
The Complex and Critical Role of Glycine 12 (G12) in Beta-Connexins of Human Skin

Abstract
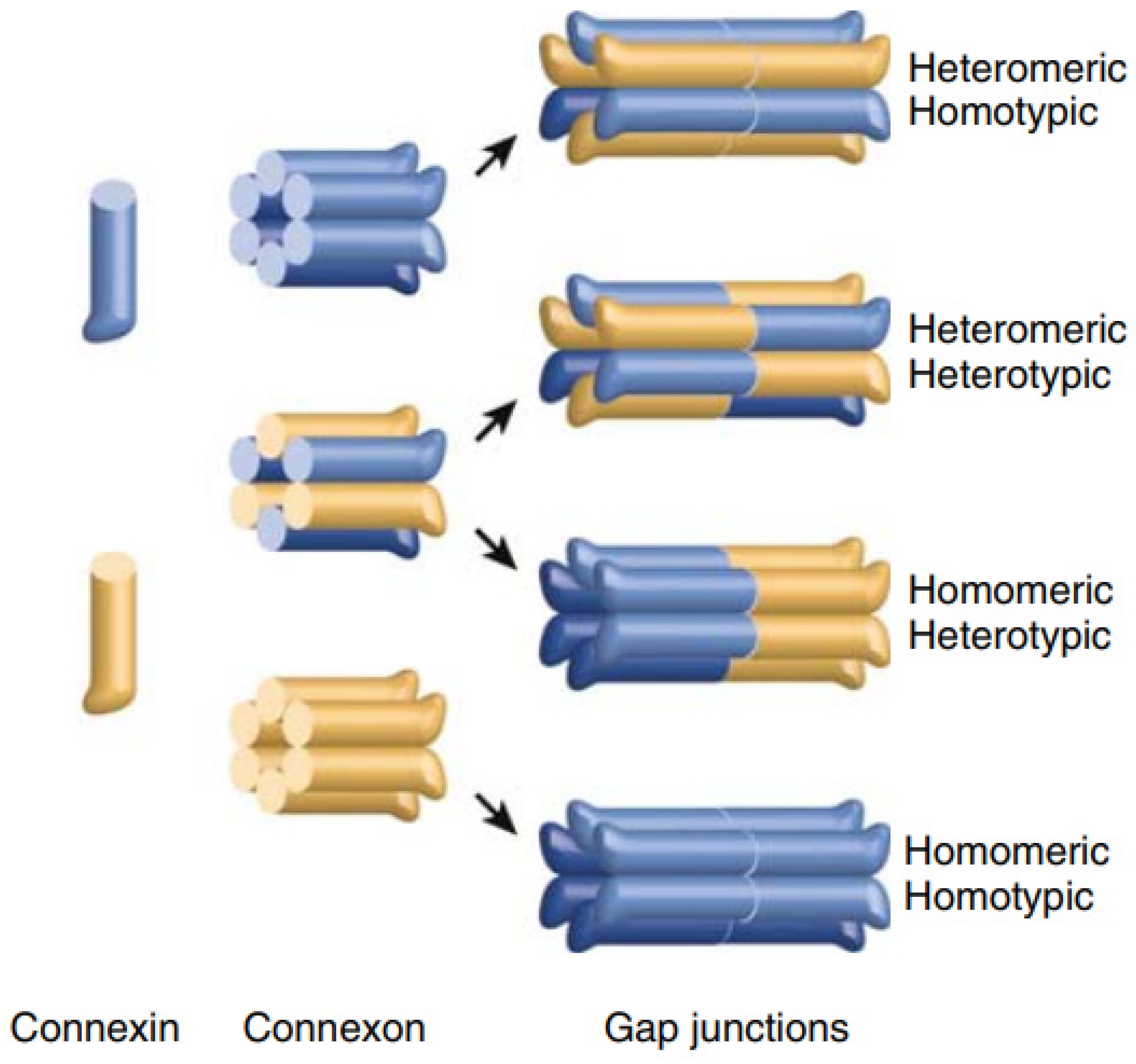
1. Genes, Proteins and Gap Junction Channels
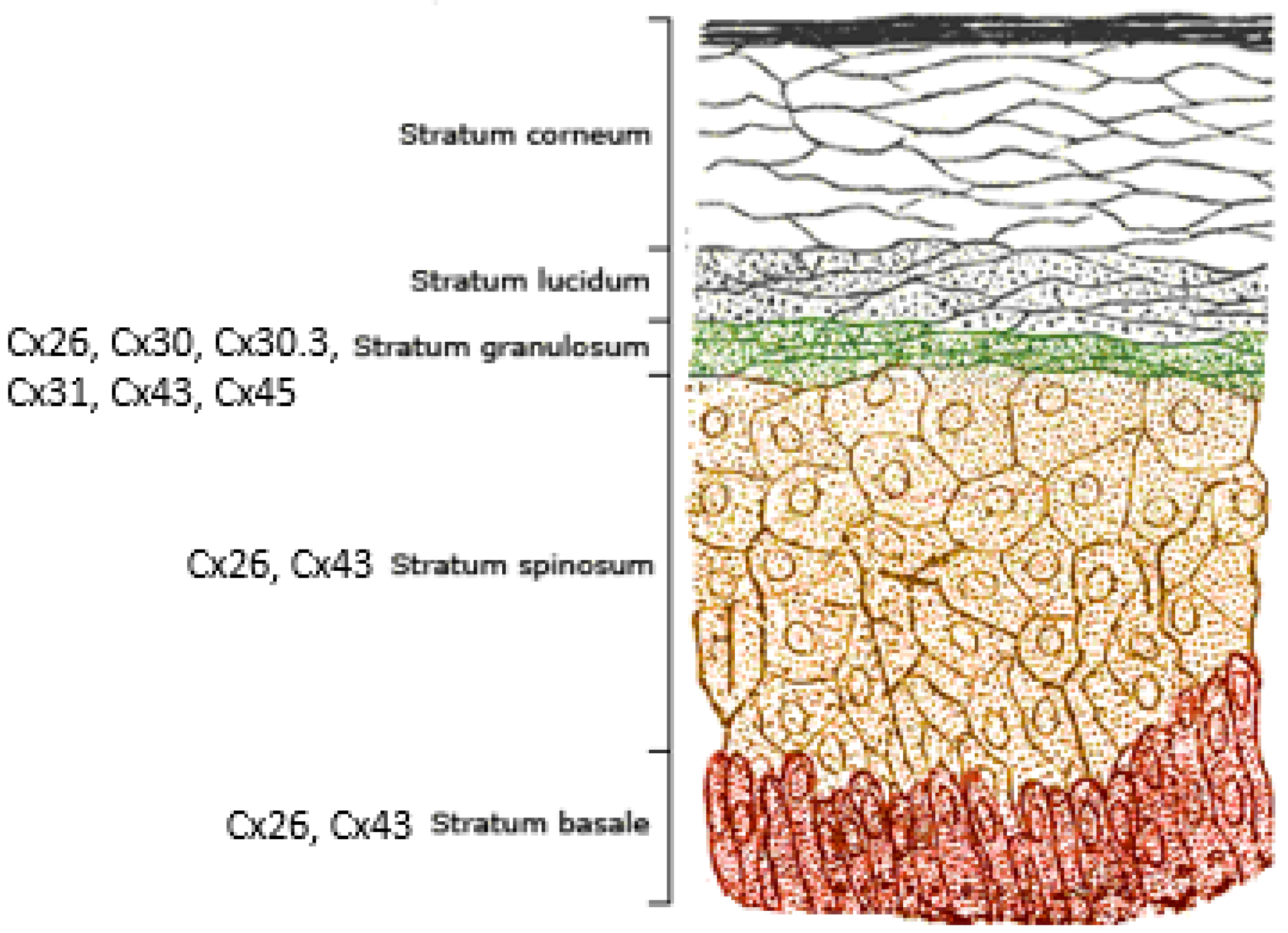
2. Connexins and the Human Epidermis
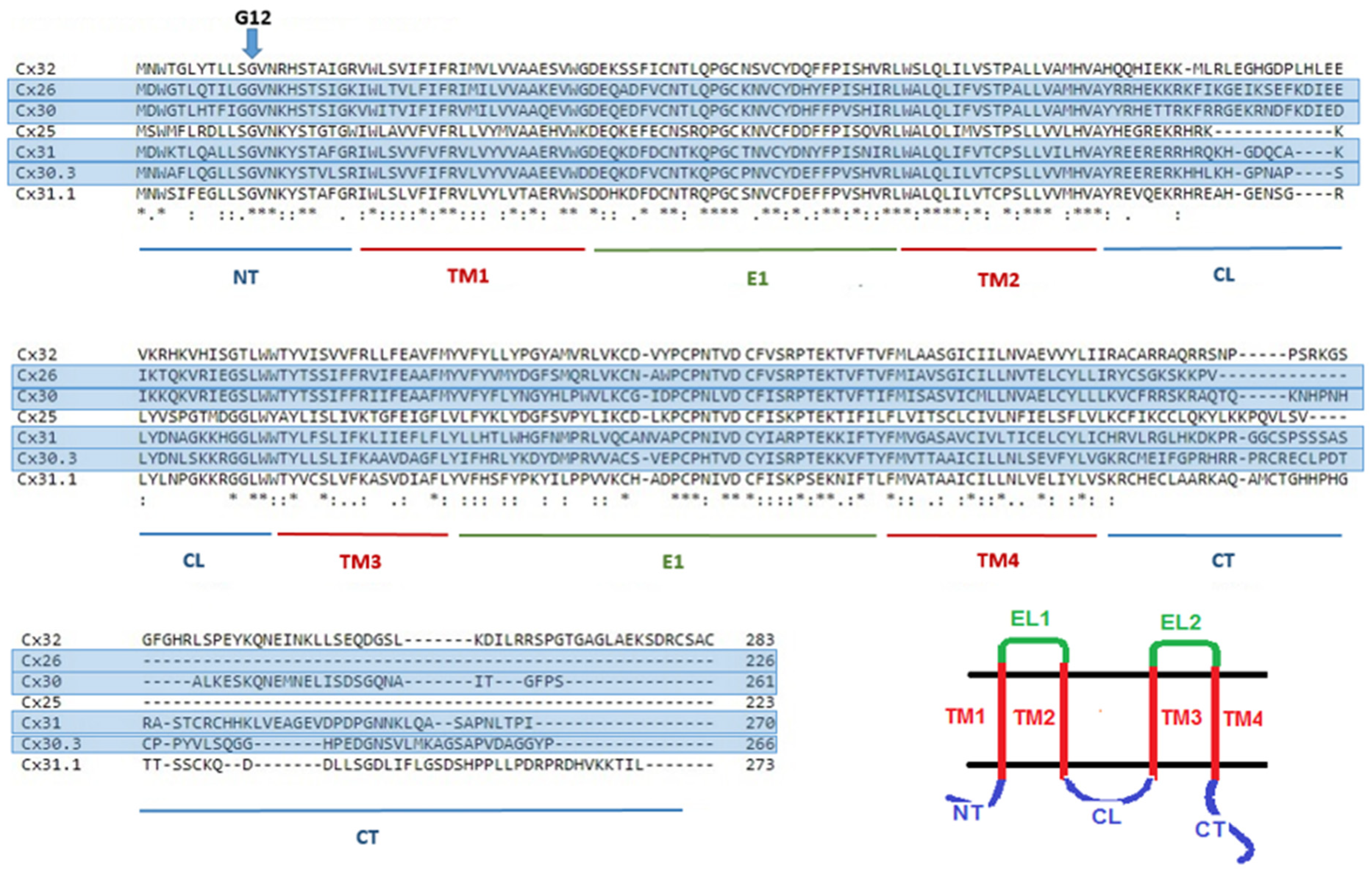
3. Glycine 12 in Beta-Connexins Is Conserved and Essential
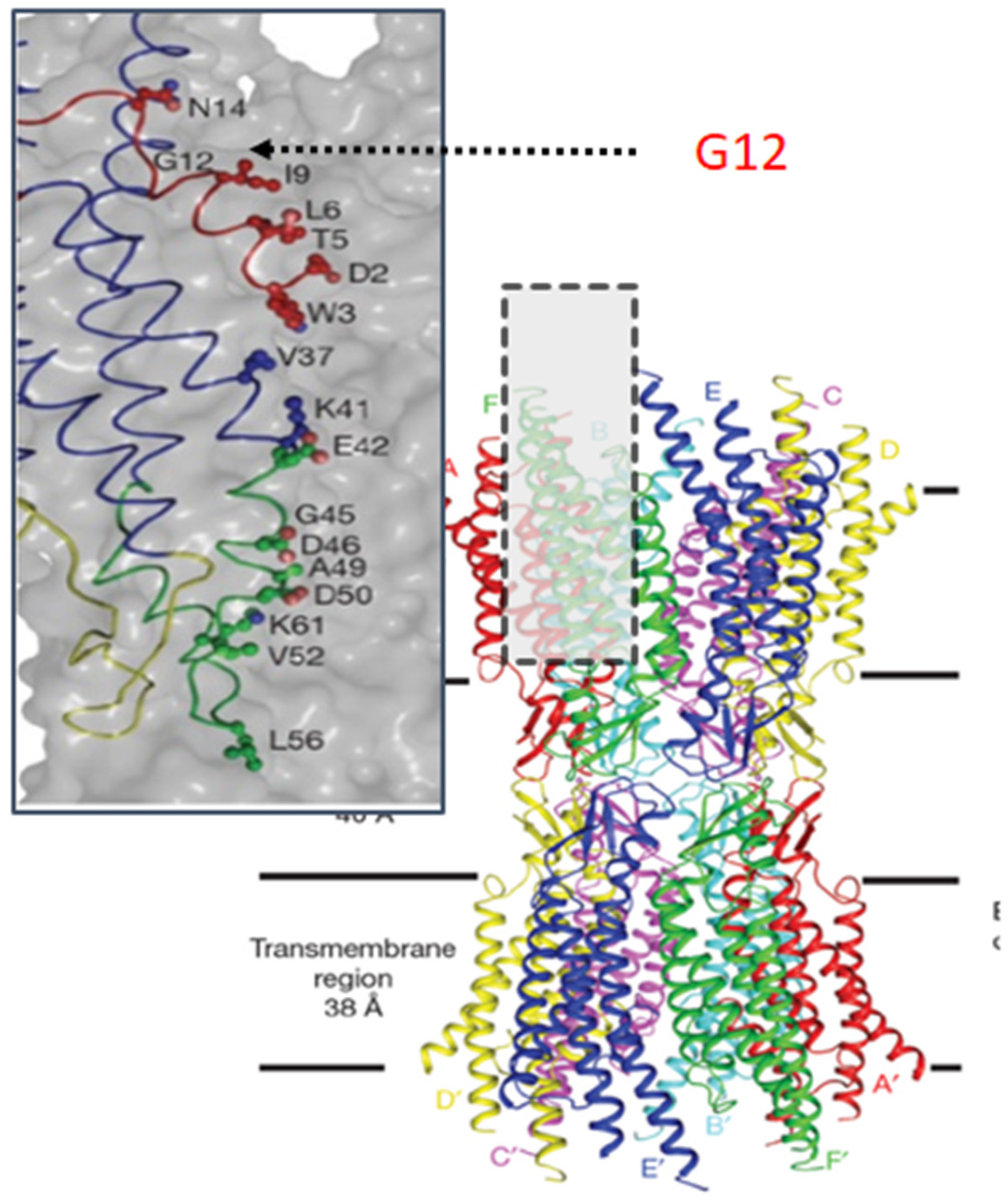
4. Understanding the Role of G12
5. N-Terminal Peptides
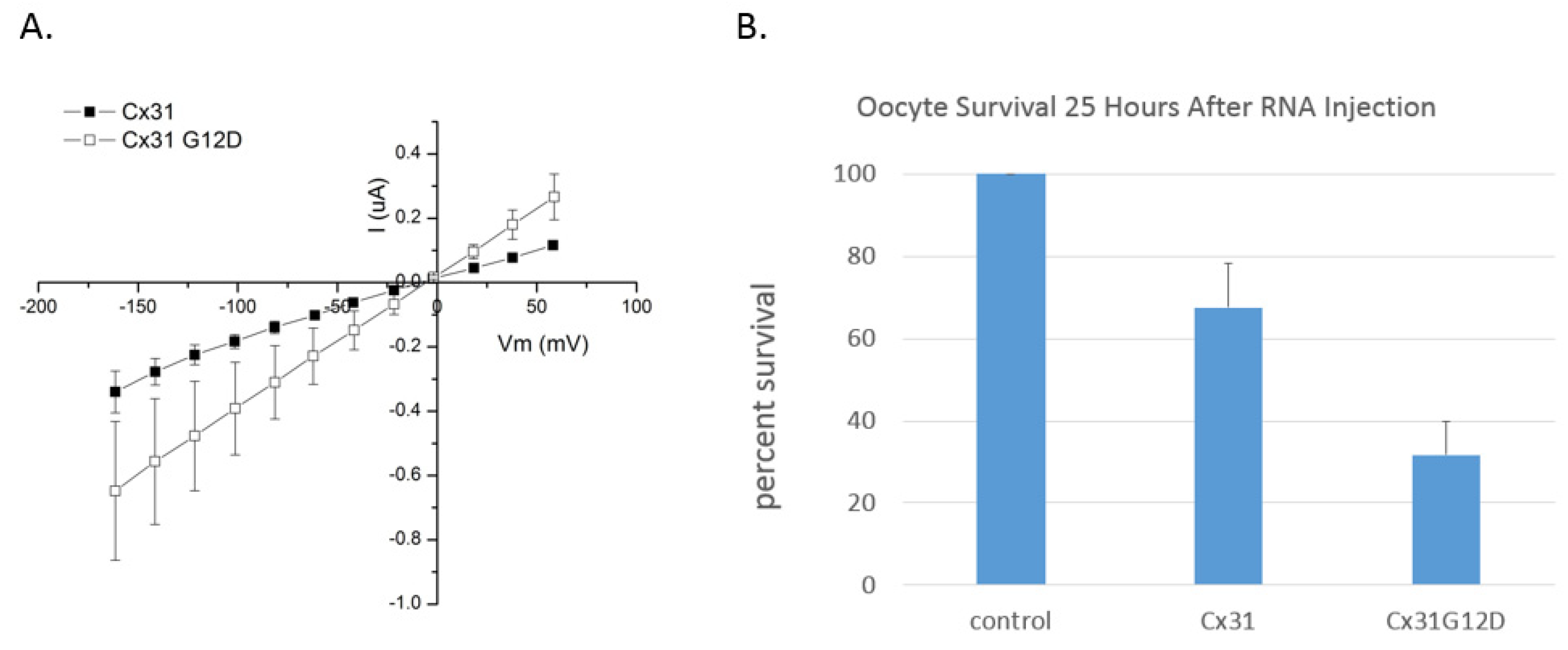
6. Functional Analysis of G12 Mutants
7. Connexin 26 (GJB2)
8. Connexin 30 (GJB6)
9. Connexin 30.3 (GJB4)
10. Connexin 31 (GJB3)
11. Connexin 31.1 (GJB5)
12. Connexin 32 (GJB1)
13. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Söhl, G.; Willecke, K. Gap junctions and the connexin protein family. Cardiovasc. Res. 2004, 62, 228–232. [Google Scholar] [CrossRef]
- Bondarev, I.; Vine, A.; Bertram, J.S. Cloning and Functional Expression of a Novel Human Connexin-25 Gene. Cell Commun. Adhes. 2001, 8, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Di, W.-L.; Rugg, E.L.; Leigh, I.M.; Kelsell, D.P. Multiple Epidermal Connexins are Expressed in Different Keratinocyte Subpopulations Including Connexin 31. J. Investig. Dermatol. 2001, 117, 958–964. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Das Sarma, J.; Wang, F.; Koval, M. Targeted gap junction protein constructs reveal connexin-specific differences in oligomerization. J. Biol. Chem. 2002, 277, 20911–20918. [Google Scholar] [CrossRef]
- Laird, D.W. Life cycle of connexins in health and disease. Biochem. J. 2006, 394, 527–543. [Google Scholar] [CrossRef]
- Jensen, J.M.; Proksch, E. The skin’s barrier. G Ital. Dermatol. Venereol. 2009, 144, 689–700. [Google Scholar] [PubMed]
- Belokhvostova, D.; Berzanskyte, I.; Cujba, A.-M.; Jowett, G.; Marshall, L.; Prueller, J.; Watt, F.M. Homeostasis, regeneration and tumour formation in the mammalian epidermis. Int. J. Dev. Biol. 2018, 62, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Proksch, E.; Brandner, J.M.; Jensen, J.-M. The skin: An indispensable barrier. Exp. Dermatol. 2008, 17, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Lilly, E.; Sellitto, C.; Milstone, L.M.; White, T.W. Connexin channels in congenital skin disorders. Semin. Cell Dev. Biol. 2016, 50, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.; Tan, T.; Chan, C.; Laxton, V.; Chan, Y.W.F.; Liu, T.; Wong, W.T.; Tse, G. The Role of Connexins in Wound Healing and Repair: Novel Therapeutic Approaches. Front. Physiol. 2016, 7, 596. [Google Scholar] [CrossRef]
- Martin, P.E.M.; Van Steensel, M. Connexins and skin disease: Insights into the role of beta connexins in skin homeostasis. Cell Tissue Res. 2015, 360, 645–658. [Google Scholar] [CrossRef]
- Richard, G.; Rouan, F.; Willoughby, C.E.; Brown, N.; Chung, P.; Ryynänen, M.; Jabs, E.W.; Bale, S.J.; DiGiovanna, J.J.; Uitto, J.; et al. Missense Mutations in GJB2 Encoding Connexin-26 Cause the Ectodermal Dysplasia Keratitis-Ichthyosis-Deafness Syndrome. Am. J. Hum. Genet. 2002, 70, 1341–1348. [Google Scholar] [CrossRef]
- Lamartine, J.; Laoudj, D.; Blanchet-Bardon, C.; Kibar, Z.; Soularue, P.; Ridoux, V.; Dubertret, L.; Rouleau, G.; Waksman, G. Refined localization of the gene for Clouston syndrome (hidrotic ectodermal dysplasia) in a large French family. Br. J. Dermatol. 2000, 142, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Richard, G.; Brown, N.; Rouan, F.; Campanelli, C.; Uitto, J.; Van Der Schroeff, J.-G.; Bijlsma, E.; Eichenfield, L.F.; Sybert, V.P.; Greer, K.E.; et al. Genetic Heterogeneity in Erythrokeratodermia Variabilis: Novel Mutations in the Connexin Gene GJB4 (Cx30.3) and Genotype-Phenotype Correlations. J. Investig. Dermatol. 2003, 120, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Richard, G.; Smith, L.E.; Bailey, R.A.; Itin, P.; Hohl, D.; Epstein, E.H., Jr.; DiGiovanna, J.J.; Compton, J.G.; Bale, S.J. Mutations in the human connexin gene GJB3 cause erythrokeratodermia variabilis. Nat. Genet. 1998, 20, 366–369. [Google Scholar] [CrossRef]
- Pizzuti, A.; Flex, E.; Mingarelli, R.; Salpietro, C.; Zelante, L.; Dallapiccola, B. A homozygousGJA1 gene mutation causes a Hallermann-Streiff/ODDD spectrum phenotype. Hum. Mutat. 2004, 23, 286. [Google Scholar] [CrossRef]
- Vreeburg, M.; Schouten, M.; Nellen, R.; Devies, M.; Van Geel, M.; Van Steensel, M.; De Zwart-Storm, E.; Marcus-Soekarman, D. Skin changes in oculo-dento-digital dysplasia are correlated with C-terminal truncations of connexin 43. Am. J. Med. Genet. Part A 2007, 143, 360–363. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Zytsar, M.V.; Barashkov, N.A.; Bady-Khoo, M.S.; Shubina-Olejnik, O.A.; Danilenko, N.G.; Bondar, A.A.; Morozov, I.V.; Solovyev, A.V.; Danilchenko, V.Y.; Maximov, V.N.; et al. Updated carrier rates for c.35delG (GJB2) associated with hearing loss in Russia and common c.35delG haplotypes in Siberia. BMC Med. Genet. 2018, 19, 138. [Google Scholar] [CrossRef]
- Kenna, M.A.; Wu, B.-L.; Cotanche, D.A.; Korf, B.R.; Rehm, H.L. Connexin 26 Studies in Patients with Sensorineural Hearing Loss. Arch. Otolaryngol. Head Neck Surg. 2001, 127, 1037–1042. [Google Scholar] [CrossRef][Green Version]
- D’Andrea, P.; Veronesi, V.; Bicego, M.; Melchionda, S.; Zelante, L.; Di Iorio, E.; Bruzzone, R.; Gasparini, P. Hearing loss: Frequency and functional studies of the most common connexin26 alleles. Biochem. Biophys. Res. Commun. 2002, 296, 685–691. [Google Scholar] [CrossRef]
- García, I.E.; Maripillán, J.; Jara, O.; Ceriani, R.; Palacios-Muñoz, A.; Ramachandran, J.; Olivero, P.; Perez-Acle, T.; González, C.; Sáez, J.C.; et al. Keratitis-Ichthyosis-Deafness Syndrome-Associated Cx26 Mutants Produce Nonfunctional Gap Junctions but Hyperactive Hemichannels When Co-Expressed With Wild Type Cx43. J. Investig. Dermatol. 2015, 135, 1338–1347. [Google Scholar] [CrossRef]
- García, I.E.; Bosen, F.; Mujica, P.; Pupo, A.; Flores-Muñoz, C.; Jara, O.; Gonzalez, C.; Willecke, K.; Martínez, A.D. From Hyperactive Connexin26 Hemichannels to Impairments in Epidermal Calcium Gradient and Permeability Barrier in the Keratitis-Ichthyosis-Deafness Syndrome. J. Investig. Dermatol. 2016, 136, 574–583. [Google Scholar] [CrossRef]
- Lee, J.R.; Derosa, A.M.; White, T.W. Connexin mutations causing skin disease and deafness increase hemichannel activity and cell death when expressed in Xenopus oocytes. J. Investig. Dermatol. 2009, 129, 870–878. [Google Scholar] [CrossRef]
- Lazic, T.; Frank, M.; Zhou, L.H.; Li, Q.; Uitto, J. Extending the Phenotypic Spectrum of Keratitis-Ichthyosis-Deafness Syndrome: Report of a Patient with GJB2 (G12R) Connexin 26 Mutation and Unusual Clinical Findings. Pediatr. Dermatol. 2012, 29, 349–357. [Google Scholar] [CrossRef]
- Taki, T.; Takeichi, T.; Sugiura, K.; Akiyama, M. Roles of aberrant hemichannel activities due to mutant connexin26 in the pathogenesis of KID syndrome. Sci. Rep. 2018, 8, 12824. [Google Scholar] [CrossRef]
- Scott, C.A.; Kelsell, D.P. Key functions for gap junctions in skin and hearing. Biochem. J. 2011, 438, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Van Steensel, M.A.; Oranje, A.P.; van der Schroeff, J.G.; Wagner, A.; van Geel, M. The missense mutation G12D in connexin30.3 can cause both erythrokeratodermia variabilis of Mendes da Costa and progressive symmetric erythrokeratodermia of Gottron. Am. J. Med. Genet. 2009, 149A, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Di, W.-L.; Monypenny, J.; Common, J.E.; Kennedy, C.T.; Holland, K.A.; Leigh, I.M.; Rugg, E.L.; Zicha, D.; Kelsell, D.P. Defective trafficking and cell death is characteristic of skin disease-associated connexin 31 mutations. Hum. Mol. Genet. 2002, 11, 2005–2014. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Wang, H.; Mou, Y.; Zeng, Q.; Xiong, X. Exome sequencing identifies novel compound heterozygous mutations in GJB3 gene that cause erythrokeratodermia variabilis et progressiva. Australas. J. Dermatol. 2019, 60, e87–e89. [Google Scholar] [CrossRef]
- Rouan, F.; Lo, C.W.; Fertala, A.; Wahl, M.; Jost, M.; Rodeck, U.; Uitto, J.; Richard, G. Divergent effects of two sequence variants of GJB3 (G12D and R32W) on the function of connexin 31 in vitro. Exp. Dermatol. 2003, 12, 191–197. [Google Scholar] [CrossRef] [PubMed]
- He, L.-Q.; Liu, Y.; Cai, F.; Tan, Z.-P.; Pan, Q.; Liang, D.-S.; Long, Z.-G.; Wu, L.-Q.; Huang, L.-Q.; Dai, H.-P.; et al. Intracellular Distribution, Assembly and Effect of Disease-associated Connexin 31 Mutants in HeLa Cells. Acta Biochim. Biophys. Sin. 2005, 37, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Scherer, S.S.; Kleopa, K.A. X-linked Charcot-Marie-Tooth disease. J. Peripher. Nerv. Syst. 2012, 17 (Suppl. S3), 9–13. [Google Scholar] [CrossRef] [PubMed]
- Deschênes, S.M.; Walcott, J.L.; Wexler, T.L.; Scherer, S.S.; Fischbeck, K.H. Altered Trafficking of Mutant Connexin32. J. Neurosci. 1997, 17, 9077–9084. [Google Scholar] [CrossRef]
- Bone, L.J.; Deschenes, S.M.; Balice-Gordon, R.J.; Fischbeck, K.H.; Scherer, S.S. Connexin32 and X-linked Charcot–Marie–Tooth Disease. Neurobiol. Dis. 1997, 4, 221–230. [Google Scholar] [CrossRef]
- Abrams, C.K.; Freidin, M.M.; Verselis, V.K.; Bennett, M.V.; Bargiello, T.A. Functional alterations in gap junction channels formed by mutant forms of connexin 32: Evidence for loss of function as a pathogenic mechanism in the X-linked form of Charcot-Marie-Tooth disease. Brain Res. 2001, 900, 9–25. [Google Scholar] [CrossRef]
- Unger, V.M.; Kumar, N.M.; Gilula, N.B.; Yeager, M. Three-dimensional structure of a recombinant gap junction membrane channel. Science 1999, 283, 1176–1180. [Google Scholar] [CrossRef]
- Oshima, A.; Tani, K.; Hiroaki, Y.; Fujiyoshi, Y.; Sosinsky, G.E. Three-dimensional structure of a human connexin26 gap junction channel reveals a plug in the vestibule. Proc. Natl. Acad. Sci. USA 2007, 104, 10034–10039. [Google Scholar] [CrossRef]
- Maeda, S.; Nakagawa, S.; Suga, M.; Yamashita, E.; Oshima, A.; Fujiyoshi, Y.; Tsukihara, T. Structure of the connexin 26 gap junction channel at 3.5 A resolution. Nature 2009, 458, 597–602. [Google Scholar] [CrossRef]
- Bennett, B.C.; Purdy, M.D.; Baker, K.A.; Acharya, C.; McIntire, W.E.; Stevens, R.C.; Zhang, Q.; Harris, A.L.; Abagyan, R.; Yeager, M. An electrostatic mechanism for Ca(2+)-mediated regulation of gap junction channels. Nat. Commun. 2016, 7, 8770. [Google Scholar] [CrossRef]
- Myers, J.B.; Haddad, B.G.; O’Neill, S.E.; Chorev, D.S.; Yoshioka, C.C.; Robinson, C.V.; Zuckerman, D.M.; Reichow, S.L. Structure of native lens connexin 46/50 intercellular channels by cryo-EM. Nature 2018, 564, 372–377. [Google Scholar] [CrossRef]
- Skerrett, I.M.; Smith, J.F.; Nicholson, B.J. Mechanistic Differences Between Chemical and Electrical Gating of Gap Junctions. In Current Topics in Membranes; Peracchia, C., Ed.; Academic Press: Cambridge, MA, USA, 1999; Volume 49, pp. 249–269. [Google Scholar]
- Marcelino, A.M.; Gierasch, L.M. Roles of beta-turns in protein folding: From peptide models to protein engineering. Biopolymers 2008, 89, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Verselis, V.K.; Ginter, C.S.; Bargiello, T.A. Opposite voltage gating polarities of two closely related connexins. Nature 1994, 368, 348–351. [Google Scholar] [CrossRef]
- Purnick, P.E.; Oh, S.; Abrams, C.K.; Verselis, V.K.; Bargiello, T.A. Reversal of the Gating Polarity of Gap Junctions by Negative Charge Substitutions in the N-Terminus of Connexin 32. Biophys. J. 2000, 79, 2403–2415. [Google Scholar] [CrossRef]
- Kyle, J.W.; Minogue, P.J.; Thomas, B.C.; Domowicz, D.A.L.; Berthoud, V.M.; Hanck, R.A.; Beyer, E.C. An intact connexin N-terminus is required for function but not gap junction formation. J. Cell Sci. 2008, 121, 2744–2750. [Google Scholar] [CrossRef]
- Beyer, E.C.; Lipkind, G.M.; Kyle, J.W.; Berthoud, V.M. Structural organization of intercellular channels II. Amino terminal domain of the connexins: Sequence, functional roles, and structure. Biochim. Biophys. Acta 2012, 1818, 1823–1830. [Google Scholar] [CrossRef] [PubMed]
- Kalmatsky, B.; Batir, Y.; Bargiello, T.A.; Dowd, T. Structural studies of N-terminal mutants of Connexin 32 using (1)H NMR spectroscopy. Arch. Biochem. Biophys. 2012, 526, 1–8. [Google Scholar] [CrossRef]
- Xu, Q.; Lin, X.; Matiukas, A.; Zhang, X.; Veenstra, R.D. Specificity of the connexin W3/4 locus for functional gap junction formation. Channels 2016, 10, 453–465. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bargiello, T.A.; Tang, Q.; Oh, S.; Kwon, T. Voltage-dependent conformational changes in connexin channels. Biochim. Biophys. Acta 2012, 1818, 1807–1822. [Google Scholar] [CrossRef] [PubMed]
- Villanelo, F.; Escalona, Y.; Pareja-Barrueto, C.; Garate, J.A.; Skerrett, I.M.; Perez-Acle, T. Accessing gap-junction channel structure-function relationships through molecular modeling and simulations. BMC Cell Biol. 2017, 18, 5. [Google Scholar] [CrossRef]
- Purnick, P.E.; Benjamin, D.C.; Verselis, V.K.; Bargiello, T.A.; Dowd, T.L. Structure of the Amino Terminus of a Gap Junction Protein. Arch. Biochem. Biophys. 2000, 381, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Batir, Y.; Bargiello, T.A.; Dowd, T.L. Structural studies of N-terminal mutants of Connexin 26 and Connexin 32 using (1)H NMR spectroscopy. Arch. Biochem. Biophys. 2016, 608, 8–19. [Google Scholar] [CrossRef]
- Montgomery, J.R.; White, T.W.; Martin, B.L.; Turner, M.L.; Holland, S.M. A novel connexin 26 gene mutation associated with features of the keratitis-ichthyosis-deafness syndrome and the follicular occlusion triad. J. Am. Acad. Dermatol. 2004, 51, 377–382. [Google Scholar] [CrossRef]
- Saez, J.C.; Berthoud, V.M.; Branes, M.C.; Martinez, A.D.; Beyer, E.C. Plasma membrane channels formed by connexins: Their Regulation and Functions. Physiol. Rev. 2003, 83, 1359–1400. [Google Scholar] [CrossRef] [PubMed]
- Meşe, G.; Richard, G.; White, T.W. Gap Junctions: Basic Structure and Function. J. Investig. Dermatol. 2007, 127, 2516–2524. [Google Scholar] [CrossRef] [PubMed]
- Skinner, B.A.; Greist, M.C.; Norins, A.L. The Keratitis, Ichthyosis, and Deafness (KID) Syndrome. Arch. Dermatol. 1981, 117, 285–289. [Google Scholar] [CrossRef]
- Kelsell, D.P.; Dunlop, J.; Stevens, H.P.; Lench, N.J.; Liang, J.N.; Parry, G.; Mueller, R.F.; Leigh, I.M. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 1997, 387, 80–83. [Google Scholar] [CrossRef]
- Denoyelle, F.; Marlin, S.; Weil, D.; Moatti, L.; Chauvin, P.; Garabédian, É.-N.; Petit, C. Clinical features of the prevalent form of childhood deafness, DFNB1, due to a connexin-26 gene defect: Implications for genetic counselling. Lancet 1999, 353, 1298–1303. [Google Scholar] [CrossRef]
- Neoh, C.Y.; Chen, H.; Ng, S.K.; Lane, E.B.; Common, J.E.A. A rare connexin 26 mutation in a patient with a forme fruste of keratitis-ichthyosis-deafness (KID) syndrome. Int. J. Dermatol. 2009, 48, 1078–1081. [Google Scholar] [CrossRef]
- Rabionet, R.; Zelante, L.; López-Bigas, N.; D’Agruma, L.; Melchionda, S.; Restagno, G.; Arbonés, M.L.; Gasparini, P.; Estivill, X. Molecular basis of childhood deafness resulting from mutations in the GJB2 (connexin 26) gene. Hum. Genet. 2000, 106, 40–44. [Google Scholar] [PubMed]
- Donnelly, S.; English, G.; De Zwart-Storm, E.A.; Lang, S.; van Steensel, M.A.M.; Martin, P.E. Differential susceptibility of Cx26 mutations associated with epidermal dysplasias to peptidoglycan derived from Staphylococcus aureus and Staphylococcus epidermidis. Exp. Dermatol. 2012, 21, 592–598. [Google Scholar] [CrossRef]
- Lagrée, V.; Brunschwig, K.; Lopez, P.; Gilula, N.B.; Richard, G.; Falk, M.M. Specific amino-acid residues in the N-terminus and TM3 implicated in channel function and oligomerization compatibility of connexin43. J. Cell Sci. 2003, 116, 3189–3201. [Google Scholar] [CrossRef]
- García, I.E.; Villanelo, F.; Contreras, G.F.; Pupo, A.; Pinto, B.I.; Contreras, J.E.; Pérez-Acle, T.; Alvarez, O.; Latorre, R.; Martínez, A.D.; et al. The syndromic deafness mutation G12R impairs fast and slow gating in Cx26 hemichannels. J. Gen. Physiol. 2018, 150, 697–711. [Google Scholar] [CrossRef]
- Snoeckx, R.L.; Huygen, P.L.; Feldmann, D.; Marlin, S.; Denoyelle, F.; Waligora, J.; Mueller-Malesinska, M.; Pollak, A.; Ploski, R.; Murgia, A.; et al. GJB2 Mutations and Degree of Hearing Loss: A Multicenter Study. Am. J. Hum. Genet. 2005, 77, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, T.; Kimura, R.; Paul, D.; Adams, J. Gap junctions in the rat cochlea: Immunohistochemical and ultrastructural analysis. Anat. Embryol. 1995, 191, 101–118. [Google Scholar] [CrossRef]
- Bitner-Glindzicz, M. Hereditary deafness and phenotyping in humans. Br. Med. Bull. 2002, 63, 73–94. [Google Scholar] [CrossRef]
- Chen, N.; Xu, C.; Han, B.; Wang, Z.-Y.; Song, Y.-L.; Li, S.; Zhang, R.-L.; Pan, C.-M.; Zhang, L. G11R mutation in GJB6 gene causes hidrotic ectodermal dysplasia involving only hair and nails in a Chinese family. J. Dermatol. 2010, 37, 559–561. [Google Scholar] [CrossRef]
- Essenfelder, G.M.; Bruzzone, R.; Lamartine, J.; Charollais, A.; Blanchet-Bardon, C.; Barbe, M.T.; Meda, P.; Waksman, G. Connexin30 mutations responsible for hidrotic ectodermal dysplasia cause abnormal hemichannel activity. Hum. Mol. Genet. 2004, 13, 1703–1714. [Google Scholar] [CrossRef] [PubMed]
- Plantard, L.; Huber, M.; Macari, F.; Meda, P.; Hohl, D. Molecular interaction of connexin 30.3 and connexin 31 suggests a dominant-negative mechanism associated with erythrokeratodermia variabilis. Hum. Mol. Genet. 2003, 12, 3287–3294. [Google Scholar] [CrossRef]
- Lopez-Bigas, N.; Olive, M.; Rabionet, R.; Ben-David, O.; Martinez-Matos, J.A.; Bravo, O.; Banchs, I.; Volpini, V.; Gasparini, P.; Avraham, K.B.; et al. Connexin 31 (GJB3) is expressed in the peripheral and auditory nerves and causes neuropathy and hearing impairment. Hum. Mol. Genet. 2001, 10, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Diestel, S.; Richard, G.; Döring, B.; Traub, O. Expression of a connexin31 mutation causing erythrokeratodermia variabilis is lethal for HeLa cells. Biochem. Biophys. Res. Commun. 2002, 296, 721–728. [Google Scholar] [CrossRef]
- Tattersall, D.; Scott, C.A.; Gray, C.; Zicha, D.; Kelsell, D.P. EKV mutant connexin 31 associated cell death is mediated by ER stress. Hum. Mol. Genet. 2009, 18, 4734–4745. [Google Scholar] [CrossRef]
- Chi, J.; Li, L.; Liu, M.; Tan, J.; Tang, C.; Pan, Q.; Wang, D.; Zhang, Z. Pathogenic Connexin-31 Forms Constitutively Active Hemichannels to Promote Necrotic Cell Death. PLoS ONE 2012, 7, e32531. [Google Scholar] [CrossRef]
- Bailey, R. Functional Analysis of a Critical Glycine (Glycine 12) in Beta-type Connexins of Human Skin. Biology. Master’s Thesis, The State University of New York College at Buffalo–Buffalo State Collage, Buffalo, NY, USA, May 2000; 41p. Available online: https://digitalcommons.buffalostate.edu/biology_theses/41 (accessed on 3 March 2021).
- Xia, J.H.; Liu, C.Y.; Tang, B.S.; Pan, Q.; Huang, L.; Dai, H.P.; Zhang, B.R.; Xie, W.; Hu, D.X.; Zheng, D.; et al. Mutations in the gene encoding gap junction protein beta-3 associated with autosomal dominant hearing impairment. Nat. Genet. 1998, 20, 270–370. [Google Scholar] [CrossRef]
- Bergoffen, J.; Scherer, S.S.; Wang, S.; Scott, M.O.; Bone, L.J.; Paul, D.L.; Chen, K.; Lensch, M.W.; Chance, P.F.; Fischbeck, K.H. Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science 1993, 262, 2039–2042. [Google Scholar] [CrossRef] [PubMed]
- Yum, S.W.; Kleopa, K.A.; Shumas, S.; Scherer, S.S. Diverse Trafficking Abnormalities of Connexin32 Mutants Causing CMTX. Neurobiol. Dis. 2002, 11, 43–52. [Google Scholar] [CrossRef]
- García, I.E.; Prado, P.; Pupo, A.; Jara, O.; Rojas-Gómez, D.; Mujica, P.; Flores-Muñoz, C.; González-Casanova, J.; Soto-Riveros, C.; Pinto, B.I.; et al. Connexinopathies: A structural and functional glimpse. BMC Cell Biol. 2016, 17 (Suppl. S1). [Google Scholar] [CrossRef]
- Terrinoni, A.; Codispoti, A.; Serra, V.; Didona, B.; Bruno, E.; Nisticò, R.; Giustizieri, M.; Alessandrini, M.; Campione, E.; Melino, G. Connexin 26 (GJB2) mutations, causing KID Syndrome, are associated with cell death due to calcium gating deregulation. Biochem. Biophys. Res. Commun. 2010, 394, 909–914. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Connexin (Gene) | Disorder | Background | G12 Mutation |
|---|---|---|---|
| Cx26 (GJB2) | Deafness Non-syndromic | Over 300 recessive mutations, mostly point mutations. 35DelG with truncation at amino acid 13 is most prevalent [19,20] | G12V [21,22,23] |
| Skin Disease with Deafness Keratitis ichthyosis and deafness (KID) syndrome Rare skin disorders with or without inflammatory response | Numerous dominant point mutations, sporadic and hereditary. Focused in NT and E1 domains [12,24] Dominant point mutations throughout coding region [12] | G12R [24,25,26,27] | |
| Cx30 (GJB6) | Skin Disease Clouston syndrome | A few point mutations resulting in amino acid substitutions in various regions [12] | |
| Cx30.3 (GJB4) | Skin Disease erythrokeratodermia variabilis et progressiva (EKV) | A few point mutations resulting in amino acid substitutions in various regions [15,28] | G12D [15,29] |
| Cx31 (GJB3) | Skin Disease erythrokeratodermia variabilis et progressiva (EKV) Hearing and neurological disorders | At least 20 mutations, most dominant and appear to result in trafficking defects and cell death [30] Several mutations associated with deafness and one with neurological disease [31] | G12D [32] G12R [33] G12S [31] |
| Cx32 (GJB1) | Neurodegenerative X-linked Charcot-Marie-Tooth (CMTX) disease | Hundreds of recessive mutations, mostly point mutations in all regions of protein [34] | G12S [35,36,37] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bailey, R.A.; Beahm, D.L.; Skerrett, I.M. The Complex and Critical Role of Glycine 12 (G12) in Beta-Connexins of Human Skin. Int. J. Mol. Sci. 2021, 22, 2615. https://doi.org/10.3390/ijms22052615
Bailey RA, Beahm DL, Skerrett IM. The Complex and Critical Role of Glycine 12 (G12) in Beta-Connexins of Human Skin. International Journal of Molecular Sciences. 2021; 22(5):2615. https://doi.org/10.3390/ijms22052615
Chicago/Turabian StyleBailey, Rasheed A., Derek L. Beahm, and I. Martha Skerrett. 2021. "The Complex and Critical Role of Glycine 12 (G12) in Beta-Connexins of Human Skin" International Journal of Molecular Sciences 22, no. 5: 2615. https://doi.org/10.3390/ijms22052615
APA StyleBailey, R. A., Beahm, D. L., & Skerrett, I. M. (2021). The Complex and Critical Role of Glycine 12 (G12) in Beta-Connexins of Human Skin. International Journal of Molecular Sciences, 22(5), 2615. https://doi.org/10.3390/ijms22052615