
Molecular Regulation of Skeletal Muscle Growth and Organelle Biosynthesis: Practical Recommendations for Exercise Training




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
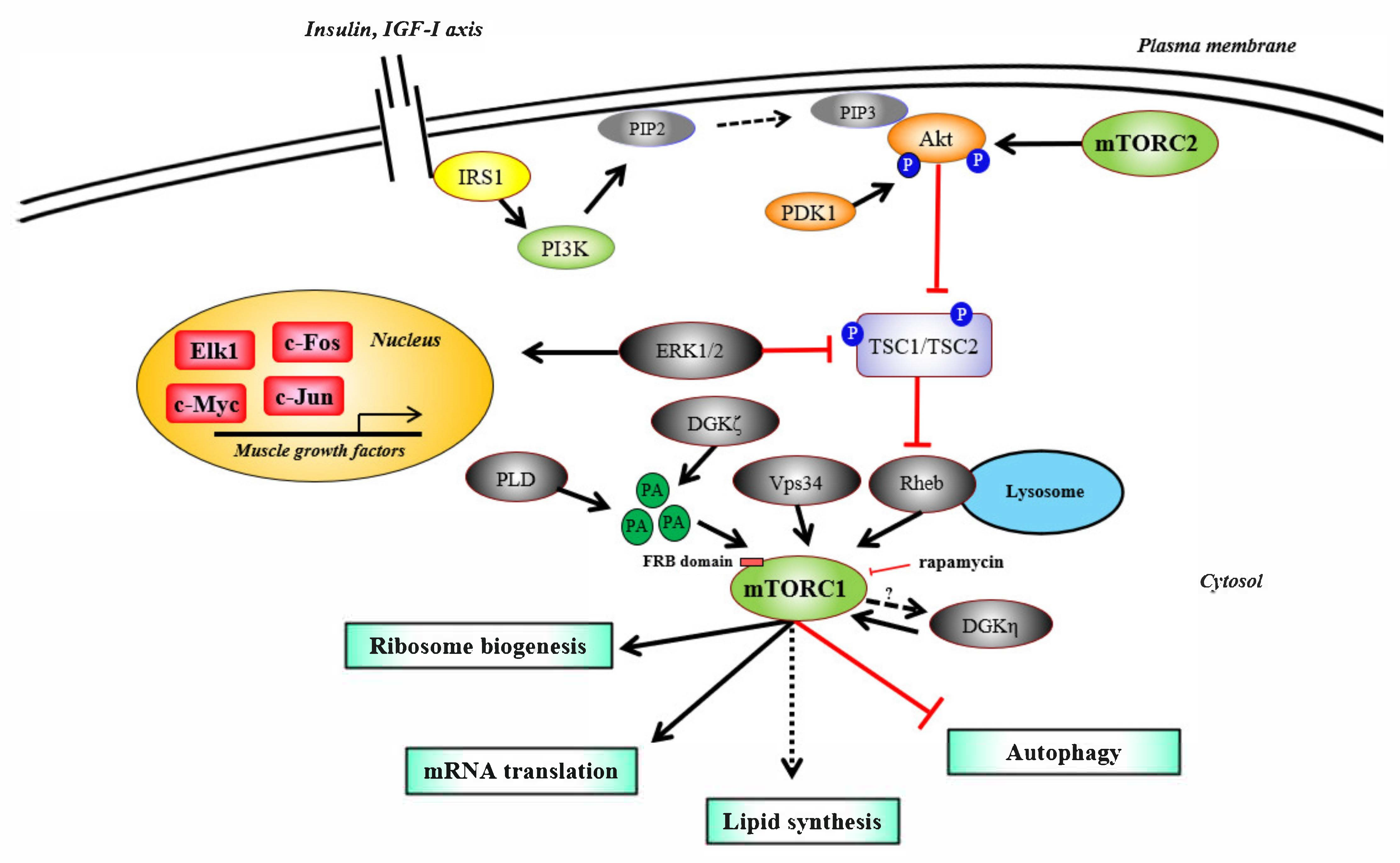
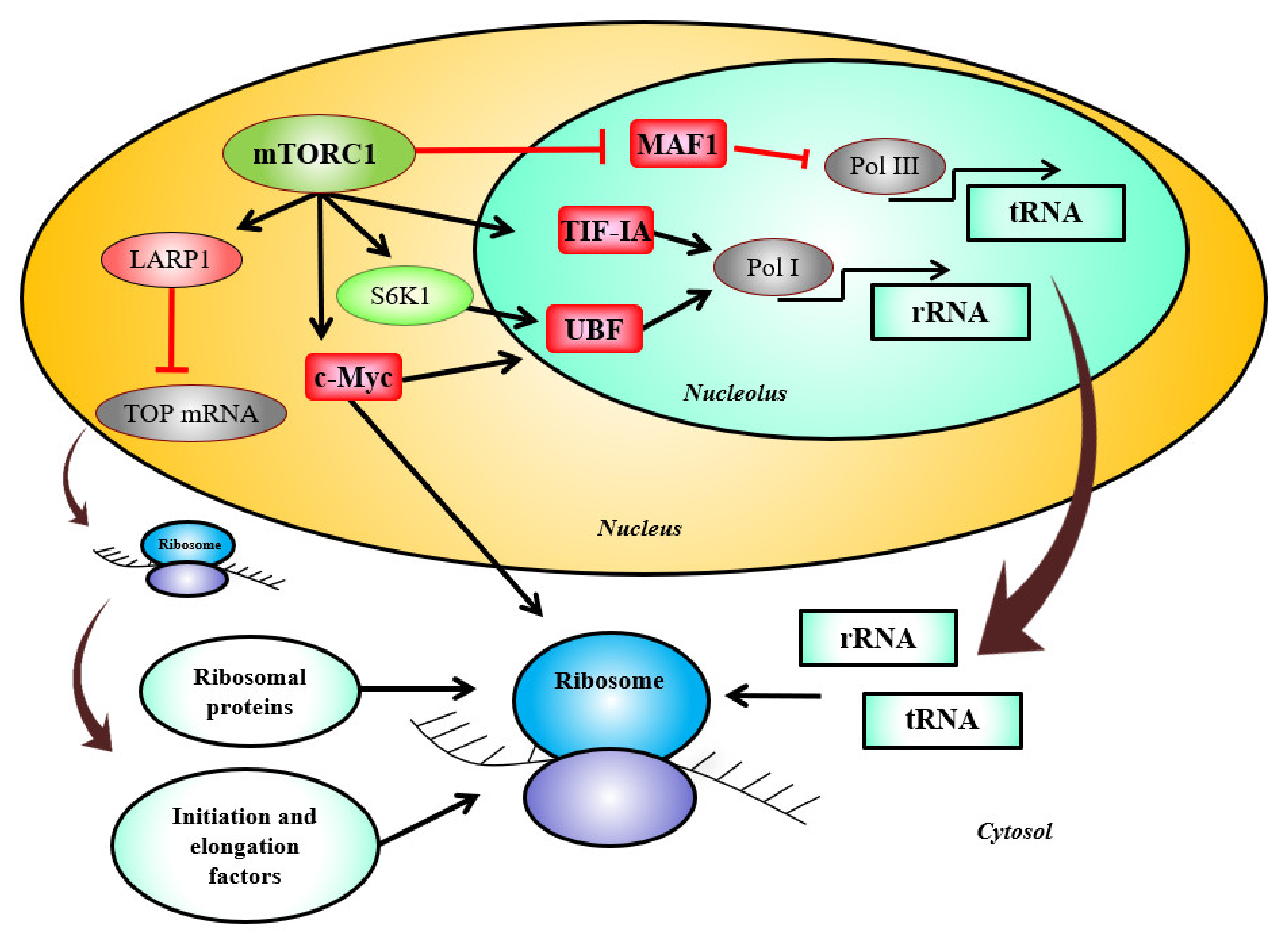
2. mTORC1 Signaling, Its Regulators, and Ribosome Biogenesis
3. eIF3f and mTORC1 in Skeletal Muscle Function
4. Implication of Satellite Cells and Myonuclear Accretion
5. Impact of Exercise Training and Practical Recommendations
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van der Meij, B.S.; Teleni, L.; McCarthy, A.L.; Isenring, E.A. Cancer Cachexia: An Overview of Diagnostic Criteria and Therapeutic Approaches for the Accredited Practicing Dietitian. J. Hum. Nutr. Diet 2020. [Google Scholar] [CrossRef]
- Qaisar, R.; Karim, A.; Muhammad, T.; Shah, I. Circulating Biomarkers of Accelerated Sarcopenia in Respiratory Diseases. Biology 2020, 9, 322. [Google Scholar] [CrossRef] [PubMed]
- Merz, K.E.; Thurmond, D.C. Role of Skeletal Muscle in Insulin Resistance and Glucose Uptake. Compr. Physiol. 2020, 10, 785–809. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.B.; Ramos, G.H.A.; Petterle, R.R.; Borba, V.Z.C. Sarcopenia as an Early Complication of Patients with Head and Neck Cancer with Dysphagia. Available online: https://pubmed.ncbi.nlm.nih.gov/33043532/ (accessed on 15 October 2020).
- Tuttle, C.S.L.; Thang, L.A.N.; Maier, A.B. Markers of Inflammation and Their Association with Muscle Strength and Mass: A Systematic Review and Meta-Analysis. Ageing Res. Rev. 2020, 64, 101185. [Google Scholar] [CrossRef]
- Avola, M.; Mangano, G.R.A.; Testa, G.; Mangano, S.; Vescio, A.; Pavone, V.; Vecchio, M. Rehabilitation Strategies for Patients with Femoral Neck Fractures in Sarcopenia: A Narrative Review. J. Clin. Med. 2020, 9, 3115. [Google Scholar] [CrossRef] [PubMed]
- Angulo, J.; El Assar, M.; Álvarez-Bustos, A.; Rodríguez-Mañas, L. Physical Activity and Exercise: Strategies to Manage Frailty. Redox Biol. 2020, 101513. [Google Scholar] [CrossRef] [PubMed]
- Borzuola, R.; Giombini, A.; Torre, G.; Campi, S.; Albo, E.; Bravi, M.; Borrione, P.; Fossati, C.; Macaluso, A. Central and Peripheral Neuromuscular Adaptations to Ageing. J. Clin. Med. 2020, 9, 741. [Google Scholar] [CrossRef]
- Baggerman, M.R.; van Dijk, D.P.J.; Winkens, B.; van Gassel, R.J.J.; Bol, M.E.; Schnabel, R.M.; Bakers, F.C.; Olde Damink, S.W.M.; van de Poll, M.C.G. Muscle Wasting Associated Co-Morbidities, Rather than Sarcopenia Are Risk Factors for Hospital Mortality in Critical Illness. J. Crit. Care 2020, 56, 31–36. [Google Scholar] [CrossRef]
- Baehr, L.M.; West, D.W.D.; Marcotte, G.; Marshall, A.G.; De Sousa, L.G.; Baar, K.; Bodine, S.C. Age-Related Deficits in Skeletal Muscle Recovery Following Disuse Are Associated with Neuromuscular Junction Instability and ER Stress, Not Impaired Protein Synthesis. Aging 2016, 8, 127–146. [Google Scholar] [CrossRef]
- Vainshtein, A.; Sandri, M. Signaling Pathways That Control Muscle Mass. Int. J. Mol. Sci. 2020, 21, 4759. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, Y.; Yoshioka, K.; Suzuki, N. The Ubiquitin-Proteasome System in Regulation of the Skeletal Muscle Homeostasis and Atrophy: From Basic Science to Disorders. J. Physiol. Sci. 2020, 70, 40. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.M.J.; Candau, R.B.; Bernardi, H. FoxO Transcription Factors: Their Roles in the Maintenance of Skeletal Muscle Homeostasis. Cell. Mol. Life Sci. 2014, 71, 1657–1671. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.M.; Candau, R.; Bernardi, H. Recent Data on Cellular Component Turnover: Focus on Adaptations to Physical Exercise. Cells 2019, 8, 542. [Google Scholar] [CrossRef] [PubMed]
- Hodson, N.; West, D.W.D.; Philp, A.; Burd, N.A.; Moore, D.R. Molecular Regulation of Human Skeletal Muscle Protein Synthesis in Response to Exercise and Nutrients: A Compass for Overcoming Age-Related Anabolic Resistance. Am. J. Physiol. Cell Physiol. 2019, 317, C1061–C1078. [Google Scholar] [CrossRef] [PubMed]
- Goodman, C.A. Role of MTORC1 in Mechanically Induced Increases in Translation and Skeletal Muscle Mass. J. Appl. Physiol. (1985) 2019, 127, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Hoppeler, H. Molecular Networks in Skeletal Muscle Plasticity. J. Exp. Biol. 2016, 219, 205–213. [Google Scholar] [CrossRef] [PubMed]
- McGlory, C.; Phillips, S.M. Exercise and the Regulation of Skeletal Muscle Hypertrophy. Prog. Mol. Biol. Transl. Sci. 2015, 135, 153–173. [Google Scholar] [CrossRef]
- Goodman, C.A. The Role of MTORC1 in Regulating Protein Synthesis and Skeletal Muscle Mass in Response to Various Mechanical Stimuli. Rev. Physiol. Biochem. Pharmacol. 2014, 166, 43–95. [Google Scholar] [CrossRef] [PubMed]
- Adegoke, O.A.J.; Abdullahi, A.; Tavajohi-Fini, P. MTORC1 and the Regulation of Skeletal Muscle Anabolism and Mass. Appl. Physiol. Nutr. Metab. 2012, 37, 395–406. [Google Scholar] [CrossRef]
- Sanchez, A.M.J.; Bernardi, H.; Py, G.; Candau, R.B. Autophagy Is Essential to Support Skeletal Muscle Plasticity in Response to Endurance Exercise. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R956–R969. [Google Scholar] [CrossRef] [PubMed]
- Hornberger, T.A. Mechanotransduction and the Regulation of MTORC1 Signaling in Skeletal Muscle. Int. J. Biochem. Cell Biol. 2011, 43, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.M.J.; Csibi, A.; Raibon, A.; Cornille, K.; Gay, S.; Bernardi, H.; Candau, R. AMPK Promotes Skeletal Muscle Autophagy through Activation of Forkhead FoxO3a and Interaction with Ulk1. J. Cell. Biochem. 2012, 113, 695–710. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.M.J.; Candau, R.B.; Csibi, A.; Pagano, A.F.; Raibon, A.; Bernardi, H. The Role of AMP-Activated Protein Kinase in the Coordination of Skeletal Muscle Turnover and Energy Homeostasis. Am. J. Physiol. Cell Physiol. 2012, 303, C475–C485. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and MTOR Regulate Autophagy through Direct Phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Guan, K.-L. Regulation of the Autophagy Initiating Kinase ULK1 by Nutrients: Roles of MTORC1 and AMPK. Cell Cycle 2011, 10, 1337–1338. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.; Kim, J.; Shaw, R.J.; Guan, K.-L. The Autophagy Initiating Kinase ULK1 Is Regulated via Opposing Phosphorylation by AMPK and MTOR. Autophagy 2011, 7, 643–644. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.M.J.; Candau, R.; Raibon, A.; Bernardi, H. Autophagy, a Highly Regulated Intracellular System Essential to Skeletal Muscle Homeostasis—Role in Disease, Exercise and Altitude Exposure. Muscle Cell Tissue 2015. [Google Scholar] [CrossRef]
- Sharples, A.P.; Hughes, D.C.; Deane, C.S.; Saini, A.; Selman, C.; Stewart, C.E. Longevity and Skeletal Muscle Mass: The Role of IGF Signalling, the Sirtuins, Dietary Restriction and Protein Intake. Aging Cell 2015, 14, 511–523. [Google Scholar] [CrossRef]
- Zhang, Q.; Duplany, A.; Moncollin, V.; Mouradian, S.; Goillot, E.; Mazelin, L.; Gauthier, K.; Streichenberger, N.; Angleraux, C.; Chen, J.; et al. Lack of Muscle MTOR Kinase Activity Causes Early Onset Myopathy and Compromises Whole-body Homeostasis. J. Cachexia Sarcopenia Muscle 2019, 10, 35–53. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Liang, X.; Shan, T.; Jiang, Q.; Deng, C.; Zheng, R.; Kuang, S. MTOR Is Necessary for Proper Satellite Cell Activity and Skeletal Muscle Regeneration. Biochem. Biophys. Res. Commun. 2015, 463, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Jash, S.; Dhar, G.; Ghosh, U.; Adhya, S. Role of the MTORC1 Complex in Satellite Cell Activation by RNA-Induced Mitochondrial Restoration: Dual Control of Cyclin D1 through MicroRNAs. Mol. Cell Biol. 2014, 34, 3594–3606. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Zhi, R.; Yang, Z.; Li, H.; Yan, H.; Wang, X. Low Dose of IGF-I Increases Cell Size of Skeletal Muscle Satellite Cells via Akt/S6K Signaling Pathway. J. Cell Biochem. 2015, 116, 2637–2648. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, V.C.; McCarthy, J.J. Regulation of Ribosome Biogenesis in Skeletal Muscle Hypertrophy. Physiology 2019, 34, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Chaillou, T.; Kirby, T.J.; McCarthy, J.J. Ribosome Biogenesis: Emerging Evidence for a Central Role in the Regulation of Skeletal Muscle Mass. J. Cell Physiol. 2014, 229, 1584–1594. [Google Scholar] [CrossRef]
- Ferrara-Romeo, I.; Martinez, P.; Saraswati, S.; Whittemore, K.; Graña-Castro, O.; Thelma Poluha, L.; Serrano, R.; Hernandez-Encinas, E.; Blanco-Aparicio, C.; Maria Flores, J.; et al. The MTOR Pathway Is Necessary for Survival of Mice with Short Telomeres. Nat. Commun. 2020, 11, 1168. [Google Scholar] [CrossRef]
- Duval, A.P.; Jeanneret, C.; Santoro, T.; Dormond, O. MTOR and Tumor Cachexia. Int. J. Mol. Sci. 2018, 19, 2225. [Google Scholar] [CrossRef] [PubMed]
- Docquier, A.; Pavlin, L.; Raibon, A.; Bertrand-Gaday, C.; Sar, C.; Leibovitch, S.; Candau, R.; Bernardi, H. EIF3f Depletion Impedes Mouse Embryonic Development, Reduces Adult Skeletal Muscle Mass and Amplifies Muscle Loss during Disuse. J. Physiol. 2019, 597, 3107–3131. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.M.J.; Csibi, A.; Raibon, A.; Docquier, A.; Lagirand-Cantaloube, J.; Leibovitch, M.-P.; Leibovitch, S.A.; Bernardi, H. EIF3f: A Central Regulator of the Antagonism Atrophy/Hypertrophy in Skeletal Muscle. Int. J. Biochem. Cell Biol. 2013, 45, 2158–2162. [Google Scholar] [CrossRef] [PubMed]
- Hornberger, T.A.; Chu, W.K.; Mak, Y.W.; Hsiung, J.W.; Huang, S.A.; Chien, S. The Role of Phospholipase D and Phosphatidic Acid in the Mechanical Activation of MTOR Signaling in Skeletal Muscle. Proc. Natl. Acad. Sci. USA 2006, 103, 4741–4746. [Google Scholar] [CrossRef] [PubMed]
- You, J.S.; Frey, J.W.; Hornberger, T.A. Mechanical Stimulation Induces MTOR Signaling via an ERK-Independent Mechanism: Implications for a Direct Activation of MTOR by Phosphatidic Acid. PLoS ONE 2012, 7, e47258. [Google Scholar] [CrossRef]
- Veverka, V.; Crabbe, T.; Bird, I.; Lennie, G.; Muskett, F.W.; Taylor, R.J.; Carr, M.D. Structural Characterization of the Interaction of MTOR with Phosphatidic Acid and a Novel Class of Inhibitor: Compelling Evidence for a Central Role of the FRB Domain in Small Molecule-Mediated Regulation of MTOR. Oncogene 2008, 27, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.-S.; Sun, Y.; Arauz, E.; Jiang, Y.; Chen, J. Phosphatidic Acid Activates Mammalian Target of Rapamycin Complex 1 (MTORC1) Kinase by Displacing FK506 Binding Protein 38 (FKBP38) and Exerting an Allosteric Effect. J. Biol. Chem. 2011, 286, 29568–29574. [Google Scholar] [CrossRef]
- You, J.-S.; Lincoln, H.C.; Kim, C.-R.; Frey, J.W.; Goodman, C.A.; Zhong, X.-P.; Hornberger, T.A. The Role of Diacylglycerol Kinase ζ and Phosphatidic Acid in the Mechanical Activation of Mammalian Target of Rapamycin (MTOR) Signaling and Skeletal Muscle Hypertrophy. J. Biol. Chem. 2014, 289, 1551–1563. [Google Scholar] [CrossRef] [PubMed]
- You, J.-S.; Dooley, M.S.; Kim, C.-R.; Kim, E.-J.; Xu, W.; Goodman, C.A.; Hornberger, T.A. A DGKζ-FoxO-Ubiquitin Proteolytic Axis Controls Fiber Size during Skeletal Muscle Remodeling. Sci. Signal 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Sakai, H.; Murakami, C.; Usuki, T.; Lu, Q.; Matsumoto, K.-I.; Urano, T.; Sakane, F. Diacylglycerol Kinase η Regulates C2C12 Myoblast Proliferation through the MTOR Signaling Pathway. Biochimie 2020, 177, 13–24. [Google Scholar] [CrossRef]
- Murach, K.A.; Mobley, C.B.; Zdunek, C.J.; Frick, K.K.; Jones, S.R.; McCarthy, J.J.; Peterson, C.A.; Dungan, C.M. Muscle Memory: Myonuclear Accretion, Maintenance, Morphology, and MiRNA Levels with Training and Detraining in Adult Mice. J. Cachexia Sarcopenia Muscle 2020. [Google Scholar] [CrossRef]
- Moro, T.; Brightwell, C.R.; Volpi, E.; Rasmussen, B.B.; Fry, C.S. Resistance Exercise Training Promotes Fiber Type-Specific Myonuclear Adaptations in Older Adults. J. Appl. Physiol. (1985) 2020, 128, 795–804. [Google Scholar] [CrossRef]
- Dungan, C.M.; Murach, K.A.; Frick, K.K.; Jones, S.R.; Crow, S.E.; Englund, D.A.; Vechetti, I.J.; Figueiredo, V.C.; Levitan, B.M.; Satin, J.; et al. Elevated Myonuclear Density during Skeletal Muscle Hypertrophy in Response to Training Is Reversed during Detraining. Am. J. Physiol. Cell Physiol. 2019, 316, C649–C654. [Google Scholar] [CrossRef]
- Widmann, M.; Nieß, A.M.; Munz, B. Physical Exercise and Epigenetic Modifications in Skeletal Muscle. Sports Med. 2019, 49, 509–523. [Google Scholar] [CrossRef]
- Turner, D.C.; Seaborne, R.A.; Sharples, A.P. Comparative Transcriptome and Methylome Analysis in Human Skeletal Muscle Anabolism, Hypertrophy and Epigenetic Memory. Sci. Rep. 2019, 9, 4251. [Google Scholar] [CrossRef] [PubMed]
- Hood, D.A.; Memme, J.M.; Oliveira, A.N.; Triolo, M. Maintenance of Skeletal Muscle Mitochondria in Health, Exercise, and Aging. Annu. Rev. Physiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Warner, S.O.; Yao, M.V.; Cason, R.L.; Winnick, J.J. Exercise-Induced Improvements to Whole Body Glucose Metabolism in Type 2 Diabetes: The Essential Role of the Liver. Front Endocrinol. 2020, 11, 567. [Google Scholar] [CrossRef] [PubMed]
- Khoshnaw, D.M.; Ghadge, A.A. Yoga as a Complementary Therapy for Metabolic Syndrome: A Narrative Review. J. Integr. Med. 2020. [Google Scholar] [CrossRef]
- Wang, X.; Kang, J.; Liu, Q.; Tong, T.; Quan, H. Fighting Diabetes Mellitus: Pharmacological and Non-Pharmacological Approaches. Curr. Pharm. Des. 2020. [Google Scholar] [CrossRef] [PubMed]
- Philippe, A.G.; Borrani, F.; Sanchez, A.M.; Py, G.; Candau, R. Modelling Performance and Skeletal Muscle Adaptations with Exponential Growth Functions during Resistance Training. J. Sports Sci. 2018, 1–8. [Google Scholar] [CrossRef]
- Stec, M.J.; Kelly, N.A.; Many, G.M.; Windham, S.T.; Tuggle, S.C.; Bamman, M.M. Ribosome Biogenesis May Augment Resistance Training-Induced Myofiber Hypertrophy and Is Required for Myotube Growth in Vitro. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E652–E661. [Google Scholar] [CrossRef]
- Stec, M.J.; Mayhew, D.L.; Bamman, M.M. The Effects of Age and Resistance Loading on Skeletal Muscle Ribosome Biogenesis. J. Appl. Physiol. 2015, 119, 851–857. [Google Scholar] [CrossRef]
- Schoenfeld, B.J.; Ogborn, D.; Krieger, J.W. Dose-Response Relationship between Weekly Resistance Training Volume and Increases in Muscle Mass: A Systematic Review and Meta-Analysis. J. Sports Sci. 2017, 35, 1073–1082. [Google Scholar] [CrossRef]
- Figueiredo, V.C.; de Salles, B.F.; Trajano, G.S. Volume for Muscle Hypertrophy and Health Outcomes: The Most Effective Variable in Resistance Training. Sports Med. 2018, 48, 499–505. [Google Scholar] [CrossRef]
- Hammarström, D.; Øfsteng, S.; Koll, L.; Hanestadhaugen, M.; Hollan, I.; Apro, W.; Whist, J.E.; Blomstrand, E.; Rønnestad, B.R.; Ellefsen, S. Benefits of Higher Resistance-Training Volume Are Related to Ribosome Biogenesis. J. Physiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Sabers, C.J.; Martin, M.M.; Brunn, G.J.; Williams, J.M.; Dumont, F.J.; Wiederrecht, G.; Abraham, R.T. Isolation of a Protein Target of the FKBP12-Rapamycin Complex in Mammalian Cells. J. Biol. Chem. 1995, 270, 815–822. [Google Scholar] [CrossRef]
- Loewith, R.; Jacinto, E.; Wullschleger, S.; Lorberg, A.; Crespo, J.L.; Bonenfant, D.; Oppliger, W.; Jenoe, P.; Hall, M.N. Two TOR Complexes, Only One of Which Is Rapamycin Sensitive, Have Distinct Roles in Cell Growth Control. Mol. Cell 2002, 10, 457–468. [Google Scholar] [CrossRef]
- Hara, K.; Maruki, Y.; Long, X.; Yoshino, K.; Oshiro, N.; Hidayat, S.; Tokunaga, C.; Avruch, J.; Yonezawa, K. Raptor, a Binding Partner of Target of Rapamycin (TOR), Mediates TOR Action. Cell 2002, 110, 177–189. [Google Scholar] [CrossRef]
- Kim, D.-H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. MTOR Interacts with Raptor to Form a Nutrient-Sensitive Complex That Signals to the Cell Growth Machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Kim, D.-H.; Sarbassov, D.D.; Ali, S.M.; Latek, R.R.; Guntur, K.V.P.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. GbetaL, a Positive Regulator of the Rapamycin-Sensitive Pathway Required for the Nutrient-Sensitive Interaction between Raptor and MTOR. Mol. Cell 2003, 11, 895–904. [Google Scholar] [CrossRef]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR Is an MTOR Inhibitor Frequently Overexpressed in Multiple Myeloma Cells and Required for Their Survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 Is an Insulin-Regulated Inhibitor of the MTORC1 Protein Kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef]
- Vander Haar, E.; Lee, S.-I.; Bandhakavi, S.; Griffin, T.J.; Kim, D.-H. Insulin Signalling to MTOR Mediated by the Akt/PKB Substrate PRAS40. Nat. Cell Biol. 2007, 9, 316–323. [Google Scholar] [CrossRef]
- Bodine, S.C.; Stitt, T.N.; Gonzalez, M.; Kline, W.O.; Stover, G.L.; Bauerlein, R.; Zlotchenko, E.; Scrimgeour, A.; Lawrence, J.C.; Glass, D.J.; et al. Akt/MTOR Pathway Is a Crucial Regulator of Skeletal Muscle Hypertrophy and Can Prevent Muscle Atrophy in Vivo. Nat. Cell Biol. 2001, 3, 1014–1019. [Google Scholar] [CrossRef]
- Rommel, C.; Bodine, S.C.; Clarke, B.A.; Rossman, R.; Nunez, L.; Stitt, T.N.; Yancopoulos, G.D.; Glass, D.J. Mediation of IGF-1-Induced Skeletal Myotube Hypertrophy by PI(3)K/Akt/MTOR and PI(3)K/Akt/GSK3 Pathways. Nat. Cell Biol. 2001, 3, 1009–1013. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.-H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a Novel Binding Partner of MTOR, Defines a Rapamycin-Insensitive and Raptor-Independent Pathway That Regulates the Cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Rüegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR Complex 2 Controls the Actin Cytoskeleton and Is Rapamycin Insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef]
- Chen, X.; Liu, M.; Tian, Y.; Li, J.; Qi, Y.; Zhao, D.; Wu, Z.; Huang, M.; Wong, C.C.L.; Wang, H.-W.; et al. Cryo-EM Structure of Human MTOR Complex 2. Cell Res. 2018, 28, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Lamming, D.W.; Ye, L.; Katajisto, P.; Goncalves, M.D.; Saitoh, M.; Stevens, D.M.; Davis, J.G.; Salmon, A.B.; Richardson, A.; Ahima, R.S.; et al. Rapamycin-Induced Insulin Resistance Is Mediated by MTORC2 Loss and Uncoupled from Longevity. Science 2012, 335, 1638–1643. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.-H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged Rapamycin Treatment Inhibits MTORC2 Assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Kleinert, M.; Parker, B.L.; Fritzen, A.M.; Knudsen, J.R.; Jensen, T.E.; Kjøbsted, R.; Sylow, L.; Ruegg, M.; James, D.E.; Richter, E.A. Mammalian Target of Rapamycin Complex 2 Regulates Muscle Glucose Uptake during Exercise in Mice. J. Physiol. 2017, 595, 4845–4855. [Google Scholar] [CrossRef]
- Hodson, N.; McGlory, C.; Oikawa, S.Y.; Jeromson, S.; Song, Z.; Rüegg, M.A.; Hamilton, D.L.; Phillips, S.M.; Philp, A. Differential Localization and Anabolic Responsiveness of MTOR Complexes in Human Skeletal Muscle in Response to Feeding and Exercise. Am. J. Physiol. Cell Physiol. 2017, 313, C604–C611. [Google Scholar] [CrossRef] [PubMed]
- Bentzinger, C.F.; Lin, S.; Romanino, K.; Castets, P.; Guridi, M.; Summermatter, S.; Handschin, C.; Tintignac, L.A.; Hall, M.N.; Rüegg, M.A. Differential Response of Skeletal Muscles to MTORC1 Signaling during Atrophy and Hypertrophy. Skeletal. Muscle 2013, 3, 6. [Google Scholar] [CrossRef]
- Dickinson, J.M.; Fry, C.S.; Drummond, M.J.; Gundermann, D.M.; Walker, D.K.; Glynn, E.L.; Timmerman, K.L.; Dhanani, S.; Volpi, E.; Rasmussen, B.B. Mammalian Target of Rapamycin Complex 1 Activation Is Required for the Stimulation of Human Skeletal Muscle Protein Synthesis by Essential Amino Acids. J. Nutr. 2011, 141, 856–862. [Google Scholar] [CrossRef]
- Drummond, M.J.; Fry, C.S.; Glynn, E.L.; Dreyer, H.C.; Dhanani, S.; Timmerman, K.L.; Volpi, E.; Rasmussen, B.B. Rapamycin Administration in Humans Blocks the Contraction-Induced Increase in Skeletal Muscle Protein Synthesis. J. Physiol. 2009, 587, 1535–1546. [Google Scholar] [CrossRef]
- Risson, V.; Mazelin, L.; Roceri, M.; Sanchez, H.; Moncollin, V.; Corneloup, C.; Richard-Bulteau, H.; Vignaud, A.; Baas, D.; Defour, A.; et al. Muscle Inactivation of MTOR Causes Metabolic and Dystrophin Defects Leading to Severe Myopathy. J. Cell Biol. 2009, 187, 859–874. [Google Scholar] [CrossRef] [PubMed]
- Bentzinger, C.F.; Romanino, K.; Cloëtta, D.; Lin, S.; Mascarenhas, J.B.; Oliveri, F.; Xia, J.; Casanova, E.; Costa, C.F.; Brink, M.; et al. Skeletal Muscle-Specific Ablation of Raptor, but Not of Rictor, Causes Metabolic Changes and Results in Muscle Dystrophy. Cell Metab. 2008, 8, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Rion, N.; Castets, P.; Lin, S.; Enderle, L.; Reinhard, J.R.; Eickhorst, C.; Rüegg, M.A. MTOR Controls Embryonic and Adult Myogenesis via MTORC1. Development 2019, 146. [Google Scholar] [CrossRef] [PubMed]
- Ham, A.S.; Chojnowska, K.; Tintignac, L.A.; Lin, S.; Schmidt, A.; Ham, D.J.; Sinnreich, M.; Rüegg, M.A. MTORC1 Signalling Is Not Essential for the Maintenance of Muscle Mass and Function in Adult Sedentary Mice. J. Cachexia Sarcopenia Muscle 2020, 11, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Goodman, C.A.; Frey, J.W.; Mabrey, D.M.; Jacobs, B.L.; Lincoln, H.C.; You, J.-S.; Hornberger, T.A. The Role of Skeletal Muscle MTOR in the Regulation of Mechanical Load-Induced Growth. J. Physiol. 2011, 589, 5485–5501. [Google Scholar] [CrossRef]
- Goodman, C.A.; Miu, M.H.; Frey, J.W.; Mabrey, D.M.; Lincoln, H.C.; Ge, Y.; Chen, J.; Hornberger, T.A. A Phosphatidylinositol 3-Kinase/Protein Kinase B-Independent Activation of Mammalian Target of Rapamycin Signaling Is Sufficient to Induce Skeletal Muscle Hypertrophy. Mol. Biol. Cell 2010, 21, 3258–3268. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Inoki, K.; Lee, M.; Wright, E.; Khuong, A.; Khuong, A.; Sugiarto, S.; Garner, M.; Paik, J.; DePinho, R.A.; et al. MTORC1 Promotes Denervation-Induced Muscle Atrophy through a Mechanism Involving the Activation of FoxO and E3 Ubiquitin Ligases. Sci. Signal 2014, 7, ra18. [Google Scholar] [CrossRef]
- Fok, W.C.; Zhang, Y.; Salmon, A.B.; Bhattacharya, A.; Gunda, R.; Jones, D.; Ward, W.; Fisher, K.; Richardson, A.; Pérez, V.I. Short-Term Treatment with Rapamycin and Dietary Restriction Have Overlapping and Distinctive Effects in Young Mice. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Neff, F.; Flores-Dominguez, D.; Ryan, D.P.; Horsch, M.; Schröder, S.; Adler, T.; Afonso, L.C.; Aguilar-Pimentel, J.A.; Becker, L.; Garrett, L.; et al. Rapamycin Extends Murine Lifespan but Has Limited Effects on Aging. J. Clin. Investig. 2013, 123, 3272–3291. [Google Scholar] [CrossRef]
- Zhang, Y.; Bokov, A.; Gelfond, J.; Soto, V.; Ikeno, Y.; Hubbard, G.; Diaz, V.; Sloane, L.; Maslin, K.; Treaster, S.; et al. Rapamycin Extends Life and Health in C57BL/6 Mice. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 119–130. [Google Scholar] [CrossRef]
- Baar, K.; Esser, K. Phosphorylation of P70(S6k) Correlates with Increased Skeletal Muscle Mass Following Resistance Exercise. Am. J. Physiol. 1999, 276, C120–C127. [Google Scholar] [CrossRef] [PubMed]
- Eliasson, J.; Elfegoun, T.; Nilsson, J.; Köhnke, R.; Ekblom, B.; Blomstrand, E. Maximal Lengthening Contractions Increase P70 S6 Kinase Phosphorylation in Human Skeletal Muscle in the Absence of Nutritional Supply. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E1197–E1205. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.A.; Nkengfac, B.; Sanchez, A.M.J.; Moroz, N.; Qureshi, S.T.; Koromilas, A.E.; Wang, S.; Burelle, Y.; Hussain, S.N.; Kristof, A.S. Regulation of ULK1 Expression and Autophagy by STAT1. J. Biol. Chem. 2017, 292, 1899–1909. [Google Scholar] [CrossRef]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.-Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.-L. ULK1 Induces Autophagy by Phosphorylating Beclin-1 and Activating VPS34 Lipid Kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Castets, P.; Lin, S.; Rion, N.; Di Fulvio, S.; Romanino, K.; Guridi, M.; Frank, S.; Tintignac, L.A.; Sinnreich, M.; Rüegg, M.A. Sustained Activation of MTORC1 in Skeletal Muscle Inhibits Constitutive and Starvation-Induced Autophagy and Causes a Severe, Late-Onset Myopathy. Cell Metab. 2013, 17, 731–744. [Google Scholar] [CrossRef] [PubMed]
- Hornberger, T.A.; Stuppard, R.; Conley, K.E.; Fedele, M.J.; Fiorotto, M.L.; Chin, E.R.; Esser, K.A. Mechanical Stimuli Regulate Rapamycin-Sensitive Signalling by a Phosphoinositide 3-Kinase-, Protein Kinase B- and Growth Factor-Independent Mechanism. Biochem. J. 2004, 380, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Hornberger, T.A.; Chien, S. Mechanical Stimuli and Nutrients Regulate Rapamycin-Sensitive Signaling through Distinct Mechanisms in Skeletal Muscle. J. Cell. Biochem. 2006, 97, 1207–1216. [Google Scholar] [CrossRef]
- West, D.W.D.; Kujbida, G.W.; Moore, D.R.; Atherton, P.; Burd, N.A.; Padzik, J.P.; De Lisio, M.; Tang, J.E.; Parise, G.; Rennie, M.J.; et al. Resistance Exercise-Induced Increases in Putative Anabolic Hormones Do Not Enhance Muscle Protein Synthesis or Intracellular Signalling in Young Men. J. Physiol. 2009, 587, 5239–5247. [Google Scholar] [CrossRef]
- Araki, E.; Lipes, M.A.; Patti, M.E.; Brüning, J.C.; Haag, B.; Johnson, R.S.; Kahn, C.R. Alternative Pathway of Insulin Signalling in Mice with Targeted Disruption of the IRS-1 Gene. Nature 1994, 372, 186–190. [Google Scholar] [CrossRef]
- Myers, M.G.; Grammer, T.C.; Wang, L.M.; Sun, X.J.; Pierce, J.H.; Blenis, J.; White, M.F. Insulin Receptor Substrate-1 Mediates Phosphatidylinositol 3’-Kinase and P70S6k Signaling during Insulin, Insulin-like Growth Factor-1, and Interleukin-4 Stimulation. J. Biol. Chem. 1994, 269, 28783–28789. [Google Scholar] [CrossRef]
- Kadowaki, T.; Tobe, K.; Honda-Yamamoto, R.; Tamemoto, H.; Kaburagi, Y.; Momomura, K.; Ueki, K.; Takahashi, Y.; Yamauchi, T.; Akanuma, Y.; et al. Signal Transduction Mechanism of Insulin and Insulin-like Growth Factor-1. Endocr. J. 1996, 43, S33–S41. [Google Scholar] [CrossRef]
- Eckstein, S.S.; Weigert, C.; Lehmann, R. Divergent Roles of IRS (Insulin Receptor Substrate) 1 and 2 in Liver and Skeletal Muscle. Curr. Med. Chem. 2017, 24, 1827–1852. [Google Scholar] [CrossRef]
- Garami, A.; Zwartkruis, F.J.T.; Nobukuni, T.; Joaquin, M.; Roccio, M.; Stocker, H.; Kozma, S.C.; Hafen, E.; Bos, J.L.; Thomas, G. Insulin Activation of Rheb, a Mediator of MTOR/S6K/4E-BP Signaling, Is Inhibited by TSC1 and 2. Mol. Cell 2003, 11, 1457–1466. [Google Scholar] [CrossRef]
- Tomasoni, R.; Mondino, A. The Tuberous Sclerosis Complex: Balancing Proliferation and Survival. Biochem. Soc. Trans. 2011, 39, 466–471. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Toker, A.; Dibble, C.C. PI 3-Kinase Signaling: AKTing up inside the Cell. Mol. Cell 2018, 71, 875–876. [Google Scholar] [CrossRef]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; Cohen, P. Characterization of a 3-Phosphoinositide-Dependent Protein Kinase Which Phosphorylates and Activates Protein Kinase Balpha. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef]
- Stokoe, D.; Stephens, L.R.; Copeland, T.; Gaffney, P.R.; Reese, C.B.; Painter, G.F.; Holmes, A.B.; McCormick, F.; Hawkins, P.T. Dual Role of Phosphatidylinositol-3,4,5-Trisphosphate in the Activation of Protein Kinase B. Science 1997, 277, 567–570. [Google Scholar] [CrossRef] [PubMed]
- Castro, A.F.; Rebhun, J.F.; Clark, G.J.; Quilliam, L.A. Rheb Binds Tuberous Sclerosis Complex 2 (TSC2) and Promotes S6 Kinase Activation in a Rapamycin- and Farnesylation-Dependent Manner. J. Biol. Chem. 2003, 278, 32493–32496. [Google Scholar] [CrossRef]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 Mediates Cellular Energy Response to Control Cell Growth and Survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and Regulation of Akt/PKB by the Rictor-MTOR Complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Spangenburg, E.E.; Le Roith, D.; Ward, C.W.; Bodine, S.C. A Functional Insulin-like Growth Factor Receptor Is Not Necessary for Load-Induced Skeletal Muscle Hypertrophy. J. Physiol. 2008, 586, 283–291. [Google Scholar] [CrossRef]
- Miyazaki, M.; Moriya, N.; Takemasa, T. Transient Activation of MTORC1 Signaling in Skeletal Muscle Is Independent of Akt1 Regulation. Physiol. Rep. 2020, 8, e14599. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, M.; McCarthy, J.J.; Fedele, M.J.; Esser, K.A. Early Activation of MTORC1 Signalling in Response to Mechanical Overload Is Independent of Phosphoinositide 3-Kinase/Akt Signalling. J. Physiol. 2011, 589, 1831–1846. [Google Scholar] [CrossRef]
- Davis, R.J. The Mitogen-Activated Protein Kinase Signal Transduction Pathway. J. Biol. Chem. 1993, 268, 14553–14556. [Google Scholar] [CrossRef]
- Ogasawara, R.; Fujita, S.; Hornberger, T.A.; Kitaoka, Y.; Makanae, Y.; Nakazato, K.; Naokata, I. The Role of MTOR Signalling in the Regulation of Skeletal Muscle Mass in a Rodent Model of Resistance Exercise. Sci. Rep. 2016, 6, 31142. [Google Scholar] [CrossRef]
- Wen, Y.; Alimov, A.; McCarthy, J. Ribosome Biogenesis Is Necessary for Skeletal Muscle Hypertrophy. Exerc. Sport Sci. Rev. 2016, 44, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Aubert, M.; O’Donohue, M.-F.; Lebaron, S.; Gleizes, P.-E. Pre-Ribosomal RNA Processing in Human Cells: From Mechanisms to Congenital Diseases. Biomolecules 2018, 8, 123. [Google Scholar] [CrossRef] [PubMed]
- Moss, T.; Stefanovsky, V.Y. Promotion and Regulation of Ribosomal Transcription in Eukaryotes by RNA Polymerase I. Prog. Nucleic Acid Res. Mol. Biol. 1995, 50, 25–66. [Google Scholar] [CrossRef] [PubMed]
- Geiss, G.K.; Radebaugh, C.A.; Paule, M.R. The Fundamental Ribosomal RNA Transcription Initiation Factor-IB (TIF-IB, SL1, Factor D) Binds to the RRNA Core Promoter Primarily by Minor Groove Contacts. J. Biol. Chem. 1997, 272, 29243–29254. [Google Scholar] [CrossRef]
- Saito, M.; Mitani, A.; Ishimori, T.; Miyashita, N.; Isago, H.; Mikami, Y.; Noguchi, S.; Tarui, M.; Nagase, T. Active MTOR in Lung Epithelium Promotes Epithelial-Mesenchymal Transition and Enhances Lung Fibrosis. Am. J. Respir Cell Mol. Biol. 2020, 62, 699–708. [Google Scholar] [CrossRef]
- Voit, R.; Grummt, I. Phosphorylation of UBF at Serine 388 Is Required for Interaction with RNA Polymerase I and Activation of RDNA Transcription. Proc. Natl. Acad. Sci. USA 2001, 98, 13631–13636. [Google Scholar] [CrossRef]
- Mayer, C.; Zhao, J.; Yuan, X.; Grummt, I. MTOR-Dependent Activation of the Transcription Factor TIF-IA Links RRNA Synthesis to Nutrient Availability. Genes Dev. 2004, 18, 423–434. [Google Scholar] [CrossRef]
- Stefanovsky, V.Y.; Langlois, F.; Bazett-Jones, D.; Pelletier, G.; Moss, T. ERK Modulates DNA Bending and Enhancesome Structure by Phosphorylating HMG1-Boxes 1 and 2 of the RNA Polymerase I Transcription Factor UBF. Biochemistry 2006, 45, 3626–3634. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, S.; Bierhoff, H.; Cado, I.; Weber, A.; Tiebe, M.; Grummt, I.; Voit, R. AMP-Activated Protein Kinase Adapts RRNA Synthesis to Cellular Energy Supply. Proc. Natl. Acad. Sci. USA 2009, 106, 17781–17786. [Google Scholar] [CrossRef]
- Iadevaia, V.; Liu, R.; Proud, C.G. MTORC1 Signaling Controls Multiple Steps in Ribosome Biogenesis. Semin. Cell Dev. Biol. 2014, 36, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Michels, A.A.; Robitaille, A.M.; Buczynski-Ruchonnet, D.; Hodroj, W.; Reina, J.H.; Hall, M.N.; Hernandez, N. MTORC1 Directly Phosphorylates and Regulates Human MAF1. Mol. Cell Biol. 2010, 30, 3749–3757. [Google Scholar] [CrossRef] [PubMed]
- Shor, B.; Wu, J.; Shakey, Q.; Toral-Barza, L.; Shi, C.; Follettie, M.; Yu, K. Requirement of the MTOR Kinase for the Regulation of Maf1 Phosphorylation and Control of RNA Polymerase III-Dependent Transcription in Cancer Cells. J. Biol. Chem. 2010, 285, 15380–15392. [Google Scholar] [CrossRef]
- Berman, A.J.; Thoreen, C.C.; Dedeic, Z.; Chettle, J.; Roux, P.P.; Blagden, S.P. Controversies around the Function of LARP1. RNA Biology 2020, 1–11. [Google Scholar] [CrossRef]
- Schiaffino, S.; Reggiani, C.; Akimoto, T.; Blaauw, B. Molecular Mechanisms of Skeletal Muscle Hypertrophy. J. Neuromuscul. Dis. 2020, 1–15. [Google Scholar] [CrossRef]
- Fonseca, B.D.; Zakaria, C.; Jia, J.-J.; Graber, T.E.; Svitkin, Y.; Tahmasebi, S.; Healy, D.; Hoang, H.-D.; Jensen, J.M.; Diao, I.T.; et al. La-Related Protein 1 (LARP1) Represses Terminal Oligopyrimidine (TOP) MRNA Translation Downstream of MTOR Complex 1 (MTORC1). J. Biol. Chem. 2015, 290, 15996–16020. [Google Scholar] [CrossRef] [PubMed]
- Van Riggelen, J.; Yetil, A.; Felsher, D.W. MYC as a Regulator of Ribosome Biogenesis and Protein Synthesis. Nat. Rev. Cancer 2010, 10, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.J.; White, R.J. MYC Regulation of Cell Growth through Control of Transcription by RNA Polymerases I and III. Cold Spring Harb. Perspect. Med. 2014, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Brook, M.S.; Wilkinson, D.J.; Smith, K.; Atherton, P.J. It’s Not Just about Protein Turnover: The Role of Ribosomal Biogenesis and Satellite Cells in the Regulation of Skeletal Muscle Hypertrophy. Eur. J. Sport Sci. 2019, 19, 952–963. [Google Scholar] [CrossRef]
- Poortinga, G.; Hannan, K.M.; Snelling, H.; Walkley, C.R.; Jenkins, A.; Sharkey, K.; Wall, M.; Brandenburger, Y.; Palatsides, M.; Pearson, R.B.; et al. MAD1 and C-MYC Regulate UBF and RDNA Transcription during Granulocyte Differentiation. EMBO J. 2004, 23, 3325–3335. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Zhou, W. TIF-IA: An Oncogenic Target of Pre-Ribosomal RNA Synthesis. Biochim. Biophys. Acta 2016, 1866, 189–196. [Google Scholar] [CrossRef]
- Grandori, C.; Gomez-Roman, N.; Felton-Edkins, Z.A.; Ngouenet, C.; Galloway, D.A.; Eisenman, R.N.; White, R.J. C-Myc Binds to Human Ribosomal DNA and Stimulates Transcription of RRNA Genes by RNA Polymerase I. Nat. Cell Biol. 2005, 7, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Fiorotto, M.L.; Davis, T.A.; Sosa, H.A.; Villegas-Montoya, C.; Estrada, I.; Fleischmann, R. Ribosome Abundance Regulates the Recovery of Skeletal Muscle Protein Mass upon Recuperation from Postnatal Undernutrition in Mice. J. Physiol. 2014, 592, 5269–5286. [Google Scholar] [CrossRef]
- Fyfe, J.J.; Bishop, D.J.; Bartlett, J.D.; Hanson, E.D.; Anderson, M.J.; Garnham, A.P.; Stepto, N.K. Enhanced Skeletal Muscle Ribosome Biogenesis, yet Attenuated MTORC1 and Ribosome Biogenesis-Related Signalling, Following Short-Term Concurrent versus Single-Mode Resistance Training. Sci. Rep. 2018, 8, 560. [Google Scholar] [CrossRef] [PubMed]
- Mobley, C.B.; Haun, C.T.; Roberson, P.A.; Mumford, P.W.; Kephart, W.C.; Romero, M.A.; Osburn, S.C.; Vann, C.G.; Young, K.C.; Beck, D.T.; et al. Biomarkers Associated with Low, Moderate, and High Vastus Lateralis Muscle Hypertrophy Following 12 Weeks of Resistance Training. PLoS ONE 2018, 13, e0195203. [Google Scholar] [CrossRef]
- Figueiredo, V.C.; Caldow, M.K.; Massie, V.; Markworth, J.F.; Cameron-Smith, D.; Blazevich, A.J. Ribosome Biogenesis Adaptation in Resistance Training-Induced Human Skeletal Muscle Hypertrophy. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E72–E83. [Google Scholar] [CrossRef]
- Figueiredo, V.C.; Roberts, L.A.; Markworth, J.F.; Barnett, M.P.G.; Coombes, J.S.; Raastad, T.; Peake, J.M.; Cameron-Smith, D. Impact of Resistance Exercise on Ribosome Biogenesis Is Acutely Regulated by Post-Exercise Recovery Strategies. Physiol. Rep. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Von Walden, F.; Casagrande, V.; Östlund Farrants, A.-K.; Nader, G.A. Mechanical Loading Induces the Expression of a Pol I Regulon at the Onset of Skeletal Muscle Hypertrophy. Am. J. Physiol. Cell Physiol. 2012, 302, C1523–C1530. [Google Scholar] [CrossRef]
- West, D.W.D.; Baehr, L.M.; Marcotte, G.R.; Chason, C.M.; Tolento, L.; Gomes, A.V.; Bodine, S.C.; Baar, K. Acute Resistance Exercise Activates Rapamycin-Sensitive and -Insensitive Mechanisms That Control Translational Activity and Capacity in Skeletal Muscle. J. Physiol. 2016, 594, 453–468. [Google Scholar] [CrossRef] [PubMed]
- Simonetti, A.; Brito Querido, J.; Myasnikov, A.G.; Mancera-Martinez, E.; Renaud, A.; Kuhn, L.; Hashem, Y. EIF3 Peripheral Subunits Rearrangement after MRNA Binding and Start-Codon Recognition. Mol. Cell 2016, 63, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Asano, K.; Vornlocher, H.P.; Richter-Cook, N.J.; Merrick, W.C.; Hinnebusch, A.G.; Hershey, J.W. Structure of CDNAs Encoding Human Eukaryotic Initiation Factor 3 Subunits. Possible Roles in RNA Binding and Macromolecular Assembly. J. Biol. Chem. 1997, 272, 27042–27052. [Google Scholar] [CrossRef]
- Benne, R.; Hershey, J.W. Purification and Characterization of Initiation Factor IF-E3 from Rabbit Reticulocytes. Proc. Natl. Acad. Sci. USA 1976, 73, 3005–3009. [Google Scholar] [CrossRef]
- Yin, Y.; Long, J.; Sun, Y.; Li, H.; Jiang, E.; Zeng, C.; Zhu, W. The Function and Clinical Significance of EIF3 in Cancer. Gene 2018, 673, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Sokabe, M.; Fraser, C.S.; Hershey, J.W.B. The Human Translation Initiation Multi-Factor Complex Promotes Methionyl-TRNAi Binding to the 40S Ribosomal Subunit. Nucleic Acids Res. 2012, 40, 905–913. [Google Scholar] [CrossRef]
- Csibi, A.; Cornille, K.; Leibovitch, M.-P.; Poupon, A.; Tintignac, L.A.; Sanchez, A.M.J.; Leibovitch, S.A. The Translation Regulatory Subunit EIF3f Controls the Kinase-Dependent MTOR Signaling Required for Muscle Differentiation and Hypertrophy in Mouse. PLoS ONE 2010, 5, e8994. [Google Scholar] [CrossRef] [PubMed]
- Schalm, S.S.; Blenis, J. Identification of a Conserved Motif Required for MTOR Signaling. Curr. Biol. 2002, 12, 632–639. [Google Scholar] [CrossRef]
- Holz, M.K.; Ballif, B.A.; Gygi, S.P.; Blenis, J. MTOR and S6K1 Mediate Assembly of the Translation Preinitiation Complex through Dynamic Protein Interchange and Ordered Phosphorylation Events. Cell 2005, 123, 569–580. [Google Scholar] [CrossRef]
- Holz, M.K.; Blenis, J. Identification of S6 Kinase 1 as a Novel Mammalian Target of Rapamycin (MTOR)-Phosphorylating Kinase. J. Biol. Chem. 2005, 280, 26089–26093. [Google Scholar] [CrossRef]
- Saitoh, M.; Pullen, N.; Brennan, P.; Cantrell, D.; Dennis, P.B.; Thomas, G. Regulation of an Activated S6 Kinase 1 Variant Reveals a Novel Mammalian Target of Rapamycin Phosphorylation Site. J. Biol. Chem. 2002, 277, 20104–20112. [Google Scholar] [CrossRef]
- Pullen, N.; Dennis, P.B.; Andjelkovic, M.; Dufner, A.; Kozma, S.C.; Hemmings, B.A.; Thomas, G. Phosphorylation and Activation of P70s6k by PDK1. Science 1998, 279, 707–710. [Google Scholar] [CrossRef]
- Chauvin, C.; Koka, V.; Nouschi, A.; Mieulet, V.; Hoareau-Aveilla, C.; Dreazen, A.; Cagnard, N.; Carpentier, W.; Kiss, T.; Meyuhas, O.; et al. Ribosomal Protein S6 Kinase Activity Controls the Ribosome Biogenesis Transcriptional Program. Oncogene 2014, 33, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Hannan, K.M.; Brandenburger, Y.; Jenkins, A.; Sharkey, K.; Cavanaugh, A.; Rothblum, L.; Moss, T.; Poortinga, G.; McArthur, G.A.; Pearson, R.B.; et al. MTOR-Dependent Regulation of Ribosomal Gene Transcription Requires S6K1 and Is Mediated by Phosphorylation of the Carboxy-Terminal Activation Domain of the Nucleolar Transcription Factor UBF. Mol. Cell Biol. 2003, 23, 8862–8877. [Google Scholar] [CrossRef] [PubMed]
- Youtani, T.; Tomoo, K.; Ishida, T.; Miyoshi, H.; Miura, K. Regulation of Human EIF4E by 4E-BP1: Binding Analysis Using Surface Plasmon Resonance. IUBMB Life 2000, 49, 27–31. [Google Scholar] [CrossRef]
- Shi, J.; Feng, Y.; Goulet, A.-C.; Vaillancourt, R.R.; Sachs, N.A.; Hershey, J.W.; Nelson, M.A. The P34cdc2-Related Cyclin-Dependent Kinase 11 Interacts with the P47 Subunit of Eukaryotic Initiation Factor 3 during Apoptosis. J. Biol. Chem. 2003, 278, 5062–5071. [Google Scholar] [CrossRef] [PubMed]
- Wen, F.; Zhou, R.; Shen, A.; Choi, A.; Uribe, D.; Shi, J. The Tumor Suppressive Role of EIF3f and Its Function in Translation Inhibition and RRNA Degradation. PLoS ONE 2012, 7, e34194. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Kim, H.-J.; Rho, S.B.; Lee, S.-H. EIF3f Reduces Tumor Growth by Directly Interrupting Clusterin with Anti-Apoptotic Property in Cancer Cells. Oncotarget 2016, 7, 18541–18557. [Google Scholar] [CrossRef]
- Xiao, H.; Xu, L.H.; Yamada, Y.; Liu, D.X. Coronavirus Spike Protein Inhibits Host Cell Translation by Interaction with EIF3f. PLoS ONE 2008, 3, e1494. [Google Scholar] [CrossRef] [PubMed]
- Bansal, M. Cardiovascular Disease and COVID-19. Diabetes Metab. Syndr. 2020, 14, 247–250. [Google Scholar] [CrossRef]
- Disser, N.P.; De Micheli, A.J.; Schonk, M.M.; Konnaris, M.A.; Piacentini, A.N.; Edon, D.L.; Toresdahl, B.G.; Rodeo, S.A.; Casey, E.K.; Mendias, C.L. Musculoskeletal Consequences of COVID-19. J. Bone Joint Surg. Am. 2020, 102, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Lagirand-Cantaloube, J.; Offner, N.; Csibi, A.; Leibovitch, M.P.; Batonnet-Pichon, S.; Tintignac, L.A.; Segura, C.T.; Leibovitch, S.A. The Initiation Factor EIF3-f Is a Major Target for Atrogin1/MAFbx Function in Skeletal Muscle Atrophy. EMBO J. 2008, 27, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Csibi, A.; Leibovitch, M.P.; Cornille, K.; Tintignac, L.A.; Leibovitch, S.A. MAFbx/Atrogin-1 Controls the Activity of the Initiation Factor EIF3-f in Skeletal Muscle Atrophy by Targeting Multiple C-Terminal Lysines. J. Biol. Chem. 2009, 284, 4413–4421. [Google Scholar] [CrossRef]
- Shin, Y.J.; Kwon, E.-S.; Lee, S.-M.; Kim, S.-K.; Min, K.-W.; Lim, J.-Y.; Lee, B.; Kang, J.S.; Kwak, J.Y.; Son, Y.H.; et al. A Subset of MicroRNAs in the Dlk1-Dio3 Cluster Regulates Age-Associated Muscle Atrophy by Targeting Atrogin-1. J. Cachexia Sarcopenia Muscle 2020. [Google Scholar] [CrossRef]
- Moretti, J.; Chastagner, P.; Gastaldello, S.; Heuss, S.F.; Dirac, A.M.; Bernards, R.; Masucci, M.G.; Israël, A.; Brou, C. The Translation Initiation Factor 3f (EIF3f) Exhibits a Deubiquitinase Activity Regulating Notch Activation. PLoS Biol. 2010, 8, e1000545. [Google Scholar] [CrossRef] [PubMed]
- Swiatek, P.J.; Lindsell, C.E.; del Amo, F.F.; Weinmaster, G.; Gridley, T. Notch1 Is Essential for Postimplantation Development in Mice. Genes Dev. 1994, 8, 707–719. [Google Scholar] [CrossRef]
- Hodson, N.; Philp, A. The Importance of MTOR Trafficking for Human Skeletal Muscle Translational Control. Exerc. Sport Sci. Rev. 2019, 47, 46–53. [Google Scholar] [CrossRef]
- Song, Z.; Moore, D.R.; Hodson, N.; Ward, C.; Dent, J.R.; O’Leary, M.F.; Shaw, A.M.; Hamilton, D.L.; Sarkar, S.; Gangloff, Y.-G.; et al. Resistance Exercise Initiates Mechanistic Target of Rapamycin (MTOR) Translocation and Protein Complex Co-Localisation in Human Skeletal Muscle. Sci. Rep. 2017, 7, 5028. [Google Scholar] [CrossRef]
- Groenewoud, M.J.; Zwartkruis, F.J.T. Rheb and Rags Come Together at the Lysosome to Activate MTORC1. Biochem. Soc. Trans. 2013, 41, 951–955. [Google Scholar] [CrossRef]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag Complex Targets MTORC1 to the Lysosomal Surface and Is Necessary for Its Activation by Amino Acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, B.L.; Goodman, C.A.; Hornberger, T.A. The Mechanical Activation of MTOR Signaling: An Emerging Role for Late Endosome/Lysosomal Targeting. J. Muscle Res. Cell Motil. 2014, 35, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Biolo, G.; Tipton, K.D.; Klein, S.; Wolfe, R.R. An Abundant Supply of Amino Acids Enhances the Metabolic Effect of Exercise on Muscle Protein. Am. J. Physiol. 1997, 273, E122–E129. [Google Scholar] [CrossRef] [PubMed]
- Børsheim, E.; Tipton, K.D.; Wolf, S.E.; Wolfe, R.R. Essential Amino Acids and Muscle Protein Recovery from Resistance Exercise. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E648–E657. [Google Scholar] [CrossRef]
- Van Vliet, S.; Shy, E.L.; Abou Sawan, S.; Beals, J.W.; West, D.W.; Skinner, S.K.; Ulanov, A.V.; Li, Z.; Paluska, S.A.; Parsons, C.M.; et al. Consumption of Whole Eggs Promotes Greater Stimulation of Postexercise Muscle Protein Synthesis than Consumption of Isonitrogenous Amounts of Egg Whites in Young Men. Am. J. Clin. Nutr. 2017, 106, 1401–1412. [Google Scholar] [CrossRef]
- Abou Sawan, S.; van Vliet, S.; West, D.W.D.; Beals, J.W.; Paluska, S.A.; Burd, N.A.; Moore, D.R. Whole Egg, but Not Egg White, Ingestion Induces MTOR Colocalization with the Lysosome after Resistance Exercise. Am. J. Physiol.-Cell Physiol. 2018, 315, C537–C543. [Google Scholar] [CrossRef]
- Yamamoto, I.; Mazumi, T.; Handa, T.; Miyajima, K. Effects of 1,2-Diacylglycerol and Cholesterol on the Hydrolysis Activity of Phospholipase D in Egg-Yolk Phosphatidylcholine Bilayers. Biochim. Biophys. Acta (BBA) Biomembr. 1993, 1145, 293–297. [Google Scholar] [CrossRef]
- Yamamoto, I.; Konto, A.; Handa, T. Regulation of Phospholipase D Activity by Neutral Lipids in Egg-Yolk Phosphatidylcholine Small Unilamellar Vesicles and by Calcium Ion in Aqueous Medium. Biochim. Biophys. Acta (BBA) Biomembr. 1995, 1233, 21–26. [Google Scholar] [CrossRef][Green Version]
- Castellano, B.M.; Thelen, A.M.; Moldavski, O.; Feltes, M.; van der Welle, R.E.N.; Mydock-McGrane, L.; Jiang, X.; van Eijkeren, R.J.; Davis, O.B.; Louie, S.M.; et al. Lysosomal Cholesterol Activates MTORC1 via an SLC38A9–Niemann-Pick C1 Signaling Complex. Science 2017, 355, 1306–1311. [Google Scholar] [CrossRef]
- Bagheri, R.; Hooshmand Moghadam, B.; Jo, E.; Tinsley, G.M.; Stratton, M.T.; Ashtary-Larky, D.; Eskandari, M.; Wong, A. Comparison of Whole Egg v. Egg White Ingestion during 12 Weeks of Resistance Training on Skeletal Muscle Regulatory Markers in Resistance-Trained Men. Br. J. Nutr. 2020, 1–9. [Google Scholar] [CrossRef]
- Ato, S.; Maruyama, Y.; Yoshizato, H.; Ogasawara, R. Habitual High-Protein Diet Does Not Influence Muscle Protein Synthesis in Response to Acute Resistance Exercise in Rats. Nutrition 2020, 78, 110795. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Rasmussen, M.L.; Li, J.; Olguín, C.H.; Knudsen, J.R.; Søgaard, O.; Madsen, A.B.; Jensen, T.E. Low- and High-protein Diets Do Not Alter Ex Vivo Insulin Action in Skeletal Muscle. Physiol. Rep. 2018, 6. [Google Scholar] [CrossRef]
- Mauro, A. Satellite Cell of Skeletal Muscle Fibers. J. Biophys. Biochem. Cytol. 1961, 9, 493–495. [Google Scholar] [CrossRef]
- Goh, Q.; Song, T.; Petrany, M.J.; Cramer, A.A.; Sun, C.; Sadayappan, S.; Lee, S.-J.; Millay, D.P. Myonuclear Accretion Is a Determinant of Exercise-Induced Remodeling in Skeletal Muscle. Elife 2019, 8. [Google Scholar] [CrossRef]
- Morgan, J.E.; Zammit, P.S. Direct Effects of the Pathogenic Mutation on Satellite Cell Function in Muscular Dystrophy. Exp. Cell Res. 2010, 316, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Schultz, E.; Gibson, M.C.; Champion, T. Satellite Cells Are Mitotically Quiescent in Mature Mouse Muscle: An EM and Radioautographic Study. J. Exp. Zool. 1978, 206, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Seale, P.; Sabourin, L.A.; Girgis-Gabardo, A.; Mansouri, A.; Gruss, P.; Rudnicki, M.A. Pax7 Is Required for the Specification of Myogenic Satellite Cells. Cell 2000, 102, 777–786. [Google Scholar] [CrossRef]
- Bazgir, B.; Fathi, R.; Rezazadeh Valojerdi, M.; Mozdziak, P.; Asgari, A. Satellite Cells Contribution to Exercise Mediated Muscle Hypertrophy and Repair. Cell J. 2017, 18, 473–484. [Google Scholar] [CrossRef]
- Murgia, M.; Toniolo, L.; Nagaraj, N.; Ciciliot, S.; Vindigni, V.; Schiaffino, S.; Reggiani, C.; Mann, M. Single Muscle Fiber Proteomics Reveals Fiber-Type-Specific Features of Human Muscle Aging. Cell Rep. 2017, 19, 2396–2409. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Bormioli, S.P.; Aloisi, M. The Fate of Newly Formed Satellite Cells during Compensatory Muscle Hypertrophy. Virchows Arch. B Cell Pathol. 1976, 21, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Aagaard, P. Making Muscles “Stronger”: Exercise, Nutrition, Drugs. J. Musculoskelet Neuronal Interact 2004, 4, 165–174. [Google Scholar] [PubMed]
- Schultz, E. A Quantitative Study of Satellite Cells in Regenerated Soleus and Extensor Digitorum Longus Muscles. Anat. Rec. 1984, 208, 501–506. [Google Scholar] [CrossRef]
- Kawano, F.; Takeno, Y.; Nakai, N.; Higo, Y.; Terada, M.; Ohira, T.; Nonaka, I.; Ohira, Y. Essential Role of Satellite Cells in the Growth of Rat Soleus Muscle Fibers. Am. J. Physiol. Cell Physiol. 2008, 295, C458–C467. [Google Scholar] [CrossRef] [PubMed]
- Pallafacchina, G.; Blaauw, B.; Schiaffino, S. Role of Satellite Cells in Muscle Growth and Maintenance of Muscle Mass. Nutr. Metab. Cardiovasc Dis. 2013, 23 (Suppl. 1), S12–S18. [Google Scholar] [CrossRef]
- Darr, K.C.; Schultz, E. Exercise-Induced Satellite Cell Activation in Growing and Mature Skeletal Muscle. J. Appl. Physiol. (1985) 1987, 63, 1816–1821. [Google Scholar] [CrossRef] [PubMed]
- Adams, G.R.; Caiozzo, V.J.; Haddad, F.; Baldwin, K.M. Cellular and Molecular Responses to Increased Skeletal Muscle Loading after Irradiation. Am. J. Physiol. Cell Physiol. 2002, 283, C1182–C1195. [Google Scholar] [CrossRef] [PubMed]
- Bruusgaard, J.C.; Egner, I.M.; Larsen, T.K.; Dupre-Aucouturier, S.; Desplanches, D.; Gundersen, K. No Change in Myonuclear Number during Muscle Unloading and Reloading. J. Appl. Physiol. (1985) 2012, 113, 290–296. [Google Scholar] [CrossRef]
- Egner, I.M.; Bruusgaard, J.C.; Gundersen, K. Satellite Cell Depletion Prevents Fiber Hypertrophy in Skeletal Muscle. Development 2016, 143, 2898–2906. [Google Scholar] [CrossRef]
- Martin, N.R.; Lewis, M.P. Satellite Cell Activation and Number Following Acute and Chronic Exercise: A Mini Review. Cell. Mol. Exerc. Physiol. 2012, 1, e3. [Google Scholar] [CrossRef]
- Parise, G.; McKinnell, I.W.; Rudnicki, M.A. Muscle Satellite Cell and Atypical Myogenic Progenitor Response Following Exercise. Muscle Nerve 2008, 37, 611–619. [Google Scholar] [CrossRef]
- Smith, H.K.; Maxwell, L.; Rodgers, C.D.; McKee, N.H.; Plyley, M.J. Exercise-Enhanced Satellite Cell Proliferation and New Myonuclear Accretion in Rat Skeletal Muscle. J. Appl. Physiol. (1985) 2001, 90, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Crameri, R.M.; Aagaard, P.; Qvortrup, K.; Langberg, H.; Olesen, J.; Kjaer, M. Myofibre Damage in Human Skeletal Muscle: Effects of Electrical Stimulation versus Voluntary Contraction. J. Physiol. 2007, 583, 365–380. [Google Scholar] [CrossRef] [PubMed]
- Crameri, R.M.; Langberg, H.; Magnusson, P.; Jensen, C.H.; Schrøder, H.D.; Olesen, J.L.; Suetta, C.; Teisner, B.; Kjaer, M. Changes in Satellite Cells in Human Skeletal Muscle after a Single Bout of High Intensity Exercise. J. Physiol. 2004, 558, 333–340. [Google Scholar] [CrossRef]
- Dreyer, H.C.; Blanco, C.E.; Sattler, F.R.; Schroeder, E.T.; Wiswell, R.A. Satellite Cell Numbers in Young and Older Men 24 Hours after Eccentric Exercise. Muscle Nerve 2006, 33, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, K.; Thompson, J.L.; Norenberg, K.M.; Fitts, R.H.; Riley, D.A. Fiber-Type Susceptibility to Eccentric Contraction-Induced Damage of Hindlimb-Unloaded Rat AL Muscles. J. Appl. Physiol. (1985) 2001, 90, 770–776. [Google Scholar] [CrossRef]
- Kurosaka, M.; Naito, H.; Ogura, Y.; Machida, S.; Katamoto, S. Satellite Cell Pool Enhancement in Rat Plantaris Muscle by Endurance Training Depends on Intensity Rather than Duration. Acta Physiol. 2012, 205, 159–166. [Google Scholar] [CrossRef]
- Verney, J.; Kadi, F.; Charifi, N.; Féasson, L.; Saafi, M.A.; Castells, J.; Piehl-Aulin, K.; Denis, C. Effects of Combined Lower Body Endurance and Upper Body Resistance Training on the Satellite Cell Pool in Elderly Subjects. Muscle Nerve 2008, 38, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.K.; Merry, T.L. Voluntary Resistance Wheel Exercise during Post-Natal Growth in Rats Enhances Skeletal Muscle Satellite Cell and Myonuclear Content at Adulthood. Acta Physiol. 2012, 204, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Burd, N.A.; West, D.W.D.; Staples, A.W.; Atherton, P.J.; Baker, J.M.; Moore, D.R.; Holwerda, A.M.; Parise, G.; Rennie, M.J.; Baker, S.K.; et al. Low-Load High Volume Resistance Exercise Stimulates Muscle Protein Synthesis More Than High-Load Low Volume Resistance Exercise in Young Men. PLoS ONE 2010, 5, e12033. [Google Scholar] [CrossRef]
- Masschelein, E.; D’Hulst, G.; Zvick, J.; Hinte, L.; Soro-Arnaiz, I.; Gorski, T.; von Meyenn, F.; Bar-Nur, O.; De Bock, K. Exercise Promotes Satellite Cell Contribution to Myofibers in a Load-Dependent Manner. Skeletal. Muscle 2020, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Verdijk, L.B.; Gleeson, B.G.; Jonkers, R.A.M.; Meijer, K.; Savelberg, H.H.C.M.; Dendale, P.; van Loon, L.J.C. Skeletal Muscle Hypertrophy Following Resistance Training Is Accompanied by a Fiber Type-Specific Increase in Satellite Cell Content in Elderly Men. J. Gerontol. A Biol. Sci. Med. Sci. 2009, 64, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.L.; Smith, S.; Morrell, G.; Addison, O.; Dibble, L.E.; Wahoff-Stice, D.; Lastayo, P.C. Comparison of Combined Aerobic and High-Force Eccentric Resistance Exercise with Aerobic Exercise Only for People with Type 2 Diabetes Mellitus. Phys. Ther. 2008, 88, 1345–1354. [Google Scholar] [CrossRef]
- Petrella, J.K.; Kim, J.-S.; Mayhew, D.L.; Cross, J.M.; Bamman, M.M. Potent Myofiber Hypertrophy during Resistance Training in Humans Is Associated with Satellite Cell-Mediated Myonuclear Addition: A Cluster Analysis. J. Appl. Physiol. (1985) 2008, 104, 1736–1742. [Google Scholar] [CrossRef]
- Hoppeler, H.; Howald, H.; Conley, K.; Lindstedt, S.L.; Claassen, H.; Vock, P.; Weibel, E.R. Endurance Training in Humans: Aerobic Capacity and Structure of Skeletal Muscle. J. Appl. Physiol. (1985) 1985, 59, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Egner, I.M.; Bruusgaard, J.C.; Eftestøl, E.; Gundersen, K. A Cellular Memory Mechanism Aids Overload Hypertrophy in Muscle Long after an Episodic Exposure to Anabolic Steroids. J. Physiol. 2013, 591, 6221–6230. [Google Scholar] [CrossRef]
- Englund, D.A.; Peck, B.D.; Murach, K.A.; Neal, A.C.; Caldwell, H.A.; McCarthy, J.J.; Peterson, C.A.; Dupont-Versteegden, E.E. Resident Muscle Stem Cells Are Not Required for Testosterone-Induced Skeletal Muscle Hypertrophy. Am. J. Physiol. Cell Physiol. 2019, 317, C719–C724. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-J.; Huynh, T.V.; Lee, Y.-S.; Sebald, S.M.; Wilcox-Adelman, S.A.; Iwamori, N.; Lepper, C.; Matzuk, M.M.; Fan, C.-M. Role of Satellite Cells versus Myofibers in Muscle Hypertrophy Induced by Inhibition of the Myostatin/Activin Signaling Pathway. Proc. Natl. Acad. Sci. USA 2012, 109, E2353–E2360. [Google Scholar] [CrossRef]
- Rehfeldt, C.; Weikard, R.; Reichel, K. [The effect of the beta-adrenergic agonist clenbuterol on the growth of skeletal muscles of rats]. Arch Tierernahr. 1994, 45, 333–344. [Google Scholar] [CrossRef]
- Blaauw, B.; Canato, M.; Agatea, L.; Toniolo, L.; Mammucari, C.; Masiero, E.; Abraham, R.; Sandri, M.; Schiaffino, S.; Reggiani, C. Inducible Activation of Akt Increases Skeletal Muscle Mass and Force without Satellite Cell Activation. FASEB J. 2009, 23, 3896–3905. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.-L.; Zhang, Z.-M.; Song, Z.-W.; Gao, C.-Q.; Yan, H.-C.; Wang, X.-Q. MTORC1-Mediated Satellite Cell Differentiation Is Required for Lysine-Induced Skeletal Muscle Growth. J. Agric. Food Chem. 2020, 68, 4884–4892. [Google Scholar] [CrossRef]
- Jin, C.-L.; Ye, J.-L.; Yang, J.; Gao, C.-Q.; Yan, H.-C.; Li, H.-C.; Wang, X.-Q. MTORC1 Mediates Lysine-Induced Satellite Cell Activation to Promote Skeletal Muscle Growth. Cells 2019, 8, 1549. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, J.J.; Mula, J.; Miyazaki, M.; Erfani, R.; Garrison, K.; Farooqui, A.B.; Srikuea, R.; Lawson, B.A.; Grimes, B.; Keller, C.; et al. Effective Fiber Hypertrophy in Satellite Cell-Depleted Skeletal Muscle. Development 2011, 138, 3657–3666. [Google Scholar] [CrossRef]
- Fry, C.S.; Lee, J.D.; Jackson, J.R.; Kirby, T.J.; Stasko, S.A.; Liu, H.; Dupont-Versteegden, E.E.; McCarthy, J.J.; Peterson, C.A. Regulation of the Muscle Fiber Microenvironment by Activated Satellite Cells during Hypertrophy. FASEB J. 2014, 28, 1654–1665. [Google Scholar] [CrossRef]
- Randrianarison-Huetz, V.; Papaefthymiou, A.; Herledan, G.; Noviello, C.; Faradova, U.; Collard, L.; Pincini, A.; Schol, E.; Decaux, J.F.; Maire, P.; et al. Srf Controls Satellite Cell Fusion through the Maintenance of Actin Architecture. J. Cell Biol. 2018, 217, 685–700. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Tanaka, T.; Mulati, M.; Ochi, H.; Sato, S.; Kaldis, P.; Yoshii, T.; Okawa, A.; Inose, H. Cyclin-Dependent Kinase 1 Is Essential for Muscle Regeneration and Overload Muscle Fiber Hypertrophy. Front. Cell Dev. Biol. 2020, 8, 564581. [Google Scholar] [CrossRef] [PubMed]
- Goh, Q.; Millay, D.P. Requirement of Myomaker-Mediated Stem Cell Fusion for Skeletal Muscle Hypertrophy. Elife 2017, 6. [Google Scholar] [CrossRef]
- Murach, K.A.; White, S.H.; Wen, Y.; Ho, A.; Dupont-Versteegden, E.E.; McCarthy, J.J.; Peterson, C.A. Differential Requirement for Satellite Cells during Overload-Induced Muscle Hypertrophy in Growing versus Mature Mice. Skelet. Muscle 2017, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Bachman, J.F.; Klose, A.; Liu, W.; Paris, N.D.; Blanc, R.S.; Schmalz, M.; Knapp, E.; Chakkalakal, J.V. Prepubertal Skeletal Muscle Growth Requires Pax7-Expressing Satellite Cell-Derived Myonuclear Contribution. Development 2018, 145. [Google Scholar] [CrossRef]
- Jackson, J.R.; Mula, J.; Kirby, T.J.; Fry, C.S.; Lee, J.D.; Ubele, M.F.; Campbell, K.S.; McCarthy, J.J.; Peterson, C.A.; Dupont-Versteegden, E.E. Satellite Cell Depletion Does Not Inhibit Adult Skeletal Muscle Regrowth Following Unloading-Induced Atrophy. Am. J. Physiol. Cell Physiol. 2012, 303, C854–C861. [Google Scholar] [CrossRef]
- Murach, K.A.; Vechetti, I.J.; Van Pelt, D.W.; Crow, S.E.; Dungan, C.M.; Figueiredo, V.C.; Kosmac, K.; Fu, X.; Richards, C.I.; Fry, C.S.; et al. Fusion-Independent Satellite Cell Communication to Muscle Fibers During Load-Induced Hypertrophy. Function 2020, 1, zqaa009. [Google Scholar] [CrossRef] [PubMed]
- Fry, C.S.; Kirby, T.J.; Kosmac, K.; McCarthy, J.J.; Peterson, C.A. Myogenic Progenitor Cells Control Extracellular Matrix Production by Fibroblasts during Skeletal Muscle Hypertrophy. Cell Stem Cell 2017, 20, 56–69. [Google Scholar] [CrossRef]
- Gundersen, K. Muscle Memory and a New Cellular Model for Muscle Atrophy and Hypertrophy. J. Exp. Biol. 2016, 219, 235–242. [Google Scholar] [CrossRef]
- Snijders, T.; Aussieker, T.; Holwerda, A.; Parise, G.; van Loon, L.J.C.; Verdijk, L.B. The Concept of Skeletal Muscle Memory: Evidence from Animal and Human Studies. Acta Physiol. 2020, 229, e13465. [Google Scholar] [CrossRef]
- Psilander, N.; Eftestøl, E.; Cumming, K.T.; Juvkam, I.; Ekblom, M.M.; Sunding, K.; Wernbom, M.; Holmberg, H.-C.; Ekblom, B.; Bruusgaard, J.C.; et al. Effects of Training, Detraining, and Retraining on Strength, Hypertrophy, and Myonuclear Number in Human Skeletal Muscle. J. Appl. Physiol. (1985) 2019, 126, 1636–1645. [Google Scholar] [CrossRef] [PubMed]
- Borja-Gonzalez, M.; Casas-Martinez, J.C.; McDonagh, B.; Goljanek-Whysall, K. Inflamma-MiR-21 Negatively Regulates Myogenesis during Ageing. Antioxidants 2020, 9, 345. [Google Scholar] [CrossRef] [PubMed]
- Silva, W.J.; Graça, F.A.; Cruz, A.; Silvestre, J.G.; Labeit, S.; Miyabara, E.H.; Yan, C.Y.I.; Wang, D.Z.; Moriscot, A.S. MiR-29c Improves Skeletal Muscle Mass and Function throughout Myocyte Proliferation and Differentiation and by Repressing Atrophy-Related Genes. Acta Physiol. 2019, 226, e13278. [Google Scholar] [CrossRef] [PubMed]
- Wada, S.; Kato, Y.; Okutsu, M.; Miyaki, S.; Suzuki, K.; Yan, Z.; Schiaffino, S.; Asahara, H.; Ushida, T.; Akimoto, T. Translational Suppression of Atrophic Regulators by MicroRNA-23a Integrates Resistance to Skeletal Muscle Atrophy. J. Biol. Chem. 2011, 286, 38456–38465. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, C.; Vajjala, A.; Arigela, H.; Lokireddy, S.; Ge, X.; Bonala, S.; Manickam, R.; Kambadur, R.; Sharma, M. Negative Auto-Regulation of Myostatin Expression Is Mediated by Smad3 and MicroRNA-27. PLoS ONE 2014, 9, e87687. [Google Scholar] [CrossRef]
- Li, J.; Chan, M.C.; Yu, Y.; Bei, Y.; Chen, P.; Zhou, Q.; Cheng, L.; Chen, L.; Ziegler, O.; Rowe, G.C.; et al. MiR-29b Contributes to Multiple Types of Muscle Atrophy. Nat. Commun. 2017, 8, 15201. [Google Scholar] [CrossRef]
- Pagano, A.F.; Py, G.; Bernardi, H.; Candau, R.B.; Sanchez, A.M.J. Autophagy and Protein Turnover Signaling in Slow-Twitch Muscle during Exercise. Med. Sci. Sports Exerc. 2014, 46, 1314–1325. [Google Scholar] [CrossRef] [PubMed]
- Merle, A.; Jollet, M.; Britto, F.A.; Goustard, B.; Bendridi, N.; Rieusset, J.; Ollendorff, V.; Favier, F.B. Endurance Exercise Decreases Protein Synthesis and ER-Mitochondria Contacts in Mouse Skeletal Muscle. J. Appl. Physiol. (1985) 2019, 127, 1297–1306. [Google Scholar] [CrossRef] [PubMed]
- Takegaki, J.; Sase, K.; Fujita, S. Repeated Bouts of Resistance Exercise Attenuate Mitogen-Activated Protein-Kinase Signal Responses in Rat Skeletal Muscle. Biochem. Biophys. Res. Commun. 2019, 520, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Takegaki, J.; Ogasawara, R.; Tamura, Y.; Takagi, R.; Arihara, Y.; Tsutaki, A.; Nakazato, K.; Ishii, N. Repeated Bouts of Resistance Exercise with Short Recovery Periods Activates MTOR Signaling, but Not Protein Synthesis, in Mouse Skeletal Muscle. Physiol. Rep. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Ato, S.; Kido, K.; Sase, K.; Fujita, S. Response of Resistance Exercise-Induced Muscle Protein Synthesis and Skeletal Muscle Hypertrophy Are Not Enhanced After Disuse Muscle Atrophy in Rat. Front. Physiol. 2020, 11, 469. [Google Scholar] [CrossRef] [PubMed]
- Miyatake, S.; Hino, K.; Natsui, Y.; Ebisu, G.; Fujita, S. Protein Supplementation Enhances the Effects of Intermittent Loading on Skeletal Muscles by Activating the MTORC1 Signaling Pathway in a Rat Model of Disuse Atrophy. Nutrients 2020, 12, 2729. [Google Scholar] [CrossRef]
- Watier, T.; Sanchez, A.M. Micro-RNAs, Exercise and Cellular Plasticity in Humans: The Impact of Dietary Factors and Hypoxia. Microrna 2017, 6, 110–124. [Google Scholar] [CrossRef]
- Fernandez-Gonzalo, R.; Lundberg, T.R.; Tesch, P.A. Acute Molecular Responses in Untrained and Trained Muscle Subjected to Aerobic and Resistance Exercise Training versus Resistance Training Alone. Acta Physiol. 2013, 209, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Fyfe, J.J.; Bishop, D.J.; Stepto, N.K. Interference between Concurrent Resistance and Endurance Exercise: Molecular Bases and the Role of Individual Training Variables. Sports Med. 2014, 44, 743–762. [Google Scholar] [CrossRef]
- Fyfe, J.J.; Loenneke, J.P. Interpreting Adaptation to Concurrent Compared with Single-Mode Exercise Training: Some Methodological Considerations. Sports Med. 2018, 48, 289–297. [Google Scholar] [CrossRef]
- Coffey, V.G.; Zhong, Z.; Shield, A.; Canny, B.J.; Chibalin, A.V.; Zierath, J.R.; Hawley, J.A. Early Signaling Responses to Divergent Exercise Stimuli in Skeletal Muscle from Well-Trained Humans. FASEB J. 2006, 20, 190–192. [Google Scholar] [CrossRef]
- Ogasawara, R.; Kobayashi, K.; Tsutaki, A.; Lee, K.; Abe, T.; Fujita, S.; Nakazato, K.; Ishii, N. MTOR Signaling Response to Resistance Exercise Is Altered by Chronic Resistance Training and Detraining in Skeletal Muscle. J. Appl. Physiol. 2013, 114, 934–940. [Google Scholar] [CrossRef] [PubMed]
- Bagley, J.R.; Burghardt, K.J.; McManus, R.; Howlett, B.; Costa, P.B.; Coburn, J.W.; Arevalo, J.A.; Malek, M.H.; Galpin, A.J. Epigenetic Responses to Acute Resistance Exercise in Trained vs. Sedentary Men. J. Strength Cond. Res. 2020, 34, 1574–1580. [Google Scholar] [CrossRef]
- Haun, C.T.; Vann, C.G.; Osburn, S.C.; Mumford, P.W.; Roberson, P.A.; Romero, M.A.; Fox, C.D.; Johnson, C.A.; Parry, H.A.; Kavazis, A.N.; et al. Muscle Fiber Hypertrophy in Response to 6 Weeks of High-Volume Resistance Training in Trained Young Men Is Largely Attributed to Sarcoplasmic Hypertrophy. PLoS ONE 2019, 14, e0215267. [Google Scholar] [CrossRef] [PubMed]
- Pickering, C.; Kiely, J. Do Non-Responders to Exercise Exist—and If So, What Should We Do About Them? Sports Med. 2019, 49, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Solsona, R.; Sanchez, A.M.J. Ribosome Biogenesis and Resistance Training Volume in Human Skeletal Muscle. J. Physiol. 2020, 598, 1121–1122. [Google Scholar] [CrossRef] [PubMed]
- Brook, M.S.; Wilkinson, D.J.; Mitchell, W.K.; Lund, J.N.; Phillips, B.E.; Szewczyk, N.J.; Greenhaff, P.L.; Smith, K.; Atherton, P.J. Synchronous Deficits in Cumulative Muscle Protein Synthesis and Ribosomal Biogenesis Underlie Age-Related Anabolic Resistance to Exercise in Humans. J. Physiol. 2016, 594, 7399–7417. [Google Scholar] [CrossRef] [PubMed]
- Chaillou, T. Impaired Ribosome Biogenesis Could Contribute to Anabolic Resistance to Strength Exercise in the Elderly. J. Physiol. 2017, 595, 1447–1448. [Google Scholar] [CrossRef] [PubMed]
- Kirby, T.J.; Lee, J.D.; England, J.H.; Chaillou, T.; Esser, K.A.; McCarthy, J.J. Blunted Hypertrophic Response in Aged Skeletal Muscle Is Associated with Decreased Ribosome Biogenesis. J. Appl. Physiol. (1985) 2015, 119, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.K.; Kirdi, N.; Bozoglu, E.; Meric, A.; Buyukturan, G.; Ozturk, A.; Doruk, H. Effect of Low-Intensity versus High-Intensity Resistance Training on the Functioning of the Institutionalized Frail Elderly. Int. J. Rehabil. Res. 2018, 41, 211–217. [Google Scholar] [CrossRef]
- Taaffe, D.R.; Pruitt, L.; Pyka, G.; Guido, D.; Marcus, R. Comparative Effects of High- and Low-Intensity Resistance Training on Thigh Muscle Strength, Fiber Area, and Tissue Composition in Elderly Women. Clin. Physiol. 1996, 16, 381–392. [Google Scholar] [CrossRef]
- Hortobágyi, T.; Tunnel, D.; Moody, J.; Beam, S.; DeVita, P. Low- or High-Intensity Strength Training Partially Restores Impaired Quadriceps Force Accuracy and Steadiness in Aged Adults. J. Gerontol. A Biol. Sci. Med. Sci. 2001, 56, B38–B47. [Google Scholar] [CrossRef]
- Vincent, K.R.; Braith, R.W.; Feldman, R.A.; Kallas, H.E.; Lowenthal, D.T. Improved Cardiorespiratory Endurance Following 6 Months of Resistance Exercise in Elderly Men and Women. Arch. Intern. Med. 2002, 162, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Berg, O.K.; Kwon, O.S.; Hureau, T.J.; Clifton, H.L.; Thurston, T.S.; Le Fur, Y.; Jeong, E.-K.; Trinity, J.D.; Richardson, R.S.; Wang, E.; et al. Skeletal Muscle Mitochondrial Adaptations to Maximal Strength Training in Older Adults. J. Gerontol. A Biol. Sci. Med. Sci. 2020. [Google Scholar] [CrossRef]
- Zhang, Y.; Oliveira, A.N.; Hood, D.A. The Intersection of Exercise and Aging on Mitochondrial Protein Quality Control. Exp. Gerontol. 2020, 131, 110824. [Google Scholar] [CrossRef] [PubMed]
- Skulachev, V.P.; Longo, V.D. Aging as a Mitochondria-Mediated Atavistic Program: Can Aging Be Switched Off? Ann. N. Y. Acad. Sci. 2005, 1057, 145–164. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Shen, L.; Hu, P.; Huang, R.; Cao, Y.; Deng, J.; Yuan, W.; Liu, D.; Yang, J.; Gu, H.; et al. Aging-Associated Mitochondrial DNA Mutations Alter Oxidative Phosphorylation Machinery and Cause Mitochondrial Dysfunctions. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2266–2273. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.M.; Johannsen, D.L.; Ravussin, E. Skeletal Muscle Mitochondria and Aging: A Review. J. Aging Res. 2012, 2012, 194821. [Google Scholar] [CrossRef] [PubMed]
- Rezuş, E.; Burlui, A.; Cardoneanu, A.; Rezuş, C.; Codreanu, C.; Pârvu, M.; Rusu Zota, G.; Tamba, B.I. Inactivity and Skeletal Muscle Metabolism: A Vicious Cycle in Old Age. Int. J. Mol. Sci. 2020, 21, 592. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.M.J. Mitophagy Flux in Skeletal Muscle during Chronic Contractile Activity and Ageing. J. Physiol. 2018, 596, 3461–3462. [Google Scholar] [CrossRef] [PubMed]
- Carter, H.N.; Chen, C.C.W.; Hood, D.A. Mitochondria, Muscle Health, and Exercise with Advancing Age. Physiology 2015, 30, 208–223. [Google Scholar] [CrossRef] [PubMed]
- Carter, H.N.; Kim, Y.; Erlich, A.T.; Zarrin-Khat, D.; Hood, D.A. Autophagy and Mitophagy Flux in Young and Aged Skeletal Muscle Following Chronic Contractile Activity. J. Physiol. 2018, 596, 3567–3584. [Google Scholar] [CrossRef] [PubMed]
- Ades, P.A.; Ballor, D.L.; Ashikaga, T.; Utton, J.L.; Nair, K.S. Weight Training Improves Walking Endurance in Healthy Elderly Persons. Ann. Intern. Med. 1996, 124, 568–572. [Google Scholar] [CrossRef] [PubMed]
- Frontera, W.R.; Meredith, C.N.; O’Reilly, K.P.; Knuttgen, H.G.; Evans, W.J. Strength Conditioning in Older Men: Skeletal Muscle Hypertrophy and Improved Function. J. Appl. Physiol. 1988, 64, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Frontera, W.R.; Meredith, C.N.; O’Reilly, K.P.; Evans, W.J. Strength Training and Determinants of VO2max in Older Men. J. Appl. Physiol. 1990, 68, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Karlsen, A.; Soendenbroe, C.; Malmgaard-Clausen, N.M.; Wagener, F.; Moeller, C.E.; Senhaji, Z.; Damberg, K.; Andersen, J.L.; Schjerling, P.; Kjaer, M.; et al. Preserved Capacity for Satellite Cell Proliferation, Regeneration, and Hypertrophy in the Skeletal Muscle of Healthy Elderly Men. FASEB J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Parry, H.A.; Roberts, M.D.; Kavazis, A.N. Human Skeletal Muscle Mitochondrial Adaptations Following Resistance Exercise Training. Int. J. Sports Med. 2020. [Google Scholar] [CrossRef]
- Bullo, V.; Gobbo, S.; Vendramin, B.; Duregon, F.; Cugusi, L.; Di Blasio, A.; Bocalini, D.S.; Zaccaria, M.; Bergamin, M.; Ermolao, A. Nordic Walking Can Be Incorporated in the Exercise Prescription to Increase Aerobic Capacity, Strength, and Quality of Life for Elderly: A Systematic Review and Meta-Analysis. Rejuvenation Res. 2018, 21, 141–161. [Google Scholar] [CrossRef] [PubMed]
- Rahbek, S.K.; Farup, J.; Møller, A.B.; Vendelbo, M.H.; Holm, L.; Jessen, N.; Vissing, K. Effects of Divergent Resistance Exercise Contraction Mode and Dietary Supplementation Type on Anabolic Signalling, Muscle Protein Synthesis and Muscle Hypertrophy. Amino Acids 2014, 46, 2377–2392. [Google Scholar] [CrossRef] [PubMed]
- Roig, M.; O’Brien, K.; Kirk, G.; Murray, R.; McKinnon, P.; Shadgan, B.; Reid, W.D. The Effects of Eccentric versus Concentric Resistance Training on Muscle Strength and Mass in Healthy Adults: A Systematic Review with Meta-Analysis. Br. J. Sports Med. 2009, 43, 556–568. [Google Scholar] [CrossRef] [PubMed]
- Norrbrand, L.; Fluckey, J.D.; Pozzo, M.; Tesch, P.A. Resistance Training Using Eccentric Overload Induces Early Adaptations in Skeletal Muscle Size. Eur. J. Appl. Physiol. 2008, 102, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Ato, S.; Makanae, Y.; Kido, K.; Fujita, S. Contraction Mode Itself Does Not Determine the Level of MTORC1 Activity in Rat Skeletal Muscle. Physiol. Rep. 2016, 4. [Google Scholar] [CrossRef]
- Ato, S.; Makanae, Y.; Kido, K.; Sase, K.; Yoshii, N.; Fujita, S. The Effect of Different Acute Muscle Contraction Regimens on the Expression of Muscle Proteolytic Signaling Proteins and Genes. Physiol. Rep. 2017, 5. [Google Scholar] [CrossRef]
- Garma, T.; Kobayashi, C.; Haddad, F.; Adams, G.R.; Bodell, P.W.; Baldwin, K.M. Similar Acute Molecular Responses to Equivalent Volumes of Isometric, Lengthening, or Shortening Mode Resistance Exercise. J. Appl. Physiol. 2007, 102, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Franchi, M.V.; Reeves, N.D.; Narici, M.V. Skeletal Muscle Remodeling in Response to Eccentric vs. Concentric Loading: Morphological, Molecular, and Metabolic Adaptations. Front. Physiol. 2017, 8. [Google Scholar] [CrossRef]
- Mallinson, J.E.; Taylor, T.; Constantin-Teodosiu, D.; Billeter-Clark, R.; Constantin, D.; Franchi, M.V.; Narici, M.V.; Auer, D.; Greenhaff, P.L. Longitudinal Hypertrophic and Transcriptional Responses to High-Load Eccentric-Concentric vs Concentric Training in Males. Scand. J. Med. Sci. Sports 2020, 30, 2101–2115. [Google Scholar] [CrossRef]
- Flaherty, G.; O’Connor, R.; Johnston, N. Altitude Training for Elite Endurance Athletes: A Review for the Travel Medicine Practitioner. Travel. Med. Infect. Dis. 2016, 14, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Katayama, K.; Sato, Y.; Ishida, K.; Mori, S.; Miyamura, M. The Effects of Intermittent Exposure to Hypoxia during Endurance Exercise Training on the Ventilatory Responses to Hypoxia and Hypercapnia in Humans. Eur. J. Appl. Physiol. Occup. Physiol. 1998, 78, 189–194. [Google Scholar] [CrossRef]
- Brocherie, F.; Girard, O.; Faiss, R.; Millet, G.P. High-Intensity Intermittent Training in Hypoxia: A Double-Blinded, Placebo-Controlled Field Study in Youth Football Players. J. Strength Cond. Res. 2015, 29, 226–237. [Google Scholar] [CrossRef]
- Allsopp, G.L.; Hoffmann, S.M.; Feros, S.A.; Pasco, J.A.; Russell, A.P.; Wright, C.R. The Effect of Normobaric Hypoxia on Resistance Training Adaptations in Older Adults. J. Strength Cond. Res. 2020. [Google Scholar] [CrossRef]
- Chobanyan-Jürgens, K.; Scheibe, R.J.; Potthast, A.B.; Hein, M.; Smith, A.; Freund, R.; Tegtbur, U.; Das, A.M.; Engeli, S.; Jordan, J.; et al. Influences of Hypoxia Exercise on Whole-Body Insulin Sensitivity and Oxidative Metabolism in Older Individuals. J. Clin. Endocrinol. Metab. 2019, 104, 5238–5248. [Google Scholar] [CrossRef] [PubMed]
- Schega, L.; Peter, B.; Törpel, A.; Mutschler, H.; Isermann, B.; Hamacher, D. Effects of Intermittent Hypoxia on Cognitive Performance and Quality of Life in Elderly Adults: A Pilot Study. GER 2013, 59, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Klarod, K.; Philippe, M.; Gatterer, H.; Burtscher, M. Different Training Responses to Eccentric Endurance Exercise at Low and Moderate Altitudes in Pre-Diabetic Men: A Pilot Study. Sport Sci. Health 2017, 13, 615–623. [Google Scholar] [CrossRef]
- Wortman, R.J.; Brown, S.M.; Savage-Elliott, I.; Finley, Z.J.; Mulcahey, M.K. Blood Flow Restriction Training for Athletes: A Systematic Review. Am. J. Sports Med. 2020. [Google Scholar] [CrossRef]
- Nielsen, J.L.; Aagaard, P.; Bech, R.D.; Nygaard, T.; Hvid, L.G.; Wernbom, M.; Suetta, C.; Frandsen, U. Proliferation of Myogenic Stem Cells in Human Skeletal Muscle in Response to Low-Load Resistance Training with Blood Flow Restriction. J. Physiol. 2012, 590, 4351–4361. [Google Scholar] [CrossRef] [PubMed]
- Larkin, K.A.; Macneil, R.G.; Dirain, M.; Sandesara, B.; Manini, T.M.; Buford, T.W. Blood Flow Restriction Enhances Post–Resistance Exercise Angiogenic Gene Expression. Med. Sci. Sports Exerc. 2012, 44, 2077. [Google Scholar] [CrossRef] [PubMed]
- Kakehi, S.; Tamura, Y.; Kubota, A.; Takeno, K.; Kawaguchi, M.; Sakuraba, K.; Kawamori, R.; Watada, H. Effects of Blood Flow Restriction on Muscle Size and Gene Expression in Muscle during Immobilization: A Pilot Study. Physiol. Rep. 2020, 8, e14516. [Google Scholar] [CrossRef] [PubMed]
- Kubota, A.; Sakuraba, K.; Koh, S.; Ogura, Y.; Tamura, Y. Blood Flow Restriction by Low Compressive Force Prevents Disuse Muscular Weakness. J. Sci. Med. Sport 2011, 14, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Lopes, K.G.; Bottino, D.A.; Farinatti, P.; de Souza, M.D.; Maranhão, P.A.; de Araujo, C.M.; Bouskela, E.; Lourenço, R.A.; de Oliveira, R.B. Strength Training with Blood Flow Restriction-a Novel Therapeutic Approach for Older Adults with Sarcopenia? A Case Report. Clin. Interv. Aging 2019, 14, 1461–1469. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, H.; Miyachi, M.; Nakajima, T.; Abe, T. Effects of 10 Weeks Walk Training with Leg Blood Flow Reduction on Carotid Arterial Compliance and Muscle Size in the Elderly Adults. Angiology 2011, 62, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Takarada, Y.; Takazawa, H.; Sato, Y.; Takebayashi, S.; Tanaka, Y.; Ishii, N. Effects of Resistance Exercise Combined with Moderate Vascular Occlusion on Muscular Function in Humans. J. Appl. Physiol. (1985) 2000, 88, 2097–2106. [Google Scholar] [CrossRef] [PubMed]
- Iida, H.; Nakajima, T.; Kurano, M.; Yasuda, T.; Sakamaki, M.; Sato, Y.; Yamasoba, T.; Abe, T. Effects of Walking with Blood Flow Restriction on Limb Venous Compliance in Elderly Subjects. Clin. Physiol. Funct. Imaging 2011, 31, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Sakamaki, M.; Fujita, S.; Ozaki, H.; Sugaya, M.; Sato, Y.; Nakajima, T. Effects of Low-Intensity Walk Training with Restricted Leg Blood Flow on Muscle Strength and Aerobic Capacity in Older Adults. J. Geriatr. Phys. Ther. 2010, 33, 34–40. [Google Scholar] [PubMed]
- Linero, C.; Choi, S.-J. Effect of Blood Flow Restriction during Low-Intensity Resistance Training on Bone Markers and Physical Functions in Postmenopausal Women. J. Exerc. Sci. Fit. 2020. [Google Scholar] [CrossRef]
- Preobrazenski, N.; Islam, H.; Drouin, P.J.; Bonafiglia, J.T.; Tschakovsky, M.E.; Gurd, B.J. A Novel Gravity-Induced Blood Flow Restriction Model Augments ACC Phosphorylation and PGC-1α MRNA in Human Skeletal Muscle Following Aerobic Exercise: A Randomized Crossover Study. Appl. Physiol. Nutr. Metab. 2020, 45, 641–649. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solsona, R.; Pavlin, L.; Bernardi, H.; Sanchez, A.M. Molecular Regulation of Skeletal Muscle Growth and Organelle Biosynthesis: Practical Recommendations for Exercise Training. Int. J. Mol. Sci. 2021, 22, 2741. https://doi.org/10.3390/ijms22052741
Solsona R, Pavlin L, Bernardi H, Sanchez AM. Molecular Regulation of Skeletal Muscle Growth and Organelle Biosynthesis: Practical Recommendations for Exercise Training. International Journal of Molecular Sciences. 2021; 22(5):2741. https://doi.org/10.3390/ijms22052741
Chicago/Turabian StyleSolsona, Robert, Laura Pavlin, Henri Bernardi, and Anthony MJ Sanchez. 2021. "Molecular Regulation of Skeletal Muscle Growth and Organelle Biosynthesis: Practical Recommendations for Exercise Training" International Journal of Molecular Sciences 22, no. 5: 2741. https://doi.org/10.3390/ijms22052741
APA StyleSolsona, R., Pavlin, L., Bernardi, H., & Sanchez, A. M. (2021). Molecular Regulation of Skeletal Muscle Growth and Organelle Biosynthesis: Practical Recommendations for Exercise Training. International Journal of Molecular Sciences, 22(5), 2741. https://doi.org/10.3390/ijms22052741