
Oncolytic Adenoviruses for Cancer Therapy


Abstract
1. Introduction: A Journey in the Adenovirus World
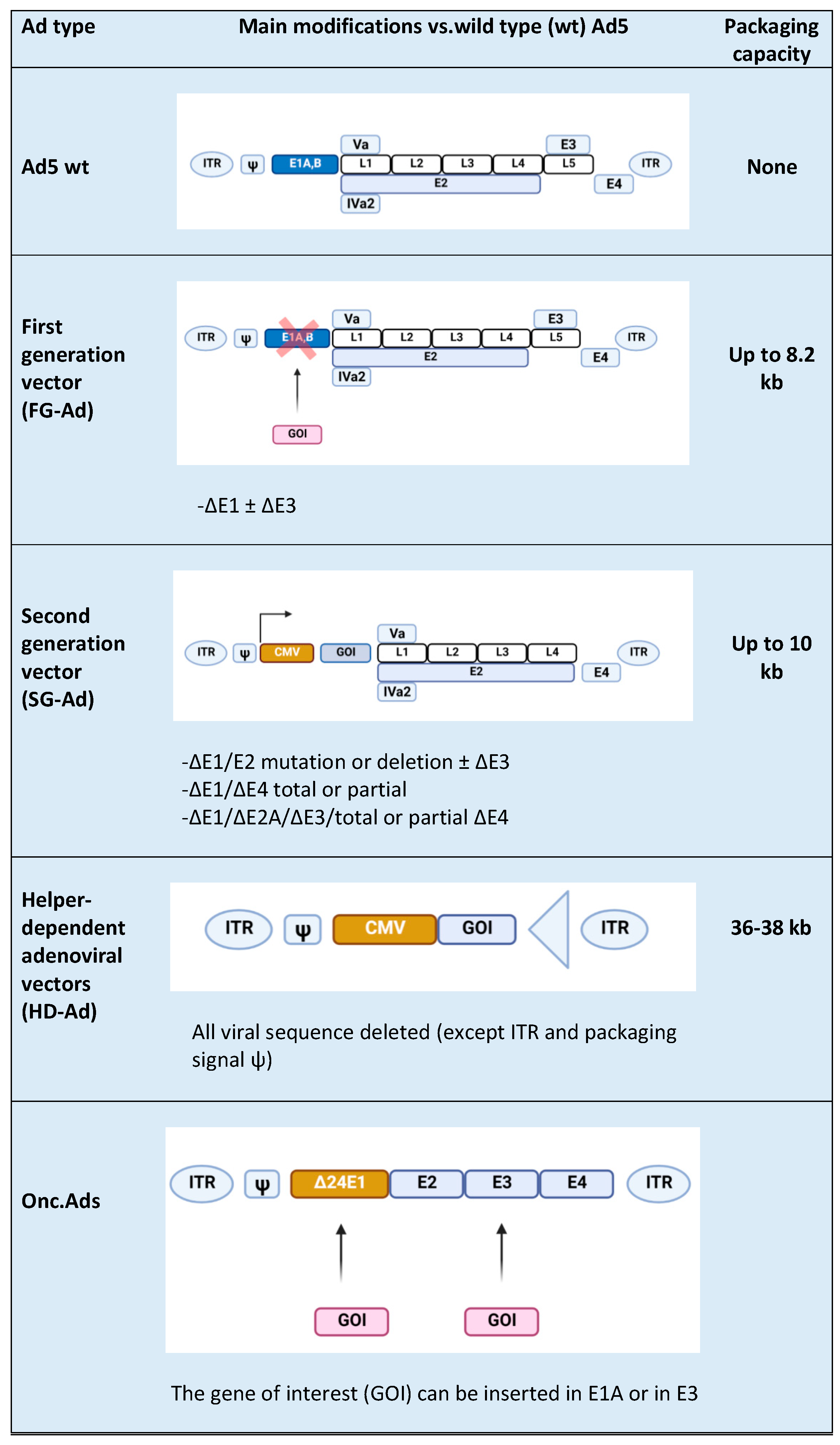
2. Adenovirus (Ads) Vector Design
3. Exploring the Tumor Microenvironment and Its Modulation by Onc.Ads
3.1. Armed Oncolytic Adenoviruses with Immunostimulatory Cytokines and Chemokines
3.2. Arming OVs with Immune-Activating Ligands and Bispecific T-Cell Engager (BiTE) Molecules
4. Oncolytic Adenoviruses and Immunotherapy
5. Limitations of Onc.Ads
6. Future Outlook: A Challenge for Oncolytic Viro-Immunotherapy
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cervera-Carrascon, V.; Havunen, R.; Hemminki, A. Oncolytic adenoviruses: A game changer approach in the battle between cancer and the immune system. Expert Opin. Biol. Ther. 2019, 19, 1–13. [Google Scholar] [CrossRef]
- Heise, C.; Kirn, D.H. Replication-selective adenoviruses as oncolytic agents. J. Clin. Investig. 2000, 105, 847–851. [Google Scholar] [CrossRef] [PubMed]
- Leggiero, E.; Labruna, G.; Iaffaldano, L.; Lombardo, B.; Greco, A.; Fiorenza, D.; Gramanzini, M.; Montanaro, D.; Baldi, A.; Cerullo, V.; et al. Helper-dependent adenovirus-mediated gene transfer of a secreted LDL receptor/transferrin chimeric protein reduces aortic atherosclerosis in ldl receptor-deficient mice. Gene Ther. 2019, 26, 121–130. [Google Scholar] [CrossRef]
- Feola, S.; Capasso, C.; Fusciello, M.; Martins, B.; Tähtinen, S.; Medeot, M.; Carpi, S.; Frascaro, F.; Ylosmäki, E.; Peltonen, K.; et al. Oncolytic vaccines increase the response to pd-l1 blockade in immunogenic and poorly immunogenic tumors. Oncoimmunology 2018, 7, 1–32. [Google Scholar] [CrossRef]
- Leggiero, E.; Astone, D.; Cerullo, V.; Lombardo, B.; Mazzaccara, C.; Labruna, G.; Sacchetti, L.; Salvatore, F.; Croyle, M.; Pastore, L. PEGylated helper-dependent adenoviral vector expressing human Apo A-I for gene therapy in ldlr-deficient mice. Gene Ther. 2013, 20, 1124–1130. [Google Scholar] [CrossRef]
- Niemann, J.; Kühnel, F. Oncolytic Viruses: Adenoviruses. Virus Genes 2017, 53, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Pastore, L.; Beaudet, A.L. Helper-dependent adenoviral vectors. Methods Enzymol. 2002, 346, 177–198. [Google Scholar] [CrossRef]
- Cassese, A.; Raciti, G.A.; Fiory, F.; Nigro, C.; Ulianich, L.; Castanò, I.; D’Esposito, V.; Terracciano, D.; Pastore, L.; Formisano, P.; et al. Adenoviral gene transfer of PLD1-D4 enhances insulin sensitivity in mice by disrupting phospholipase D1 interaction with PED/PEA. PLoS ONE 2013, 8, e60555. [Google Scholar] [CrossRef] [PubMed]
- Ylösmäki, E.; Malorzo, C.; Capasso, C.; Honkasalo, O.; Fusciello, M.; Martins, B.; Ylösmäki, L.; Louna, A.; Feola, S.; Paavilainen, H.; et al. Personalized cancer vaccine platform for clinically relevantoncolytic enveloped viruses. Mol. Ther. 2018, 26, 2315–2325. [Google Scholar] [CrossRef]
- Tähtinen, S.; Feola, S.; Capasso, C.; Laustio, N.; Groeneveldt, C.; Ylösmäki, E.O.; Ylösmäki, L.; Martins, B.; Fusciello, M.; Medeot, M.; et al. Exploiting preexisting immunity to enhance oncolytic cancer immunotherapy. Cancer Res. 2020, 80, 2575–2585. [Google Scholar] [CrossRef]
- Davola, M.E.; Mossman, K.L. Oncolytic viruses: How “Lytic” must they be for therapeutic efficacy? Oncoimmunology 2019, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ylösmäki, E.; Cerullo, V. Design and application of oncolytic viruses for cancer immunotherapy. Curr. Opin. Biotech. 2020, 65, 25–36. [Google Scholar] [CrossRef]
- King, A.M.Q.; Adams, M.J.; Carstens, E.B.; Lefkowitz, E.J. (Eds.) Adenoviridae—Ninth Report of the International Committee on Taxonomy of Viruses; Elsevier: Oxford, UK, 2012; p. 125. [Google Scholar]
- Esposito, S.; Preti, V.; Consolo, S.; Nazzari, E.; Principi, N. Adenovirus 36 infection and obesity. J. Clin. Virol. 2012, 55, 95–100. [Google Scholar] [CrossRef]
- Lion, T. Adenovirus infections in immunocompetent and immunocompromised patients. Clin. Microbiol. Rev. 2014, 27, 441–462. [Google Scholar] [CrossRef] [PubMed]
- Alemany, R. Chapter four design of improved oncolytic adenoviruses. Adv. Cancer Res. 2012, 115, 93–114. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Deng, Z.-L.; Luo, X.; Tang, N.; Song, W.-X.; Chen, J.; Sharff, K.A.; Luu, H.H.; Haydon, R.C.; Kinzler, K.W.; et al. A protocol for rapid generation of recombinant adenoviruses using the adeasy system. Nat. Protoc. 2007, 2, 1236–1247. [Google Scholar] [CrossRef]
- Dormond, E.; Perrier, M.; Kamen, A. From the first to the third generation adenoviral vector: What parameters are governing the production yield? Biotechnol. Adv. 2009, 27, 133–144. [Google Scholar] [CrossRef]
- Pastore, L.; Belalcazar, L.M.; Oka, K.; Cela, R.; Lee, B.; Chan, L.; Beaudet, A.L. Helper-dependent adenoviral vector-mediated long-term expression of human apolipoprotein A-I reduces atherosclerosis in apo E-deficient mice. Gene 2004, 327, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Farzad, L.; Cerullo, V.; Yagyu, S.; Bertin, T.; Hemminki, A.; Rooney, C.; Lee, B.; Suzuki, M. Combinatorial treatment with oncolytic adenovirus and helper-dependent adenovirus augments adenoviral cancer gene therapy. Mol. Ther. Oncolytics 2014, 1, 14008. [Google Scholar] [CrossRef] [PubMed]
- Farzad, L.M.; Suzuki, M. Feasibility of applying helper-dependent adenoviral vectors for cancer immunotherapy. BioMed 2014, 2, 110–131. [Google Scholar] [CrossRef]
- Muruve, D.A.; Cotter, M.J.; Zaiss, A.K.; White, L.R.; Liu, Q.; Chan, T.; Clark, S.A.; Ross, P.J.; Meulenbroek, R.A.; Maelandsmo, G.M.; et al. Helper-dependent adenovirus vectors elicit intact innate but attenuated adaptive host immune responses in vivo. J. Virol. 2004, 78, 5966–5972. [Google Scholar] [CrossRef] [PubMed]
- Cerullo, V.; Capasso, C.; Vaha-Koskela, M.; Hemminki, O.; Hemminki, A. Cancer-targeted oncolytic adenoviruses for modulation of the immune system. Curr. Cancer Drug Targets 2018, 18. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Buoncervello, M.; Gabriele, L.; Toschi, E. The janus face of tumor microenvironment targeted by immunotherapy. Int. J. Mol. Sci. 2019, 20, 4320. [Google Scholar] [CrossRef]
- Najafi, M.; Goradel, N.H.; Farhood, B.; Salehi, E.; Solhjoo, S.; Toolee, H.; Kharazinejad, E.; Mortezaee, K. Tumor microenvironment: Interactions and therapy. J. Cell Physiol. 2018, 234, 5700–5721. [Google Scholar] [CrossRef]
- Zhao, P.; Wang, Y.; Kang, X.; Wu, A.; Yin, W.; Tang, Y.; Wang, J.; Zhang, M.; Duan, Y.; Huang, Y. Dual-targeting biomimetic delivery for anti-glioma activity via remodeling the tumor microenvironment and directing macrophage-mediated immunotherapy. Chem. Sci. 2018, 9, 2674–2689. [Google Scholar] [CrossRef]
- Albini, A.; Bruno, A.; Noonan, D.M.; Mortara, L. Contribution to tumor angiogenesis from innate immune cells within the tumor microenvironment: Implications for immunotherapy. Front. Immunol. 2018, 9, 527. [Google Scholar] [CrossRef] [PubMed]
- de Matos, A.L.; Franco, L.S.; McFadden, G. Oncolytic viruses and the immune system: The dynamic duo. Mol. Ther. Methods Clin. Dev. 2020, 17, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Platanias, L.C. Mechanisms of Type-I- and Type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Marelli, G.; Howells, A.; Lemoine, N.R.; Wang, Y. Oncolytic viral therapy and the immune system: A double-edged sword against cancer. Front. Immunol. 2018, 9, 866. [Google Scholar] [CrossRef] [PubMed]
- de Graaf, J.F.; de Vor, L.; Fouchier, R.A.M.; Hoogen, B.G. Armed oncolytic viruses: A kick-start for anti-tumor immunity. Cytokine Growth Factor Rev. 2018, 41, 28–39. [Google Scholar] [CrossRef]
- Li, X.; Wang, P.; Li, H.; Du, X.; Liu, M.; Huang, Q.; Wang, Y.; Wang, S. The efficacy of oncolytic adenovirus is mediated by t-cell responses against virus and tumor in syrian hamster model. Clin. Cancer Res. 2016, 23, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Harrington, K.; Freeman, D.J.; Kelly, B.; Harper, J.; Soria, J.-C. Optimizing oncolytic virotherapy in cancer treatment. Nat. Rev. Drug Discov. 2019, 18, 689–706. [Google Scholar] [CrossRef]
- Escutenaire, S.; Cerullo, V.; Diaconu, I.; Ahtiainen, L.; Hannuksela, P.; Oksanen, M.; Haavisto, E.; Karioja-Kallio, A.; Holm, S.-L.; Kangasniemi, L.; et al. In vivo and in vitro distribution of type 5 and fiber-modified oncolytic adenoviruses in human blood compartments. Ann. Med. 2011, 43, 151–163. [Google Scholar] [CrossRef]
- Bhattacharya, P.; Budnick, I.; Singh, M.; Thiruppathi, M.; Alharshawi, K.; Elshabrawy, H.; Holterman, M.J.; Prabhakar, B.S. Dual role of GM-CSF as a pro-inflammatory and a regulatory cytokine: Implications for immune therapy. J. Interf. Cytokine Res. 2015, 35, 585–599. [Google Scholar] [CrossRef] [PubMed]
- Bommareddy, P.K.; Patel, A.; Hossain, S.; Kaufman, H.L. Talimogene Laherparepvec (T-VEC) and other oncolytic viruses for the treatment of melanoma. Am. J. Clin. Dermatol. 2017, 18, 1–15. [Google Scholar] [CrossRef]
- Kuryk, L.; Møller, A.-S.W.; Jaderberg, M. Combination of immunogenic oncolytic adenovirus ONCOS-102 with Anti-PD-1 pembrolizumab exhibits synergistic antitumor effect in humanized A2058 melanoma HuNOG mouse model. Oncoimmunology 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Tanaka, K.; Towata, S.; Nakao, K.; Mizuguchi, H.; Hayakawa, T.; Niwa, M.; Ishii, N.; Nagayama, Y. Thyroid cancer immuno-therapy with retroviral and adenoviral vectors expressing granulocyte macrophage colony stimulating factor and interleukin-12 in a rat model. Clin. Endocrinol. 2003, 59, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G. Interleukin-12: A proinflammatory cytokine with immunoregulatory functions that bridge innate resistance and antigen-specific adaptive immunity. Annu. Rev. Immunol. 1995, 13, 251–276. [Google Scholar] [CrossRef] [PubMed]
- Markel, J.E.; Lacinski, R.A.; Lindsey, B.A. Current Advances in Osteosarcoma, Clinical Perspectives: Past, Present and Future; Springer: Berlin, Germany, 2020; pp. 155–168. [Google Scholar] [CrossRef]
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Immunology 2014, 32, 659–702. [Google Scholar] [CrossRef] [PubMed]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Si, C.; Zhu, Y.; Jin, Y.; Zhu, T.; Liu, M.; Liu, G. CCL21/IL21-armed oncolytic adenovirus enhances antitumor activity against TERT-positive tumor cells. Virus Res. 2016, 220, 172–178. [Google Scholar] [CrossRef]
- Liu, G.; Li, Z.; Li, Q.; Jin, Y.; Zhu, Y.; Wang, Y.; Liu, M.; Li, Y.; Li, Y. Enhanced growth suppression of TERT-positive tumor cells by oncolytic adenovirus armed with CCL20 and CD40L. Int. Immunopharmacol. 2015, 28, 487–493. [Google Scholar] [CrossRef]
- Cerullo, V.; Pesonen, S.; Diaconu, I.; Escutenaire, S.; Arstila, P.T.; Ugolini, M.; Nokisalmi, P.; Raki, M.; Laasonen, L.; Särkioja, M.; et al. Oncolytic adenovirus coding for granulocyte macrophage colony-stimulating factor induces antitumoral immunity in cancer patients. Cancer Res. 2010, 70, 4297–4309. [Google Scholar] [CrossRef] [PubMed]
- Tuve, S.; Liu, Y.; Tragoolpua, K.; Jacobs, J.D.; Yumul, R.C.; Li, Z.-Y.; Strauss, R.; Hellström, K.-E.; Disis, M.L.; Roffler, S.; et al. In situ adenovirus vaccination engages T effector cells against cancer. Vaccine 2009, 27, 4225–4239. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Godfrey, D.I.; Trapani, J.A. A fresh look at tumor immunosurveillance and immunotherapy. Nat. Immunol. 2001, 2, 293–299. [Google Scholar] [CrossRef]
- Eriksson, E.; Moreno, R.; Milenova, I.; Liljenfeldt, L.; Dieterich, L.C.; Christiansson, L.; Karlsson, H.; Ullenhag, G.; Mangsbo, S.M.; Dimberg, A.; et al. Activation of myeloid and endothelial cells by CD40L gene therapy supports T-cell expansion and migration into the tumor microenvironment. Gene Ther. 2016, 24, 92–103. [Google Scholar] [CrossRef]
- Piechutta, M.; Berghoff, A.S. New emerging targets in cancer immunotherapy: The role of cluster of differentiation 40 (CD40/TNFR5). ESMO Open 2019, 4, e000510. [Google Scholar] [CrossRef]
- Schoenberger, S.P.; Toes, R.E.M.; van der Voort, E.I.H.; Offringa, R.; Melief, C.J.M. T-cell help for cytotoxic t lymphocytes is mediated by CD40–CD40L interactions. Nature 1998, 393, 480–483. [Google Scholar] [CrossRef]
- Pesonen, S.; Diaconu, I.; Kangasniemi, L.; Ranki, T.; Kanerva, A.; Pesonen, S.K.; Gerdemann, U.; Leen, A.M.; Kairemo, K.; Oksanen, M.; et al. Oncolytic immunotherapy of advanced solid tumors with a CD40L-expressing replicating adenovirus: Assessment of safety and immunologic responses in patients. Cancer Res. 2012, 72, 1621–1631. [Google Scholar] [CrossRef]
- Malmström, P.-U.; Loskog, A.S.I.; Lindqvist, C.A.; Mangsbo, S.M.; Fransson, M.; Wanders, A.; Gårdmark, T.; Tötterman, T.H. AdCD40L immunogene therapy for bladder carcinoma—The first phase I/IIa trial. Clin. Cancer Res. 2010, 16, 3279–3287. [Google Scholar] [CrossRef] [PubMed]
- Loskog, A.; Maleka, A.; Mangsbo, S.; Svensson, E.; Lundberg, C.; Nilsson, A.; Krause, J.; Agnarsdóttir, M.; Sundin, A.; Ahlström, H.; et al. Immunostimulatory AdCD40L gene therapy combined with low-dose cyclophosphamide in metastatic melanoma patients. Brit. J. Cancer 2016, 114, 872–880. [Google Scholar] [CrossRef]
- Schiza, A.; Wenthe, J.; Mangsbo, S.; Eriksson, E.; Nilsson, A.; Tötterman, T.H.; Loskog, A.; Ullenhag, G. Adenovirus-mediated CD40L gene transfer increases teffector/tregulatory cell ratio and upregulates death receptors in Metastatic Melanoma Patients. J. Transl. Med. 2017, 15, 79. [Google Scholar] [CrossRef]
- Diaconu, I.; Cerullo, V.; Hirvinen, M.L.M.; Escutenaire, S.; Ugolini, M.; Pesonen, S.K.; Bramante, S.; Parviainen, S.; Kanerva, A.; Loskog, A.S.I.; et al. Immune response is an important aspect of the antitumor effect produced by a CD40L-encoding oncolytic adenovirus. Cancer Res. 2012, 72, 2327–2338. [Google Scholar] [CrossRef] [PubMed]
- Liljenfeldt, L.; Gkirtzimanaki, K.; Vyrla, D.; Svensson, E.; Loskog, A.S.; Eliopoulos, A.G. Enhanced therapeutic anti-tumor immunity induced by Co-administration of 5-fluorouracil and adenovirus expressing CD40 ligand. Cancer Immunol. Immunother. 2014, 63, 273–282. [Google Scholar] [CrossRef]
- Ishii, N.; Takahashi, T.; Soroosh, P.; Sugamura, K. Chapter 3 OX40–OX40 ligand interaction in T-cell-mediated immunity and immunopathology. Adv. Immunol. 2010, 105, 63–98. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Shin, D.H.; Nguyen, T.T.; Fueyo, J.; Fan, X.; Henry, V.; Carrillo, C.C.; Yi, Y.; Alonso, M.M.; Collier, T.L.; et al. Localized treatment with oncolytic adenovirus delta-24-RGDOX induces systemic immunity against disseminated subcutaneous and intracranial melanomas. Clin. Cancer Res. 2019, 25, 6801–6814. [Google Scholar] [CrossRef]
- Eriksson, E.; Milenova, I.; Wenthe, J.; Ståhle, M.; Leja-Jarblad, J.; Ullenhag, G.; Dimberg, A.; Moreno, R.; Alemany, R.; Loskog, A. Shaping the tumor stroma and sparking immune activation by CD40 and 4-1BB signaling induced by an armed oncolytic virus. Clin. Cancer Res. 2017, 23, 5846–5857. [Google Scholar] [CrossRef]
- Martínez-Vélez, N.; Garcia-Moure, M.; Marigil, M.; González-Huarriz, M.; Puigdelloses, M.; Pérez-Larraya, J.G.; Zalacaín, M.; Marrodán, L.; Varela-Guruceaga, M.; Laspidea, V.; et al. The oncolytic virus delta-24-RGD elicits an antitumor effect in Pediatric Glioma and DIPG Mouse Models. Nat. Commun. 2019, 10, 2235. [Google Scholar] [CrossRef] [PubMed]
- Calmels, B.; Paul, S.; Futin, N.; Ledoux, C.; Stoeckel, F.; Acres, B. Bypassing tumor-associated immune suppression with recombinant adenovirus constructs expressing membrane bound or secreted GITR-L. Cancer Gene Ther. 2005, 12, 198–205. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Scott, E.M.; Duffy, M.R.; Freedman, J.D.; Fisher, K.D.; Seymour, L.W. Solid tumor immunotherapy with T cell engager-armed oncolytic viruses. Macromol. Biosci. 2018, 18, 1700187. [Google Scholar] [CrossRef]
- Freedman, J.D.; Hagel, J.; Scott, E.M.; Psallidas, I.; Gupta, A.; Spiers, L.; Miller, P.; Kanellakis, N.; Ashfield, R.; Fisher, K.D.; et al. Oncolytic adenovirus expressing bispecific antibody targets T-cell cytotoxicity in cancer biopsies. EMBO Mol. Med. 2017, 9, 1067–1087. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Song, X.; Wang, Y.; Liu, F.; Wei, J. Combining oncolytic viruses with cancer immunotherapy: Establishing a new generation of cancer treatment. Front. Immunol. 2020, 11, 683. [Google Scholar] [CrossRef] [PubMed]
- Samson, A.; Scott, K.J.; Taggart, D.; West, E.J.; Wilson, E.; Nuovo, G.J.; Thomson, S.; Corns, R.; Mathew, R.K.; Fuller, M.J.; et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci. Transl. Med. 2018, 10, eaam7577. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois-Daigneault, M.-C.; Roy, D.G.; Aitken, A.S.; Sayes, N.E.; Martin, N.T.; Varette, O.; Falls, T.; St-Germain, L.E.; Pelin, A.; Lichty, B.D.; et al. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci. Transl. Med. 2018, 10, eaao1641. [Google Scholar] [CrossRef]
- Cervera-Carrascon, V.; Siurala, M.; Santos, J.M.; Havunen, R.; Tähtinen, S.; Karell, P.; Sorsa, S.; Kanerva, A.; Hemminki, A. TNFa and IL-2 Armed adenoviruses enable complete responses by anti-PD-1 checkpoint blockade. Oncoimmunology 2018, 7, e1412902. [Google Scholar] [CrossRef]
- Havunen, R.; Siurala, M.; Sorsa, S.; Grönberg-Vähä-Koskela, S.; Behr, M.; Tähtinen, S.; Santos, J.M.; Karell, P.; Rusanen, J.; Nettelbeck, D.M.; et al. Oncolytic adenoviruses armed with tumor necrosis factor alpha and interleukin-2 enable successful adoptive cell therapy. Mol. Ther. Oncolytics 2017, 4, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Capasso, C.; Hirvinen, M.; Garofalo, M.; Romaniuk, D.; Kuryk, L.; Sarvela, T.; Vitale, A.; Antopolsky, M.; Magarkar, A.; Viitala, T.; et al. Oncolytic adenoviruses coated with MHC-I tumor epitopes increase the antitumor immunity and efficacy against melanoma. Oncoimmunology 2015, 5, e1105429. [Google Scholar] [CrossRef]
- Belcaid, Z.; Berrevoets, C.; Choi, J.; van Beelen, E.; Stavrakaki, E.; Pierson, T.; Kloezeman, J.; Routkevitch, D.; van der Kaaij, M.; van der Ploeg, A.; et al. Low-dose oncolytic adenovirus therapy overcomes tumor-induced immune suppression and sensitizes intracranial gliomas to anti-PD-1 therapy. Neuro Oncol. Adv. 2020, 2. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Woller, N.; Gürlevik, E.; Fleischmann-Mundt, B.; Schumacher, A.; Knocke, S.; Kloos, A.M.; Saborowski, M.; Geffers, R.; Manns, M.P.; Wirth, T.C.; et al. Viral infection of tumors overcomes resistance to PD-1-immunotherapy by broadening neoantigenome-directed T-cell responses. Mol. Ther. 2015, 23, 1630–1640. [Google Scholar] [CrossRef] [PubMed]
- Chaurasiya, S.; Fong, Y.; Warner, S.G. Optimizing oncolytic viral design to enhance antitumor efficacy: Progress and challenges. Cancers 2020, 12, 1699. [Google Scholar] [CrossRef]
- Agrawal, P.; Nawadkar, R.; Ojha, H.; Kumar, J.; Sahu, A. Complement evasion strategies of viruses: An overview. Front. Microbiol. 2017, 8, 1117. [Google Scholar] [CrossRef]
- Kirn, D. Clinical research results with Dl1520 (Onyx-015), a replication-selective adenovirus for the treatment of cancer: What have we learned? Gene Ther. 2001, 8, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Taipale, K.; Liikanen, I.; Koski, A.; Heiskanen, R.; Kanerva, A.; Hemminki, O.; Oksanen, M.; Grönberg-Vähä-Koskela, S.; Hemminki, K.; Joensuu, T.; et al. Predictive and prognostic clinical variables in cancer patients treated with adenoviral oncolytic immunotherapy. Mol. Ther. 2016, 24, 1323–1332. [Google Scholar] [CrossRef]
- Niemann, J.; Woller, N.; Brooks, J.; Fleischmann-Mundt, B.; Martin, N.T.; Kloos, A.; Knocke, S.; Ernst, A.M.; Manns, M.P.; Kubicka, S.; et al. Molecular retargeting of antibodies converts immune defense against oncolytic viruses into cancer immunotherapy. Nat. Commun. 2019, 10, 3236. [Google Scholar] [CrossRef]
- Mok, W.; Boucher, Y.; Jain, R.K. Matrix metalloproteinases-1 and -8 improve the distribution and efficacy of an oncolytic virus. Cancer Res. 2007, 67, 10664–10668. [Google Scholar] [CrossRef]
- Shen, B.H.; Hermiston, T.W. Effect of hypoxia on Ad5 infection, transgene expression and replication. Gene Ther. 2005, 12, 902–910. [Google Scholar] [CrossRef]
- Zheng, M.; Huang, J.; Tong, A.; Yang, H. Oncolytic viruses for cancer therapy: Barriers and recent advances. Mol. Ther. Oncolytics 2019, 15, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, S.; Kagawa, S.; Fujiwara, T. Gene therapy of cancer. Sect. Oncolytic Viruses 2014, 171–183. [Google Scholar] [CrossRef]
- Vähä-Koskela, M.; Hinkkanen, A. Tumor restrictions to oncolytic virus. BioMed 2014, 2, 163–194. [Google Scholar] [CrossRef]
- Guedan, S.; Rojas, J.J.; Gros, A.; Mercade, E.; Cascallo, M.; Alemany, R. Hyaluronidase expression by an oncolytic adenovirus enhances its intratumoral spread and suppresses tumor growth. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 1275–1283. [Google Scholar] [CrossRef]
- Zhu, F.-C.; Li, Y.-H.; Guan, X.-H.; Hou, L.-H.; Wang, W.-J.; Li, J.-X.; Wu, S.-P.; Wang, B.-S.; Wang, Z.; Wang, L.; et al. Safety, tolerability, and immunogenicity of a recombinant adenovirus type-5 vectored COVID-19 vaccine: A dose-escalation, open-label, non-randomised, first-in-human trial. Lancet 2020, 395, 1845–1854. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Romero, P. Metabolic control of CD8+ T cell fate decisions and antitumor immunity. Trends Mol. Med. 2018, 24, 30–48. [Google Scholar] [CrossRef]
- Vito, A.; El-Sayes, N.; Mossman, K. Hypoxia-driven immune escape in the tumor microenvironment. Cells 2020, 9, 992. [Google Scholar] [CrossRef]
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning cold into hot: Firing up the tumor microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Marijt, K.A.; Sluijter, M.; Blijleven, L.; Tolmeijer, S.H.; Scheeren, F.A.; van der Burg, S.H.; van Hall, T. Metabolic stress in cancer cells induces immune escape through a pi3k-dependent blockade of IFNγ receptor signaling. J. Immunother. Cancer 2019, 7, 152. [Google Scholar] [CrossRef]
- Kovács, T.; Mikó, E.; Ujlaki, G.; Sári, Z.; Bai, P. Advances in experimental medicine and biology. Adv. Exp. Med. Biol. 2020, 1225, 137–153. [Google Scholar] [CrossRef]
- Zitvogel, L.; Ayyoub, M.; Routy, B.; Kroemer, G. Microbiome and anticancer immunosurveillance. Cell 2016, 165, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Eslami-S, Z.; Majidzadeh-A, K.; Halvaei, S.; Babapirali, F.; Esmaeili, R. Microbiome and breast cancer: New role for an ancient population. Front. Oncol. 2020, 10, 120. [Google Scholar] [CrossRef] [PubMed]
- Bindels, L.B.; Porporato, P.; Dewulf, E.M.; Verrax, J.; Neyrinck, A.M.; Martin, J.C.; Scott, K.P.; Calderon, P.B.; Feron, O.; Muccioli, G.G.; et al. Gut microbiota-derived propionate reduces cancer cell proliferation in the liver. Br. J. Cancer 2012, 107, 1337–1344. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| OV Type | Genetic Modification | Checkpoint Inhibitor | Indication | Clinical Phase | NCT Number |
|---|---|---|---|---|---|
| Herpes simplex virus 1 | Deletions in ICP34.5 and ICP47 and transgenic expression of GM-CSF | Pembrolizumab (anti-PD1) | Unresectable Stage IIIB–IV melanoma | III | NCT02263508 |
| T-VEC | Nivolumab (anti-PD1) | Lymphomas and some rare cutaneous tumors | II | NCT02978625 | |
| Pembrolizumab | Advanced melanoma progressed on anti-PD1/L1 based therapy | II | NCT02965716 | ||
| Pembrolizumab | Metastatic squamous cell carcinoma of the head and neck | I | NCT02626000 | ||
| Ipilimumab (anti-CTLA4) | Melanoma | I/II | NCT01740297 | ||
| Atezolizumab (anti-PDL1) | Breast cancer | I | NCT03802604 | ||
| Ipilimumab and nivolumab | Before surgery of localized breast cancer | I | NCT04185311 | ||
| Vaccinia virus Pexa-Vec | TK deletion and expression of GM-CSF and β-galactosidase | Ipilimumab | Metastatic solid tumors | I | NCT02977156 |
| Durvalumab (anti-PD1)-Tremelimumab | CRC | I/II | NCT03206073 | ||
| (Anti-CTLA4) nivolumab | HCC | I/II | NCT03071094 | ||
| Cemiplimab (anti-PD1) | RCC | I | NCT03294083 | ||
| Vesicular stomatitis virus (VSV) | Engineered to express Na+/I− symporter (NIS) and human | Avelumab | Refractory solid tumors | I | NCT02923466 |
| Interferon Beta (VSV-IFNβ-NIS) | Pembrolizumab | Refractory NSCLC and HCC | I | NCT03647163 | |
| Reovirus reolysin | None | Nivolumab | Relapsed/refractory multiple myeloma | I | NCT03605719 |
| Adenovirus (Ad) ONCOS-102 | Onc.Ad expressing GM-CSF | Pembrolizumab | Advanced or unresectable melanoma | I | NCT03003676 |
| CG0070 | Onc.Ad with a tumor specific promoter expressing GM-CSF | Pembrolizumab | NMIBC | II | NCT04387461 |
| Ad-p53 | Ad. expressing p53 | Pembrolizumab | HNSCC | I/II | NCT02842125 |
| PD-1/PD-L1 Inhibitors | Lymphoma | II | NCT03544723 | ||
| Ad-MAGEA3 | Ad. expressing MAGE-A3 with MG1-MAGEA3 | Pembrolizumab | NSCLC | I/II | NCT02879760 |
| Pembrolizumab | Metastatic melanoma squamous cell skin carcinoma | I | NCT03773744 | ||
| Ad5-DNX-2401 or Delta-24-RGD | Ad. expressing an Integrin-binding RGD-4C motif | Pembrolizumab | GBM and GS | II | NCT02798406 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tripodi, L.; Vitale, M.; Cerullo, V.; Pastore, L. Oncolytic Adenoviruses for Cancer Therapy. Int. J. Mol. Sci. 2021, 22, 2517. https://doi.org/10.3390/ijms22052517
Tripodi L, Vitale M, Cerullo V, Pastore L. Oncolytic Adenoviruses for Cancer Therapy. International Journal of Molecular Sciences. 2021; 22(5):2517. https://doi.org/10.3390/ijms22052517
Chicago/Turabian StyleTripodi, Lorella, Maria Vitale, Vincenzo Cerullo, and Lucio Pastore. 2021. "Oncolytic Adenoviruses for Cancer Therapy" International Journal of Molecular Sciences 22, no. 5: 2517. https://doi.org/10.3390/ijms22052517
APA StyleTripodi, L., Vitale, M., Cerullo, V., & Pastore, L. (2021). Oncolytic Adenoviruses for Cancer Therapy. International Journal of Molecular Sciences, 22(5), 2517. https://doi.org/10.3390/ijms22052517