska-Owsiak, D.; Hardman, C.S.; Westmoreland, M.; MacKenzie, T.; Cifuentes, L.; Waithe, D.; Lloyd-Lavery, A.; Marquette, A.; Londei, M.; et al. Proof-of-concept clinical trial of etokimab shows a key role for IL-33 in atopic dermatitis pathogenesis. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Nakatsuji, T.; Chen, T.H.; Narala, S.; Chun, K.A.; Two, A.M.; Yun, T.; Shafiq, F.; Kotol, P.F.; Bouslimani, A.; Melnik, A.V.; et al. Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.R.; Costa, S.K.; Zaramela, L.S.; Khalil, S.; Todd, D.A.; Winter, H.L.; Sanford, J.A.; O’Neill, A.M.; Liggins, M.C.; Nakatsuji, T.; et al. Quorum sensing between bacterial species on the skin protects against epidermal injury in atopic dermatitis. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Myles, I.A.; Williams, K.W.; Reckhow, J.D.; Jammeh, M.L.; Pincus, N.B.; Sastalla, I.; Saleem, D.; Stone, K.D.; Datta, S.K. Transplantation of human skin microbiota in models of atopic dermatitis. JCI Insight 2016, 1, e86955. [Google Scholar] [CrossRef]
- Myles, I.; Earland, N.; Anderson, E.; Moore, I.; Kieh, M.; Williams, K. First-in-human topical microbiome transplantation with Roseomonas mucosa for atopic dermatitis. JCI Insight 2018, 3, e120608. [Google Scholar] [CrossRef]
- Itano, A.; Cormack, T.; Ramani, K.; Barth, K.; Wang, I.; Mukherjee, A.; Ponichtera, H.; McKenna, C.; Jahic, M.; Bodmer, M. Orally-Administered EDP1815, a Single Strain of Prevotella histicola, Has Potent Systemic Anti-Inflammatory Effects in Type 1, Type 2, and Type 3 Inflammatory Models; Evelo Biosciences: Cambridge, MA, USA, 2020. [Google Scholar]
- Lee, W.-R.; Shen, S.-C.; Chen, W.-Y.; Aljuffali, I.A.; Suen, S.-Y.; Fang, J.-Y. Noninvasive delivery of siRNA and plasmid DNA into skin by fractional ablation: Erbium: YAG laser versus CO2 laser. Eur. J. Pharm. Biopharm. 2014, 86, 315–323. [Google Scholar] [CrossRef]
- Ibaraki, H.; Kanazawa, T.; Kurano, T.; Oogi, C.; Takashima, Y.; Seta, Y. Anti-RelA siRNA-Encapsulated Flexible Liposome with Tight Junction-Opening Peptide as a Non-invasive Topical Therapeutic for Atopic Dermatitis. Biol. Pharm. Bull. 2019, 42, 1216–1225. [Google Scholar] [CrossRef] [PubMed]
- Ibaraki, H.; Kanazawa, T.; Takashima, Y.; Okada, H.; Seta, Y. Transdermal anti-nuclear kappaB siRNA therapy for atopic dermatitis using a combination of two kinds of functional oligopeptide. Int. J. Pharm. 2018, 542, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Ibaraki, H.; Kanazawa, T.; Takashima, Y.; Okada, H.; Seta, Y. Development of an Innovative Intradermal siRNA Delivery System Using a Combination of a Functional Stearylated Cytoplasm-Responsive Peptide and a Tight Junction-Opening Peptide. Molecules 2016, 21, 1279. [Google Scholar] [CrossRef]
- Liew, W.C.; Sundaram, G.M.; Quah, S.; Lum, G.G.; Tan, J.S.; Ramalingam, R.; Common, J.E.; Tang, M.B.; Lane, E.B.; Thng, S.T.G. Belinostat resolves skin barrier defects in atopic dermatitis by targeting the dysregulated miR-335: SOX6 axis. J. Allergy Clin. Immunol. 2020, 146, 606–620. [Google Scholar] [CrossRef]
- Baumgart, D.C.; Sandborn, W.J. Crohn’s disease. Lancet 2012, 380, 1590–1605. [Google Scholar] [CrossRef]
- Feuerstein, J.D.; Cheifetz, A.S. Crohn Disease: Epidemiology, Diagnosis, and Management. Mayo Clin. Proc. 2017, 92, 1088–1103. [Google Scholar] [CrossRef]
- Leung, Y.; Panaccione, R. Anti-adhesion molecule strategies for Crohn disease. BioDrugs 2008, 22, 259–264. [Google Scholar] [CrossRef]
- Zundler, S.; Becker, E.; Weidinger, C.; Siegmund, B. Anti-Adhesion Therapies in Inflammatory Bowel Disease-Molecular and Clinical Aspects. Front. Immunol. 2017, 8, 891. [Google Scholar] [CrossRef] [PubMed]
- Pagnini, C.; Arseneau, K.O.; Cominelli, F. Natalizumab in the treatment of Crohn’s disease patients. Expert Opin. Biol. Ther. 2017, 17, 1433–1438. [Google Scholar] [CrossRef]
- Dotan, I.; Allez, M.; Danese, S.; Keir, M.; Tole, S.; McBride, J. The role of integrins in the pathogenesis of inflammatory bowel disease: Approved and investigational anti-integrin therapies. Med. Res. Rev. 2020, 40, 245–262. [Google Scholar] [CrossRef]
- Protagonist Therapeutics Inc. Oral Alpha-4-Beta-7 Integrin Antagonist PN-943 Demonstrates Sustained Dose-Related Target Engagement Activity in a Multiple Ascending Dose Phase 1 Study. 2019. Available online: www.prnewswire.com (accessed on 20 August 2020).
- Sandborn, W.J.; Lee, S.D.; Tarabar, D.; Louis, E.; Klopocka, M.; Klaus, J.; Reinisch, W.; Hébuterne, X.; Park, D.I.; Schreiber, S.; et al. Phase II evaluation of anti-MAdCAM antibody PF-00547659 in the treatment of Crohn’s disease: Report of the OPERA study. Gut 2018, 67, 1824–1835. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Furfaro, F.; Vetrano, S. Targeting S1P in Inflammatory bowel disease: New avenues for modulating intestinal leukocyte migration. J. Crohns Colitis 2018, 12, S678–S686. [Google Scholar] [CrossRef]
- Peyrin-Biroulet, L.; Christopher, R.; Behan, D.; Lassen, C. Modulation of sphingosine-1-phosphate in inflammatory bowel disease. Autoimmun. Rev. 2017, 16, 495–503. [Google Scholar] [CrossRef]
- Ishii, I.; Ye, X.; Friedman, B.; Kawamura, S.; Contos, J.J.; Kingsbury, M.A.; Yang, A.H.; Zhang, G.; Brown, J.H.; Chun, J. Marked perinatal lethality and cellular signaling deficits in mice null for the two sphingosine 1-phosphate (S1P) receptors, S1P2/LPB2/EDG-5 and S1P3/LPB3/EDG-3. J. Biol. Chem. 2002, 277, 25152–25159. [Google Scholar] [CrossRef] [PubMed]
- Feagan, B.G.; Sandborn, W.J.; Danese, S.; Wolf, D.C.; Liu, W.J.; Hua, S.Y.; Minton, N.; Olson, A.; D’Haens, G. Ozanimod induction therapy for patients with moderate to severe Crohn’s disease: A single-arm, phase 2, prospective observer-blinded endpoint study. Lancet Gastroenterol. Hepatol. 2020, 5, 819–828. [Google Scholar] [CrossRef]
- Gerstenberger, B.S.; Banker, M.E.; Clark, J.D.; Dowty, M.E.; Fensome, A.; Gifford, R.; Griffor, M.C.; Hegen, M.; Hollingshead, B.D.; Knafels, J.D.; et al. Demonstration of In Vitro to In Vivo Translation of a TYK2 Inhibitor That Shows Cross Species Potency Differences. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Benson, J.M.; Peritt, D.; Scallon, B.J.; Heavner, G.A.; Shealy, D.J.; Giles-Komar, J.M.; Mascelli, M.A. Discovery and mechanism of ustekinumab: A human monoclonal antibody targeting interleukin-12 and interleukin-23 for treatment of immune-mediated disorders. MAbs 2011, 3, 535–545. [Google Scholar] [CrossRef]
- Feagan, B.G.; Panés, J.; Ferrante, M.; Kaser, A.; D’Haens, G.R.; Sandborn, W.J.; Louis, E.; Neurath, M.F.; Franchimont, D.; Dewit, O.; et al. Risankizumab in patients with moderate to severe Crohn’s disease: An open-label extension study. Lancet Gastroenterol. Hepatol. 2018, 3, 671–680. [Google Scholar] [CrossRef]
- Xiao, Q.; Boushey, R.P.; Cino, M.; Drucker, D.J.; Brubaker, P.L. Circulating levels of glucagon-like peptide-2 in human subjects with inflammatory bowel disease. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 278, R1057–R1063. [Google Scholar] [CrossRef]
- Benjamin, M.; McKay, D.; Yang, P.; Cameron, H.; Perdue, M. Glucagon-like peptide-2 enhances intestinal epithelial barrier function of both transcellular and paracellular pathways in the mouse. Gut 2000, 47, 112–119. [Google Scholar] [CrossRef]
- Buchman, A.L.; Katz, S.; Fang, J.C.; Bernstein, C.N.; Abou-Assi, S.G.; Group, T.S. Teduglutide, a novel mucosally active analog of glucagon-like peptide-2 (GLP-2) for the treatment of moderate to severe Crohn’s disease. Inflamm. Bowel Dis. 2010, 16, 962–973. [Google Scholar] [CrossRef]
- Khan, I.; Ullah, N.; Zha, L.; Bai, Y.; Khan, A.; Zhao, T.; Che, T.; Zhang, C. Alteration of Gut Microbiota in Inflammatory Bowel Disease (IBD): Cause or Consequence? IBD Treatment Targeting the Gut Microbiome. Pathogens 2019, 8, 126. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.J.; Sartor, R.B. Therapeutic Manipulation of the Microbiome in IBD: Current Results and Future Approaches. Curr. Treat. Options Gastroenterol. 2015, 13, 105–120. [Google Scholar] [CrossRef]
- Durack, J.; Lynch, S.V. The gut microbiome: Relationships with disease and opportunities for therapy. J. Exp. Med. 2019, 216, 20–40. [Google Scholar] [CrossRef] [PubMed]
- Agus, A.; Massier, S.; Darfeuille-Michaud, A.; Billard, E.; Barnich, N. Understanding host-adherent-invasive Escherichia coli interaction in Crohn’s disease: Opening up new therapeutic strategies. BioMed. Res. Int. 2014, 2014, 567929. [Google Scholar] [CrossRef] [PubMed]
- Sivignon, A.; Bouckaert, J.; Bernard, J.; Gouin, S.G.; Barnich, N. The potential of FimH as a novel therapeutic target for the treatment of Crohn’s disease. Expert Opin. Ther. Targets 2017, 21, 837–847. [Google Scholar] [CrossRef]
- Lopetuso, L.R.; Giorgio, M.E.; Saviano, A.; Scaldaferri, F.; Gasbarrini, A.; Cammarota, G. Bacteriocins and bacteriophages: Therapeutic weapons for gastrointestinal diseases? Int. J. Mol. Sci. 2019, 20, 183. [Google Scholar] [CrossRef]
- Lin, D.M.; Koskella, B.; Lin, H.C. Phage therapy: An alternative to antibiotics in the age of multi-drug resistance. World J. Gastrointest. Pharmacol. Ther. 2017, 8, 162. [Google Scholar] [CrossRef] [PubMed]
- Galtier, M.; De Sordi, L.; Sivignon, A.; de Vallée, A.; Maura, D.; Neut, C.; Rahmouni, O.; Wannerberger, K.; Darfeuille-Michaud, A.; Desreumaux, P.; et al. Bacteriophages Targeting Adherent Invasive Escherichia coli Strains as a Promising New Treatment for Crohn’s Disease. J. Crohns Colitis 2017, 11, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Laveissière, A.; Morra, R.; Paillarse, J.M.; Bonny, C.; Patel, J. EB8018, a First-In-Class Fimh Blocker, Reduces Proinflammatory Cytokines TNFÎʻ, Il6 and Il8 in Surgical Explants and May Represent a Potential New Therapeutic Approach for the Treatment of Crohn’s Disease. Gastroenterology 2017, 152, 989. [Google Scholar] [CrossRef]
- Brown, C.L.; Smith, K.; Wall, D.M.; Walker, D. Activity of Species-specific Antibiotics Against Crohn’s Disease–Associated Adherent-invasive Escherichia coli. Inflamm. Bowel Dis. 2015, 21, 2372–2382. [Google Scholar]
- McKay, R.; Ghodasra, M.; Schardt, J.; Quan, D.; Pottash, A.E.; Shang, W.; Jay, S.M.; Payne, G.F.; Chang, M.W.; March, J.C.; et al. A platform of genetically engineered bacteria as vehicles for localized delivery of therapeutics: Toward applications for Crohn’s disease. Bioeng. Transl. Med. 2018, 3, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Suzuki, K.; Watanabe, M.; Okamoto, R. Stem cell-based therapy for inflammatory bowel disease. Intest. Res. 2019, 17, 311–316. [Google Scholar] [CrossRef]
- Barnhoorn, M.C.; Wasser, M.; Roelofs, H.; Maljaars, P.W.J.; Molendijk, I.; Bonsing, B.A.; Oosten, L.E.M.; Dijkstra, G.; van der Woude, C.J.; Roelen, D.L.; et al. Long-term Evaluation of Allogeneic Bone Marrow-derived Mesenchymal Stromal Cell Therapy for Crohn’s Disease Perianal Fistulas. J. Crohns Colitis 2020, 14, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Stubbington, M.J.T.; Rozenblatt-Rosen, O.; Regev, A.; Teichmann, S.A. Single-cell transcriptomics to explore the immune system in health and disease. Science 2017, 358, 58–63. [Google Scholar] [CrossRef]
- Martin, J.C.; Chang, C.; Boschetti, G.; Ungaro, R.; Giri, M.; Grout, J.A.; Gettler, K.; Chuang, L.S.; Nayar, S.; Greenstein, A.J.; et al. Single-Cell Analysis of Crohn’s Disease Lesions Identifies a Pathogenic Cellular Module Associated with Resistance to Anti-TNF Therapy. Cell 2019, 178, 1493–1508. [Google Scholar] [CrossRef]
- Grand View Research. Inflammatory Bowel Disease Treatment Market. Size, Share & Trends Analysis Report By Type (Ulcerative Colitis, Crohn’s Disease), By Route of Administration, By Distribution Channel, And Segment Forecasts 2019–2026. 2019. Available online: https://www.grandviewresearch.com/industry-analysis/inflammatory-bowel-disease-ibd-treatment-market (accessed on 10 September 2020).
- GlobalData. Crohn’s Disease Market Growth to 2026 Fuelled by Interleukin Inhibitor and Anti-Integrin Therapy Launches. 2017. Available online: https://www.globaldata.com/crohns-disease-market-growth-to-2026-fuelled-by-interleukin-inhibitor-and-anti-integrin-therapy-launches/ (accessed on 10 September 2020).
- Dahlhamer, J.; Lucas, J.; Zelaya, C.; Nahin, R.; Mackey, S.; DeBar, L.; Kerns, R.; Von Korff, M.; Porter, L.; Helmick, C. Prevalence of Chronic Pain and High-Impact Chronic Pain Among Adults—United States, 2016. MMWR Morb. Mortal. Wkly. Rep. 2018, 67, 1001–1006. [Google Scholar] [CrossRef]
- Raja, S.N.; Carr, D.B.; Cohen, M.; Finnerup, N.B.; Flor, H.; Gibson, S.; Keefe, F.J.; Mogil, J.S.; Ringkamp, M.; Sluka, K.A.; et al. The revised International Association for the Study of Pain definition of pain: Concepts, challenges, and compromises. Pain 2020, 161, 1976–1982. [Google Scholar] [CrossRef]
- Watson, J.C. Treatment of Pain. 2020. Available online: https://www.merckmanuals.com/home/brain,-spinal-cord,-and-nerve-disorders/pain/treatment-of-pain (accessed on 2 October 2020).
- Binder, A.; Baron, R. The Pharmacological Therapy of Chronic Neuropathic Pain. Dtsch. Arztebl. Int. 2016, 113, 616–625. [Google Scholar] [CrossRef]
- Galluzzi, K.E. Management of neuropathic pain. J. Am. Osteopath. Assoc. 2005, 105, S12–S19. [Google Scholar] [PubMed]
- Disease, G.B.D.; Injury, I.; Prevalence, C. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef]
- Bijlsma, J.W.; Berenbaum, F.; Lafeber, F.P. Osteoarthritis: An update with relevance for clinical practice. Lancet 2011, 377, 2115–2126. [Google Scholar] [CrossRef]
- Fu, K.; Robbins, S.R.; McDougall, J.J. Osteoarthritis: The genesis of pain. Rheumatology 2018, 57, iv43–iv50. [Google Scholar] [CrossRef] [PubMed]
- Kuca, B.; Silberstein, S.D.; Wietecha, L.; Berg, P.H.; Dozier, G.; Lipton, R.B.; Group, C.M.-S. Lasmiditan is an effective acute treatment for migraine: A phase 3 randomized study. Neurology 2018, 91, e2222–e2232. [Google Scholar] [CrossRef] [PubMed]
- Felson, D.T.; Zhang, Y.; Hannan, M.T.; Naimark, A.; Weissman, B.N.; Aliabadi, P.; Levy, D. The incidence and natural history of knee osteoarthritis in the elderly. The Framingham Osteoarthritis Study. Arthritis Rheum. 1995, 38, 1500–1505. [Google Scholar] [CrossRef]
- Sokolove, J.; Lepus, C.M. Role of inflammation in the pathogenesis of osteoarthritis: Latest findings and interpretations. Ther. Adv. Musculoskelet. Dis. 2013, 5, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Duarte, R.V.; Raphael, J.H.; Dimitroulas, T.; Sparkes, E.; Southall, J.L.; Ashford, R.L.; Kitas, G.D. Osteoarthritis pain has a significant neuropathic component: An exploratory in vivo patient model. Rheumatol. Int. 2014, 34, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Thakur, M.; Dickenson, A.H.; Baron, R. Osteoarthritis pain: Nociceptive or neuropathic? Nat. Rev. Rheumatol. 2014, 10, 374–380. [Google Scholar] [CrossRef]
- Chen, Q.; Heinricher, M.M. Descending Control Mechanisms and Chronic Pain. Curr. Rheumatol. Rep. 2019, 21, 1–7. [Google Scholar] [CrossRef]
- Chimenti, R.L.; Frey-Law, L.A.; Sluka, K.A. A Mechanism-Based Approach to Physical Therapist Management of Pain. Phys. Ther. 2018, 98, 302–314. [Google Scholar] [CrossRef]
- Chang, D.S.; Hsu, E.; Hottinger, D.G.; Cohen, S.P. Anti-nerve growth factor in pain management: Current evidence. J. Pain Res. 2016, 9, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Leung, Y.Y.; Huebner, J.L.; Haaland, B.; Wong, S.B.S.; Kraus, V.B. Synovial fluid pro-inflammatory profile differs according to the characteristics of knee pain. Osteoarthr. Cartil. 2017, 25, 1420–1427. [Google Scholar] [CrossRef]
- Mora, J.C.; Przkora, R.; Cruz-Almeida, Y. Knee osteoarthritis: Pathophysiology and current treatment modalities. J. Pain Res. 2018, 11, 2189–2196. [Google Scholar] [CrossRef]
- Dray, A.; Read, S.J. Arthritis and pain. Future targets to control osteoarthritis pain. Arthritis Res. Ther. 2007, 9, 1–14. [Google Scholar] [CrossRef]
- OARSI. Osteoarthritis: A Serious Disease 2016. Available online: https://oarsi.org/sites/default/files/docs/2016/oarsi_white_paper_oa-serious-disease.pdf (accessed on 10 August 2020).
- Kloppenburg, M.; Berenbaum, F. Osteoarthritis year in review 2019: Epidemiology and therapy. Osteoarthr. Cartil. 2020, 28, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Majeed, M.H.; Sherazi, S.A.A.; Bacon, D.; Bajwa, Z.H. Pharmacological Treatment of Pain in Osteoarthritis: A Descriptive Review. Curr. Rheumatol. Rep. 2018, 20, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gregori, D.; Giacovelli, G.; Minto, C.; Barbetta, B.; Gualtieri, F.; Azzolina, D.; Vaghi, P.; Rovati, L.C. Association of Pharmacological Treatments With Long-term Pain Control in Patients With Knee Osteoarthritis: A Systematic Review and Meta-analysis. JAMA 2018, 320, 2564–2579. [Google Scholar] [CrossRef]
- Sarzi-Puttini, P.; Cimmino, M.A.; Scarpa, R.; Caporali, R.; Parazzini, F.; Zaninelli, A.; Atzeni, F.; Canesi, B. Osteoarthritis: An overview of the disease and its treatment strategies. Semin. Arthritis Rheum. 2005, 35, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pertusi, R.M. Selective cyclooxygenase inhibition in pain management. J. Am. Osteopath Assoc. 2004, 104, S19–S24. [Google Scholar]
- Citrome, L.; Weiss-Citrome, A. A systematic review of duloxetine for osteoarthritic pain: What is the number needed to treat, number needed to harm, and likelihood to be helped or harmed? Postgrad. Med. 2012, 124, 83–93. [Google Scholar] [CrossRef]
- Lee, B.; Sodhi, N.; Anis, H.K.; Ehiorobo, J.O.; Mont, M.A. Injection Alternatives for the Management of Knee Osteoarthritis Pain. Surg. Technol. Int. 2019, 34, 513–519. [Google Scholar] [PubMed]
- Miller, R.E.; Block, J.A.; Malfait, A.M. What is new in pain modification in osteoarthritis? Rheumatology 2018, 57, iv99–iv107. [Google Scholar] [CrossRef] [PubMed]
- Kan, S.L.; Li, Y.; Ning, G.Z.; Yuan, Z.F.; Chen, L.X.; Bi, M.C.; Sun, J.C.; Feng, S.Q. Tanezumab for Patients with Osteoarthritis of the Knee: A Meta-Analysis. PLoS ONE 2016, 11, e0157105. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, J.; Li, R.; Wang, H.; Yang, J.; Xu, J.; Zha, Z. Efficacy and Safety of Tanezumab on Osteoarthritis Knee and Hip Pains: A Meta-Analysis of Randomized Controlled Trials. Pain Med. 2017, 18, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Schnitzer, T.J.; Ekman, E.F.; Spierings, E.L.; Greenberg, H.S.; Smith, M.D.; Brown, M.T.; West, C.R.; Verburg, K.M. Efficacy and safety of tanezumab monotherapy or combined with non-steroidal anti-inflammatory drugs in the treatment of knee or hip osteoarthritis pain. Ann. Rheum. Dis 2015, 74, 1202–1211. [Google Scholar] [CrossRef]
- Miller, R.E.; Block, J.A.; Malfait, A.M. Nerve growth factor blockade for the management of osteoarthritis pain: What can we learn from clinical trials and preclinical models? Curr. Opin. Rheumatol. 2017, 29, 110–118. [Google Scholar] [CrossRef]
- Lane, N.E.; Corr, M. Osteoarthritis in 2016: Anti-NGF treatments for pain—Two steps forward, one step back? Nat. Rev. Rheumatol. 2017, 13, 76–78. [Google Scholar] [CrossRef]
- Kloppenburg, M.; Peterfy, C.; Haugen, I.K.; Kroon, F.; Chen, S.; Wang, L.; Liu, W.; Levy, G.; Fleischmann, R.M.; Berenbaum, F.; et al. Phase IIa, placebo-controlled, randomised study of lutikizumab, an anti-interleukin-1alpha and anti-interleukin-1beta dual variable domain immunoglobulin, in patients with erosive hand osteoarthritis. Ann. Rheum. Dis. 2019, 78, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.M.; Bliddal, H.; Blanco, F.J.; Schnitzer, T.J.; Peterfy, C.; Chen, S.; Wang, L.; Feng, S.; Conaghan, P.G.; Berenbaum, F.; et al. A Phase II Trial of Lutikizumab, an Anti-Interleukin-1alpha/beta Dual Variable Domain Immunoglobulin, in Knee Osteoarthritis Patients With Synovitis. Arthritis Rheumatol. 2019, 71, 1056–1069. [Google Scholar] [CrossRef]
- Chevalier, X.; Ravaud, P.; Maheu, E.; Baron, G.; Rialland, A.; Vergnaud, P.; Roux, C.; Maugars, Y.; Mulleman, D.; Lukas, C.; et al. Adalimumab in patients with hand osteoarthritis refractory to analgesics and NSAIDs: A randomised, multicentre, double-blind, placebo-controlled trial. Ann. Rheum. Dis. 2015, 74, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Aitken, D.; Laslett, L.L.; Pan, F.; Haugen, I.K.; Otahal, P.; Bellamy, N.; Bird, P.; Jones, G. A randomised double-blind placebo-controlled crossover trial of HUMira (adalimumab) for erosive hand OsteoaRthritis—The HUMOR trial. Osteoarthr. Cartil. 2018, 26, 880–887. [Google Scholar] [CrossRef]
- Schett, G.; Bainbridge, C.; Berkowitz, M. A Phase IIa Study of Anti-GM-CSF Antibody GSK3196165 in Subjects with Inflammatory Hand Osteoarthritis [abstract]. Arthritis Rheumatol. 2018, 70 (Suppl. 10), 1365. [Google Scholar]
- Jin, Y.; Smith, C.; Monteith, D.; Brown, R.; Camporeale, A.; McNearney, T.A.; Deeg, M.A.; Raddad, E.; Xiao, N.; de la Pena, A.; et al. CGRP blockade by galcanezumab was not associated with reductions in signs and symptoms of knee osteoarthritis in a randomized clinical trial. Osteoarthr. Cartil. 2018, 26, 1609–1618. [Google Scholar] [CrossRef]
- Yekkirala, A.S.; Roberson, D.P.; Bean, B.P.; Woolf, C.J. Breaking barriers to novel analgesic drug development. Nat. Rev. Drug Discov. 2017, 16, 545–564. [Google Scholar] [CrossRef]
- Bagal, S.M.C.; Brady, P.; Stauffer, J. A Phase 2, Randomized, Double-Blind, Placebo-Controlled, Titration-to-Effect Study of Orally Administered CR845 in Patients with Osteoarthritis of the Hip or Knee [abstract]. Arthritis Rheumatol. 2017, 69 (Suppl. 10), 15L. [Google Scholar]
- Vertex. Vertex Reports Full-Year and Fourth-Quarter 2016 Financial Results. 2017. Available online: https://investors.vrtx.com/news-releases/news-release-details/vertex-reports-full-year-and-fourth-quarter-2016-financial (accessed on 22 September 2020).
- Stevens, R.M.; Ervin, J.; Nezzer, J.; Nieves, Y.; Guedes, K.; Burges, R.; Hanson, P.D.; Campbell, J.N. Randomized, Double-Blind, Placebo-Controlled Trial of Intraarticular Trans-Capsaicin for Pain Associated With Osteoarthritis of the Knee. Arthritis Rheumatol. 2019, 71, 1524–1533. [Google Scholar] [CrossRef]
- Arsenault, P.; Chiche, D.; Brown, W.; Miller, J.; Treister, R.; Leff, R.; Walker, P.; Katz, N. NEO6860, modality-selective TRPV1 antagonist: A randomized, controlled, proof-of-concept trial in patients with osteoarthritis knee pain. Pain Rep. 2018, 3, e696. [Google Scholar] [CrossRef]
- Lories, R.J.; Monteagudo, S. Review Article: Is Wnt Signaling an Attractive Target for the Treatment of Osteoarthritis? Rheumatol. Ther. 2020, 7, 259–270. [Google Scholar] [CrossRef]
- Cao, P.; Li, Y.; Tang, Y.; Ding, C.; Hunter, D.J. Pharmacotherapy for knee osteoarthritis: Current and emerging therapies. Expert Opin. Pharmacother. 2020, 21, 797–809. [Google Scholar] [CrossRef]
- Wu, Y.; Goh, E.L.; Wang, D.; Ma, S. Novel treatments for osteoarthritis: An update. Open Access Rheumatol. 2018, 10, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Ha, C.W.; In, Y.; Cho, S.D.; Choi, E.S.; Ha, J.K.; Lee, J.H.; Yoo, J.D.; Bin, S.I.; Choi, C.H.; et al. A Multicenter, Double-Blind, Phase III Clinical Trial to Evaluate the Efficacy and Safety of a Cell and Gene Therapy in Knee Osteoarthritis Patients. Hum. Gene Ther. Clin. Dev. 2018, 29, 48–59. [Google Scholar] [CrossRef] [PubMed]
- MacFarlane, E.G.; Haupt, J.; Dietz, H.C.; Shore, E.M. TGF-beta Family Signaling in Connective Tissue and Skeletal Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a022269. [Google Scholar] [CrossRef]
- Maheshwer, B.; Polce, E.M.; Paul, K.; Williams, B.T.; Wolfson, T.S.; Yanke, A.; Verma, N.N.; Cole, B.J.; Chahla, J. Regenerative Potential of Mesenchymal Stem Cells for the Treatment of Knee Osteoarthritis and Chondral Defects: A Systematic Review and Meta-Analysis. Arthroscopy 2020, 37, 362–378. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Suo, H.; Wang, Z.; Feng, W. Progress in the treatment of osteoarthritis with umbilical cord stem cells. Hum. Cell 2020, 33, 470–475. [Google Scholar] [CrossRef]
- Pourakbari, R.; Khodadadi, M.; Aghebati-Maleki, A.; Aghebati-Maleki, L.; Yousefi, M. The potential of exosomes in the therapy of the cartilage and bone complications; emphasis on osteoarthritis. Life Sci. 2019, 236, 116861. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Cai, Y.; Jiang, Y.; Lin, X. Exosomes in osteoarthritis and cartilage injury: Advanced development and potential therapeutic strategies. Int. J. Biol. Sci. 2020, 16, 1811–1820. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.H.; Gouze, J.N.; Gouze, E.; Robbins, P.D.; Ghivizzani, S.C. Osteoarthritis gene therapy. Gene Ther. 2004, 11, 379–389. [Google Scholar] [CrossRef]
- Zhao, L.; Huang, J.; Fan, Y.; Li, J.; You, T.; He, S.; Xiao, G.; Chen, D. Exploration of CRISPR/Cas9-based gene editing as therapy for osteoarthritis. Ann. Rheum. Dis. 2019, 78, 676–682. [Google Scholar] [CrossRef]
- Zhang, W. The powerful placebo effect in osteoarthritis. Clin. Exp. Rheumatol. 2019, 37 (Suppl. S120), 118–123. [Google Scholar]
- Grandi, F.C.; Bhutani, N. Epigenetic Therapies for Osteoarthritis. Trends Pharmacol. Sci. 2020, 41, 557–569. [Google Scholar] [CrossRef]
- Migraine Research Foundation. About Migraine. Available online: https://migraineresearchfoundation.org/about-migraine/migraine-facts/ (accessed on 10 October 2020).
- American Headache Society. Migraine. Available online: https://americanheadachesociety.org/wp-content/uploads/2019/03/GENERALMIGRIANE_11x14.pdf (accessed on 10 October 2020).
- Zameel Cader, M. The molecular pathogenesis of migraine: New developments and opportunities. Hum. Mol. Genet. 2013, 22, R39-44. [Google Scholar] [CrossRef]
- Andreou, A.P.; Edvinsson, L. Mechanisms of migraine as a chronic evolutive condition. J. Headache Pain 2019, 20, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Becker, W.J. Acute Migraine Treatment in Adults. Headache 2015, 55, 778–793. [Google Scholar] [CrossRef] [PubMed]
- Olesen, J.; Burstein, R.; Ashina, M.; Tfelt-Hansen, P. Origin of pain in migraine: Evidence for peripheral sensitisation. Lancet Neurol. 2009, 8, 679–690. [Google Scholar] [CrossRef]
- Charbit, A.R.; Akerman, S.; Goadsby, P.J. Dopamine: What’s new in migraine? Curr. Opin. Neurol. 2010, 23, 275–281. [Google Scholar] [CrossRef]
- Barbanti, P.; Aurilia, C.; Egeo, G.; Fofi, L.; Palmirotta, R. Serotonin receptor targeted therapy for migraine treatment: An overview of drugs in phase I and II clinical development. Expert Opin. Investig. Drugs 2017, 26, 269–277. [Google Scholar] [CrossRef]
- Taylor, F.R.; Kaniecki, R.G. Symptomatic treatment of migraine: When to use NSAIDs, triptans, or opiates. Curr. Treat. Options Neurol. 2011, 13, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Scuteri, D.; Adornetto, A.; Rombola, L.; Naturale, M.D.; Morrone, L.A.; Bagetta, G.; Tonin, P.; Corasaniti, M.T. New Trends in Migraine Pharmacology: Targeting Calcitonin Gene-Related Peptide (CGRP) With Monoclonal Antibodies. Front. Pharmacol. 2019, 10, 363. [Google Scholar] [CrossRef] [PubMed]
- Colman, I.; Brown, M.D.; Innes, G.D.; Grafstein, E.; Roberts, T.E.; Rowe, B.H. Parenteral dihydroergotamine for acute migraine headache: A systematic review of the literature. Ann. Emerg. Med. 2005, 45, 393–401. [Google Scholar] [CrossRef]
- Dahlof, C.; Maassen Van Den Brink, A. Dihydroergotamine, ergotamine, methysergide and sumatriptan—Basic science in relation to migraine treatment. Headache 2012, 52, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Andersson, K.E.; Vinge, E. Beta-adrenoceptor blockers and calcium antagonists in the prophylaxis and treatment of migraine. Drugs 1990, 39, 355–373. [Google Scholar] [CrossRef]
- Diener, H.C.; Kaube, H.; Limmroth, V. Migraine: Clinical aspects, diagnosis and pharmacotherapy. Internist 2000, 41, 390–398. [Google Scholar] [CrossRef]
- Akerman, S.; Goadsby, P.J. Dopamine and migraine: Biology and clinical implications. Cephalalgia 2007, 27, 1308–1314. [Google Scholar] [CrossRef]
- Peroutka, S.J.; Wilhoit, T.; Jones, K. Clinical susceptibility to migraine with aura is modified by dopamine D2 receptor (DRD2) NcoI alleles. Neurology 1997, 49, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Markley, H.G.; Cheronis, J.C.; Piepho, R.W. Verapamil in prophylactic therapy of migraine. Neurology 1984, 34, 973–976. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, D.; Arceri, S.; Tronconi, L.; Tassorelli, C. Chronic migraine and Botulinum Toxin Type A: Where do paths cross? Toxicon 2020, 178, 69–76. [Google Scholar] [CrossRef]
- Sprenger, T.; Viana, M.; Tassorelli, C. Current Prophylactic Medications for Migraine and Their Potential Mechanisms of Action. Neurotherapeutics 2018, 15, 313–323. [Google Scholar] [CrossRef]
- Oswald, J.C.; Schuster, N.M. Lasmiditan for the treatment of acute migraine: A review and potential role in clinical practice. J. Pain Res. 2018, 11, 2221–2227. [Google Scholar] [CrossRef]
- Clemow, D.B.; Johnson, K.W.; Hochstetler, H.M.; Ossipov, M.H.; Hake, A.M.; Blumenfeld, A.M. Lasmiditan mechanism of action—Review of a selective 5-HT1F agonist. J. Headache Pain 2020, 21, 1–13. [Google Scholar] [CrossRef]
- Deen, M.; Correnti, E.; Kamm, K.; Kelderman, T.; Papetti, L.; Rubio-Beltran, E.; Vigneri, S.; Edvinsson, L.; Maassen Van Den Brink, A.M. Blocking CGRP in migraine patients—A review of pros and cons. J. Headache Pain 2017, 18, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Rea, B.J.; Wattiez, A.S.; Waite, J.S.; Castonguay, W.C.; Schmidt, C.M.; Fairbanks, A.M.; Robertson, B.R.; Brown, C.J.; Mason, B.N.; Moldovan-Loomis, M.C.; et al. Peripherally administered calcitonin gene-related peptide induces spontaneous pain in mice: Implications for migraine. Pain 2018, 159, 2306–2317. [Google Scholar] [CrossRef] [PubMed]
- Lassen, L.H.; Haderslev, P.A.; Jacobsen, V.B.; Iversen, H.K.; Sperling, B.; Olesen, J. CGRP may play a causative role in migraine. Cephalalgia 2002, 22, 54–61. [Google Scholar] [CrossRef]
- Bucklan, J.; Ahmed, Z. CGRP antagonists for decreasing migraine frequency: New options, long overdue. Clevel. Clin. J. Med. 2020, 87, 211–218. [Google Scholar] [CrossRef]
- Moreno-Ajona, D.P.-R.A.; Goadsby, P.J. Small-molecule CGRP receptor antagonists: A new approach to the acute and preventive treatment of migraine. Med. Drug Discov. 2020, 7, 100053. [Google Scholar] [CrossRef]
- Karuna Therapeutics. Pipline. 2020. Available online: https://karunatx.com/programs/ (accessed on 12 October 2020).
- Alzheimer’s Association. Facts and Figures. Available online: https://www.alz.org/alzheimers-dementia/facts-figures (accessed on 12 September 2020).
- Parkinson’s Foundation. Statistics. Available online: https://www.parkinson.org/Understanding-Parkinsons/Statistics (accessed on 12 September 2020).
- Alzheimer’s Disease International; Patterson, C. World Alzheimer Report 2018. The State of the Art of Dementia Research: New Frontiers. 2018. Available online: https://www.alzint.org/resource/world-alzheimer-report-2018/ (accessed on 13 September 2020).
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10 (Suppl. S1), S10–S17. [Google Scholar] [CrossRef]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Jucker, M.; Walker, L.C. Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s, A. 2016 Alzheimer’s disease facts and figures. Alzheimers Dement. 2016, 12, 459–509. [Google Scholar] [CrossRef]
- Smith, A.D. Imaging the progression of Alzheimer pathology through the brain. Proc. Natl. Acad. Sci. USA 2002, 99, 4135–4137. [Google Scholar] [CrossRef] [PubMed]
- Hebert, L.E.; Scherr, P.A.; Bienias, J.L.; Bennett, D.A.; Evans, D.A. Alzheimer disease in the US population: Prevalence estimates using the 2000 census. Arch. Neurol. 2003, 60, 1119–1122. [Google Scholar] [CrossRef] [PubMed]
- Mayo Clinic. Alzheimer’s Stages: How the Disease Progresses. Available online: https://www.mayoclinic.org/diseases-conditions/alzheimers-disease/in-depth/alzheimers-stages/art-20048448#:~:text=There%20are%20five%20stages%20associated,dementia%20due%20to%20Alzheimer’s%20disease (accessed on 11 October 2020).
- Alzheimer’s Association. Alzheimer’s Disease Facts and Figures. 2020. Available online: https://www.alz.org/media/Documents/facts2020_report_1.pdf (accessed on 1 October 2020).
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.M.; Mandelkow, E. Tau in Alzheimer’s disease. Trends Cell Biol. 1998, 8, 425–427. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Malek-Ahmadi, M.; Perez, S.E.; Chen, K.; Mufson, E.J. Braak Stage, Cerebral Amyloid Angiopathy, and Cognitive Decline in Early Alzheimer’s Disease. J. Alzheimers Dis. 2020, 74, 189–197. [Google Scholar] [CrossRef]
- Markesbery, W.R. Neuropathologic alterations in mild cognitive impairment: A review. J. Alzheimers Dis. 2010, 19, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Gold, G.; Kovari, E.; Corte, G.; Herrmann, F.R.; Canuto, A.; Bussiere, T.; Hof, P.R.; Bouras, C.; Giannakopoulos, P. Clinical validity of A beta-protein deposition staging in brain aging and Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 946–952. [Google Scholar] [CrossRef]
- Sarter, M.; Bruno, J.P. Cognitive functions of cortical acetylcholine: Toward a unifying hypothesis. Brain Res. Brain Res. Rev. 1997, 23, 28–46. [Google Scholar] [CrossRef]
- Hakansson, L. Mechanism of action of cholinesterase inhibitors in Alzheimer’s disease. Acta Neurol. Scand. Suppl. 1993, 149, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Singh, B. A review on cholinesterase inhibitors for Alzheimer’s disease. Arch. Pharm. Res. 2013, 36, 375–399. [Google Scholar] [CrossRef] [PubMed]
- Lagadic-Gossmann, D.; Rissel, M.; Le Bot, M.A.; Guillouzo, A. Toxic effects of tacrine on primary hepatocytes and liver epithelial cells in culture. Cell Biol. Toxicol. 1998, 14, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Bullock, R.; Touchon, J.; Bergman, H.; Gambina, G.; He, Y.; Rapatz, G.; Nagel, J.; Lane, R. Rivastigmine and donepezil treatment in moderate to moderately-severe Alzheimer’s disease over a 2-year period. Curr. Med. Res. Opin. 2005, 21, 1317–1327. [Google Scholar] [CrossRef]
- Wilcock, G.; Howe, I.; Coles, H.; Lilienfeld, S.; Truyen, L.; Zhu, Y.; Bullock, R.; Kershaw, P.; Group, G.-G.-S. A long-term comparison of galantamine and donepezil in the treatment of Alzheimer’s disease. Drugs Aging 2003, 20, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.W.; Kotermanski, S.E. Mechanism of action of memantine. Curr. Opin. Pharmacol. 2006, 6, 61–67. [Google Scholar] [CrossRef]
- Gauthier, S.; Ng, K.P.; Pascoal, T.A.; Zhang, H.; Rosa-Neto, P. Targeting Alzheimer’s Disease at the Right Time and the Right Place: Validation of a Personalized Approach to Diagnosis and Treatment. J. Alzheimers Dis. 2018, 64, S23–S31. [Google Scholar] [CrossRef] [PubMed]
- Kurz, A. The therapeutic potential of tacrine. J. Neural. Transm. Suppl. 1998, 54, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Seltzer, B. Donepezil: A review. Expert Opin. Drug Metab. Toxicol. 2005, 1, 527–536. [Google Scholar] [CrossRef]
- Onor, M.L.; Trevisiol, M.; Aguglia, E. Rivastigmine in the treatment of Alzheimer’s disease: An update. Clin. Interv. Aging 2007, 2, 17–32. [Google Scholar] [CrossRef]
- Razay, G.; Wilcock, G.K. Galantamine in Alzheimer’s disease. Expert Rev. Neurother. 2008, 8, 9–17. [Google Scholar] [CrossRef]
- Van Marum, R.J. Update on the use of memantine in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2009, 5, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Selkoe, D.J. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498. [Google Scholar] [CrossRef]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2013, 6, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, T.; Bieger, S.C.; Bruhl, B.; Tienari, P.J.; Ida, N.; Allsop, D.; Roberts, G.W.; Masters, C.L.; Dotti, C.G.; Unsicker, K.; et al. Distinct sites of intracellular production for Alzheimer’s disease A beta40/42 amyloid peptides. Nat. Med. 1997, 3, 1016–1020. [Google Scholar] [CrossRef]
- Atri, A. Current and Future Treatments in Alzheimer’s Disease. Semin. Neurol. 2019, 39, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2019. Alzheimers Dement. 2019, 5, 272–293. [Google Scholar] [CrossRef]
- Biogen and Eisai to Discontinue Phase 3 ENGAGE and EMERGE Trials of Aducanumab in Alzheimer’s Disease. 2019. Available online: https://investors.biogen.com/news-releases/news-release-details/biogen-and-eisai-discontinue-phase-3-engage-and-emerge-trials (accessed on 22 September 2020).
- Vandenberghe, R.; Rinne, J.O.; Boada, M.; Katayama, S.; Scheltens, P.; Vellas, B.; Tuchman, M.; Gass, A.; Fiebach, J.B.; Hill, D.; et al. Bapineuzumab for mild to moderate Alzheimer’s disease in two global, randomized, phase 3 trials. Alzheimers Res. Ther. 2016, 8, 1–13. [Google Scholar] [CrossRef]
- Roche. Roche to Discontinue Phase III CREAD 1 and 2 Clinical Studies of Crenezumab in Early Alzheimer’s Disease (AD)—Other Company Programmes in AD Continue. 2019. Available online: https://www.roche.com/media/releases/med-cor-2019-01-30.htm (accessed on 10 October 2020).
- Lancet Neurology. Solanezumab: Too late in mild Alzheimer’s disease? Lancet Neurol. 2017, 16, 97. [Google Scholar] [CrossRef]
- Henley, D.; Raghavan, N.; Sperling, R.; Aisen, P.; Raman, R.; Romano, G. Preliminary Results of a Trial of Atabecestat in Preclinical Alzheimer’s Disease. N. Engl. J. Med. 2019, 380, 1483–1485. [Google Scholar] [CrossRef] [PubMed]
- Burki, T. Alzheimer’s disease research: The future of BACE inhibitors. Lancet 2018, 391, 2486. [Google Scholar] [CrossRef]
- Mullard, A. Alzheimer prevention failure rattles field, anew. Nat. Rev. Drug Discov. 2019, 18, 656–657. [Google Scholar] [CrossRef]
- Egan, M.F.; Kost, J.; Voss, T.; Mukai, Y.; Aisen, P.S.; Cummings, J.L.; Tariot, P.N.; Vellas, B.; van Dyck, C.H.; Boada, M.; et al. Randomized Trial of Verubecestat for Prodromal Alzheimer’s Disease. N. Engl. J. Med. 2019, 380, 1408–1420. [Google Scholar] [CrossRef]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Initiation of Phase III Clinical Trial of BAN2401 in Early Alzheimer’s Disease. 2019. Available online: https://www.eisai.com/news;https://www.eisai.com/news/2019/news201919.html (accessed on 22 September 2020).
- Klein, G.; Delmar, P.; Voyle, N.; Rehal, S.; Hofmann, C.; Abi-Saab, D.; Andjelkovic, M.; Ristic, S.; Wang, G.; Bateman, R.; et al. Gantenerumab reduces amyloid-beta plaques in patients with prodromal to moderate Alzheimer’s disease: A PET substudy interim analysis. Alzheimers Res. Ther. 2019, 11, 1–12. [Google Scholar] [CrossRef]
- Biogen. FDA Accepts Biogen’s Aducanumab Biologics License Application for Alzheimer’s Disease with Priority Review. Available online: http://media.biogen.com/news-releases/news-release-details/fda-accepts-biogens-aducanumab-biologics-license-application (accessed on 21 November 2020).
- Gauthier, S.; Albert, M.; Fox, N.; Goedert, M.; Kivipelto, M.; Mestre-Ferrandiz, J.; Middleton, L.T. Why has therapy development for dementia failed in the last two decades? Alzheimers Dement. 2016, 12, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Jackson, R.; Paul, G.; Shi, J.; Sabbagh, M. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010-2015. Expert Opin. Investig. Drugs 2017, 26, 735–739. [Google Scholar] [CrossRef]
- Pardridge, W.M. Alzheimer’s disease: Future drug development and the blood-brain barrier. Expert Opin. Investig. Drugs 2019, 28, 569–572. [Google Scholar] [CrossRef]
- Pardridge, W.M. Blood-Brain Barrier and Delivery of Protein and Gene Therapeutics to Brain. Front. Aging Neurosci. 2019, 11, 373. [Google Scholar] [CrossRef]
- Drummond, E.; Wisniewski, T. Alzheimer’s disease: Experimental models and reality. Acta Neuropathol. 2017, 133, 155–175. [Google Scholar] [CrossRef] [PubMed]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 1–18. [Google Scholar] [CrossRef]
- Wang, C.Y.; Wang, P.N.; Chiu, M.J.; Finstad, C.L.; Lin, F.; Lynn, S.; Tai, Y.H.; De Fang, X.; Zhao, K.; Hung, C.H.; et al. UB-311, a novel UBITh((R)) amyloid beta peptide vaccine for mild Alzheimer’s disease. Alzheimers Dement. 2017, 3, 262–272. [Google Scholar] [CrossRef]
- Inacio, P. UB-311 Vaccine Safe in Mild Alzheimer’s Patients, Phase 2a Trial Shows. 2019. Available online: https://alzheimersnewstoday.com/2019/04/03/ub-311-vaccine-safe-mild-alzheimers-patients-phase-2-study/ (accessed on 2 September 2020).
- Salloway, S.P.; Sevingy, J.; Budur, K.; Pederson, J.T.; DeMattos, R.B.; Von Rosenstiel, P.; Paez, A.; Evans, R.; Weber, C.J.; Hendrix, J.A.; et al. Advancing combination therapy for Alzheimer’s disease. Alzheimers Dement. 2020, 6, e12073. [Google Scholar] [CrossRef] [PubMed]
- Sasaguri, H.; Nilsson, P.; Hashimoto, S.; Nagata, K.; Saito, T.; De Strooper, B.; Hardy, J.; Vassar, R.; Winblad, B.; Saido, T.C. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 2017, 36, 2473–2487. [Google Scholar] [CrossRef]
- Pardridge, W.M. Re-engineering therapeutic antibodies for Alzheimer’s disease as blood-brain barrier penetrating bi-specific antibodies. Expert Opin. Biol. Ther. 2016, 16, 1455–1468. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Targeted delivery of protein and gene medicines through the blood-brain barrier. Clin. Pharmacol. Ther. 2015, 97, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Weber, F.; Bohrmann, B.; Niewoehner, J.; Fischer, J.A.A.; Rueger, P.; Tiefenthaler, G.; Moelleken, J.; Bujotzek, A.; Brady, K.; Singer, T.; et al. Brain Shuttle Antibody for Alzheimer’s Disease with Attenuated Peripheral Effector Function due to an Inverted Binding Mode. Cell Rep. 2018, 22, 149–162. [Google Scholar] [CrossRef]
- Golde, T.E. Open questions for Alzheimer’s disease immunotherapy. Alzheimers Res. Ther. 2014, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kingston, A.; Comas-Herrera, A.; Jagger, C.; MODEM Project. Forecasting the care needs of the older population in England over the next 20 years: Estimates from the Population Ageing and Care Simulation (PACSim) modelling study. Lancet Public Health 2018, 3, e447–e455. [Google Scholar] [CrossRef]
- Gauthier, S.; Feldman, H.H.; Schneider, L.S.; Wilcock, G.K.; Frisoni, G.B.; Hardlund, J.H.; Moebius, H.J.; Bentham, P.; Kook, K.A.; Wischik, D.J.; et al. Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: A randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet 2016, 388, 2873–2884. [Google Scholar] [CrossRef]
- Wilcock, G.K.; Gauthier, S.; Frisoni, G.B.; Jia, J.; Hardlund, J.H.; Moebius, H.J.; Bentham, P.; Kook, K.A.; Schelter, B.O.; Wischik, D.J.; et al. Potential of Low Dose Leuco-Methylthioninium Bis(Hydromethanesulphonate) (LMTM) Monotherapy for Treatment of Mild Alzheimer’s Disease: Cohort Analysis as Modified Primary Outcome in a Phase III Clinical Trial. J. Alzheimers Dis. 2018, 61, 435–457. [Google Scholar] [CrossRef] [PubMed]
- Billingsley, M.L.; Kincaid, R.L. Regulated phosphorylation and dephosphorylation of tau protein: Effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochem J. 1997, 323 Pt 3, 577–591. [Google Scholar] [CrossRef]
- Buee, L.; Bussiere, T.; Buee-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef]
- Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Hart, G.W.; Gong, C.X. O-GlcNAcylation regulates phosphorylation of tau: A mechanism involved in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 10804–10809. [Google Scholar] [CrossRef] [PubMed]
- Yuzwa, S.A.; Shan, X.; Macauley, M.S.; Clark, T.; Skorobogatko, Y.; Vosseller, K.; Vocadlo, D.J. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat. Chem. Biol. 2012, 8, 393–399. [Google Scholar] [CrossRef]
- Yuzwa, S.A.; Cheung, A.H.; Okon, M.; McIntosh, L.P.; Vocadlo, D.J. O-GlcNAc modification of tau directly inhibits its aggregation without perturbing the conformational properties of tau monomers. J. Mol. Biol. 2014, 426, 1736–1752. [Google Scholar] [CrossRef]
- Duits, F.H.; Martinez-Lage, P.; Paquet, C.; Engelborghs, S.; Lleo, A.; Hausner, L.; Molinuevo, J.L.; Stomrud, E.; Farotti, L.; Ramakers, I.; et al. Performance and complications of lumbar puncture in memory clinics: Results of the multicenter lumbar puncture feasibility study. Alzheimers Dement. 2016, 12, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Wittenberg, R.; Knapp, M.; Karagiannidou, M.; Dickson, J.; Schott, J. Economic impacts of introducing diagnostics for mild cognitive impairment Alzheimer’s disease patients. Alzheimers Dement. 2019, 5, 382–387. [Google Scholar] [CrossRef]
- Janelidze, S.; Mattsson, N.; Palmqvist, S.; Smith, R.; Beach, T.G.; Serrano, G.E.; Chai, X.; Proctor, N.K.; Eichenlaub, U.; Zetterberg, H.; et al. Plasma P-tau181 in Alzheimer’s disease: Relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat. Med. 2020, 26, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Palmqvist, S.; Janelidze, S.; Quiroz, Y.T.; Zetterberg, H.; Lopera, F.; Stomrud, E.; Su, Y.; Chen, Y.; Serrano, G.E.; Leuzy, A.; et al. Discriminative Accuracy of Plasma Phospho-tau217 for Alzheimer Disease vs. Other Neurodegenerative Disorders. JAMA 2020, 324, 772–781. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s disease. J. Clin. Investig. 2017, 127, 3240–3249. [Google Scholar] [CrossRef]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef]
- Zhang, C.; Griciuc, A.; Hudry, E.; Wan, Y.; Quinti, L.; Ward, J.; Forte, A.M.; Shen, X.; Ran, C.; Elmaleh, D.R.; et al. Cromolyn Reduces Levels of the Alzheimer’s Disease-Associated Amyloid beta-Protein by Promoting Microglial Phagocytosis. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef]
- Vlad, S.C.; Miller, D.R.; Kowall, N.W.; Felson, D.T. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology 2008, 70, 1672–1677. [Google Scholar] [CrossRef] [PubMed]
- AZTherapies. Advancing A Broad Pipeline Targeting Neurodegenerative Disease. Available online: https://aztherapies.com/pipeline/ (accessed on 9 November 2020).
- Cai, Z.; Liu, N.; Wang, C.; Qin, B.; Zhou, Y.; Xiao, M.; Chang, L.; Yan, L.J.; Zhao, B. Role of RAGE in Alzheimer’s Disease. Cell Mol. Neurobiol. 2016, 36, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Lue, L.F.; Yan, S.D.; Stern, D.M.; Walker, D.G. Preventing activation of receptor for advanced glycation endproducts in Alzheimer’s disease. Curr. Drug Targets CNS Neurol. Disord. 2005, 4, 249–266. [Google Scholar] [CrossRef]
- Burstein, A.H.; Sabbagh, M.; Andrews, R.; Valcarce, C.; Dunn, I.; Altstiel, L. Development of Azeliragon, an Oral Small Molecule Antagonist of the Receptor for Advanced Glycation Endproducts, for the Potential Slowing of Loss of Cognition in Mild Alzheimer’s Disease. J. Prev. Alzheimers Dis. 2018, 5, 149–154. [Google Scholar] [CrossRef]
- Folch, J.; Petrov, D.; Ettcheto, M.; Pedros, I.; Abad, S.; Beas-Zarate, C.; Lazarowski, A.; Marin, M.; Olloquequi, J.; Auladell, C.; et al. Masitinib for the treatment of mild to moderate Alzheimer’s disease. Expert Rev. Neurother. 2015, 15, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.B.; Siman, R.; Iqbal, M.A.; Potter, H. Identification of a chymotrypsin-like mast cell protease in rat brain capable of generating the N-terminus of the Alzheimer amyloid beta-protein. J. Neurochem. 1993, 61, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Tengfei Lia, E.M.; Abada, Y.; Boucher, C.; Ces, A.; Youssef, I.; Fenaux, G.; Forand, Y.; Legrand, A.; Nachiket, N.; Dhenain, M.; et al. Effects of Chronic Masitinib Treatment in APPswe/PSEN1dE9 Transgenic Mice Modeling Alzheimer’s Disease. J. Alzheimers Dis. 2020, 76, 1339–1345. [Google Scholar]
- Piette, F.; Belmin, J.; Vincent, H.; Schmidt, N.; Pariel, S.; Verny, M.; Marquis, C.; Mely, J.; Hugonot-Diener, L.; Kinet, J.P.; et al. Masitinib as an adjunct therapy for mild-to-moderate Alzheimer’s disease: A randomised, placebo-controlled phase 2 trial. Alzheimers Res. Ther. 2011, 3, 1–11. [Google Scholar] [CrossRef]
- Minter, M.R.; Zhang, C.; Leone, V.; Ringus, D.L.; Zhang, X.; Oyler-Castrillo, P.; Musch, M.W.; Liao, F.; Ward, J.F.; Holtzman, D.M.; et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sun, G.; Feng, T.; Zhang, J.; Huang, X.; Wang, T.; Xie, Z.; Chu, X.; Yang, J.; Wang, H.; et al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 2019, 29, 787–803. [Google Scholar] [CrossRef]
- NIH U.S. National Library of Medicine. A Study of Sodium Oligomannate (GV-971) in Participants With Mild to Moderate Alzheimer’s Disease (GREEN MEMORY). 2020. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04520412 (accessed on 12 November 2020).
- Zheng, H.; Cheng, B.; Li, Y.; Li, X.; Chen, X.; Zhang, Y.W. TREM2 in Alzheimer’s Disease: Microglial Survival and Energy Metabolism. Front. Aging Neurosci. 2018, 10, 395. [Google Scholar] [CrossRef]
- Wang, S.; Mustafa, M.; Yuede, C.M.; Salazar, S.V.; Kong, P.; Long, H.; Ward, M.; Siddiqui, O.; Paul, R.; Gilfillan, S.; et al. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef]
- Steed, P.M.; Tansey, M.G.; Zalevsky, J.; Zhukovsky, E.A.; Desjarlais, J.R.; Szymkowski, D.E.; Abbott, C.; Carmichael, D.; Chan, C.; Cherry, L.; et al. Inactivation of TNF signaling by rationally designed dominant-negative TNF variants. Science 2003, 301, 1895–1898. [Google Scholar] [CrossRef]
- Sama, D.M.; Mohmmad Abdul, H.; Furman, J.L.; Artiushin, I.A.; Szymkowski, D.E.; Scheff, S.W.; Norris, C.M. Inhibition of soluble tumor necrosis factor ameliorates synaptic alterations and Ca2+ dysregulation in aged rats. PLoS ONE 2012, 7, e38170. [Google Scholar] [CrossRef]
- McAlpine, F.E.; Lee, J.K.; Harms, A.S.; Ruhn, K.A.; Blurton-Jones, M.; Hong, J.; Das, P.; Golde, T.E.; LaFerla, F.M.; Oddo, S.; et al. Inhibition of soluble TNF signaling in a mouse model of Alzheimer’s disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol. Dis. 2009, 34, 163–177. [Google Scholar] [CrossRef]
- Cavanagh, C.; Tse, Y.C.; Nguyen, H.B.; Krantic, S.; Breitner, J.C.; Quirion, R.; Wong, T.P. Inhibiting tumor necrosis factor-alpha before amyloidosis prevents synaptic deficits in an Alzheimer’s disease model. Neurobiol. Aging 2016, 47, 41–49. [Google Scholar] [CrossRef] [PubMed]
- MacPherson, K.P.; Sompol, P.; Kannarkat, G.T.; Chang, J.; Sniffen, L.; Wildner, M.E.; Norris, C.M.; Tansey, M.G. Peripheral administration of the soluble TNF inhibitor XPro1595 modifies brain immune cell profiles, decreases beta-amyloid plaque load, and rescues impaired long-term potentiation in 5xFAD mice. Neurobiol. Dis. 2017, 102, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Aziz, N.A.; van der Marck, M.A.; Pijl, H.; Olde Rikkert, M.G.; Bloem, B.R.; Roos, R.A. Weight loss in neurodegenerative disorders. J. Neurol. 2008, 255, 1872–1880. [Google Scholar] [CrossRef] [PubMed]
- Papapetropoulos, S.; Ellul, J.; Argyriou, A.A.; Talelli, P.; Chroni, E.; Papapetropoulos, T. The effect of vascular disease on late onset Parkinson’s disease. Eur. J. Neurol. 2004, 11, 231–235. [Google Scholar] [CrossRef]
- Fukumoto, H.; Cheung, B.S.; Hyman, B.T.; Irizarry, M.C. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch. Neurol. 2002, 59, 1381–1389. [Google Scholar] [CrossRef]
- Li, R.; Lindholm, K.; Yang, L.B.; Yue, X.; Citron, M.; Yan, R.; Beach, T.; Sue, L.; Sabbagh, M.; Cai, H.; et al. Amyloid beta peptide load is correlated with increased beta-secretase activity in sporadic Alzheimer’s disease patients. Proc. Natl. Acad. Sci. USA 2004, 101, 3632–3637. [Google Scholar] [CrossRef]
- Meakin, P.J.; Harper, A.J.; Hamilton, D.L.; Gallagher, J.; McNeilly, A.D.; Burgess, L.A.; Vaanholt, L.M.; Bannon, K.A.; Latcham, J.; Hussain, I.; et al. Reduction in BACE1 decreases body weight, protects against diet-induced obesity and enhances insulin sensitivity in mice. Biochem. J. 2012, 441, 285–296. [Google Scholar] [CrossRef]
- Thirumangalakudi, L.; Prakasam, A.; Zhang, R.; Bimonte-Nelson, H.; Sambamurti, K.; Kindy, M.S.; Bhat, N.R. High cholesterol-induced neuroinflammation and amyloid precursor protein processing correlate with loss of working memory in mice. J. Neurochem. 2008, 106, 475–485. [Google Scholar] [CrossRef]
- Ng, R.C.; Jian, M.; Ma, O.K.; Bunting, M.; Kwan, J.S.; Zhou, G.J.; Senthilkumar, K.; Iyaswamy, A.; Chan, P.K.; Li, M.; et al. Chronic oral administration of adipoRon reverses cognitive impairments and ameliorates neuropathology in an Alzheimer’s disease mouse model. Mol. Psychiatry 2020, 1–21. [Google Scholar] [CrossRef]
- Liu, B.; Liu, J.; Wang, J.G.; Liu, C.L.; Yan, H.J. AdipoRon improves cognitive dysfunction of Alzheimer’s disease and rescues impaired neural stem cell proliferation through AdipoR1/AMPK pathway. Exp. Neurol. 2020, 327, 113249. [Google Scholar] [CrossRef]
- De la Monte, S.M.; Wands, J.R. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar] [CrossRef] [PubMed]
- Kandimalla, R.; Thirumala, V.; Reddy, P.H. Is Alzheimer’s disease a Type 3 Diabetes? A critical appraisal. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1078–1089. [Google Scholar] [CrossRef]
- Clarke, D.W.; Boyd, F.T., Jr.; Kappy, M.S.; Raizada, M.K. Insulin binds to specific receptors and stimulates 2-deoxy-D-glucose uptake in cultured glial cells from rat brain. J. Biol. Chem. 1984, 259, 11672–11675. [Google Scholar] [CrossRef]
- Raizada, M.K.; Shemer, J.; Judkins, J.H.; Clarke, D.W.; Masters, B.A.; LeRoith, D. Insulin receptors in the brain: Structural and physiological characterization. Neurochem. Res. 1988, 13, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Kellar, D.; Craft, S. Brain insulin resistance in Alzheimer’s disease and related disorders: Mechanisms and therapeutic approaches. Lancet Neurol. 2020, 19, 758–766. [Google Scholar] [CrossRef]
- Born, J.; Lange, T.; Kern, W.; McGregor, G.P.; Bickel, U.; Fehm, H.L. Sniffing neuropeptides: A transnasal approach to the human brain. Nat. Neurosci. 2002, 5, 514–516. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Tramutola, A.; Triani, F.; Calcagnini, S.; Di Domenico, F.; Ripoli, C.; Gaetani, S.; Grassi, C.; Butterfield, D.A.; Cassano, T.; et al. Biliverdin Reductase-A Mediates the Beneficial Effects of Intranasal Insulin in Alzheimer Disease. Mol. Neurobiol. 2019, 56, 2922–2943. [Google Scholar] [CrossRef]
- Claxton, A.; Baker, L.D.; Hanson, A.; Trittschuh, E.H.; Cholerton, B.; Morgan, A.; Callaghan, M.; Arbuckle, M.; Behl, C.; Craft, S. Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer’s disease dementia. J. Alzheimers Dis. 2015, 44, 897–906. [Google Scholar] [CrossRef]
- Craft, S.; Claxton, A.; Baker, L.D.; Hanson, A.J.; Cholerton, B.; Trittschuh, E.H.; Dahl, D.; Caulder, E.; Neth, B.; Montine, T.J.; et al. Effects of Regular and Long-Acting Insulin on Cognition and Alzheimer’s Disease Biomarkers: A Pilot Clinical Trial. J. Alzheimers Dis. 2017, 57, 1325–1334. [Google Scholar] [CrossRef]
- Craft, S.; Baker, L.D.; Montine, T.J.; Minoshima, S.; Watson, G.S.; Claxton, A.; Arbuckle, M.; Callaghan, M.; Tsai, E.; Plymate, S.R.; et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: A pilot clinical trial. Arch. Neurol. 2012, 69, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Raman, R.; Chow, T.W.; Rafii, M.S.; Sun, C.K.; Rissman, R.A.; Donohue, M.C.; Brewer, J.B.; Jenkins, C.; Harless, K.; et al. Safety, Efficacy, and Feasibility of Intranasal Insulin for the Treatment of Mild Cognitive Impairment and Alzheimer Disease Dementia: A Randomized Clinical Trial. JAMA Neurol. 2020, 77, 1099–1109. [Google Scholar] [CrossRef]
- Kirpichnikov, D.; McFarlane, S.I.; Sowers, J.R. Metformin: An update. Ann. Intern. Med. 2002, 137, 25–33. [Google Scholar] [CrossRef]
- Craig, A.; Parvez, F.; Issberner, J. A systematic literature review of the effect of insulin sensitizers on the cognitive symptoms of Alzheimer’s Disease in transgenic mice. Behav. Brain Res. 2019, 372, 112015. [Google Scholar] [CrossRef]
- Luchsinger, J.A.; Perez, T.; Chang, H.; Mehta, P.; Steffener, J.; Pradabhan, G.; Ichise, M.; Manly, J.; Devanand, D.P.; Bagiella, E. Metformin in Amnestic Mild Cognitive Impairment: Results of a Pilot Randomized Placebo Controlled Clinical Trial. J. Alzheimers Dis. 2016, 51, 501–514. [Google Scholar] [CrossRef]
- Batista, A.F.; Forny-Germano, L.; Clarke, J.R.; Lyra, E.S.N.M.; Brito-Moreira, J.; Boehnke, S.E.; Winterborn, A.; Coe, B.C.; Lablans, A.; Vital, J.F.; et al. The diabetes drug liraglutide reverses cognitive impairment in mice and attenuates insulin receptor and synaptic pathology in a non-human primate model of Alzheimer’s disease. J. Pathol. 2018, 245, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Perna, S.; Mainardi, M.; Astrone, P.; Gozzer, C.; Biava, A.; Bacchio, R.; Spadaccini, D.; Solerte, S.B.; Rondanelli, M. 12-month effects of incretins versus SGLT2-Inhibitors on cognitive performance and metabolic profile. A randomized clinical trial in the elderly with Type-2 diabetes mellitus. Clin. Pharmacol. 2018, 10, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Michalik, L.; Auwerx, J.; Berger, J.P.; Chatterjee, V.K.; Glass, C.K.; Gonzalez, F.J.; Grimaldi, P.A.; Kadowaki, T.; Lazar, M.A.; O’Rahilly, S.; et al. International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol. Rev. 2006, 58, 726–741. [Google Scholar] [CrossRef]
- D’Orio, B.; Fracassi, A.; Ceru, M.P.; Moreno, S. Targeting PPARalpha in Alzheimer’s Disease. Curr. Alzheimer Res. 2018, 15, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Reyes-Irisarri, E.; Hull, M.; Kummer, M.P. Impact and Therapeutic Potential of PPARs in Alzheimer’s Disease. Curr. Neuropharmacol. 2011, 9, 643–650. [Google Scholar] [CrossRef]
- Kumar, V.; Jahan, S.; Singh, S.; Khanna, V.K.; Pant, A.B. Progress toward the development of in vitro model system for chemical-induced developmental neurotoxicity: Potential applicability of stem cells. Arch. Toxicol. 2015, 89, 265–267. [Google Scholar] [CrossRef]
- Liu, Y.; Weick, J.P.; Liu, H.; Krencik, R.; Zhang, X.; Ma, L.; Zhou, G.M.; Ayala, M.; Zhang, S.C. Medial ganglionic eminence-like cells derived from human embryonic stem cells correct learning and memory deficits. Nat. Biotechnol. 2013, 31, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Lunn, J.S.; Sakowski, S.A.; Hur, J.; Feldman, E.L. Stem cell technology for neurodegenerative diseases. Ann. Neurol. 2011, 70, 353–361. [Google Scholar] [CrossRef]
- Choi, S.S.; Lee, S.R.; Kim, S.U.; Lee, H.J. Alzheimer’s disease and stem cell therapy. Exp. Neurobiol. 2014, 23, 45–52. [Google Scholar] [CrossRef]
- Medipost. NEUROSTEM. Available online: http://www.medi-post.com/neurostem/ (accessed on 13 September 2020).
- Stemedica. Stemedica Begins First Clinical Trials In The U.S. Using Adult Allogeneic Stem Cells To Treat Alzheimer’s Disease. 2016. Available online: https://www.stemedica.com/stemedica-begins-first-clinical-trials-in-the-u-s-using-adult-allogeneic-stem-cells-to-treat-alzheimers-disease/ (accessed on 10 August 2020).
- Longeveron. Open Clinical Trials: Allogeneic Human Mesenchymal Stem Cell Infusion Versus Placebo in Patients With Alzheimer’s Disease. Available online: http://longeveron.com/clinical-trials/ (accessed on 11 October 2020).
- Klug, B.; Celis, P.; Carr, M.; Reinhardt, J. Regulatory structures for gene therapy medicinal products in the European Union. Methods Enzymol. 2012, 507, 337–354. [Google Scholar] [CrossRef]
- Loera-Valencia, R.; Piras, A.; Ismail, M.A.M.; Manchanda, S.; Eyjolfsdottir, H.; Saido, T.C.; Johansson, J.; Eriksdotter, M.; Winblad, B.; Nilsson, P. Targeting Alzheimer’s disease with gene and cell therapies. J. Intern. Med. 2018, 284, 2–36. [Google Scholar] [CrossRef] [PubMed]
- Penaud-Budloo, M.; Francois, A.; Clement, N.; Ayuso, E. Pharmacology of Recombinant Adeno-associated Virus Production. Mol. Ther. Methods Clin. Dev. 2018, 8, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Scoles, D.R.; Minikel, E.V.; Pulst, S.M. Antisense oligonucleotides: A primer. Neurol. Genet. 2019, 5, e323. [Google Scholar] [CrossRef]
- Rafii, M.S.; Baumann, T.L.; Bakay, R.A.; Ostrove, J.M.; Siffert, J.; Fleisher, A.S.; Herzog, C.D.; Barba, D.; Pay, M.; Salmon, D.P.; et al. A phase1 study of stereotactic gene delivery of AAV2-NGF for Alzheimer’s disease. Alzheimers Dement. 2014, 10, 571–581. [Google Scholar] [CrossRef]
- Fischer, W.; Wictorin, K.; Bjorklund, A.; Williams, L.R.; Varon, S.; Gage, F.H. Amelioration of cholinergic neuron atrophy and spatial memory impairment in aged rats by nerve growth factor. Nature 1987, 329, 65–68. [Google Scholar] [CrossRef]
- Ionis. Ionis Innovation Pipeline. Available online: https://www.ionispharma.com/ionis-innovation/pipeline/ (accessed on 14 October 2020).
- Rosenberg, J.B.; Kaplitt, M.G.; De, B.P.; Chen, A.; Flagiello, T.; Salami, C.; Pey, E.; Zhao, L.; Ricart Arbona, R.J.; Monette, S.; et al. AAVrh.10-Mediated APOE2 Central Nervous System Gene Therapy for APOE4-Associated Alzheimer’s Disease. Hum. Gene Ther. Clin. Dev. 2018, 29, 24–47. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Lane, H.Y.; Lin, C.H. Brain Stimulation in Alzheimer’s Disease. Front. Psychiatry 2018, 9, 201. [Google Scholar] [CrossRef]
- Yu, D.; Yan, H.; Zhou, J.; Yang, X.; Lu, Y.; Han, Y. A circuit view of deep brain stimulation in Alzheimer’s disease and the possible mechanisms. Mol. Neurodegener. 2019, 14, 1–12. [Google Scholar] [CrossRef]
- Tarsy, D. Treatment of Parkinson disease: A 64-year-old man with motor complications of advanced Parkinson disease. JAMA 2012, 307, 2305–2314. [Google Scholar] [CrossRef] [PubMed]
- Antony, P.M.; Diederich, N.J.; Kruger, R.; Balling, R. The hallmarks of Parkinson’s disease. FEBS J. 2013, 280, 5981–5993. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Parkinson’s Foundation. Understanding Parkinson’s. Available online: https://www.parkinson.org/understanding-parkinsons/what-is-parkinsons (accessed on 8 September 2020).
- Goedert, M. NEURODEGENERATION. Alzheimer’s and Parkinson’s Diseases: The prion concept in relation to assembled Abeta, tau, and alpha-synuclein. Science 2015, 349. [Google Scholar] [CrossRef] [PubMed]
- Schulz-Schaeffer, W.J. The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol. 2010, 120, 131–143. [Google Scholar] [CrossRef]
- Davie, C.A. A review of Parkinson’s disease. Br. Med. Bull. 2008, 86, 109–127. [Google Scholar] [CrossRef]
- Mahul-Mellier, A.L.; Burtscher, J.; Maharjan, N.; Weerens, L.; Croisier, M.; Kuttler, F.; Leleu, M.; Knott, G.W.; Lashuel, H.A. The process of Lewy body formation, rather than simply alpha-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 4971–4982. [Google Scholar] [CrossRef]
- Goedert, M. Parkinson’s disease and other alpha-synucleinopathies. Clin. Chem. Lab. Med. 2001, 39, 308–312. [Google Scholar] [CrossRef]
- Sonne, J.; Goyal, A.; Bansal, P.; Lopez-Ojeda, W. Dopamine. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Boshes, B. Sinemet and the treatment of Parkinsonism. Ann. Intern. Med. 1981, 94, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Thanvi, B.R.; Lo, T.C. Long term motor complications of levodopa: Clinical features, mechanisms, and management strategies. Postgrad. Med. J. 2004, 80, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Borovac, J.A. Side effects of a dopamine agonist therapy for Parkinson’s disease: A mini-review of clinical pharmacology. Yale J. Biol. Med. 2016, 89, 37–47. [Google Scholar]
- Clarke, C.E.; Guttman, M. Dopamine agonist monotherapy in Parkinson’s disease. Lancet 2002, 360, 1767–1769. [Google Scholar] [CrossRef]
- MAO-B inhibitors for the treatment of Parkinson’s disease. Mov. Disord. 2002, 17 (Suppl. S4), S38–S44. [CrossRef]
- Kaakkola, S. Clinical pharmacology, therapeutic use and potential of COMT inhibitors in Parkinson’s disease. Drugs 2000, 59, 1233–1250. [Google Scholar] [CrossRef] [PubMed]
- Connolly, B.S.; Lang, A.E. Pharmacological treatment of Parkinson disease: A review. JAMA 2014, 311, 1670–1683. [Google Scholar] [CrossRef]
- Blanpied, T.A.; Clarke, R.J.; Johnson, J.W. Amantadine inhibits NMDA receptors by accelerating channel closure during channel block. J. Neurosci. 2005, 25, 3312–3322. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, S.; Fu, P.; Zhang, Z.; Lin, K.; Ko, J.K.; Yung, K.K. Roles of Glutamate Receptors in Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 4391. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, Y.; Kondo, T.; Japanese Istradefylline Study Group. Adenosine A2A receptor antagonist istradefylline reduces daily OFF time in Parkinson’s disease. Mov. Disord. 2013, 28, 1138–1141. [Google Scholar] [CrossRef]
- Groiss, S.J.; Wojtecki, L.; Sudmeyer, M.; Schnitzler, A. Deep brain stimulation in Parkinson’s disease. Ther. Adv. Neurol. Disord. 2009, 2, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Hamani, C.; Florence, G.; Heinsen, H.; Plantinga, B.R.; Temel, Y.; Uludag, K.; Alho, E.; Teixeira, M.J.; Amaro, E.; Fonoff, E.T. Subthalamic Nucleus Deep Brain Stimulation: Basic Concepts and Novel Perspectives. eNeuro 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, C.C.; Thakor, N.V. Uncovering the mechanisms of deep brain stimulation for Parkinson’s disease through functional imaging, neural recording, and neural modeling. Crit. Rev. Biomed. Eng. 2002, 30, 249–281. [Google Scholar] [CrossRef] [PubMed]
- Dostrovsky, J.O.; Levy, R.; Wu, J.P.; Hutchison, W.D.; Tasker, R.R.; Lozano, A.M. Microstimulation-induced inhibition of neuronal firing in human globus pallidus. J. Neurophysiol. 2000, 84, 570–574. [Google Scholar] [CrossRef]
- Beurrier, C.; Bioulac, B.; Audin, J.; Hammond, C. High-frequency stimulation produces a transient blockade of voltage-gated currents in subthalamic neurons. J. Neurophysiol. 2001, 85, 1351–1356. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, A.; Mehdizadeh, A.R. Deep Brain Stimulation and Gene Expression Alterations in Parkinson’s Disease. J. Biomed. Phys. Eng. 2016, 6, 47–50. [Google Scholar] [PubMed]
- Maiti, P.; Manna, J.; Dunbar, G.L. Current understanding of the molecular mechanisms in Parkinson’s disease: Targets for potential treatments. Transl. Neurodegener. 2017, 6, 1–35. [Google Scholar] [CrossRef]
- Zeng, X.S.; Geng, W.S.; Jia, J.J.; Chen, L.; Zhang, P.P. Cellular and Molecular Basis of Neurodegeneration in Parkinson Disease. Front. Aging Neurosci. 2018, 10, 109. [Google Scholar] [CrossRef]
- Shihabuddin, L.S.; Brundin, P.; Greenamyre, J.T.; Stephenson, D.; Sardi, S.P. New Frontiers in Parkinson’s Disease: From Genetics to the Clinic. J. Neurosci. 2018, 38, 9375–9382. [Google Scholar] [CrossRef]
- Raza, C.; Anjum, R.; Shakeel, N.U.A. Parkinson’s disease: Mechanisms, translational models and management strategies. Life Sci. 2019, 226, 77–90. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Zunke, F.; Tsunemi, T.; Toker, N.J.; Jeon, S.; Burbulla, L.F.; Patnaik, S.; Sidransky, E.; Marugan, J.J.; Sue, C.M.; et al. Activation of beta-Glucocerebrosidase Reduces Pathological alpha-Synuclein and Restores Lysosomal Function in Parkinson’s Patient Midbrain Neurons. J. Neurosci. 2016, 36, 7693–7706. [Google Scholar] [CrossRef]
- Sardi, S.P.; Clarke, J.; Kinnecom, C.; Tamsett, T.J.; Li, L.; Stanek, L.M.; Passini, M.A.; Grabowski, G.A.; Schlossmacher, M.G.; Sidman, R.L.; et al. CNS expression of glucocerebrosidase corrects alpha-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc. Natl. Acad. Sci. USA 2011, 108, 12101–12106. [Google Scholar] [CrossRef] [PubMed]
- Silveira, C.R.A.; MacKinley, J.; Coleman, K.; Li, Z.; Finger, E.; Bartha, R.; Morrow, S.A.; Wells, J.; Borrie, M.; Tirona, R.G.; et al. Ambroxol as a novel disease-modifying treatment for Parkinson’s disease dementia: Protocol for a single-centre, randomized, double-blind, placebo-controlled trial. BMC Neurol. 2019, 19, 20. [Google Scholar] [CrossRef]
- Prevail Therapeutics. PR001. 2020. Available online: https://www.prevailtherapeutics.com/programs/#:~:text=PR001,the%20same%20gene%2C%20called%20GBA1. (accessed on 20 September 2020).
- Weihofen, A.; Liu, Y.; Arndt, J.W.; Huy, C.; Quan, C.; Smith, B.A.; Baeriswyl, J.L.; Cavegn, N.; Senn, L.; Su, L.; et al. Development of an aggregate-selective, human-derived alpha-synuclein antibody BIIB054 that ameliorates disease phenotypes in Parkinson’s disease models. Neurobiol. Dis. 2019, 124, 276–288. [Google Scholar] [CrossRef]
- Price, D.L.; Koike, M.A.; Khan, A.; Wrasidlo, W.; Rockenstein, E.; Masliah, E.; Bonhaus, D. The small molecule alpha-synuclein misfolding inhibitor, NPT200-11, produces multiple benefits in an animal model of Parkinson’s disease. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Neuropore. Inhibit the Formation of Toxic Aggregates of Misfolded Proteins. 2020. Available online: https://www.neuropore.com/programs/anti-oligomerization.htm (accessed on 21 September 2020).
- Levin, J.; Schmidt, F.; Boehm, C.; Prix, C.; Botzel, K.; Ryazanov, S.; Leonov, A.; Griesinger, C.; Giese, A. The oligomer modulator anle138b inhibits disease progression in a Parkinson mouse model even with treatment started after disease onset. Acta Neuropathol. 2014, 127, 779–780. [Google Scholar] [CrossRef]
- Wegrzynowicz, M.; Bar-On, D.; Calo, L.; Anichtchik, O.; Iovino, M.; Xia, J.; Ryazanov, S.; Leonov, A.; Giese, A.; Dalley, J.W.; et al. Depopulation of dense alpha-synuclein aggregates is associated with rescue of dopamine neuron dysfunction and death in a new Parkinson’s disease model. Acta Neuropathol. 2019, 138, 575–595. [Google Scholar] [CrossRef] [PubMed]
- MODAG Neuroscience Solutions. MODAG Successfully Completes Phase 1 Study of their Lead Candidate Anle138b and Receives Additional USD 1.4 Million from Michael J. Fox Foundation. Available online: https://www.modag.net/index.php/en/press-releases (accessed on 21 September 2020).
- Smith, S.B. Introduction to Sigma Receptors: Their Role in Disease and as Therapeutic Targets. Adv. Exp. Med. Biol. 2017, 964, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Alessi, D.R. Advances in elucidating the function of leucine-rich repeat protein kinase-2 in normal cells and Parkinson’s disease. Curr. Opin. Cell Biol. 2020, 63, 102–113. [Google Scholar] [CrossRef]
- Rui, Q.; Ni, H.; Li, D.; Gao, R.; Chen, G. The Role of LRRK2 in Neurodegeneration of Parkinson Disease. Curr. Neuropharmacol. 2018, 16, 1348–1357. [Google Scholar] [CrossRef]
- Huttunen, H.J.; Saarma, M. CDNF Protein Therapy in Parkinson’s Disease. Cell Transplant. 2019, 28, 349–366. [Google Scholar] [CrossRef] [PubMed]
- Albert, K.; Renko, J.M.; Matlik, K.; Airavaara, M.; Voutilainen, M.H. Cerebral Dopamine Neurotrophic Factor Diffuses Around the Brainstem and Does Not Undergo Anterograde Transport After Injection to the Substantia Nigra. Front. Neurosci. 2019, 13, 590. [Google Scholar] [CrossRef]
- Chen, Y.C.; Baronio, D.; Semenova, S.; Abdurakhmanova, S.; Panula, P. Cerebral Dopamine Neurotrophic Factor regulates multiple neuronal subtypes and behavior. J. Neurosci. 2020, 40, 6146–6164. [Google Scholar] [CrossRef] [PubMed]
- Abi Hussein, H.; Geneix, C.; Petitjean, M.; Borrel, A.; Flatters, D.; Camproux, A.C. Global vision of druggability issues: Applications and perspectives. Drug Discov. Today 2017, 22, 404–415. [Google Scholar] [CrossRef] [PubMed]


| Year | Total Approval | Fast Track (% of Total) | Breakthrough (% of Total) | Priority Review (% of Total) | Accelerated Approval (% of Total) | Used One or More Expedited Pathway (% of Total) |
|---|---|---|---|---|---|---|
| 2011 | 30 | 14 (47%) | N/A | 15 (50%) | 3 (10%) | 17 (57%) |
| 2012 | 39 | 14 (36%) | N/A | 16 (41%) | 4 (10%) | 22 (56%) |
| 2013 | 27 | 10 (37%) | 3 (11%) | 10 (37%) | 2 (7%) | 13 (48%) |
| 2014 | 41 | 17 (41%) | 9 (22%) | 25 (61%) | 8 (20%) | 27 (66%) |
| 2015 | 45 | 14 (31%) | 10 (22%) | 24 (53%) | 6 (13%) | 27 (60%) |
| 2016 | 22 | 8 (36%) | 7 (32%) | 15 (68%) | 6 (27%) | 16 (73%) |
| 2017 | 46 | 18 (39%) | 17 (37%) | 28 (61%) | 6 (13%) | 28 (61%) |
| 2018 | 59 | 24 (41%) | 14 (24%) | 43 (73%) | 4 (7%) | 43 (73%) |
| 2019 | 48 | 17 (35%) | 13 (27%) | 28 (58%) | 9 (19%) | 29 (60%) |
| 9/2020 | 40 | 0 | 0 | 19 (48%) | 0 | 17 (48%) |
| Modality | Target | Target Site | Delivery | ||||||
|---|---|---|---|---|---|---|---|---|---|
| DNA | RNA | Protein | Extracellular | Plasma Membrane | Intracellular | Oral | Injection | Inhaled | |
| Small molecules | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Antibody-based agents | Yes | Yes | Yes | Yes | |||||
| Peptides | Yes | Yes | Yes | Yes | Yes | Yes | |||
| Oligonucleotide therapy | Yes | Yes | Yes | Yes | |||||
| Gene and Cell therapy | Yes | Yes | Yes | Yes | Yes | ||||
| Year | Small Molecule | rProtein | mAb/bsAb/Nanobody | ADC | Peptide | Oligonucleotide | Cellular & Gene Therapy | Other |
|---|---|---|---|---|---|---|---|---|
| 2015 | 31 | 3 | 8 (mAb), 1 (Fab) | 0 | 1 | 0 | 1 (oncolytic virus) | 1 (oligosaccharides) |
| 2016 | 11 | 0 | 7 (mAb) | 0 | 1 | 3 | 0 | 0 |
| 2017 | 29 | 2 | 8 (mAb), 1 (bsAb) | 1 | 5 | 0 | 2 (CAR T), 1 (gene) | 0 |
| 2018 | 38 | 5 | 11 (mAb) | 1 | 1 | 2 | 0 | 1 (diagnostic agent) |
| 2019 | 32 | 2 | 3 (mAb), 1 (scFv), 1 (NB) | 3 | 3 | 2 | 1 (gene) | 1 (fatty acid) |
| 9/2020 | 28 | 1 | 6 (mAb) | 2 | 1 | 1 | 1 (CAR T) | 1 (fatty acid) |
| Total | 169 | 13 | 47 | 7 | 12 | 8 | 6 | 4 |
| Strategy | Approval Year | Trade Name | Drug Name | Sponsor | Properties | Indication for Use |
|---|---|---|---|---|---|---|
| Viral vectors | 2015 | Imlygic | talimogene laherparepvec | Amgen | Genetically modified oncolytic virus | Melanoma |
| 2017 | Luxturna | voretigene neparvovec-rzyl | Spark Therapeutics | AAV-based RPE65 gene therapy | Confirmed biallelic RPE65 mutation-associated retinal dystrophy | |
| 2019 | Zolgensma | onasemnogene abeparvovec-xioi | AveXis/Novartis | AAV- based SMN gene therapy | Spinal muscular atrophy (SMA) with bi-allelic mutations in the survival motor neuron 1 (SMN1) gene | |
| Oligonucleotides | 1998 | Vitravene * | fomivirsen | Novartis | ASO designed to inhibit human cytomegalovirus replication | Cytomegalovirus (CMV) retinitis |
| 2004 | Macugen * | pegaptanib | Valeant Pharmas | Aptamar designed to target VEGF | Neovascular age-related macular degeneration | |
| 2013 | Kynamro * | mipomersen | Kastle Theraps | Oligonucleotide inhibitor of apolipoprotein B-100 synthesis | Homozygous familial hypercholesterolemia | |
| 2016 | Defitelio | defibrotide | Gentium | Oligonucleotide mixture with Profibrinolytic properties | Hepatic veno-occlusive disease with additional kidney or lung abnormalities after receiving a hematopoietic stem cell transplantation | |
| 2016 | Exondys 51 | eteplirsen | Sarepta Therapeutics | ASO designed to target dystrophin pre-mRNA | Duchenne muscular dystrophy | |
| 2016 | Spinraza | nusinersen | Biogen/Ionis Pharmaceuticals | ASO designed to target SMN2 pre-mRNA | Spinal muscular atrophy (SMA) | |
| 2018 | Onpattro | patisiran | Alnylam Pharmaceuticals | TTR-directed siRNA | Polyneuropathy of hereditary transthyretin-mediated amyloidosis | |
| 2018 | Tegsedi | inotersen | Ionis Pharmaceuticals | TTR-directed antisense oligonucleotide | Polyneuropathy of hereditary transthyretin-mediated amyloidosis | |
| 2019 | Givlaari | givosiran sodium | Alnylam | AL AS1-directed siRNA (GalNac conjugation) | Acute hepatic porphyria | |
| 2019 | Vyondys 53 | golodirsen | Sarepta | Exon 53 skipping antisense | Duchenne muscular dystrophy |
| Target | Modality | Drug Name | Sponsor | Status | NCT | Route of Administration |
|---|---|---|---|---|---|---|
| TNF | mAb | Enbrel (etanercept) | Amgen | Approved, 2002 | Subcutaneous | |
| mAb | Remicade (infliximab) | Janssen Biotech | Approved, 1999 | Intravenous | ||
| mAb | Humira (adalimumab) | AbbVie Inc | Approved, 2002 | Subcutaneous | ||
| mAb | CIMZIA (certolizumab pegol) | UCB | Aproved, 2009 | Subcutaneous | ||
| mAb | Simponi (golimumab) | Centocor, Inc. | Aproved, 2009 | Subcutaneous | ||
| mAb | ABBV-154 | AbbVie | Phase II | n/a | Intravenous | |
| mAb | ABBV-3373 | AbbVie | Phase II | NCT03823391 | Intravenous | |
| IL-6 | mAb | Actemra (tocilizumab) | Genentech | Approved, 2010 | Intravenous or Subcutaneous | |
| mAb | Plivensia (sirukumab) | Janssen Biotech | Withdrawn | Subcutaneous | ||
| mAb | Kevzara (sarilumab) | Sanofi and Regeneron Pharmaceuticals | Approved, 2017 | Subcutaneous | ||
| CD20 | mAb | Rituxan (rituximab) | Genentech, Inc. | Approved, 2006 | Subcutaneous | |
| mAb | Ocrelizumab | Genentech, Inc. | Phase III (terminated) | NCT02720120 | Intravenous | |
| mAb | Veltuzumab | Takeda | Phase II (terminated) | NCT01390545 | Subcutaneous | |
| mAb | Ofatumumab | GlaxoSmithKline | Phase III (terminated) | NCT00611455 | Intravenous | |
| CD80/CD86 | mAb | Orencia (abatacept), | Bristol-Myers Squibb | Approved, 2011 | Intravenous | |
| JAK | SM | XELJANZ (tofacitinib) | Pfizer | Approved, 2016 | Oral | |
| SM | Olumiant (Baricitinib) | Eli Lilly | Approved, 2018 | Oral | ||
| SM | Rinvoq (upadacitinib) | AbbVie | Approved, 2019 | Oral | ||
| SM | Abrocitinib | Pfizer | Phase III | n/a | Oral | |
| SM | Filgotinib | Galapagos NV/Gilead | Phase III | NCT02886728 (request additional data by FDA, approved in Japan) | Oral | |
| SM | Decernotinib | Aclaris Therapeutics | Phase II | n/a | Oral | |
| GM-CSF | mAb | Mavrilimumab | Kiniksa | Phase II | NCT01715896 | Subcutaneous |
| mAb | Namilumab | Takeda | Phase | NCT02379091, NCT02393378 | Subcutaneous | |
| mAb | Otilimab | GSK | Phase III | NCT04134728 | Subcutaneous | |
| BTK | SM | Spebrutinib (CC-292) | Celgene | Phase IIb | NCT01975610 | Oral |
| SM | BMS-986142 | Bristol-Myers Squibb | Phase II | NCT02638948 | Oral | |
| SM | Branebrutinib | Bristol-Myers Squibb | Phase IIb | NCT04186871 | Oral | |
| SM | Evobrutinib | Merk | Phase IIb | NCT03233230 | Oral | |
| SM | TK-020 | Takeda | Phase I | NCT02413255 | Oral | |
| SM | HM71224 | Eli Lilly/Hamni | Phase II (terminated) | NCT01765478 | Oral | |
| SM | Fenebrutinib | Roche | Phase II | n/a | ||
| BTK/JAK1 | SM | ABBV-599 | AbbVie | Phase II (terminated) | NCT03823378 | Oral |
| Target | Modality | Drug | Sponsor | Status | NCT | Route of Admiration |
|---|---|---|---|---|---|---|
| PDE4 | SM | Eucrisa (crisaborole) | Pfizer | Approved, 2016 | Topical Ointment | |
| SM | Apremilast | Amgen | Phase II | NCT02087943 | Oral | |
| SM | E6005 | Elsai | Phase II | NCT01461941 | Ointment | |
| SM | DRM02 | QLT | Phase II | NCT01993420 | Ointment | |
| IL-13 and IL-4 | mAb | Dupixent (dupilumab) | Sanofi/Regeneron | Approved, 2017 | Subcutaneous | |
| mAb | Tralokinumab | LEO Pharma | phase IIb | NCT03562377 | Subcutaneous | |
| mAb | Lebrikizumab | Eli Lilly | Phase III | NCT04392154 | Subcutaneous | |
| IL-5 | mAb | Mepolizumab | GlaxoSmithKline | Phase II | NCT03055195 | Subcutaneous |
| mAb | benralizumab | AstraZeneca | Phase II | NCT03563066 | Subcutaneous | |
| IL-31 | mAb | Nemolizumab | Chugai Pharmaceutical Company | Phase III | NCT03985943 | n/a |
| IL-23 | mAb | Risankizumab | AbbVie | Phase II | NCT03706040 | Subcutaneous |
| IL-22 | mAb | Fezakinumab | Pfizer | Phase Iia | n/a | |
| IL-17C | mAb | MOR106 | MorphoSys and Galapagos | phase II (terminated) | NCT03864627 | Subcutaneous |
| IL-17A | mAb | Secukinumab | GWT-TUD GmbH | Phase II | NCT03568136 | subcutaneous |
| JAK | SM | Olumiant (Baricitinib) | Eli Lilly | Phase III | NCT03334422 | Oral |
| SM | ruxolitinib | Incyte Corporation | Phase II | NCT03011892 | Ointment | |
| SM | RINVOQ (upadacitinib) | AbbVie | Phase III | NCT03569293 | Oral | |
| SM | abrocitinib | Pfizer | Phase III | NCT04345367 | Oral | |
| SM | Gusacitinib | Asana Biosciences | Phase IIb (terminated) | NCT03654755 | Oral | |
| HRH4 | SM | ZPL-3893787 | Ziarco Pharma | Phase II | NCT02424253 | Oral |
| SM | JNJ39758979 | Janssen Pharmaceutical | Phase II (terminated) | NCT01497119 | Oral | |
| TSLP | mAb | Tezepelumab | AstraZeneca | Phase IIa | NCT03809663 | Subcutaneous |
| mAb | MK8226 | Merck Sharp & Dohme Corp. | Phase I (terminated) | NCT01732510 | Intravenous | |
| OX40 | mAb | GBR 830 KHK4083 | Glenmark Pharmaceuticals Kyowa Kirin Pharmaceutical Development, Inc. | Phase II | NCT02683928 NCT03703102 | Intravenous |
| IL-33 | mAb | Etokinumab | AnaptysBio | Phase II (failed) | NCT03533751 | n/a |
| mAb | PF-06817024 | Pfizer | Phase I | NCT02743871 | Intravenous | |
| mAb | REGN3500 | Regeneron/Sanofi | Phase II | NCT03736967 | Subcutaneous |
| Target | Modality | Drug | Sponsor | Status | NCT | Route of Admiration |
|---|---|---|---|---|---|---|
| Integrin | mAb | Natalizumab | Biogen Idec/Elan Corporation | Approved, 2008 | Intravenous | |
| mAb | Entyvio (vedolizumab) | Takeda | Approved, 2014 | Subcutaneous | ||
| mAb | Etrolizumab | Roche | Phase III | NCT02394028 | Subcutaneous | |
| S1P1 | SM | ZEPOSIA (ozanimod) | Bristol-Myers Squibb | Phase III | NCT03440372 | Oral |
| SM | Amiselim (MT-1303) | Biogen | Phase II (terminated) | NCT02378688 | Oral | |
| MAdCAM | mAb | PF-00547659 | Pfizer | Phase II | NCT03283085 | Subcutaneous |
| TYK2 | SM | BMS-986165 | Bristol-Myers Squibb | Phase II | NCT03599622 | Oral |
| TYK2/JAK1 | SM | PF-06700841 | Pfizer | Phase II | NCT03395184 | Oral |
| Pan-JAK/TYK2 | SM | TD-1473 | J&J/Theravance | Phase II | NCT03635112 | Oral |
| IL-23 and IL-12 | mAb | Ustekinumab | Janssen Biotech | Phase I | NCT02968108 | Intravenous |
| mAb | Risankizumab | AbbVie | Phase III | NCT03105128 | Subcutaneous | |
| IL-23R | peptide | PTG-200 | Protagonist Therapeutics/J&J | Phase II | Oral |
| Category | Drugs | Mechanism of Action | Analgesic Action Level |
|---|---|---|---|
| NSAIDs | Aspirin, naproxen, ibuprofen, diclofenac, celecoxib, piroxicam, indomethacin, meloxicam, ketoprofen, sulindac, diflunisal, nabumetone, oxaprozin, tolmetin, salsalate, etodolac, fenoprofen, flurbiprofen, ketorolac, meclofenamate, mefenamic acid, etoricoxib, and rofecoxib. | inhibit COX enzymes leading to decreased prostaglandin synthesis. increasing serotonin in central sites | Peripheral and central effects |
| Analgesics | Acetaminophen | Peripheral: COX1 and 2 inhibition Central: descending serotonergic neuronal pathways, inhibition of L-arginine/NO pathway, stimulation of endocannabinoid system, and anti-nociception mechanisms | Peripheral and central effects |
| Duloxetine | Serotonin and nonrepinephrine reuptake inhibitor | Central effects | |
| Opioids and opioid receptor ligands | Morphine, codeine, acetaminophen with codeine, fentanyl, hydrocodone, acetaminophen with hydrocodone, hydromorphone, meperidine, oxycodone | Activate opioids receptors to hyperpolarizes sensory neurons and attenuate nerve hyperexcitability | Peripheral and central effects |
| Corticosteroids | Prednisone, betamethasone, cortisone, dexamethasone, hydrocortisone, methylprednisolone, prednisolone, triamcinolone acetonide | Immunosuppressive and anti-inflammatory by interrupting the inflammatory cascade | Peripheral |
| Joint modifying treatments | Chondroitin and glucosamine | Increase proteoglycan synthesis in articular cartilage | Peripheral/local |
| Hyaluronic acid | Enhance chondrocyte synthesis of endogenous hyaluronic acid and proteoglycans | Peripheral/local |
| Class | Drugs | Mechanism of Action |
|---|---|---|
| NSAIDs | Aspirin, naproxen, ibuprofen, tolfenamic acid, diclofenac, piroxicam, ketoprofen, and ketorolac | Inhibit prostaglandin synthesis |
| Analgesics | Acetaminophen | Inhibit prostaglandin synthesis |
| Triptans | Sumatriptan, eletriptan, naratriptan, zolmitriptan, rizatriptan, frovatriptan, and almotriptan | Serotonin 5-HT1B/1D receptor agonists |
| Ergotamines | Ergotamines, Dihydroergotamine | Serotonin 5-HT1B/1D/1F receptor agonists |
| β-blockers | Propranolol, timolol | Unclear, inhibit noradrenaline release or serotonergic blockade |
| Anti-depressants | Tricyclic antidepressant (TCA) amitriptyline and the selective serotonin reuptake inhibitor (SSRI) fluoxetine | Increase amounts of serotonin and norepinephrine. |
| Anti-emetics | Metoclopramide, prochlorperazine, Domperidone, promethazine, chlorpromazine | Dopamine antagonists |
| Calcium channel blockers | Verapamil, cinnarizine | Unclear, preventing the constriction of the blood vessels prior to the migraine attack. |
| Botox | Botulinum toxin A | Unclear, peripheral and central system sensitization, inactivation of trigeminovascular system |
| Anti-epileptics | Topiramate and divalproex | Unclear, inhibit glutamate-mediated excitation, GABAergic inhibition and reduce CGRP. |
| Ditans | Lasmiditan | Serotonin 5-HT1F receptor agonists |
| Anti-CGRP peptide | mAbs: erenumab, fremanezumab, galcanezumab, eptinezumabSM: ubrogepant, rimegepant | Blocks CGRP binding to receptors |
| Drug | Sponsor | Modality | Mechanism of Action | Stage | ClinicalTrials.gov Identifier |
|---|---|---|---|---|---|
| AAB-003 | Janssen, Pfizer | mAb | Anti-Aβ antibody | Phase I (terminated) | NCT01193608 |
| Aducanumab | Biogen, Neurimmune | mAb | Anti-Aβ antibody | Phase III (terminated) Phase IIIc | NCT02484547 NCT04241068 |
| BAN2401 | Biogen, Eisai Co., Ltd. | mAb | Anti-Aβ antibody | Phase III | NCT03887455 |
| Bapineuzumab | Janssen, r | mAb | Anti-Aβ antibody | Phase III (terminated) | NCT00998764 |
| Crenezumab | AC Immune SA, Genentech, Hoffmann-La Roche | mAb | Anti-Aβ antibody | Phase III (terminated) | NCT02670083 |
| Donanemab | Eli Lilly and Co. | mAb | Anti-Aβ antibody | Phase II | NCT03367403 |
| GSK933776 | GlaxoSmithKline (GSK) | mAb | Anti-Aβ antibody | Phase I (terminated) | NCT00459550 |
| Gantenerumab | Chugai Pharmaceutical Co., Ltd., Hoffmann-La Roche | mAb | Anti-Aβ antibody | Phase III | NCT03444870 |
| LY2599666 | Eli Lilly and Co. | Fc-less, antigen-binding fragment of a monoclonal anti-Aβ antibody linked to polyethylene glycol | Anti-Aβ antibody | Phase I (terminated) | NCT02614131 |
| LY3372993 | Eli Lilly and Co. | mAb | Anti-Aβ antibody | Phase I | NCT03720548 |
| MEDI1814 | AstraZeneca, Eli Lilly and Co. | mAb | Anti-Aβ antibody | Phase I | NCT02036645 |
| Ponezumab | Pfizer | mAb | Anti-Aβ antibody | Phase II (terminated) | NCT00945672 |
| RO7126209 | Hoffmann-La Roche | mAb with “brain shuttle” technology | Anti-Aβ antibody | Phase I | NCT04023994 |
| SAR228810 | Sanofi | mAb | Anti-Aβ antibody | Phase I | NCT01485302 |
| Solanezumab | Eli Lilly and Co. | mAb | Anti-Aβ antibody | Phase III (terminated) | NCT02760602 |
| Atabecestat | Janssen, Shionogi Pharma | SM | BACE inhibitor | Phase III (terminated) | NCT02569398 |
| BI 1181181 | Boehringer Ingelheim, Vitae Pharmaceuticals | SM | BACE inhibitor | Phase I (terminated) | NCT02106247 |
| Elenbecestat | Biogen, Eisai Co., Ltd. | SM | BACE inhibitor | Phase I (terminated) | NCT01600859 |
| LY2886721 | Eli Lilly and Co. | SM | BACE inhibitor | Phase II (terminated) | NCT01561430 |
| Lanabecestat | AstraZeneca, Eli Lilly & Co. | SM | BACE inhibitor | Phase III (terminated) | NCT02783573 |
| PF-06751979 | Pfizer | SM | BACE inhibitor | Phase I (terminated) | NCT02509117 |
| RG7129 | Roche | SM | BACE inhibitor | Phase I (terminated) | NCT01461967 |
| Umibecestat | Amgen, Inc., Novartis Pharmaceuticals Corporation | SM | BACE inhibitor | Phase II/III (terminated) | NCT02565511 |
| Verubecestat | Merck | SM | BACE inhibitor | Phase II/III (terminated | NCT01739348 |
| Avagacestat | Bristol-Myers Squibb | SM | γ-secretase inhibitor | Phase II (terminated) | NCT00890890 |
| Semagacestat | Eli Lilly and Co. | SM | γ-secretase inhibitor | Phase III (terminated) | NCT01035138 |
| ABBV-8E12 | AbbVie, C2N Diagnostics, LLC | mAb | Anti-tau antibody | Phase II (terminated) | NCT02880956 |
| BIIB076 | Biogen, Neurimmune | mAb | Anti-tau antibody | Phase II | NCT03056729 |
| Gosuranemab | Biogen, Bristol-Myers Squibb | mAb | Anti-tau antibody | Phase II | NCT03352557 |
| JNJ-63733657 | Janssen | mAb | Anti-tau antibody | Phase I | NCT03375697 |
| Lu AF87908 | H. Lundbeck | mAb | Anti-tau antibody | Phase I | NCT04149860 |
| PNT001 | Pinteon Therapeutics | mAb | Anti-tau antibody | Phase I | NCT04096287 |
| RG7345 | Roche | mAb | Anti-tau antibody | Phase I (terminated) | NCT02281786 |
| Semorinemab | AC Immune SA, Genentech, Hoffmann-La Roche | mAb | Anti-tau antibody | Phase II | NCT03828747 |
| Zagotenemab | Eli Lilly and Co. | mAb | Anti-tau antibody | Phase II | NCT03518073 |
| LMTM | TauRx Therapeutics Ltd. | SM | tau protein aggregation inhibitor | Phase III | NCT01689246 |
| Epothilone D | Bristol-Myers Squibb | SM | microtubule stabilizer | Phase I (terminated) | NCT01492374 |
| TPI 287 | Cortice Biosciences | SM | microtubule stabilizer | Phase I (terminated) | NCT01966666 |
| Tideglusib | Zeltia Group | SM | glycogen synthase kinase 3 (GSK3-β) inhibitor | Phase II (terminated) | NCT01350362 |
| AL002 | AbbVie, Alector | mAb | TREM2 agonist | Phase I | NCT03635047 |
| AL003 | AbbVie, Alector | mAb | SIGLEC3 antagonist | Phase I | NCT03822208 |
| ALZT-OP1 | AZTherapies, Inc. | SM | NSAID, anti-inflammatory | Phase III | NCT02547818 |
| Azeliragon | Pfizer, TransTech Pharma, Inc., vTv Therapeutics LLC | SM | RAGE antagonist | Phase II/III | NCT03980730 |
| Etanercept | Amgen, Inc., Pfizer | Receptor-Fc fusion | TNF-α antagonist | Phase II | NCT01068353 |
| Masitinib | AB Science | SM | Protein tyrosine kinase antagonist | Phase III | NCT01872598 |
| XPro1595 | INmune Bio Inc. | Heterotrimer biologic | TNF-α antagonist | Phase I | NCT03943264 |
| Dapagliflozin | AstraZeneca, Bristol-Myers Squibb | SM | SGLT2 inhibitor | Phase I/II | NCT04120623 |
| Empagliflozin | Boehringer Ingelheim, Eli Lilly and Co. | SM | SGLT2 inhibitor | Phase I | NCT03852901 |
| Gemfibrozil | Gregory Jicha, 323–5550 | SM | PPARα agonist | Phase I | NCT02045056 |
| Liraglutide | Novo Nordisk A/S | SM | GLP-1R agonist | Phase II | NCT01843075 |
| Metformin | Columbia University | SM | Glucose lowering | Phase II/III | NCT04098666 |
| Nasal Insulin | University of Southern California | SM | unknown | Phase II/III | NCT01767909 |
| T3D-959 | T3D Therapeutics, Inc. | SM | PPARδ/γ agonist | Phase II | NCT04251182 |
| Drug | Sponsor | Mechanism of Action | Stage | ClinicalTrials.gov Identifier |
|---|---|---|---|---|
| AstroStem | Nature Cell Co. | Regenerative | Phase II | NCT03117738 |
| hUCB-MSCs | Medipost Co | Regenerative | Phase II | NCT02054208 |
| hUCB-MSCs | Medipost Co. | Regenerative | Phase II | NCT03172117 |
| hUCB-MSCs | South China Research Center | Regenerative | Phase II | NCT02513706 |
| hUCB-MSCs | South China Research Center | Regenerative | Phase II | NCT02672306 |
| hMSCs | Stemedica Cell | Regenerative | Phase II | NCT02833792 |
| LMSCs | Longeveron | Regenerative | Phase I | NCT02600130 |
| Drug | Sponsor | Mechanism of Action | Stage | ClinicalTrials.gov Identifier |
|---|---|---|---|---|
| CERE-110 | Sangamo Therapeutics | Adeno-associated virus-based gene delivery of NGF | Phase II (terminated) | NCT00876863 |
| IONIS MAPTRX (BIIB080) | Ioni Pharmaceuticals, Biogen | MAPt RNA inhibitor ASO | Phase II | NCT03186989 |
| AAVrh.10hAPOE2 | Cornell University | Serotype rh. 10 adeno-associated virus gene delivery of ApoE2 | Phase I | NCT03634007 |
| Drug | Brand Name/FDA Approval | Modality | MOA |
|---|---|---|---|
| Levodopa and Carbidopa | Sinemet/1975, Parcopa/2004, Rytary/2015 | SM | Dopamine precursor-dopamine decarboxylase inhibitor |
| Selegiline | Eldepryl/1989, Emsam/2006, Zelapar/2006 | SM | MAO B inhibitors |
| Rasagiline | Azilect/2006 | SM | MAO B inhibitors |
| Safinamide | Xadago/2017 | SM | MAO B inhibitors |
| Bromocriptine | Parlodel/2005 | SM | Dopamine agonist |
| Pramipexole | Mirapex/1997 | SM | Dopamine agonist |
| Ropinirole | Requip/1997 | SM | Dopamine agonist |
| Rotigotine | Neupro/2007 | SM | Dopamine agonist |
| Apomorphine | Apokyn/2004 | SM | Dopamine agonist |
| Tolcapone | Tasmar/1998 | SM | COMT inhibitors |
| Entacapone | Comtan/1999 | SM | COMT inhibitors |
| Opicapone | Ongentys/2020 | SM | COMT inhibitors |
| Amantadine | Symmetrel/2003 | SM | Weak, non-competitive NMDA receptor antagonist |
| Istradefylline | Nourianz/2019 | SM | Adenosine receptor antagonist (A2A) |
| Deep brain stimulation | n/a/1997, 2002, 2003 | Device | Electric stimulation |
| Drug | Sponsor | Modality | Mechanism of Action | Stage | ClinicalTrials.gov Identifier |
|---|---|---|---|---|---|
| Ambroxol | University College, London | SM | GCase activation | Phase II | NCT02914366 |
| Anavex 2-73 (blarcamesine) | Anavex Life Science Corp. | SM | Sigma-1 receptor (SIGMAR1) agonist | Phase II | NCT03774459 |
| DNL151 | Denali Therapeutics Inc. | SM | LRRK2 inhibitor | Phase I | NCT04056689 |
| DNL201 | Denali Therapeutics Inc. | SM | LRRK2 inhibitor | Phase I | NCT03710707 |
| Nilotinib | Novartis Pharmaceuticals Corporation | SM | c-Abl kinase inhibitor | Phase II | NCT02954978 |
| K0706 | Sun Pharma Advanced Research Company | SM | c-Abl kinase inhibitor | Phase II | NCT03655236 |
| FB-101 | 1ST Biotherapeutics, Inc. | SM | c-Abl kinase inhibitor | Phase I | NCT04165837 |
| BIIB094 | Biogen, IONIS Pharmaceuticals | Gene Therapy | LRRK2 inhibitor | Phase I | NCT03976349 |
| PR001 | Prevail Therapeutics | Gene Therapy | GBA1 (encodes for Gcase) | Phase II | NCT04127578 |
| ABBV-0805 | AbbVie, BioArctic AB | mAb | α-synuclein | Phase I (withdrawn due to strategic considerations) | NCT04127695 |
| Cinpanemab | Biogen, Neurimmune | mAb | α-synuclein | Phase II | NCT03318523 |
| LU AF82422 | Genmab A/S, H. Lundbeck | mAb | α-synuclein | Phase I | NCT03611569 |
| MEDI1341 | AstraZeneca, Takeda Pharmaceutical Company | mAb | α-synuclein | Phase I | NCT04449484 |
| Prasinezumab | Hoffmann-La Roche, Prothena | mAb | α-synuclein | Phase II | NCT02157714 |
| NPT200-11 | Neuropore Therapies Inc. | SM | α-synuclein | Phase I | NCT02606682 |
| anle138b | MODAG GmbH | SM | α-synuclein | Phase I | NCT04208152 |
| CDNF (cerebral dopamine neurotrophic factor) | Herantis Pharma Plc, Renishaw plc. | Peptide | promotes survival of midbrain dopaminergic neurons | Phase II | NCT03295786 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Ting, J.P.; Al-Azzam, S.; Ding, Y.; Afshar, S. Therapeutic Advances in Diabetes, Autoimmune, and Neurological Diseases. Int. J. Mol. Sci. 2021, 22, 2805. https://doi.org/10.3390/ijms22062805
Liu J, Ting JP, Al-Azzam S, Ding Y, Afshar S. Therapeutic Advances in Diabetes, Autoimmune, and Neurological Diseases. International Journal of Molecular Sciences. 2021; 22(6):2805. https://doi.org/10.3390/ijms22062805
Chicago/Turabian StyleLiu, Jinsha, Joey Paolo Ting, Shams Al-Azzam, Yun Ding, and Sepideh Afshar. 2021. "Therapeutic Advances in Diabetes, Autoimmune, and Neurological Diseases" International Journal of Molecular Sciences 22, no. 6: 2805. https://doi.org/10.3390/ijms22062805
APA StyleLiu, J., Ting, J. P., Al-Azzam, S., Ding, Y., & Afshar, S. (2021). Therapeutic Advances in Diabetes, Autoimmune, and Neurological Diseases. International Journal of Molecular Sciences, 22(6), 2805. https://doi.org/10.3390/ijms22062805