
Understanding the Pathophysiology of Thrombotic APS through Animal Models


Abstract
1. Introduction
2. Venous APS Models
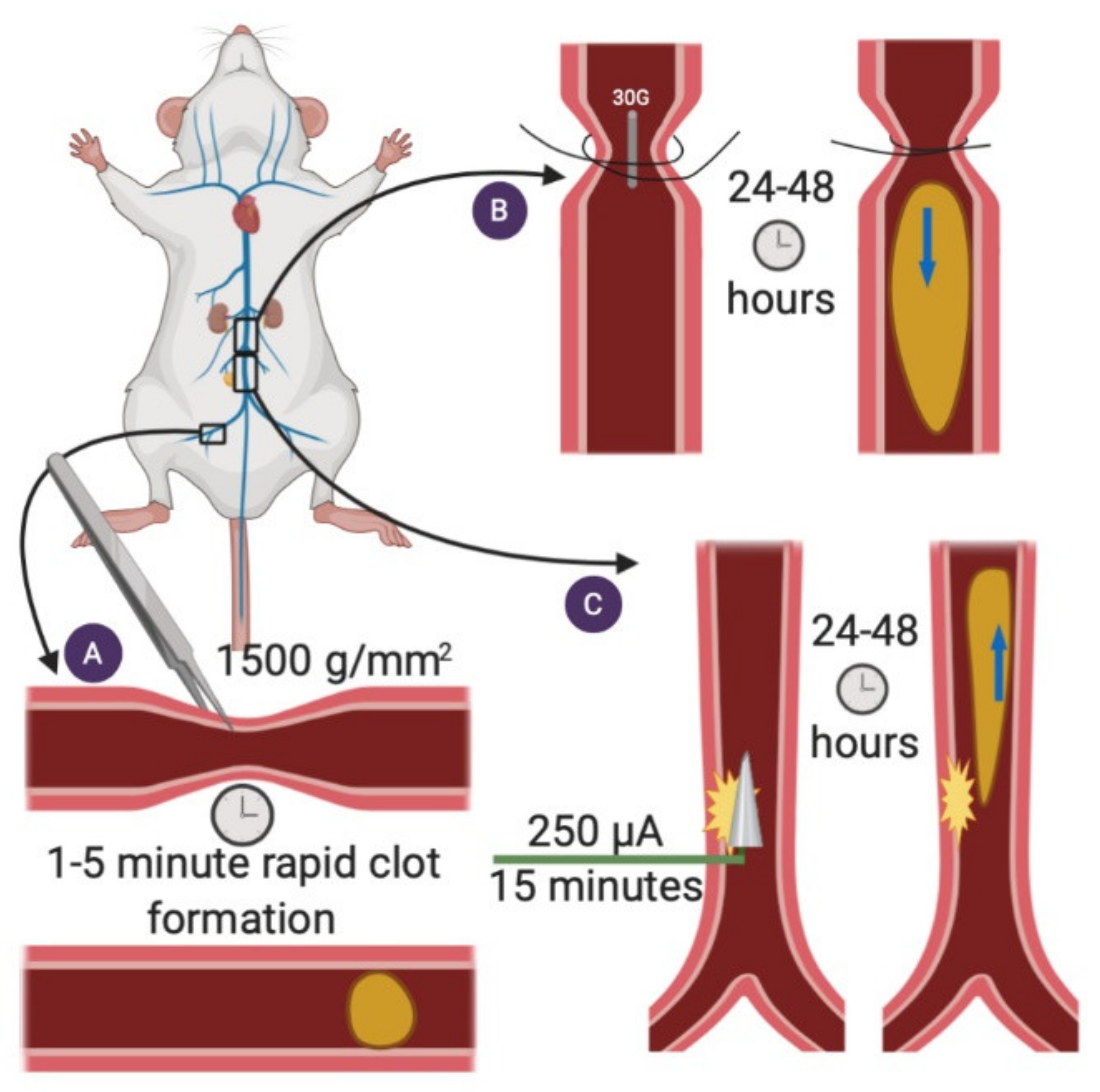
2.1. Femoral Vein Pinch
2.2. Stenosis
2.3. Electrolytic IVC
2.4. Ferric Chloride Injury Model—Femoral Vein
2.5. What Have We Learned about APS-Associated Venous Thrombosis from Animal Models?
3. Arterial APS Models
3.1. Ferric Chloride Injury Model—Carotid Artery
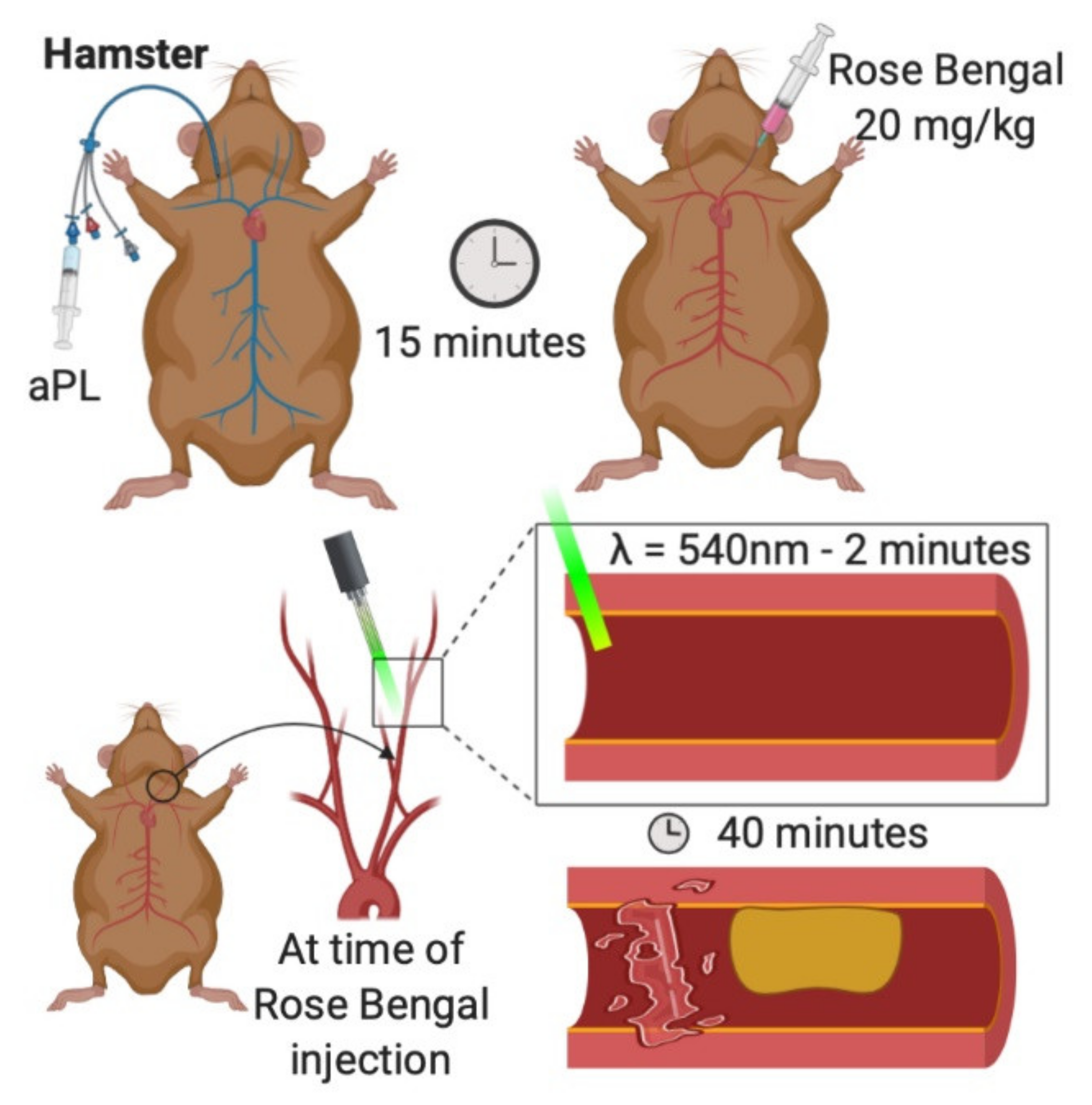
3.2. Photochemical Carotid
3.3. What Have We Learned about APS-Associated Arterial Thrombosis from Animal Models?
4. Microvascular APS Models
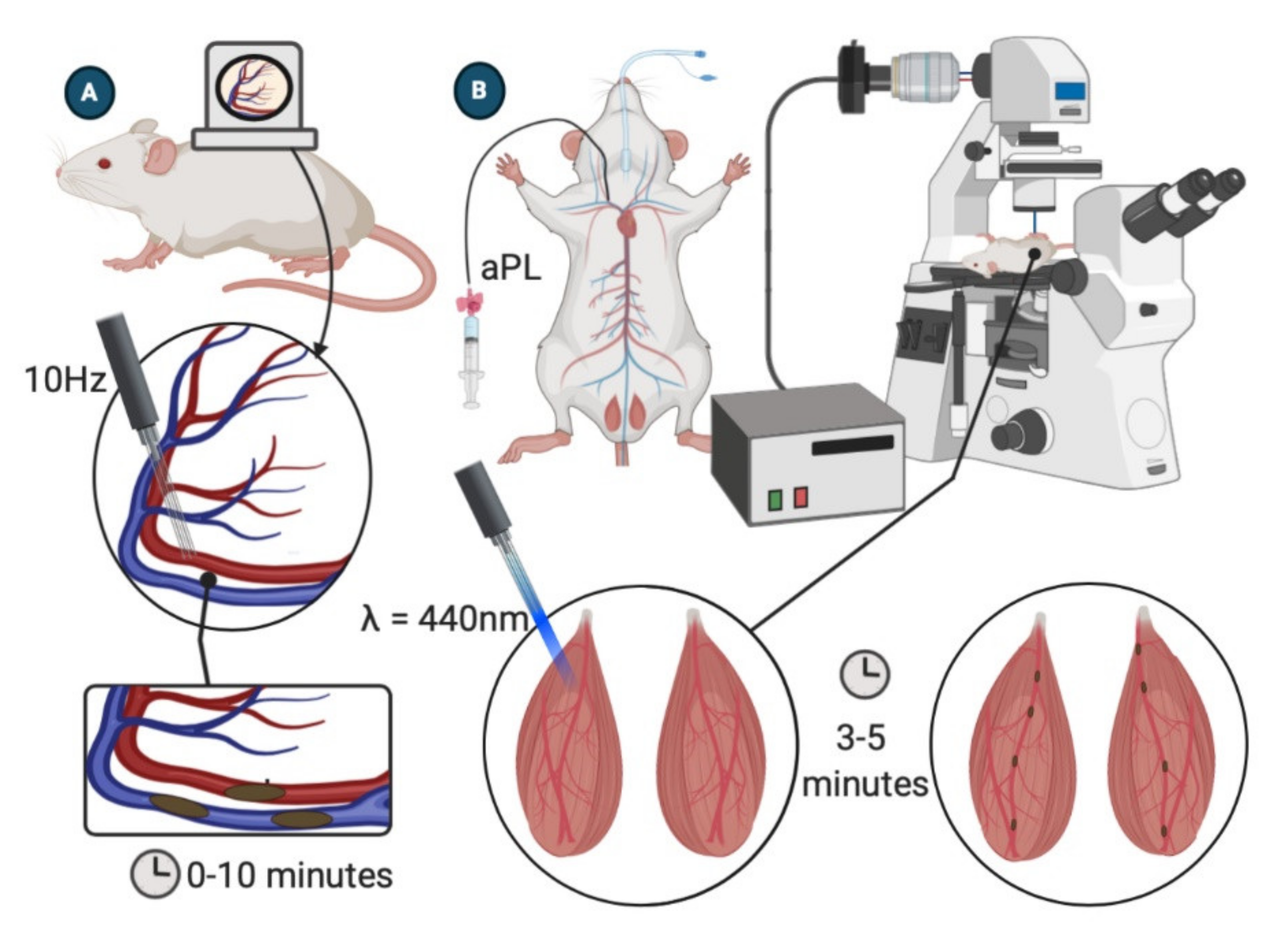
4.1. Dorsal Skinfold Chamber
4.2. Laser-Induced Injury of Cremaster Arterioles
4.3. Ferric Chloride Injury Model—Mesenteric Microcirculation
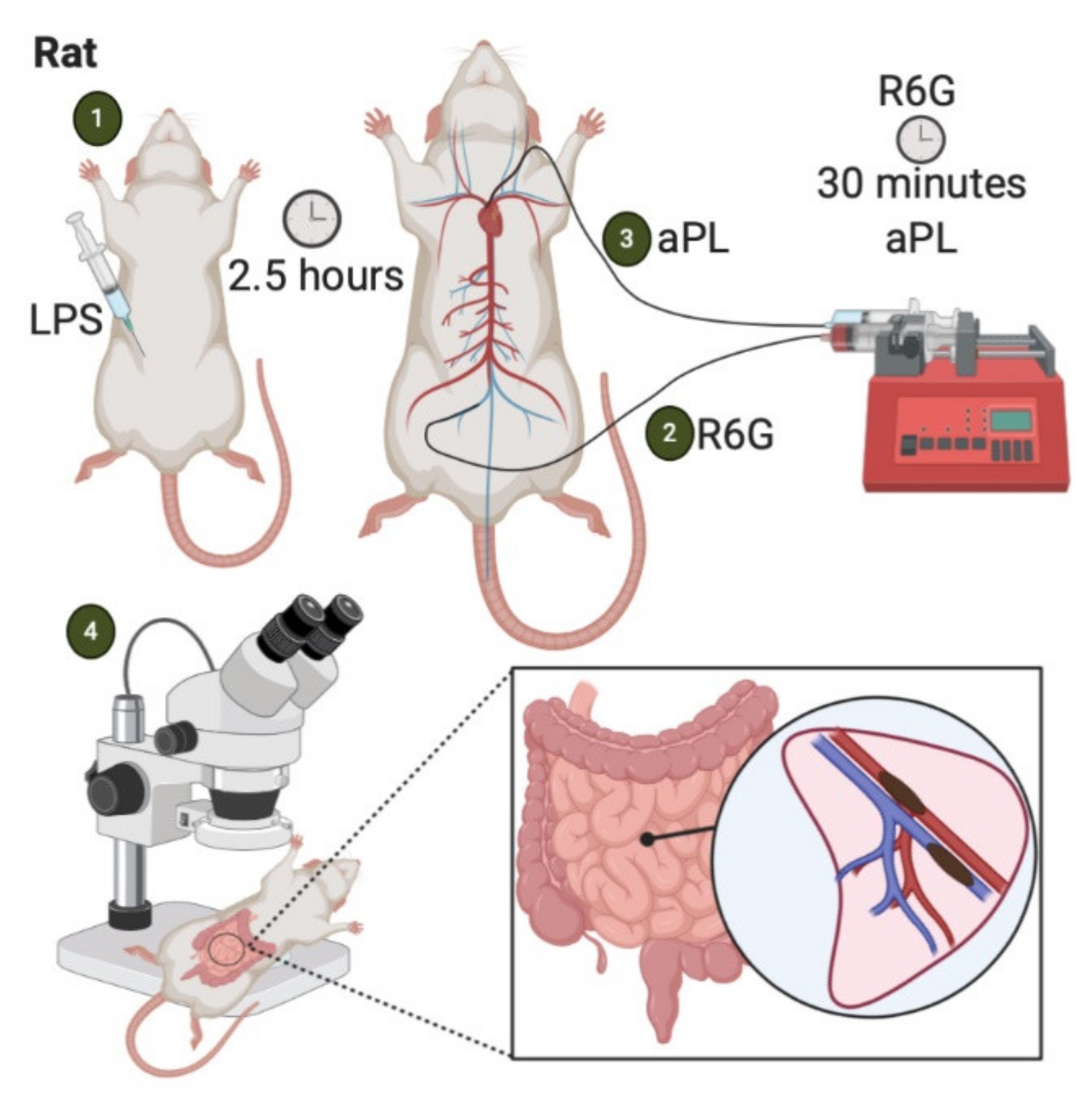
4.4. LPS-Priming
4.5. Histone Priming
4.6. What Have We Learned about APS-Associated Microvascular Thrombosis from Animal Models?
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Cervera, R.; Piette, J.C.; Font, J.; Khamashta, M.A.; Shoenfeld, Y.; Camps, M.T.; Jacobsen, S.; Lakos, G.; Tincani, A.; Kontopoulou-Griva, I.; et al. Antiphospholipid syndrome: Clinical and immunologic manifestations and patterns of disease expression in a cohort of 1000 patients. Arthritis Rheumatol. 2002, 46, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Duarte-Garcia, A.; Pham, M.M.; Crowson, C.S.; Amin, S.; Moder, K.G.; Pruthi, R.K.; Warrington, K.J.; Matteson, E.L. The Epidemiology of Antiphospholipid Syndrome: A Population-Based Study. Arthritis Rheumatol. 2019, 71, 1545–1552. [Google Scholar] [CrossRef]
- Cervera, R. Antiphospholipid syndrome. Thromb. Res. 2017, 151 (Suppl. 1), S43–S47. [Google Scholar] [CrossRef]
- Abreu, M.M.; Danowski, A.; Wahl, D.G.; Amigo, M.C.; Tektonidou, M.; Pacheco, M.S.; Fleming, N.; Domingues, V.; Sciascia, S.; Lyra, J.O.; et al. The relevance of “non-criteria” clinical manifestations of antiphospholipid syndrome: 14th International Congress on Antiphospholipid Antibodies Technical Task Force Report on Antiphospholipid Syndrome Clinical Features. Autoim. Rev. 2015. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Simantov, R.; Zhang, J.C.; Silverstein, R.; Hajjar, K.A.; McCrae, K.R. High affinity binding of beta 2-glycoprotein I to human endothelial cells is mediated by annexin II. J. Biol. Chem. 2000, 275, 15541–15548. [Google Scholar] [CrossRef] [PubMed]
- Allen, K.L.; Fonseca, F.V.; Betapudi, V.; Willard, B.; Zhang, J.; McCrae, K.R. A novel pathway for human endothelial cell activation by antiphospholipid/anti-beta2 glycoprotein I antibodies. Blood 2012, 119, 884–893. [Google Scholar] [CrossRef] [PubMed]
- Sorice, M.; Longo, A.; Capozzi, A.; Garofalo, T.; Misasi, R.; Alessandri, C.; Conti, F.; Buttari, B.; Rigano, R.; Ortona, E.; et al. Anti-beta2-glycoprotein I antibodies induce monocyte release of tumor necrosis factor alpha and tissue factor by signal transduction pathways involving lipid rafts. Arthritis Rheumatol. 2007, 56, 2687–2697. [Google Scholar] [CrossRef]
- Lutters, B.C.; Derksen, R.H.; Tekelenburg, W.L.; Lenting, P.J.; Arnout, J.; de Groot, P.G. Dimers of beta 2-glycoprotein I increase platelet deposition to collagen via interaction with phospholipids and the apolipoprotein E receptor 2’. J. Biol. Chem. 2003, 278, 33831–33838. [Google Scholar] [CrossRef]
- De Groot, P.G.; Urbanus, R.T.; Derksen, R.H. Pathophysiology of thrombotic APS: Where do we stand? Lupus 2012, 21, 704–707. [Google Scholar] [CrossRef]
- Salmon, J.E.; Girardi, G. Antiphospholipid antibodies and pregnancy loss: A disorder of inflammation. J. Reprod. Immunol. 2008, 77, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Miyakis, S.; Lockshin, M.D.; Atsumi, T.; Branch, D.W.; Brey, R.L.; Cervera, R.; Derksen, R.H.; PG, D.E.G.; Koike, T.; Meroni, P.L.; et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J. Thromb. Haemost. 2006, 4, 295–306. [Google Scholar] [CrossRef]
- Shi, H.; Zheng, H.; Yin, Y.F.; Hu, Q.Y.; Teng, J.L.; Sun, Y.; Liu, H.L.; Cheng, X.B.; Ye, J.N.; Su, Y.T.; et al. Antiphosphatidylserine/prothrombin antibodies (aPS/PT) as potential diagnostic markers and risk predictors of venous thrombosis and obstetric complications in antiphospholipid syndrome. Clin. Chem. Lab. Med. 2018, 56, 614–624. [Google Scholar] [CrossRef]
- Pierangeli, S.S.; Barker, J.H.; Stikovac, D.; Ackerman, D.; Anderson, G.; Barquinero, J.; Acland, R.; Harris, E.N. Effect of human IgG antiphospholipid antibodies on an in vivo thrombosis model in mice. Thromb. Haemostol. 1994, 71, 670–674. [Google Scholar] [CrossRef]
- Pierangeli, S.S.; Colden-Stanfield, M.; Liu, X.; Barker, J.H.; Anderson, G.L.; Harris, E.N. Antiphospholipid antibodies from antiphospholipid syndrome patients activate endothelial cells in vitro and in vivo. Circulation 1999, 99, 1997–2002. [Google Scholar] [CrossRef]
- Payne, H.; Brill, A. Stenosis of the Inferior Vena Cava: A Murine Model of Deep Vein Thrombosis. J. Vis. Exp. 2017. [Google Scholar] [CrossRef]
- Von Bruhl, M.L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Kollnberger, M.; et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012, 209, 819–835. [Google Scholar] [CrossRef]
- Diaz, J.A.; Obi, A.T.; Myers, D.D., Jr.; Wrobleski, S.K.; Henke, P.K.; Mackman, N.; Wakefield, T.W. Critical review of mouse models of venous thrombosis. Arterioscl. Thromb. Vasc. Biol. 2012, 32, 556–562. [Google Scholar] [CrossRef]
- Brandt, M.; Schonfelder, T.; Schwenk, M.; Becker, C.; Jackel, S.; Reinhardt, C.; Stark, K.; Massberg, S.; Munzel, T.; von Bruhl, M.L.; et al. Deep vein thrombus formation induced by flow reduction in mice is determined by venous side branches. Clin. Hemorheol. Microcirc. 2014, 56, 145–152. [Google Scholar] [CrossRef]
- Campos, J.; Brill, A. By word of mouse: Using animal models in venous thrombosis research. Platelets 2020, 31, 447–454. [Google Scholar] [CrossRef]
- Diaz, J.A.; Wrobleski, S.K.; Hawley, A.E.; Lucchesi, B.R.; Wakefield, T.W.; Myers, D.D., Jr. Electrolytic inferior vena cava model (EIM) of venous thrombosis. J. Vis. Exp. 2011, e2737. [Google Scholar] [CrossRef]
- Palmer, O.R.; Shaydakov, M.E.; Rainey, J.P.; Lawrence, D.A.; Greve, J.M.; Diaz, J.A. Update on the electrolytic IVC model for pre-clinical studies of venous thrombosis. Res. Pract. Thromb. Haemostol. 2018, 2, 266–273. [Google Scholar] [CrossRef]
- Alvarado, C.M.; Diaz, J.A.; Hawley, A.E.; Wrobleski, S.K.; Sigler, R.E.; Myers, D.D. Male mice have increased thrombotic potential: Sex differences in a mouse model of venous thrombosis. Thromb. Res. 2011, 127, 478–486. [Google Scholar] [CrossRef]
- Xie, H.; Kong, X.; Zhou, H.; Xie, Y.; Sheng, L.; Wang, T.; Xia, L.; Yan, J. TLR4 is involved in the pathogenic effects observed in a murine model of antiphospholipid syndrome. Clin. Immunol. 2015, 160, 198–210. [Google Scholar] [CrossRef]
- Li, W.; Nieman, M.; Sen Gupta, A. Ferric Chloride-induced Murine Thrombosis Models. J. Vis. Exp. 2016. [Google Scholar] [CrossRef]
- Bonnard, T.; Hagemeyer, C.E. Ferric Chloride-induced Thrombosis Mouse Model on Carotid Artery and Mesentery Vessel. J. Vis. Exp. 2015, e52838. [Google Scholar] [CrossRef]
- Jankowski, M.; Vreys, I.; Wittevrongel, C.; Boon, D.; Vermylen, J.; Hoylaerts, M.F.; Arnout, J. Thrombogenicity of beta 2-glycoprotein I-dependent antiphospholipid antibodies in a photochemically induced thrombosis model in the hamster. Blood 2003, 101, 157–162. [Google Scholar] [CrossRef]
- Ushiyama, A.; Yamada, S.; Ohkubo, C. Microcirculatory parameters measured in subcutaneous tissue of the mouse using a novel dorsal skinfold chamber. Microvasc. Res. 2004, 68, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, M.; Nii, T.; Trimova, G.; Miura, S.; Umezawa, K.; Ushiyama, A.; Kubota, T. The NF-κB specific inhibitor DHMEQ prevents thrombus formation in a mouse model of antiphospholipid syndrome. J. Nephropathol. 2013, 2, 114–121. [Google Scholar] [CrossRef][Green Version]
- Arad, A.; Proulle, V.; Furie, R.A.; Furie, B.C.; Furie, B. β₂-Glycoprotein-1 autoantibodies from patients with antiphospholipid syndrome are sufficient to potentiate arterial thrombus formation in a mouse model. Blood 2011, 117, 3453–3459. [Google Scholar] [CrossRef]
- Proulle, V.; Furie, R.A.; Merrill-Skoloff, G.; Furie, B.C.; Furie, B. Platelets are required for enhanced activation of the endothelium and fibrinogen in a mouse thrombosis model of APS. Blood 2014, 124, 611–622. [Google Scholar] [CrossRef]
- Kolyada, A.; Porter, A.; Beglova, N. Inhibition of thrombotic properties of persistent autoimmune anti-β2GPI antibodies in the mouse model of antiphospholipid syndrome. Blood 2014, 123, 1090–1097. [Google Scholar] [CrossRef]
- Falati, S.; Liu, Q.; Gross, P.; Merrill-Skoloff, G.; Chou, J.; Vandendries, E.; Celi, A.; Croce, K.; Furie, B.C.; Furie, B. Accumulation of tissue factor into developing thrombi in vivo is dependent upon microparticle P-selectin glycoprotein ligand 1 and platelet P-selectin. J. Exp. Med. 2003, 197, 1585–1598. [Google Scholar] [CrossRef]
- Fischetti, F.; Durigutto, P.; Pellis, V.; Debeus, A.; Macor, P.; Bulla, R.; Bossi, F.; Ziller, F.; Sblattero, D.; Meroni, P.; et al. Thrombus formation induced by antibodies to beta2-glycoprotein I is complement dependent and requires a priming factor. Blood 2005, 106, 2340–2346. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, S.; Brodsky, R.A.; McCrae, K.R. Complement in the Pathophysiology of the Antiphospholipid Syndrome. Front. Immunol. 2019, 10, 449. [Google Scholar] [CrossRef]
- Yamada, M.; Kawakami, T.; Takashima, K.; Nishioka, Y.; Nishibata, Y.; Masuda, S.; Yoshida, S.; Tomaru, U.; Ishizu, A. Establishment of a rat model of thrombosis induced by intravenous injection of anti-phosphatidylserine-prothrombin complex antibody. Rheumatology 2017, 56, 1013–1018. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Diaz, J.A.; Saha, P.; Cooley, B.; Palmer, O.R.; Grover, S.P.; Mackman, N.; Wakefield, T.W.; Henke, P.K.; Smith, A.; Lal, B.K. Choosing a Mouse Model of Venous Thrombosis. Arterioscl. Thromb. Vasc. Biol. 2019, 39, 311–318. [Google Scholar] [CrossRef] [PubMed]
- De Laat, B.; Pengo, V.; Pabinger, I.; Musial, J.; Voskuyl, A.E.; Bultink, I.E.; Ruffatti, A.; Rozman, B.; Kveder, T.; de Moerloose, P.; et al. The association between circulating antibodies against domain I of beta2-glycoprotein I and thrombosis: An international multicenter study. J. Thromb. Haemostol. 2009, 7, 1767–1773. [Google Scholar] [CrossRef]
- Pericleous, C.; Ferreira, I.; Borghi, O.; Pregnolato, F.; McDonnell, T.; Garza-Garcia, A.; Driscoll, P.; Pierangeli, S.; Isenberg, D.; Ioannou, Y.; et al. Measuring IgA Anti-β2-Glycoprotein I and IgG/IgA Anti-Domain I Antibodies Adds Value to Current Serological Assays for the Antiphospholipid Syndrome. PLoS ONE 2016, 11, e0156407. [Google Scholar] [CrossRef] [PubMed]
- Pericleous, C.; Ruiz-Limón, P.; Romay-Penabad, Z.; Marín, A.C.; Garza-Garcia, A.; Murfitt, L.; Driscoll, P.C.; Latchman, D.S.; Isenberg, D.A.; Giles, I.; et al. Proof-of-concept study demonstrating the pathogenicity of affinity-purified IgG antibodies directed to domain I of β2-glycoprotein I in a mouse model of anti-phospholipid antibody-induced thrombosis. Rheumatology 2015, 54, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Pierangeli, S.S.; Espinola, R.G.; Liu, X.; Harris, E.N. Thrombogenic effects of antiphospholipid antibodies are mediated by intercellular cell adhesion molecule-1, vascular cell adhesion molecule-1, and P-selectin. Circ. Res. 2001, 88, 245–250. [Google Scholar] [CrossRef]
- Knight, J.S.; Meng, H.; Coit, P.; Yalavarthi, S.; Sule, G.; Gandhi, A.A.; Grenn, R.C.; Mazza, L.F.; Ali, R.A.; Renauer, P.; et al. Activated signature of antiphospholipid syndrome neutrophils reveals potential therapeutic target. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Tambralli, A.; Gockman, K.; Knight, J.S. NETs in APS: Current Knowledge and Future Perspectives. Curr. Rheumatol. Rep. 2020, 22, 67. [Google Scholar] [CrossRef]
- Yalavarthi, S.; Gould, T.J.; Rao, A.N.; Mazza, L.F.; Morris, A.E.; Nunez-Alvarez, C.; Hernandez-Ramirez, D.; Bockenstedt, P.L.; Liaw, P.C.; Cabral, A.R.; et al. Release of neutrophil extracellular traps by neutrophils stimulated with antiphospholipid antibodies: A newly identified mechanism of thrombosis in the antiphospholipid syndrome. Arthritis Rheumatol. 2015, 67, 2990–3003. [Google Scholar] [CrossRef]
- Meng, H.; Yalavarthi, S.; Kanthi, Y.; Mazza, L.F.; Elfline, M.A.; Luke, C.E.; Pinsky, D.J.; Henke, P.K.; Knight, J.S. In Vivo Role of Neutrophil Extracellular Traps in Antiphospholipid Antibody-Mediated Venous Thrombosis. Arthritis Rheumatol. 2017, 69, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.A.; Gandhi, A.A.; Meng, H.; Yalavarthi, S.; Vreede, A.P.; Estes, S.K.; Palmer, O.R.; Bockenstedt, P.L.; Pinsky, D.J.; Greve, J.M.; et al. Adenosine receptor agonism protects against NETosis and thrombosis in antiphospholipid syndrome. Nat. Commun. 2019, 10, 1916. [Google Scholar] [CrossRef]
- Ali, R.A.; Gandhi, A.A.; Dai, L.; Weiner, J.K.; Estes, S.K.; Yalavarthi, S.; Gockman, K.; Sun, D.; Knight, J.S. Anti-neutrophil properties of natural gingerols in models of lupus. JCI Insight 2020. [Google Scholar] [CrossRef]
- Manukyan, D.; Müller-Calleja, N.; Jäckel, S.; Luchmann, K.; Mönnikes, R.; Kiouptsi, K.; Reinhardt, C.; Jurk, K.; Walter, U.; Lackner, K.J. Cofactor-independent human antiphospholipid antibodies induce venous thrombosis in mice. J. Thromb. Haemostol. 2016, 14, 1011–1020. [Google Scholar] [CrossRef]
- Pierangeli, S.S.; Girardi, G.; Vega-Ostertag, M.; Liu, X.; Espinola, R.G.; Salmon, J. Requirement of activation of complement C3 and C5 for antiphospholipid antibody-mediated thrombophilia. Arthritis Rheumatol. 2005, 52, 2120–2124. [Google Scholar] [CrossRef] [PubMed]
- Laplante, P.; Fuentes, R.; Salem, D.; Subang, R.; Gillis, M.A.; Hachem, A.; Farhat, N.; Qureshi, S.T.; Fletcher, C.A.; Roubey, R.A.; et al. Antiphospholipid antibody-mediated effects in an arterial model of thrombosis are dependent on Toll-like receptor 4. Lupus 2016, 25, 162–176. [Google Scholar] [CrossRef]
- Qiao, J.; Arthur, J.F.; Gardiner, E.E.; Andrews, R.K.; Zeng, L.; Xu, K. Regulation of platelet activation and thrombus formation by reactive oxygen species. Redox Biol. 2018, 14, 126–130. [Google Scholar] [CrossRef]
- Miranda, S.; Billoir, P.; Damian, L.; Thiebaut, P.A.; Schapman, D.; Le Besnerais, M.; Jouen, F.; Galas, L.; Levesque, H.; Le Cam-Duchez, V.; et al. Hydroxychloroquine reverses the prothrombotic state in a mouse model of antiphospholipid syndrome: Role of reduced inflammation and endothelial dysfunction. PLoS ONE 2019, 14, e0212614. [Google Scholar] [CrossRef]
- Ramesh, S.; Morrell, C.N.; Tarango, C.; Thomas, G.D.; Yuhanna, I.S.; Girardi, G.; Herz, J.; Urbanus, R.T.; de Groot, P.G.; Thorpe, P.E.; et al. Antiphospholipid antibodies promote leukocyte-endothelial cell adhesion and thrombosis in mice by antagonizing eNOS via β2GPI and apoER2. J. Clin. Investig. 2011, 121, 120–131. [Google Scholar] [CrossRef]
- Talley Watts, L.; Zheng, W.; Garling, R.J.; Frohlich, V.C.; Lechleiter, J.D. Rose Bengal Photothrombosis by Confocal Optical Imaging In Vivo: A Model of Single Vessel Stroke. J. Vis. Exp. 2015, e52794. [Google Scholar] [CrossRef] [PubMed]
- Nanda, S.K.; Hatchell, D.L.; Tiedeman, J.S.; Dutton, J.J.; Hatchell, M.C.; McAdoo, T. A new method for vascular occlusion. Photochemical initiation of thrombosis. Arch. Ophthalmol. 1987, 105, 1121–1124. [Google Scholar] [CrossRef] [PubMed]
- Kuroiwa, T.; Xi, G.; Hua, Y.; Nagaraja, T.N.; Fenstermacher, J.D.; Keep, R.F. Development of a rat model of photothrombotic ischemia and infarction within the caudoputamen. Stroke 2009, 40, 248–253. [Google Scholar] [CrossRef]
- Bray, M.A.; Sartain, S.E.; Gollamudi, J.; Rumbaut, R.E. Microvascular thrombosis: Experimental and clinical implications. Transl. Res. 2020, 225, 105–130. [Google Scholar] [CrossRef]
- Grover, S.P.; Mackman, N. How useful are ferric chloride models of arterial thrombosis? Platelets 2020, 31, 432–438. [Google Scholar] [CrossRef]
- Wang, X.; Xu, L. An optimized murine model of ferric chloride-induced arterial thrombosis for thrombosis research. Thromb. Res. 2005, 115, 95–100. [Google Scholar] [CrossRef]
- Dubois, C.; Panicot-Dubois, L.; Merrill-Skoloff, G.; Furie, B.; Furie, B.C. Glycoprotein VI-dependent and -independent pathways of thrombus formation in vivo. Blood 2006, 107, 3902–3906. [Google Scholar] [CrossRef]
- Litvinova, E.; Darnige, L.; Kirilovsky, A.; Burnel, Y.; de Luna, G.; Dragon-Durey, M.A. Prevalence and Significance of Non-conventional Antiphospholipid Antibodies in Patients With Clinical APS Criteria. Front. Immunol. 2018, 9, 2971. [Google Scholar] [CrossRef]
- Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Thomas, G.M.; Martinod, K.; De Meyer, S.F.; Bhandari, A.A.; Wagner, D.D. Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thromb. Haemostol. 2012, 10, 136–144. [Google Scholar] [CrossRef]
- Kazzaz, N.M.; McCune, W.J.; Knight, J.S. Treatment of catastrophic antiphospholipid syndrome. Curr. Opin. Rheumatol. 2016, 28, 218–227. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Thrombosis Model | Strengths | Limitations |
|---|---|---|
| Venous Models | ||
| Femoral Vein Pinch [13,14] | Well suited for the study of thrombus propagation and resolution; enables real-time visualization of thrombus formation. | Thrombus propagates against the direction of blood flow. |
| Stenosis IVC [15,16,17,18,19] | Variable thrombus generation enables study of thrombus initiation in prothrombotic conditions; thrombi produced are structurally similar to humans, thrombus experiences constant blood flow which supports the study of therapeutics; induces thrombosis without endothelial damage. | Variable thrombus size necessitates larger experimental group sizes; thrombus propagates against the direction of blood flow; challenging to observe thrombus formation in real-time. |
| Electrolytic IVC [20,21,22] | Thrombus experiences constant blood flow; suited for study of antithrombotic or thrombolytic agents; thrombi form in the direction of blood flow. | Longer operative time; physical damage to IVC vein lumen; can cause necrosis in the female reproductive organs of C57BL/6 mice; challenging to observe thrombus formation in real-time. |
| FeCl3 IVC [17,23] | Acute model suited to study early timepoints in thrombosis; thrombi form in direction of blood flow; intravital microscopy possible. | Smaller thrombus size can limit options for biochemical assays; transmural vein injury induced by FeCl3 may not mimic clinical thrombosis. |
| Arterial Models | ||
| FeCl3 Carotid [24,25] | Acute model, suited to study early timepoints in thrombosis; thrombi form in direction of blood flow. | Requires a challenging surgical procedure to isolate the carotid artery; transmural vein injury may not mimic clinical thrombosis. |
| Photochemical Carotid [26] | Intravital microscopy enables real-time observation of thrombosis; catheterization allows easy administration of therapeutics; photochemical injury is highly standardizable. | Relies on local injury to produce thrombus; Acute model not suited for chronic studies. |
| Microvascular Models | ||
| Dorsal Skinfold Chamber [27,28] | DSC device enables observation of the microcirculation for ≤3 weeks; real-time visualization of thrombosis initiation and resolution. | Requires surgery to implant DSC device and a recovery period before performing thrombosis experiments. |
| Laster-Induced Injury in the Cremaster Muscle [29,30,31,32] | Intraviral microscopy allows observation of thrombosis; easily accessible vascular bed; allows for the induction of multiple thrombi in the same mouse. | Can only be performed on male mice. |
| FeCl3 Mesenteric Microcirculation [24,25] | Easily accessible microvasculature; well suited for intravital microscopy; suited for acute study of thrombosis. | Variable vessel size and visceral fat can influence thrombus size; oxidative injury induced may limit applicability for studies of endothelial inflammation-associated thrombus. |
| LPS-Priming [33,34] | Consistent thrombus generation; real-time visualization of thrombosis with intravital microcopy; using a non-localized inflammatory stimulus, LPS, as a prothrombotic trigger might be more relevant to clinical thrombosis. | Complex surgical procedure; longer operative time. |
| Histone-Priming [35] | Has allowed study of anti-PS/PT antibodies in APS-mediated thrombosis; does not require surgical procedure. | Not suited for the study of thrombosis in specific regions; non-surgical procedure prevents real-time observation of thrombosis. |
| Pathway/Factor | Role |
|---|---|
| Venous | |
| Anti-domain I | Pathogenic aPL bind the N-terminal domain of β2GPI (DI) [39]. |
| Adhesion molecules | P-selectin, VCAM-1, PSGL-1, etc., facilitate interactions between leukocytes and the endothelium [40]. |
| Deoxyribonucleases | Degradation of DNA (a key component of NETs) decreases thrombus size and incidence [44]. |
| Adenosine receptor agonists | Adenosine receptor agonists (CGS21680, dipyridamole) suppress NETosis via stimulation of cAMP production [45]. |
| Natural gingerols | Gingerols inhibit phosphodiesterase activity and suppress proinflammatory cytokine release [46]. |
| NADPH Oxidase | NOX2-mediated tissue factor activation induces prothrombotic responses [47]. |
| TLR4 | The requirement for TLR4 may depend upon the type of aPL (anti-β2GPI versus cofactor-independent) [23,47]. |
| Complement cascade | C3 in particular has been shown to be necessary for aPL-mediated thrombosis [48]. |
| Arterial | |
| TLR4 | TLR4 acts as a trigger of innate immune responses and has been shown to be necessary for aPL-potentiated thrombosis [49]. |
| Reactive oxygen species (ROS) | ROS induce oxidative stress and exposure of subendothelial collagen, which promotes platelet adherence [50]. |
| anti-β2GPI antibodies | Anti-β2GPI antibodies induce platelet activation [29]. |
| Hydroxychloroquine | Hydroxychloroquine increases eNOS activity (other roles are also likely) [51]. |
| Microvascular | |
| Platelet-endothelial cell interactions | Binding of anti-β2GP1/β2GP1 complexes to the developing thrombus activates platelets. Platelet-derived products then activate endothelial cells [30]. |
| Complement cascade | Inhibition of membrane attack complex assembly protects against the prothrombotic effects of aPL [33]. |
| Nitric oxide synthase | Increased levels of eNOS production potentiate NO production and thereby inhibit further platelet aggregation [52]. |
| β2GPI/anti-β2GPI interaction | Disruption of the interaction between β2GPI and anti-β2GPI antibodies is protective [31,33]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gandhi, A.A.; Estes, S.K.; Rysenga, C.E.; Knight, J.S. Understanding the Pathophysiology of Thrombotic APS through Animal Models. Int. J. Mol. Sci. 2021, 22, 2588. https://doi.org/10.3390/ijms22052588
Gandhi AA, Estes SK, Rysenga CE, Knight JS. Understanding the Pathophysiology of Thrombotic APS through Animal Models. International Journal of Molecular Sciences. 2021; 22(5):2588. https://doi.org/10.3390/ijms22052588
Chicago/Turabian StyleGandhi, Alex A., Shanea K. Estes, Christine E. Rysenga, and Jason S. Knight. 2021. "Understanding the Pathophysiology of Thrombotic APS through Animal Models" International Journal of Molecular Sciences 22, no. 5: 2588. https://doi.org/10.3390/ijms22052588
APA StyleGandhi, A. A., Estes, S. K., Rysenga, C. E., & Knight, J. S. (2021). Understanding the Pathophysiology of Thrombotic APS through Animal Models. International Journal of Molecular Sciences, 22(5), 2588. https://doi.org/10.3390/ijms22052588