
Vascular Inflammation and Dysfunction in Lupus-Prone Mice-IL-6 as Mediator of Disease Initiation

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
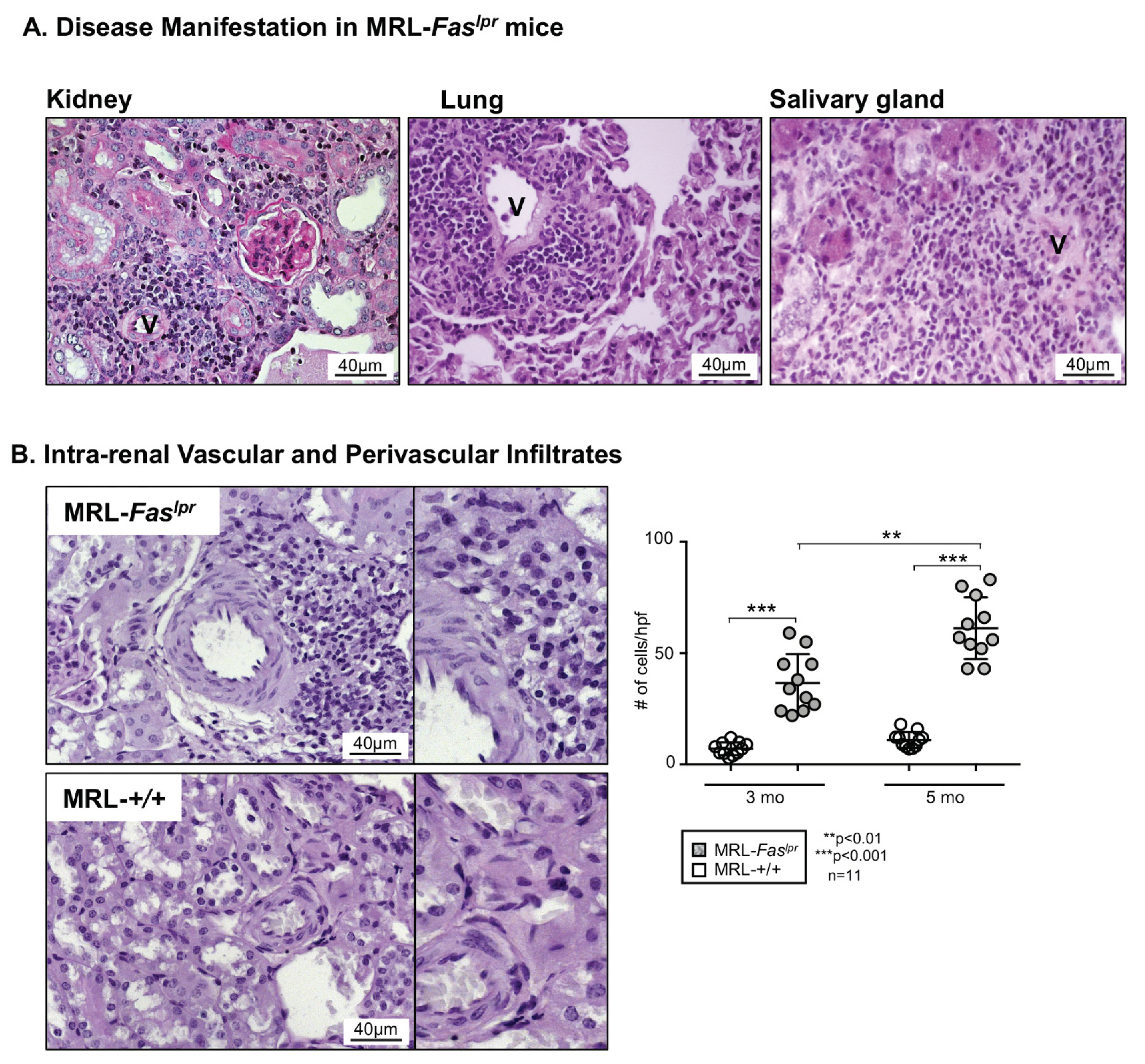
2.1. Predominantly Vascular and Perivascular Infiltrates Are Increased in MRL-Faslpr Mice
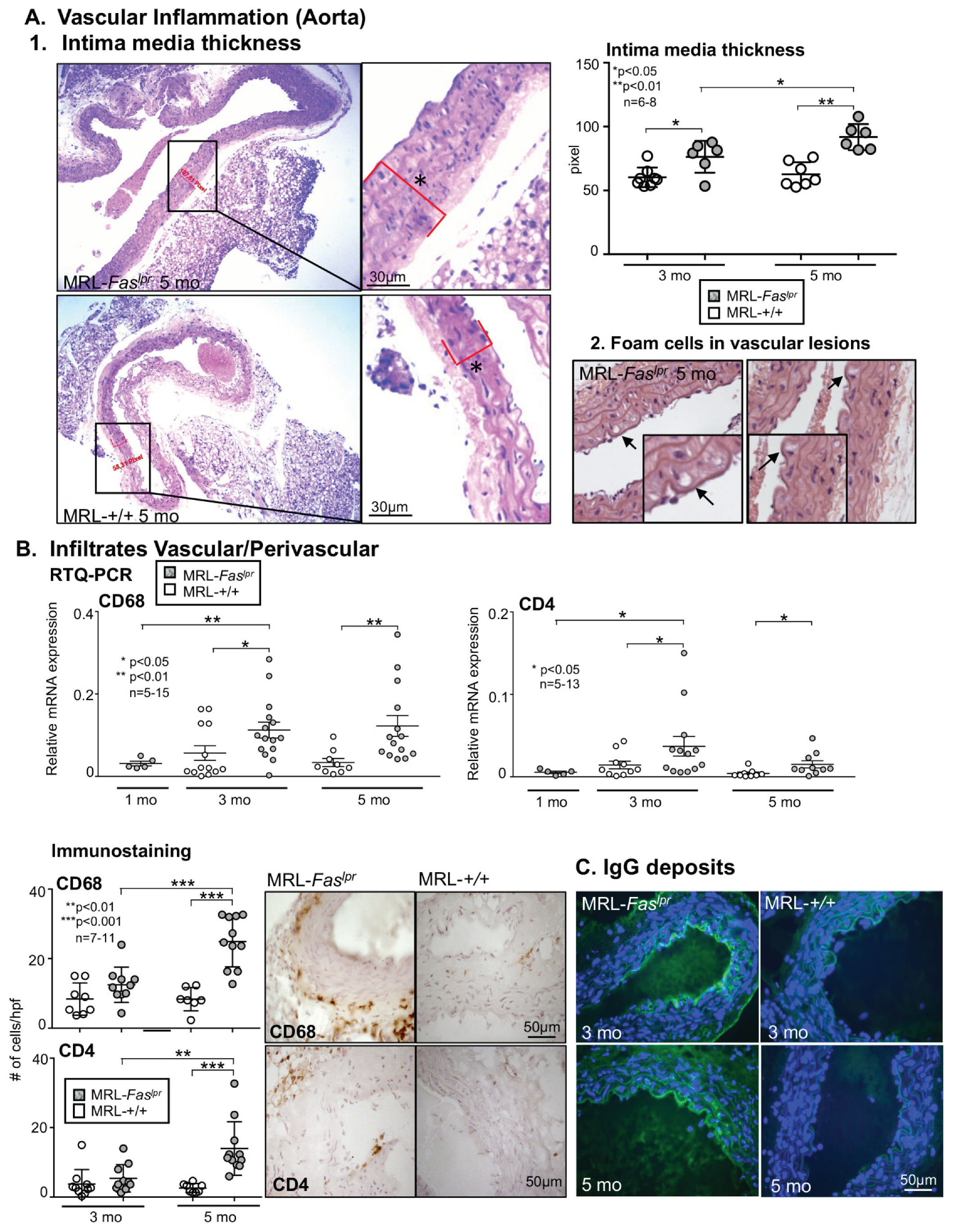
2.2. Increased Vascular and Perivascular Infiltrates of Macrophages and T cells Goes Along with an Increased Intima Media Thickness in MRL-Faslpr Mice
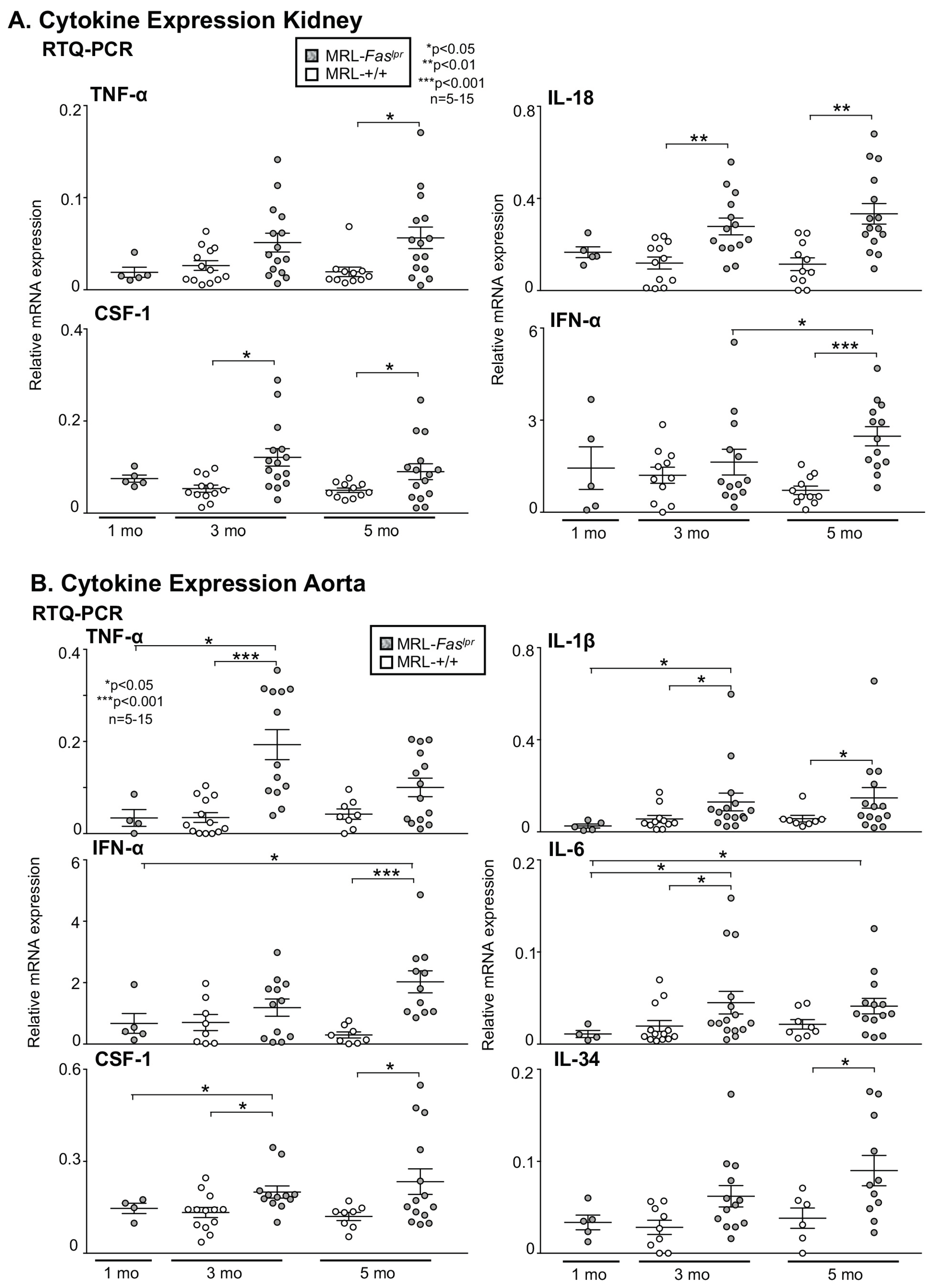
2.3. Inflammatory Markers Like TNF-α Are Not Only Expressed in the Kidney with Advancing Disease, They Are Abundantly Detectable in Aortic Tissue of MRL-Faslpr Mice
2.4. Amelioration of Vascular Inflammation in Il6−/− MRL-Faslpr Mice
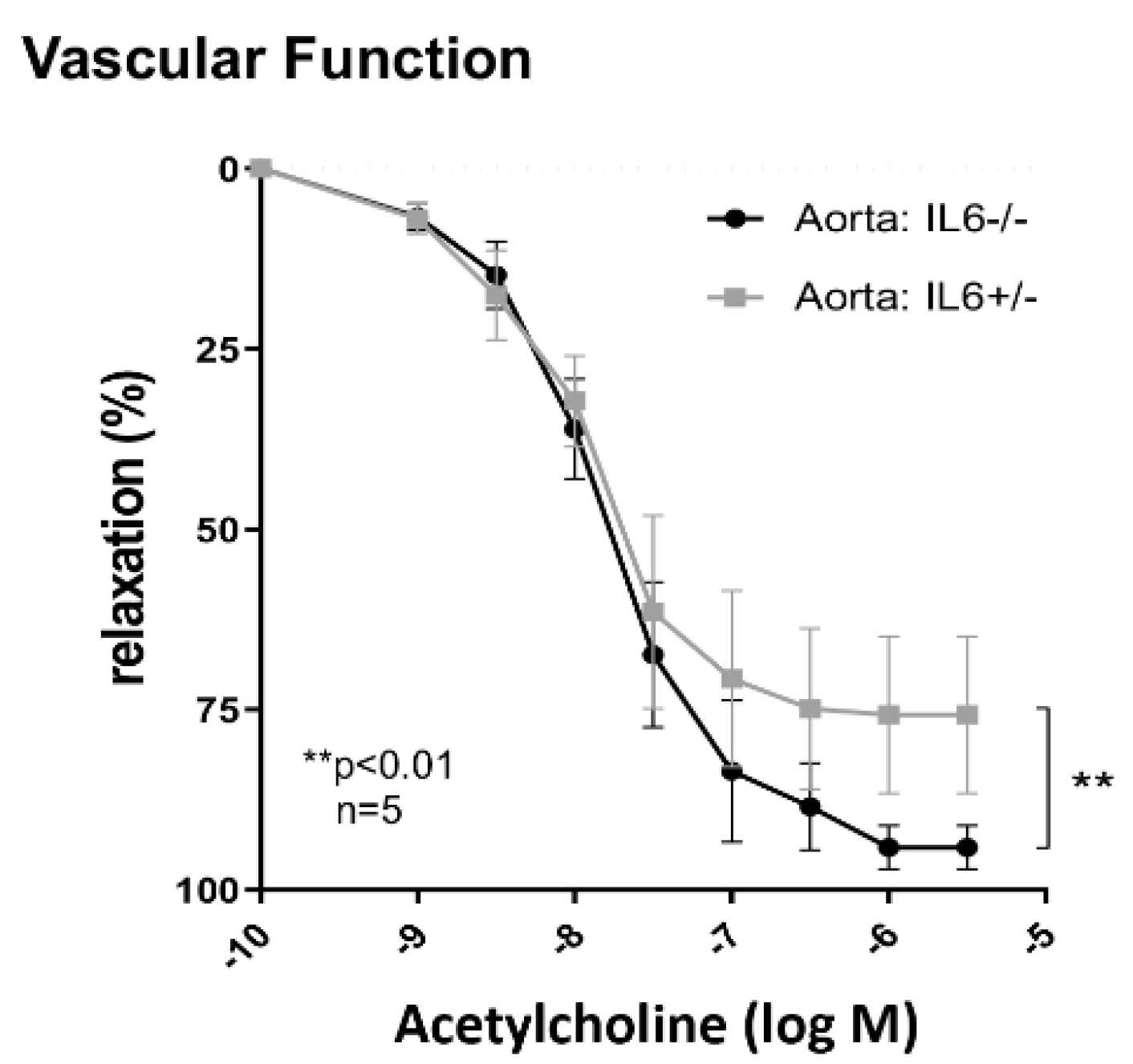
2.5. IL-6 Mediates Vascular Dysfunction in MRL-Faslpr Mice
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Renal Histopathology
4.3. Immunostaining
4.4. ELISA
4.5. Quantitative RTQ-PCR
4.6. Assessment of Vascular Function
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Interleukin-6 | IL-6 |
| high power field | HPF |
| Systemic lupus erythematosus | SLE |
| Antibody | Ab |
| Cardiovascular disease | CVD |
References
- Rees, F.; Doherty, M.; Grainge, M.J.; Lanyon, P.; Zhang, W. The worldwide incidence and prevalence of systemic lupus erythematosus: A systematic review of epidemiological studies. Rheumatology 2017, 56, 1945–1961. [Google Scholar] [CrossRef] [PubMed]
- Fors Nieves, C.E.; Izmirly, P.M. Mortality in Systemic Lupus Erythematosus: An Updated Review. Curr. Rheumatol. Rep. 2016, 18, 21. [Google Scholar] [CrossRef]
- Thomas, G.; Mancini, J.; Jourde-Chiche, N.; Sarlon, G.; Amoura, Z.; Harlé, J.R.; Jougla, E.; Chiche, L. Mortality associated with systemic lupus erythematosus in France assessed by multiple-cause-of-death analysis. Arthritis Rheumatol. 2014, 66, 2503–2511. [Google Scholar] [CrossRef]
- Yurkovich, M.; Vostretsova, K.; Chen, W.; Aviña-Zubieta, J.A. Overall and cause-specific mortality in patients with systemic lupus erythematosus: A meta-analysis of observational studies. Arthritis Care Res. 2014, 66, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Rathi, M.; Sahoo, S.; Prakash, M.; Dhir, V.; Singh, S. Assessment of premature atherosclerosis in systemic lupus erythematosus patients with and without nephritis. Lupus 2016, 25, 525–531. [Google Scholar] [CrossRef]
- Giannelou, M.; Mavragani, C.P. Cardiovascular disease in systemic lupus erythematosus: A comprehensive update. J. Autoimmun. 2017, 82, 1–12. [Google Scholar] [CrossRef]
- Bakshi, J.; Segura, B.T.; Wincup, C.; Rahman, A. Unmet Needs in the Pathogenesis and Treatment of Systemic Lupus Erythematosus. Clin. Rev. Allergy Immunol. 2018, 55, 352–367. [Google Scholar] [CrossRef]
- Menke, J.; Rabacal, W.A.; Byrne, K.T.; Iwata, Y.; Schwartz, M.M.; Stanley, E.R.; Schwarting, A.; Kelley, V.R. Circulating CSF-1 promotes monocyte and macrophage phenotypes that enhance lupus nephritis. J. Am. Soc. Nephrol. 2009, 20, 2581–2592. [Google Scholar] [CrossRef]
- Liu, Y.; Kaplan, M.J. Cardiovascular disease in systemic lupus erythematosus: An update. Curr. Opin. Rheumatol. 2018, 30, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Skaggs, B.J.; Hahn, B.H.; McMahon, M. Accelerated atherosclerosis in patients with SLE--mechanisms and management. Nat. Rev. Rheumatol. 2012, 8, 214–223. [Google Scholar] [CrossRef]
- Atehortúa, L.; Rojas, M.; Vásquez, G.M.; Castaño, D. Endothelial Alterations in Systemic Lupus Erythematosus and Rheumatoid Arthritis: Potential Effect of Monocyte Interaction. Mediators Inflamm. 2017, 2017, 9680729. [Google Scholar] [CrossRef] [PubMed]
- Nhek, S.; Clancy, R.; Lee, K.A.; Allen, N.M.; Barrett, T.J.; Marcantoni, E.; Nwaukoni, J.; Rasmussen, S.; Rubin, M.; Newman, J.D.; et al. Activated Platelets Induce Endothelial Cell Activation via an Interleukin-1beta Pathway in Systemic Lupus Erythematosus. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 707–716. [Google Scholar] [CrossRef]
- Theofilopoulos, A.N.; Dixon, F.J. Etiopathogenesis of murine SLE. Immunol. Rev. 1981, 55, 179–216. [Google Scholar] [CrossRef]
- Moyer, C.F.; Strandberg, J.D.; Reinisch, C.L. Systemic mononuclear-cell vasculitis in MRL/Mp-lpr/lpr mice. A histologic and immunocytochemical analysis. Am. J. Pathol. 1987, 127, 229–242. [Google Scholar]
- Kelley, V.E.; Roths, J.B. Interaction of mutant lpr gene with background strain influences renal disease. Clin. Immunol. Immunopathol. 1985, 37, 220–229. [Google Scholar] [CrossRef]
- Knight, J.S.; Subramanian, V.; O’Dell, A.A.; Yalavarthi, S.; Zhao, W.; Smith, C.K.; Hodgin, J.B.; Thompson, P.R.; Kaplan, M.J. Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann. Rheum. Dis. 2015, 74, 2199–2206. [Google Scholar] [CrossRef]
- Marshall, D.; Dangerfield, J.P.; Bhatia, V.K.; Larbi, K.Y.; Nourshargh, S.; Haskard, D.O. MRL/lpr lupus-prone mice show exaggerated ICAM-1-dependent leucocyte adhesion and transendothelial migration in response to TNF-alpha. Rheumatology 2003, 42, 929–934. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Hahn, B.H.; Skaggs, B.J. Systemic lupus erythematosus and cardiovascular disease: Prediction and potential for therapeutic intervention. Expert Rev. Clin. Immunol. 2011, 7, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Thacker, S.G.; Duquaine, D.; Park, J.; Kaplan, M.J. Lupus-prone New Zealand Black/New Zealand White F1 mice display endothelial dysfunction and abnormal phenotype and function of endothelial progenitor cells. Lupus 2010, 19, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Sitia, S.; Tomasoni, L.; Atzeni, F.; Ambrosio, G.; Cordiano, C.; Catapano, A.; Tramontana, S.; Perticone, F.; Naccarato, P.; Camici, P.; et al. From endothelial dysfunction to atherosclerosis. Autoimmun. Rev. 2010, 9, 830–834. [Google Scholar] [CrossRef]
- Tesauro, M.; Mauriello, A.; Rovella, V.; Annicchiarico-Petruzzelli, M.; Cardillo, C.; Melino, G.; Di Daniele, N. Arterial ageing: From endothelial dysfunction to vascular calcification. J. Intern. Med. 2017, 281, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Sciatti, E.; Cavazzana, I.; Vizzardi, E.; Bonadei, I.; Fredi, M.; Taraborelli, M.; Ferizi, R.; Metra, M.; Tincani, A.; Franceschini, F. Systemic Lupus Erythematosus and Endothelial Dysfunction: A Close Relationshi. Curr. Rheumatol. Rev. 2019, 15, 177–188. [Google Scholar] [CrossRef]
- Bitterli, L.; Afan, S.; Bühler, S.; DiSanto, S.; Zwahlen, M.; Schmidlin, K.; Yang, Z.; Baumgartner, I.; Diehm, N.; Kalka, C. Endothelial progenitor cells as a biological marker of peripheral artery disease. Vasc. Med. 2016, 21, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Altabas, V.; Altabas, K.; Kirigin, L. Endothelial progenitor cells (EPCs) in ageing and age-related diseases: How currently available treatment modalities affect EPC biology, atherosclerosis, and cardiovascular outcomes. Mech. Ageing Dev. 2016, 159, 49–62. [Google Scholar] [CrossRef]
- Denny, M.F.; Thacker, S.; Mehta, H.; Somers, E.C.; Dodick, T.; Barrat, F.J.; McCune, W.J.; Kaplan, M.J. Interferon-alpha promotes abnormal vasculogenesis in lupus: A potential pathway for premature atherosclerosis. Blood 2007, 110, 2907–2915. [Google Scholar] [CrossRef]
- Willeit, P.; Tschiderer, L.; Allara, E.; Reuber, K.; Seekircher, L.; Gao, L.; Liao, X.; Lonn, E.; Gerstein, H.C.; Yusuf, S.; et al. Carotid Intima-Media Thickness Progression as Surrogate Marker for Cardiovascular Risk: Meta-Analysis of 119 Clinical Trials Involving 100 667 Patients. Circulation 2020, 142, 621–642. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, M.W.; Polak, J.F.; Kavousi, M.; Mathiesen, E.B.; Völzke, H.; Tuomainen, T.P.; Sander, D.; Plichart, M.; Catapano, A.L.; Robertson, C.M.; et al. Carotid intima-media thickness progression to predict cardiovascular events in the general population (the PROG-IMT collaborative project): A meta-analysis of individual participant data. Lancet 2012, 379, 2053–2062. [Google Scholar] [CrossRef]
- Bots, M.L. Carotid intima-media thickness as a surrogate marker for cardiovascular disease in intervention studies. Curr. Med. Res. Opin. 2006, 22, 2181–2190. [Google Scholar] [CrossRef]
- Cash, H.; Relle, M.; Menke, J.; Brochhausen, C.; Jones, S.A.; Topley, N.; Galle, P.R.; Schwarting, A. Interleukin 6 (IL-6) deficiency delays lupus nephritis in MRL-Faslpr mice: The IL-6 pathway as a new therapeutic target in treatment of autoimmune kidney disease in systemic lupus erythematosus. J. Rheumatol. 2010, 37, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Schwarting, A.; Wada, T.; Kinoshita, K.; Tesch, G.; Kelley, V.R. IFN-gamma receptor signaling is essential for the initiation, acceleration, and destruction of autoimmune kidney disease in MRL-Fas(lpr) mice. J. Immunol. 1998, 161, 494–503. [Google Scholar]
- Yin, Z.; Bahtiyar, G.; Zhang, N.; Liu, L.; Zhu, P.; Robert, M.E.; McNiff, J.; Madaio, M.P.; Craft, J. IL-10 Regulates Murine Lupus. J. Immunol. 2002, 169, 2148–2155. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, K.; Yamagata, T.; Nozaki, Y.; Sugiyama, M.; Ikoma, S.; Funauchi, M.; Kanamaru, A. Blockade of IL-18 Receptor Signaling Delays the Onset of Autoimmune Disease in MRL-Faslpr Mice. J. Immunol. 2004, 173, 5312–5318. [Google Scholar] [CrossRef]
- Rankin, A.L.; Guay, H.; Herber, D.; Bertino, S.A.; Duzanski, T.A.; Carrier, Y.; Keegan, S.; Senices, M.; Stedman, N.; Ryan, M.; et al. IL-21 Receptor Is Required for the Systemic Accumulation of Activated B and T Lymphocytes in MRL/MpJ-Faslpr/lpr/J Mice. J. Immunol. 2012, 188, 1656–1667. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Su, S.; You, T.; Xia, T.; Lin, X.; Chen, Z.; Zhang, L. Serum interleukin-6 level is correlated with the disease activity of systemic lupus erythematosus: A meta-analysis. Clinics 2020, 75, 1801. [Google Scholar] [CrossRef]
- Wallace, D.J.; Strand, V.; Merrill, J.T.; Popa, S.; Spindler, A.J.; Eimon, A.; Petri, M.; Smolen, J.S.; Wajdula, J.; Christensen, J.; et al. Efficacy and safety of an interleukin 6 monoclonal antibody for the treatment of systemic lupus erythematosus: A phase II dose-ranging randomised controlled trial. Ann. Rheum. Dis. 2017, 76, 534–542. [Google Scholar] [CrossRef]
- Asanuma, Y.; Chung, C.P.; Oeser, A.; Shintani, A.; Stanley, E.; Raggi, P.; Stein, C.M. Increased concentration of proatherogenic inflammatory cytokines in systemic lupus erythematosus: Relationship to cardiovascular risk factors. J. Rheumatol. 2006, 33, 539–545. [Google Scholar]
- Wada, Y.; Gonzalez-Sanchez, H.M.; Weinmann-Menke, J.; Iwata, Y.; Ajay, A.K.; Meineck, M.; Kelley, V.R. IL-34-Dependent Intrarenal and Systemic Mechanisms Promote Lupus Nephritis in MRL-Fas(lpr) Mice. J. Am. Soc. Nephrol. 2019, 30, 244–259. [Google Scholar] [CrossRef]
- Iwata, Y.; Boström, E.A.; Menke, J.; Rabacal, W.A.; Morel, L.; Wada, T.; Kelley, V.R. Aberrant Macrophages Mediate Defective Kidney Repair That Triggers Nephritis in Lupus-Susceptible Mice. J. Immunol. 2012, 188, 4568–4580. [Google Scholar] [CrossRef] [PubMed]
- Xia, N.; Horke, S.; Habermeier, A.; Closs, E.I.; Reifenberg, G.; Gericke, A.; Mikhed, Y.; Münzel, T.; Daiber, A.; Förstermann, U.; et al. Uncoupling of Endothelial Nitric Oxide Synthase in Perivascular Adipose Tissue of Diet-Induced Obese Mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 78–85. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marczynski, P.; Meineck, M.; Xia, N.; Li, H.; Kraus, D.; Roth, W.; Möckel, T.; Boedecker, S.; Schwarting, A.; Weinmann-Menke, J. Vascular Inflammation and Dysfunction in Lupus-Prone Mice-IL-6 as Mediator of Disease Initiation. Int. J. Mol. Sci. 2021, 22, 2291. https://doi.org/10.3390/ijms22052291
Marczynski P, Meineck M, Xia N, Li H, Kraus D, Roth W, Möckel T, Boedecker S, Schwarting A, Weinmann-Menke J. Vascular Inflammation and Dysfunction in Lupus-Prone Mice-IL-6 as Mediator of Disease Initiation. International Journal of Molecular Sciences. 2021; 22(5):2291. https://doi.org/10.3390/ijms22052291
Chicago/Turabian StyleMarczynski, Paul, Myriam Meineck, Ning Xia, Huige Li, Daniel Kraus, Wilfried Roth, Tamara Möckel, Simone Boedecker, Andreas Schwarting, and Julia Weinmann-Menke. 2021. "Vascular Inflammation and Dysfunction in Lupus-Prone Mice-IL-6 as Mediator of Disease Initiation" International Journal of Molecular Sciences 22, no. 5: 2291. https://doi.org/10.3390/ijms22052291
APA StyleMarczynski, P., Meineck, M., Xia, N., Li, H., Kraus, D., Roth, W., Möckel, T., Boedecker, S., Schwarting, A., & Weinmann-Menke, J. (2021). Vascular Inflammation and Dysfunction in Lupus-Prone Mice-IL-6 as Mediator of Disease Initiation. International Journal of Molecular Sciences, 22(5), 2291. https://doi.org/10.3390/ijms22052291