
Apoptotic Cells induce Proliferation of Peritoneal Macrophages

 , , ,
, , , 
Abstract
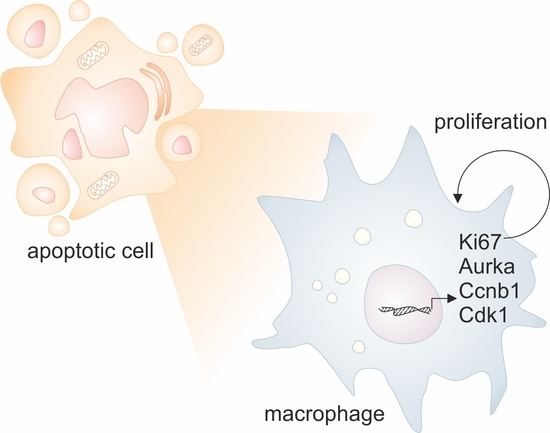
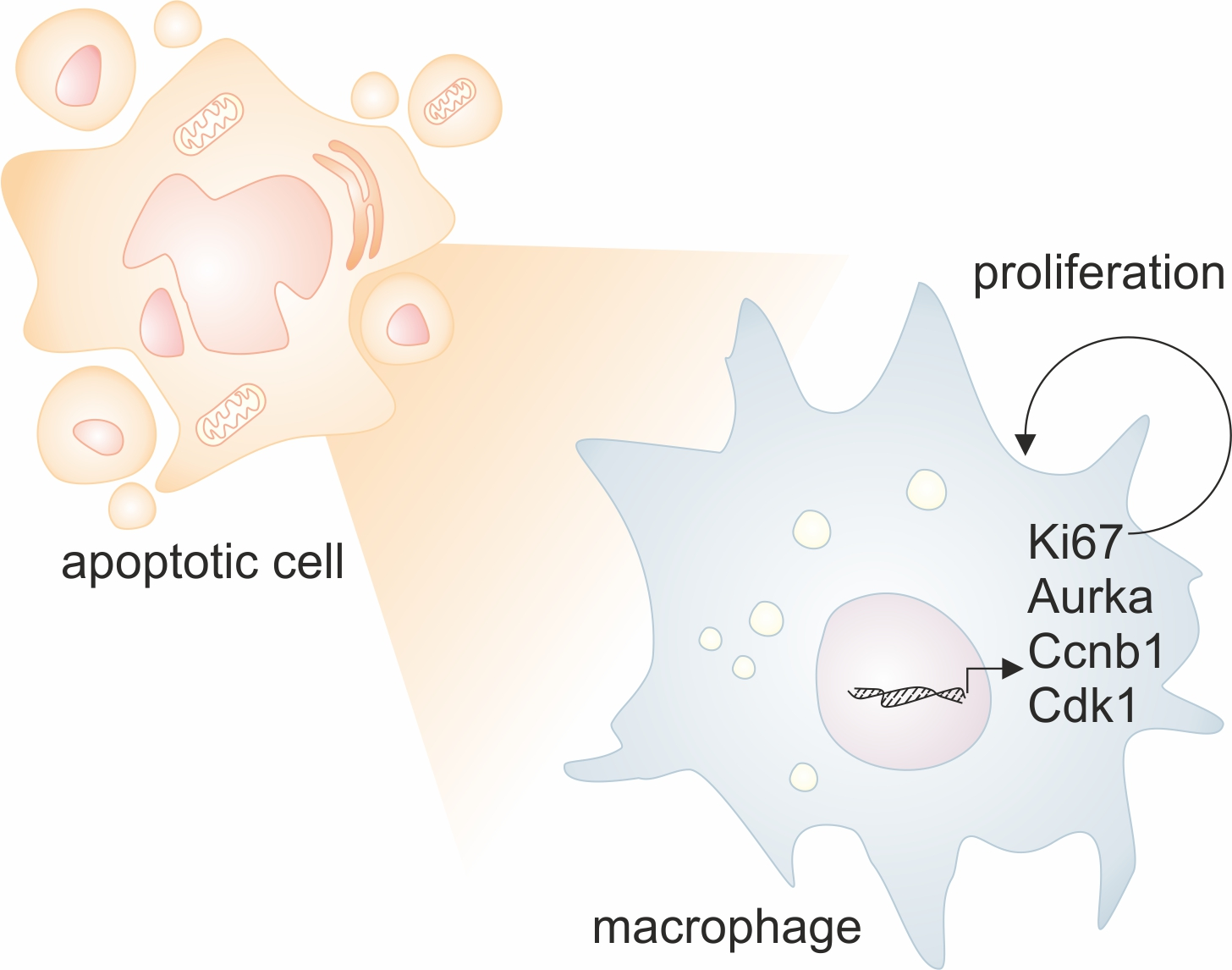
1. Introduction
2. Results
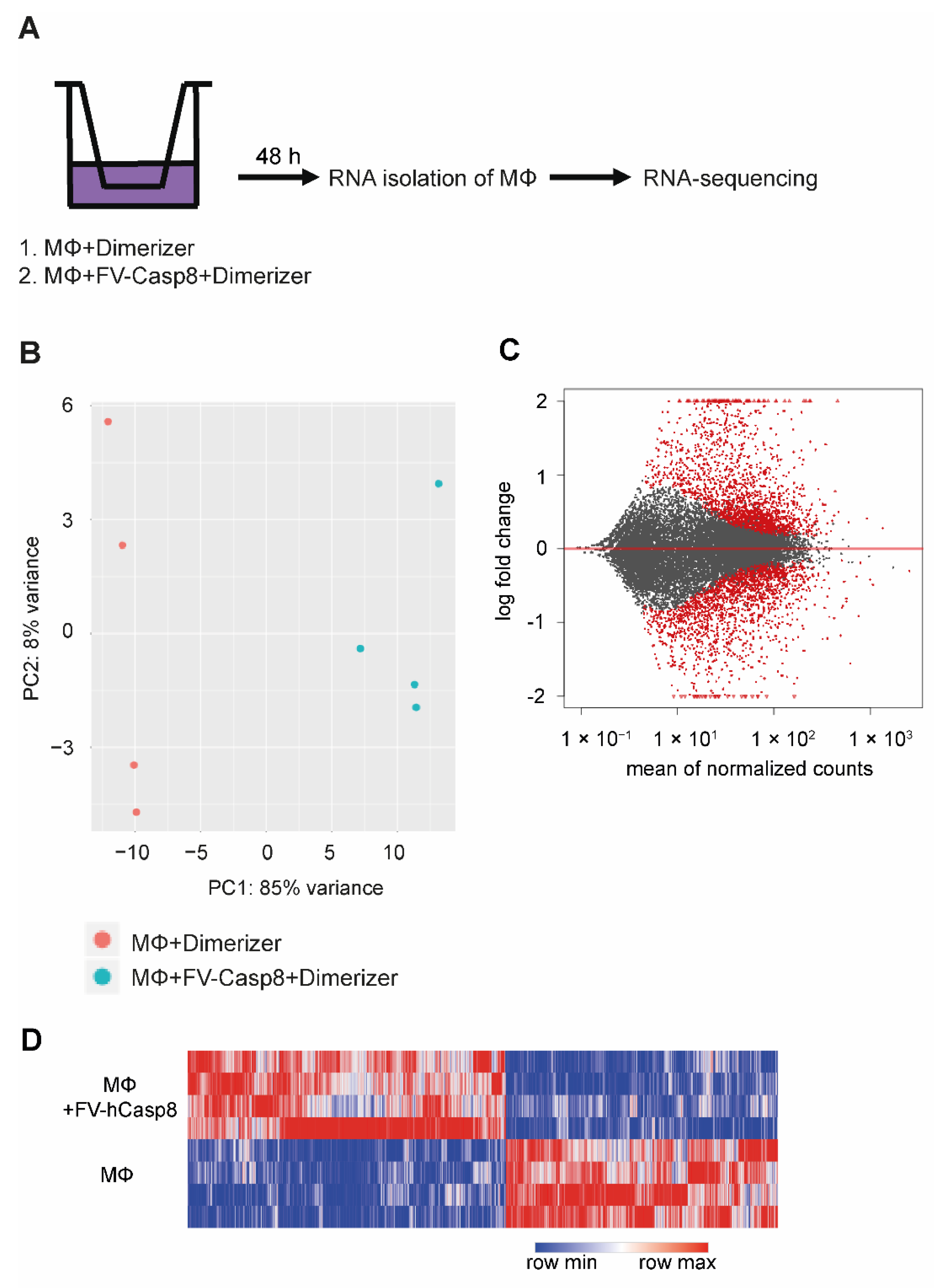
2.1. Generation of a Dimerizer-Induced Apoptotic System
2.2. Co-Culture with Apoptotic Cells Induces Macrophage Proliferation
2.3. Inhibition of Apoptosis in a Model of Self-Resolving Inflammation Reduces mRNA Expression of Cell Cycle Genes in Macrophages In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Chemicals
4.2. Generation of NIH 3T3 Cells Expressing FV-hCasp8
4.3. Co-Culture of Peritoneal Macrophages with Apoptotic Cells
4.4. Zymosan-A-Induced Peritonitis
4.5. Western Blot Analysis
4.6. Cell Death Analysis
4.7. Caspase-Activity Assay
4.8. Chemokine Analysis
4.9. Quantitative Real-Time PCR
4.10. RNA Sequencing
4.11. Proliferation of Macrophages
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| APC | Allophycocyanin |
| ATCC | American Type Culture Collection |
| CDK1 | cyclin-dependent kinase 1 |
| DAMPS | danger associated molecular patterns |
| DED | death effector domain |
| DISC | death-inducing signaling complex |
| DNA | Deoxyribonucleic acid |
| DSMZ | Deutsche Sammlung von Mikroorganismen |
| e.g., | exempli gratia |
| FADD | Fas-associating protein with death domain |
| FCS | fetal calf serum |
| FITC | Fluorescein isothiocyanate |
| FKBP | FK506 binding protein |
| GFP | green fluorescent protein |
| GM-CSF | granulocyte-macrophage colony-stimulating factor |
| GO | Gene ontology |
| GSEA | Gene set enrichment analysis |
| hCasp8 | human Caspase 8 |
| i.p. | intraperitoneal |
| IFN | interferon |
| Ig | immunoglobin |
| IL | interleukin |
| kDa | kilo Dalton |
| LPS | lipopolysaccharide |
| M-CSF | macrophage colony-stimulating factor |
| MΦ | macrophage |
| NES | normalized enrichment score |
| NO | nitric oxide |
| PBS | phosphate-buffered saline |
| PCA | principal component analysis |
| PI | propidium iodide |
| PMNs | polymorphonuclear leukocytes |
| PPAR | peroxisome proliferator-activated receptor |
| RNA | Ribonucleic acid |
| RNA-Seq | RNA sequencing |
| ROS | reactive oxygen species |
| RT-PCR | Reverse transcription polymerase chain reaction |
| SEM | standard error of the mean |
| TAM | tumor-associated macrophage |
| TGF | transforming growth factor |
| TLR2 | toll-like receptor 2 |
| TNF | tumor necrosis factor |
References
- Bonnefoy, F.; Gauthier, T.; Vallion, R.; Martin-Rodriguez, O.; Missey, A.; Daoui, A.; Valmary-Degano, S.; Saas, P.; Couturier, M.; Perruche, S. Factors Produced by Macrophages Eliminating Apoptotic Cells Demonstrate Pro-Resolutive Properties and Terminate Ongoing Inflammation. Front. Immunol. 2018, 9, 2586. [Google Scholar] [CrossRef] [PubMed]
- Kourtzelis, I.; Hajishengallis, G.; Chavakis, T. Phagocytosis of Apoptotic Cells in Resolution of Inflammation. Front. Immunol. 2020, 11, 553. [Google Scholar] [CrossRef] [PubMed]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Thapa, B.; Lee, K. Metabolic influence on macrophage polarization and pathogenesis. BMB Rep. 2019, 52, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Pluddemann, A. Macrophage Clearance of Apoptotic Cells: A Critical Assessment. Front. Immunol. 2018, 9, 127. [Google Scholar] [CrossRef]
- Weigert, A.; Jennewein, C.; Brune, B. The liaison between apoptotic cells and macrophages—The end programs the beginning. Biol. Chem. 2009, 390, 379–390. [Google Scholar] [CrossRef]
- Sieweke, M.H.; Allen, J.E. Beyond stem cells: Self-renewal of differentiated macrophages. Science 2013, 342, 1242974. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Tummers, B.; Green, D.R. Caspase-8: Regulating life and death. Immunol. Rev. 2017, 277, 76–89. [Google Scholar] [CrossRef]
- Fogarty, C.E.; Diwanji, N.; Lindblad, J.L.; Tare, M.; Amcheslavsky, A.; Makhijani, K.; Bruckner, K.; Fan, Y.; Bergmann, A. Extracellular Reactive Oxygen Species Drive Apoptosis-Induced Proliferation via Drosophila Macrophages. Curr. Biol. 2016, 26, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Perez-Garijo, A. When dying is not the end: Apoptotic caspases as drivers of proliferation. Semin. Cell Dev. Biol. 2018, 82, 86–95. [Google Scholar] [CrossRef]
- Davidovich, P.; Kearney, C.J.; Martin, S.J. Inflammatory outcomes of apoptosis, necrosis and necroptosis. Biol. Chem. 2014, 395, 1163–1171. [Google Scholar] [CrossRef]
- Yatim, N.; Jusforgues-Saklani, H.; Orozco, S.; Schulz, O.; Barreira da Silva, R.; Reis e Sousa, C.; Green, D.R.; Oberst, A.; Albert, M.L. RIPK1 and NF-kappaB signaling in dying cells determines cross-priming of CD8(+) T cells. Science 2015, 350, 328–334. [Google Scholar] [CrossRef]
- Roca, H.; Jones, J.D.; Purica, M.C.; Weidner, S.; Koh, A.J.; Kuo, R.; Wilkinson, J.E.; Wang, Y.; Daignault-Newton, S.; Pienta, K.J.; et al. Apoptosis-induced CXCL5 accelerates inflammation and growth of prostate tumor metastases in bone. J. Clin. Investig. 2018, 128, 248–266. [Google Scholar] [CrossRef] [PubMed]
- Brecht, K.; Weigert, A.; Hu, J.; Popp, R.; Fisslthaler, B.; Korff, T.; Fleming, I.; Geisslinger, G.; Brune, B. Macrophages programmed by apoptotic cells promote angiogenesis via prostaglandin E2. FASEB J. 2011, 25, 2408–2417. [Google Scholar] [CrossRef] [PubMed]
- Goodridge, H.S.; Underhill, D.M. Fungal Recognition by TLR2 and Dectin-1. Handb. Exp. Pharmacol. 2008, 183, 87–109. [Google Scholar]
- Cash, J.L.; White, G.E.; Greaves, D.R. Chapter 17. Zymosan-induced peritonitis as a simple experimental system for the study of inflammation. Methods Enzymol. 2009, 461, 379–396. [Google Scholar] [PubMed]
- Battistelli, M.; Falcieri, E. Apoptotic Bodies: Particular Extracellular Vesicles Involved in Intercellular Communication. Biology 2020, 9, 21. [Google Scholar] [CrossRef]
- Li, F.; Huang, Q.; Chen, J.; Peng, Y.; Roop, D.R.; Bedford, J.S.; Li, C.Y. Apoptotic cells activate the “phoenix rising” pathway to promote wound healing and tissue regeneration. Sci. Signal. 2010, 3, ra13. [Google Scholar] [CrossRef]
- Sergeeva, M.G.; Gonchar, M.V.; Mevkh, A.T.; Varfolomeyev, S.D. Prostaglandin E2 biphasic control of lymphocyte proliferation: Inhibition by picomolar concentrations. FEBS Lett. 1997, 418, 235–238. [Google Scholar] [CrossRef]
- Sanin, D.E.; Matsushita, M.; Klein Geltink, R.I.; Grzes, K.M.; van Teijlingen Bakker, N.; Corrado, M.; Kabat, A.M.; Buck, M.D.; Qiu, J.; Lawless, S.J.; et al. Mitochondrial Membrane Potential Regulates Nuclear Gene Expression in Macrophages Exposed to Prostaglandin E2. Immunity 2018, 49, 1021–1033.e6. [Google Scholar] [CrossRef]
- Stolberg, V.R.; Martin, B.; Mancuso, P.; Olszewski, M.A.; Freeman, C.M.; Curtis, J.L.; Chensue, S.W. Role of CC chemokine receptor 4 in natural killer cell activation during acute cigarette smoke exposure. Am. J. Pathol. 2014, 184, 454–463. [Google Scholar] [CrossRef]
- Geissmann, F.; Mass, E. A stratified myeloid system, the challenge of understanding macrophage diversity. Semin. Immunol. 2015, 27, 353–356. [Google Scholar] [CrossRef]
- Roszer, T. Understanding the Biology of Self-Renewing Macrophages. Cells 2018, 7, 103. [Google Scholar] [CrossRef]
- Gentek, R.; Molawi, K.; Sieweke, M.H. Tissue macrophage identity and self-renewal. Immunol. Rev. 2014, 262, 56–73. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.C.; Rosas, M.; Jenkins, S.J.; Liao, C.T.; Scurr, M.J.; Brombacher, F.; Fraser, D.J.; Allen, J.E.; Jones, S.A.; Taylor, P.R. Distinct bone marrow-derived and tissue-resident macrophage lineages proliferate at key stages during inflammation. Nat. Commun. 2013, 4, 1886. [Google Scholar] [CrossRef]
- Bosurgi, L.; Cao, Y.G.; Cabeza-Cabrerizo, M.; Tucci, A.; Hughes, L.D.; Kong, Y.; Weinstein, J.S.; Licona-Limon, P.; Schmid, E.T.; Pelorosso, F.; et al. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science 2017, 356, 1072–1076. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Tucker, S.C.; Casadevall, A. Fc- and complement-receptor activation stimulates cell cycle progression of macrophage cells from G1 to S. J. Immunol. 2005, 174, 7226–7233. [Google Scholar] [CrossRef]
- Robbins, C.S.; Hilgendorf, I.; Weber, G.F.; Theurl, I.; Iwamoto, Y.; Figueiredo, J.L.; Gorbatov, R.; Sukhova, G.K.; Gerhardt, L.M.; Smyth, D.; et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat. Med. 2013, 19, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Murray, L.A.; Chen, Q.; Kramer, M.S.; Hesson, D.P.; Argentieri, R.L.; Peng, X.; Gulati, M.; Homer, R.J.; Russell, T.; van Rooijen, N.; et al. TGF-beta driven lung fibrosis is macrophage dependent and blocked by Serum amyloid P. Int. J. Biochem. Cell Biol. 2011, 43, 154–162. [Google Scholar] [CrossRef]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Weigert, A.; Mora, J.; Sekar, D.; Syed, S.; Brune, B. Killing Is Not Enough: How Apoptosis Hijacks Tumor-Associated Macrophages to Promote Cancer Progression. Adv. Exp. Med. Biol. 2016, 930, 205–239. [Google Scholar]
- Fulda, S.; Sieverts, H.; Friesen, C.; Herr, I.; Debatin, K.M. The CD95 (APO-1/Fas) system mediates drug-induced apoptosis in neuroblastoma cells. Cancer Res. 1997, 57, 3823–3829. [Google Scholar]
- Knuth, A.K.; Rosler, S.; Schenk, B.; Kowald, L.; van Wijk, S.J.L.; Fulda, S. Interferons Transcriptionally Up-Regulate MLKL Expression in Cancer Cells. Neoplasia 2019, 21, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Eden, E.; Lipson, D.; Yogev, S.; Yakhini, Z. Discovering motifs in ranked lists of DNA sequences. PLoS Comput. Biol. 2007, 3, e39. [Google Scholar] [CrossRef]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinform. 2009, 10, 48. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Marker | Dye | Company |
|---|---|---|
| CD16/32 | none | BD |
| CD3 | PE-CF594 | BD |
| CD4 | BV711 | Biolegend |
| CD8 | BV650 | Biolegend |
| CD11b | BV605 | BD |
| CD11c | AlexaFluor 700 | BD |
| CD19 | APC-H7 | Biolegend |
| CD34 | FITC | BD |
| CD45 | VioBlue | Miltenyi |
| CD90.2 | PE | Miltenyi |
| CD117 | FITC | BD |
| CD140a | PE | BD |
| GITR | FITC | Biolegend |
| F4/80 | PE-Cy7 | Biolegend |
| HLA-DR (MHC II) | APC | Biolegend |
| Ly-6C | PerCP-Cy5.5 | Biolegend |
| Ly-6G | APC-Cy7 | Biolegend |
| NK1.1 | AlexaFluor 700 | BD |
| Primer | Sequence |
|---|---|
| mCD206_fwd | CCATCTCAGTTCAGACGGCA |
| mCD206_rev | ACGGAAGCCCAGTCAGTTTT |
| mMki67_fwd | AGGAATCGCGGGAGACACAGCT |
| mMki67_rev | CCATTCCACCGCGCCATCTCTC |
| mTop2a_fwd | CGCTGGTTTTGTCGCTTTCCGG |
| mTop2a_rev | TACAGGCTGCAGCGGTGACAAC |
| mAurka_fwd | AGACCACTGTTCCCTTCGGTCC |
| mAurka_rev | CTGGCCACTGCTAGCAGATCCT |
| mAurkb_fwd | GTCTGGCCTGAACACGTTGTCC |
| mAurkb_rev | GGACTGGCTGTTGAACCGGTTC |
| mCdk1_fwd | CGAGGAAGAAGGAGTGCCCAGT |
| mCdk1_rev | AGCACATCCTGCAGGCTGACTA |
| mCcnb1_fwd | CTGAGCCTGAGCCTGAACCTGA |
| mCcnb1_rev | CCATCGGGCTTGGAGAGGGATT |
| mRPLP0_fwd | GCTGATCATCCAGCAGGTGT |
| mRPLP0_rev | GGACACCCTCCAGAAAGCGA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knuth, A.-K.; Huard, A.; Naeem, Z.; Rappl, P.; Bauer, R.; Mota, A.C.; Schmid, T.; Fleming, I.; Brüne, B.; Fulda, S.; et al. Apoptotic Cells induce Proliferation of Peritoneal Macrophages. Int. J. Mol. Sci. 2021, 22, 2230. https://doi.org/10.3390/ijms22052230
Knuth A-K, Huard A, Naeem Z, Rappl P, Bauer R, Mota AC, Schmid T, Fleming I, Brüne B, Fulda S, et al. Apoptotic Cells induce Proliferation of Peritoneal Macrophages. International Journal of Molecular Sciences. 2021; 22(5):2230. https://doi.org/10.3390/ijms22052230
Chicago/Turabian StyleKnuth, Anne-Kathrin, Arnaud Huard, Zumer Naeem, Peter Rappl, Rebekka Bauer, Ana Carolina Mota, Tobias Schmid, Ingrid Fleming, Bernhard Brüne, Simone Fulda, and et al. 2021. "Apoptotic Cells induce Proliferation of Peritoneal Macrophages" International Journal of Molecular Sciences 22, no. 5: 2230. https://doi.org/10.3390/ijms22052230
APA StyleKnuth, A.-K., Huard, A., Naeem, Z., Rappl, P., Bauer, R., Mota, A. C., Schmid, T., Fleming, I., Brüne, B., Fulda, S., & Weigert, A. (2021). Apoptotic Cells induce Proliferation of Peritoneal Macrophages. International Journal of Molecular Sciences, 22(5), 2230. https://doi.org/10.3390/ijms22052230