
Peptidylarginine Deiminase Inhibitor Application, Using Cl-Amidine, PAD2, PAD3 and PAD4 Isozyme-Specific Inhibitors in Pancreatic Cancer Cells, Reveals Roles for PAD2 and PAD3 in Cancer Invasion and Modulation of Extracellular Vesicle Signatures

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
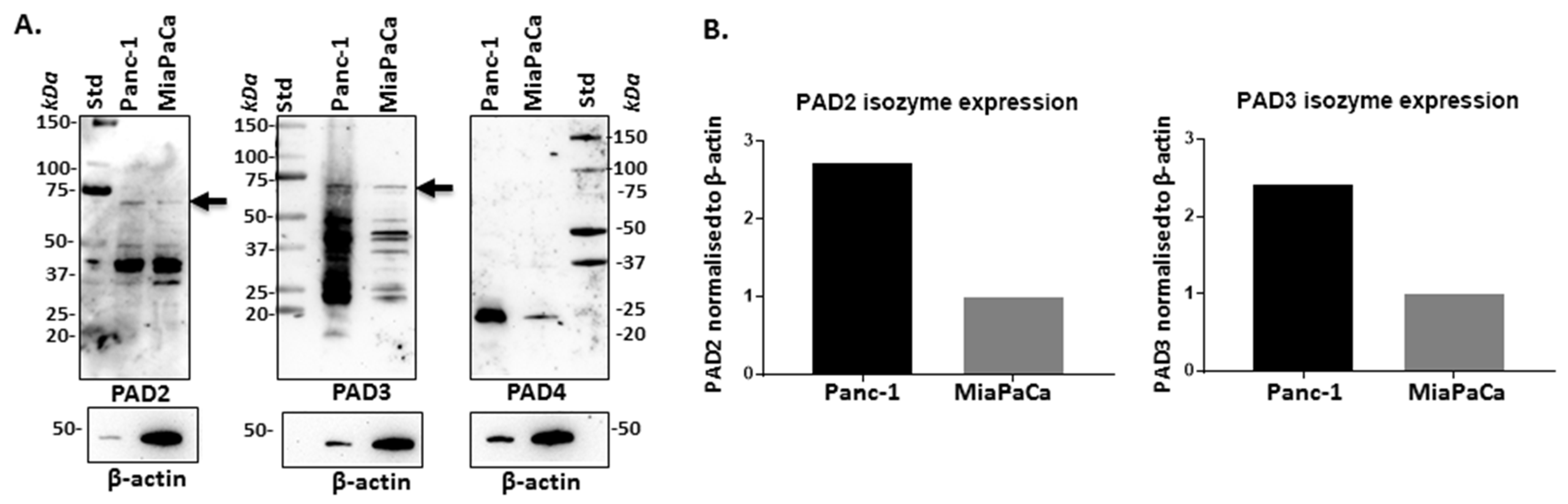
2.1. PAD Isozymes Are Differently Expressed in PDAC Cells
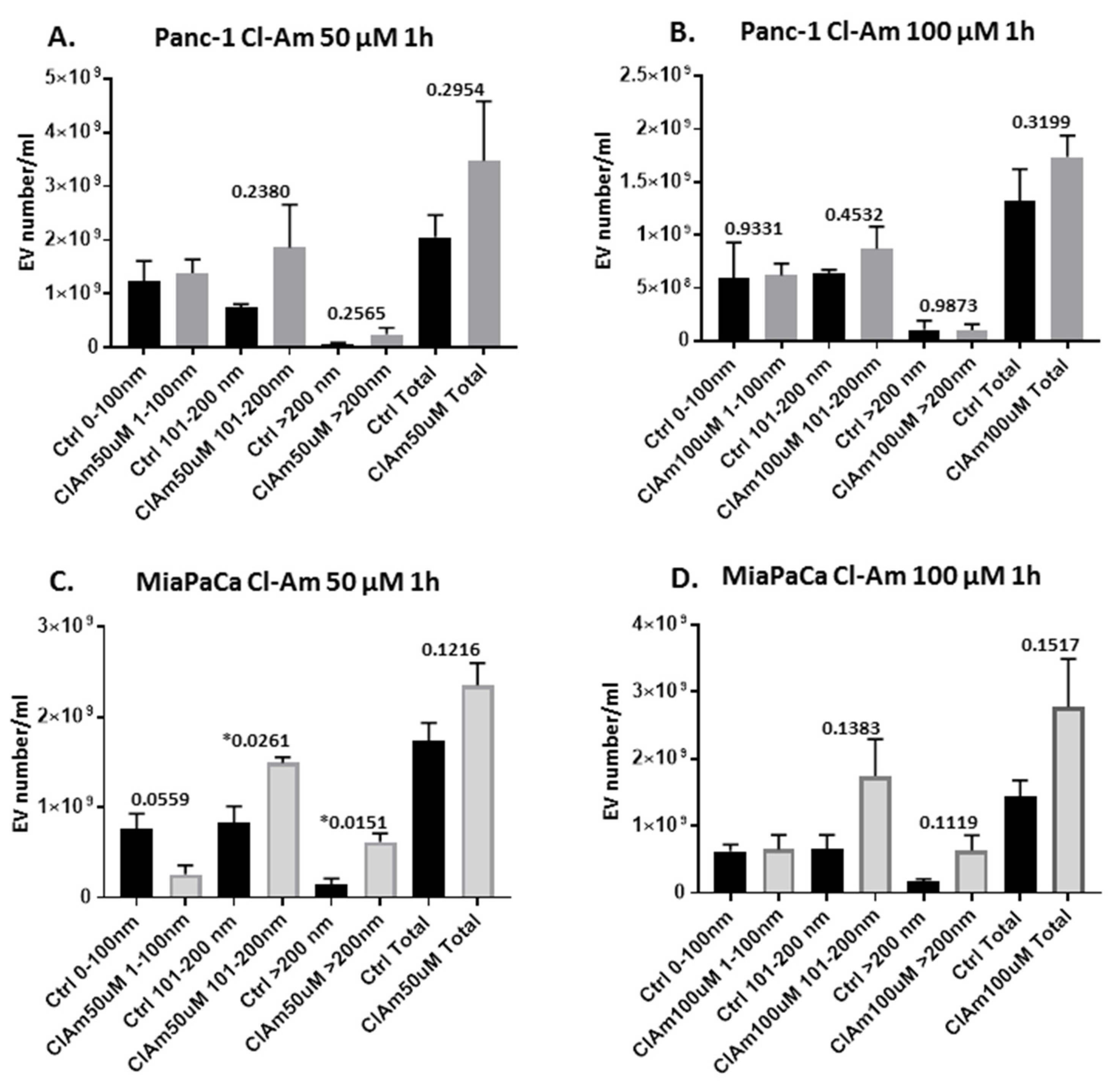
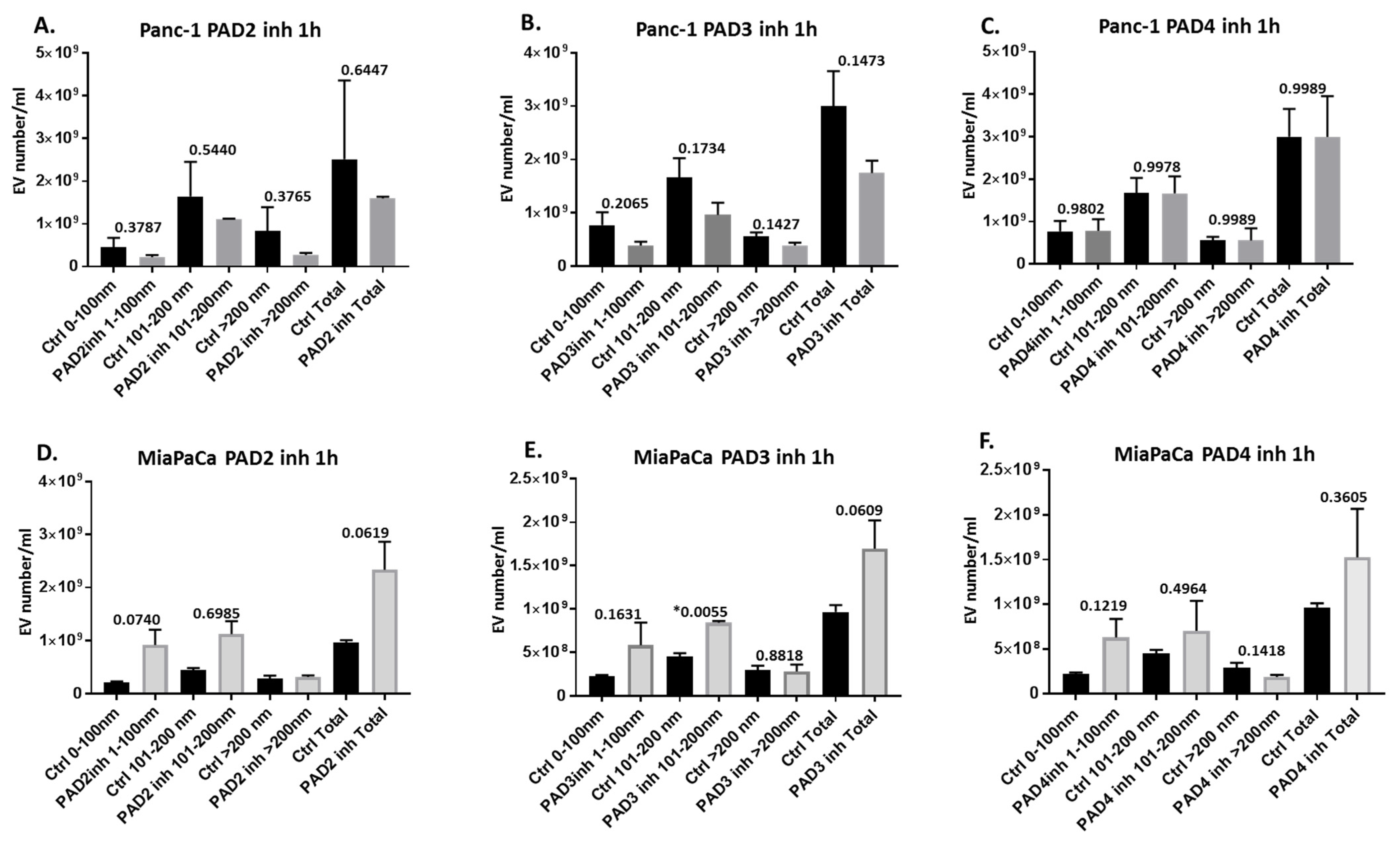
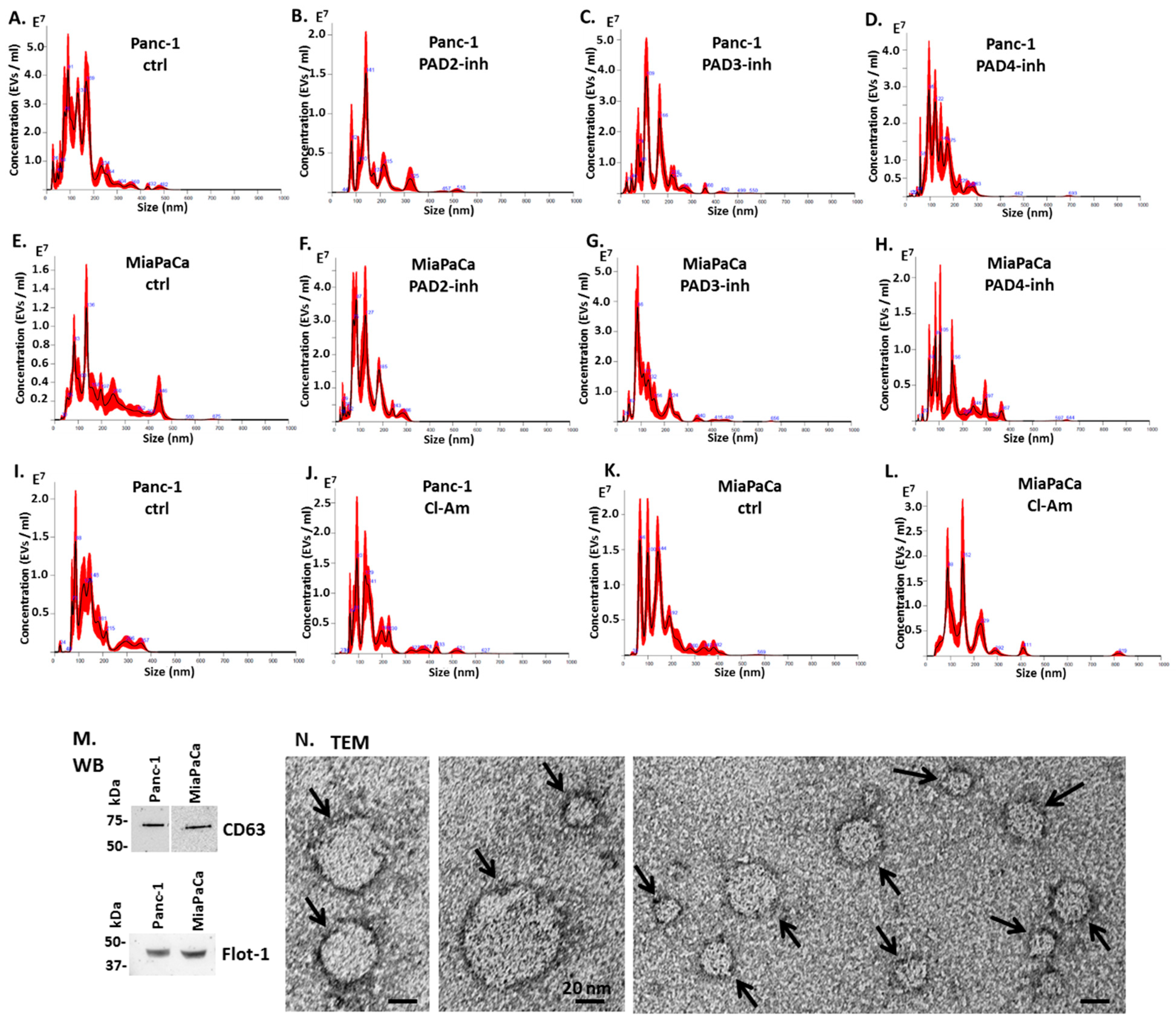
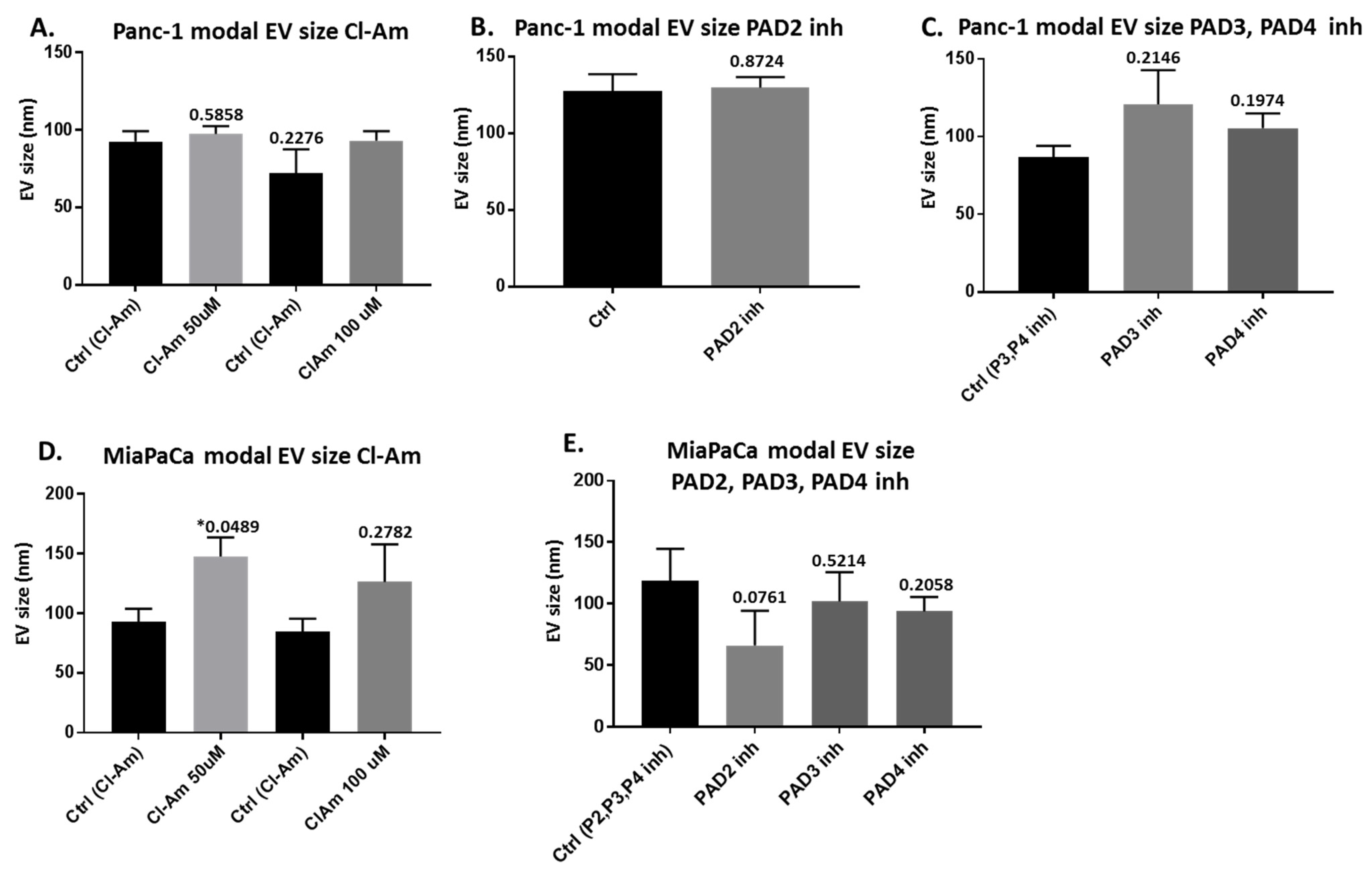
2.2. Pan-PAD and PAD Isozyme-Specific Inhibitors Differently Modulate EV Release in Pancreatic Cancer Cells Following 1 h Treatment
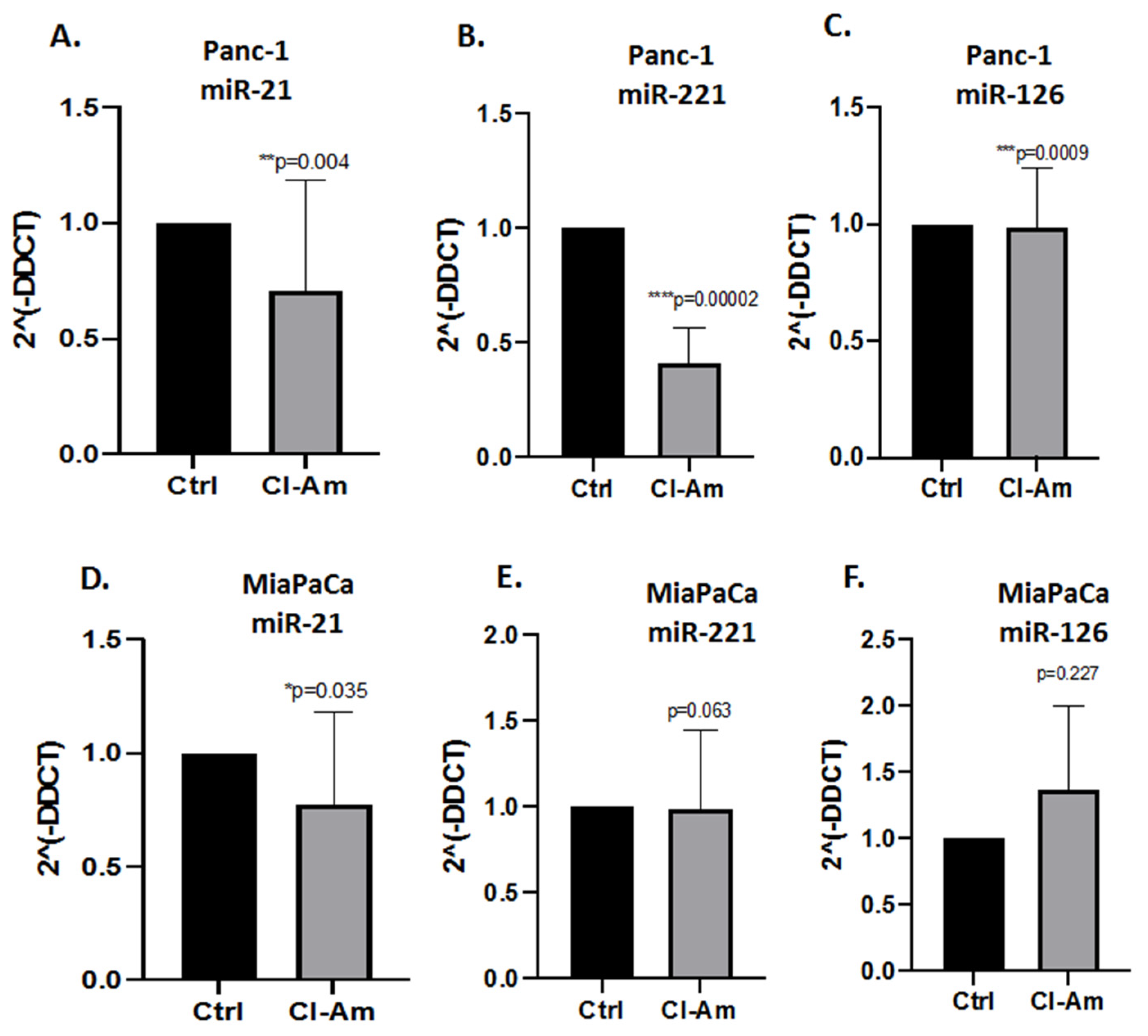
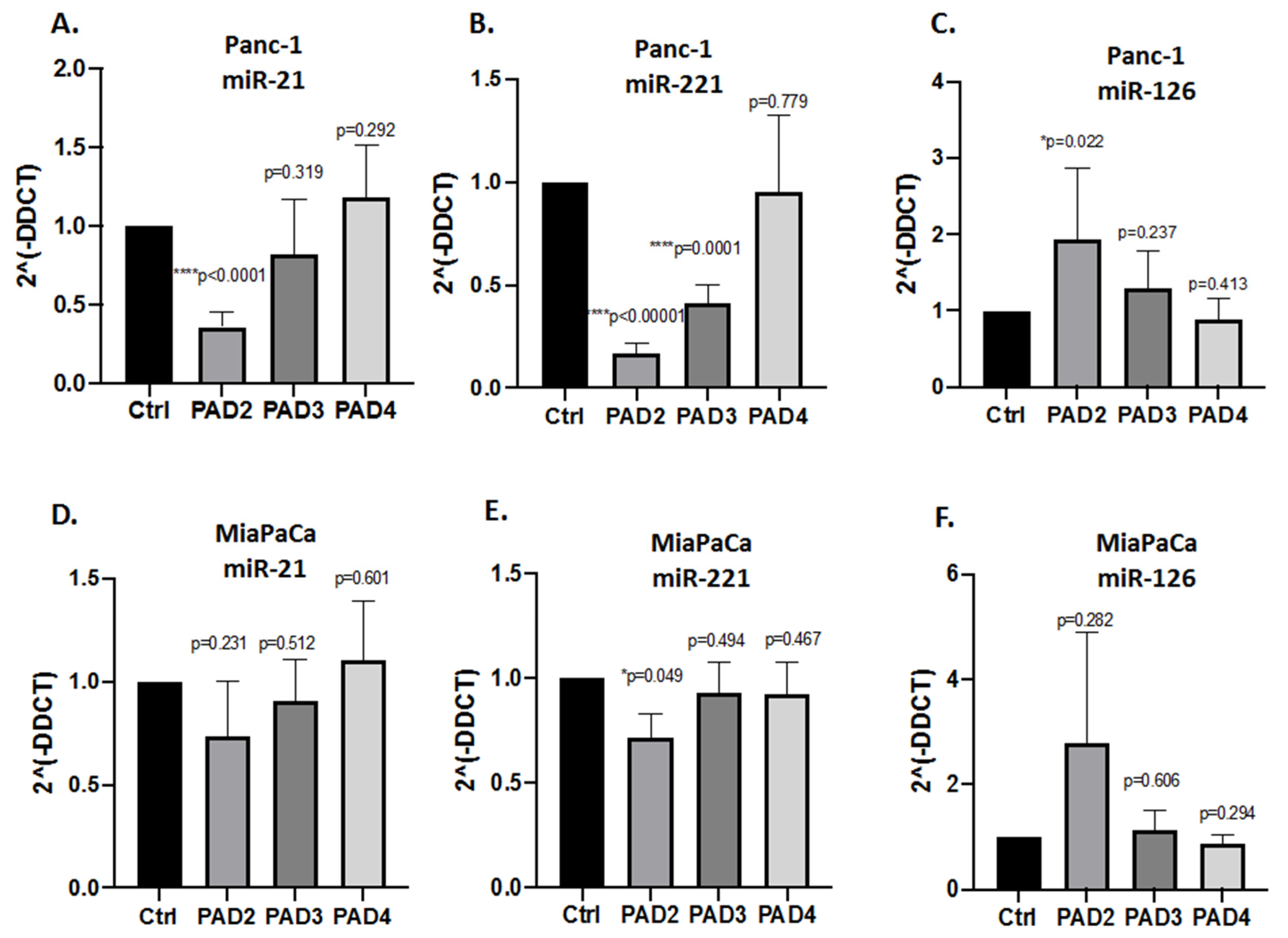
2.3. MicroRNA EV Cargo Is Differently Modulated in Response to 1 h PAD Inhibitor Treatments in PDAC Cells
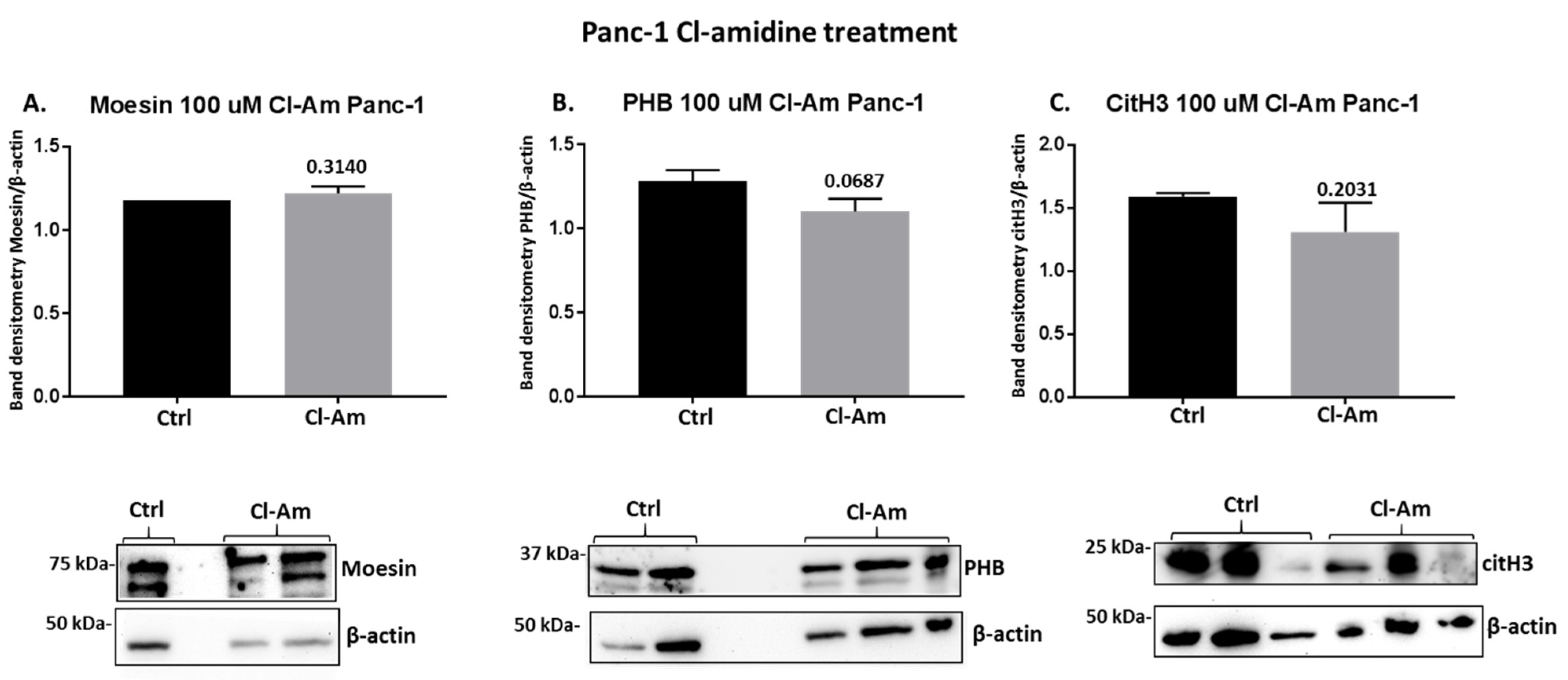
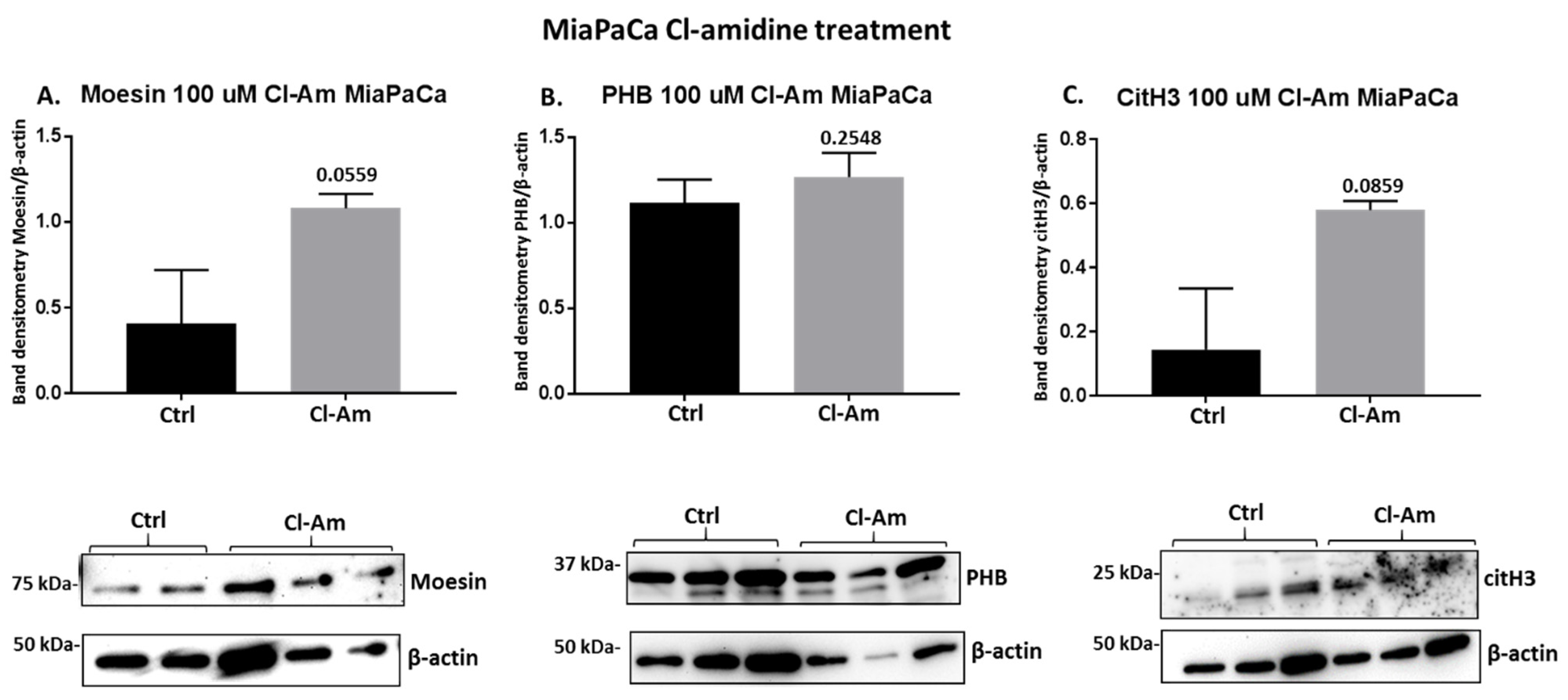
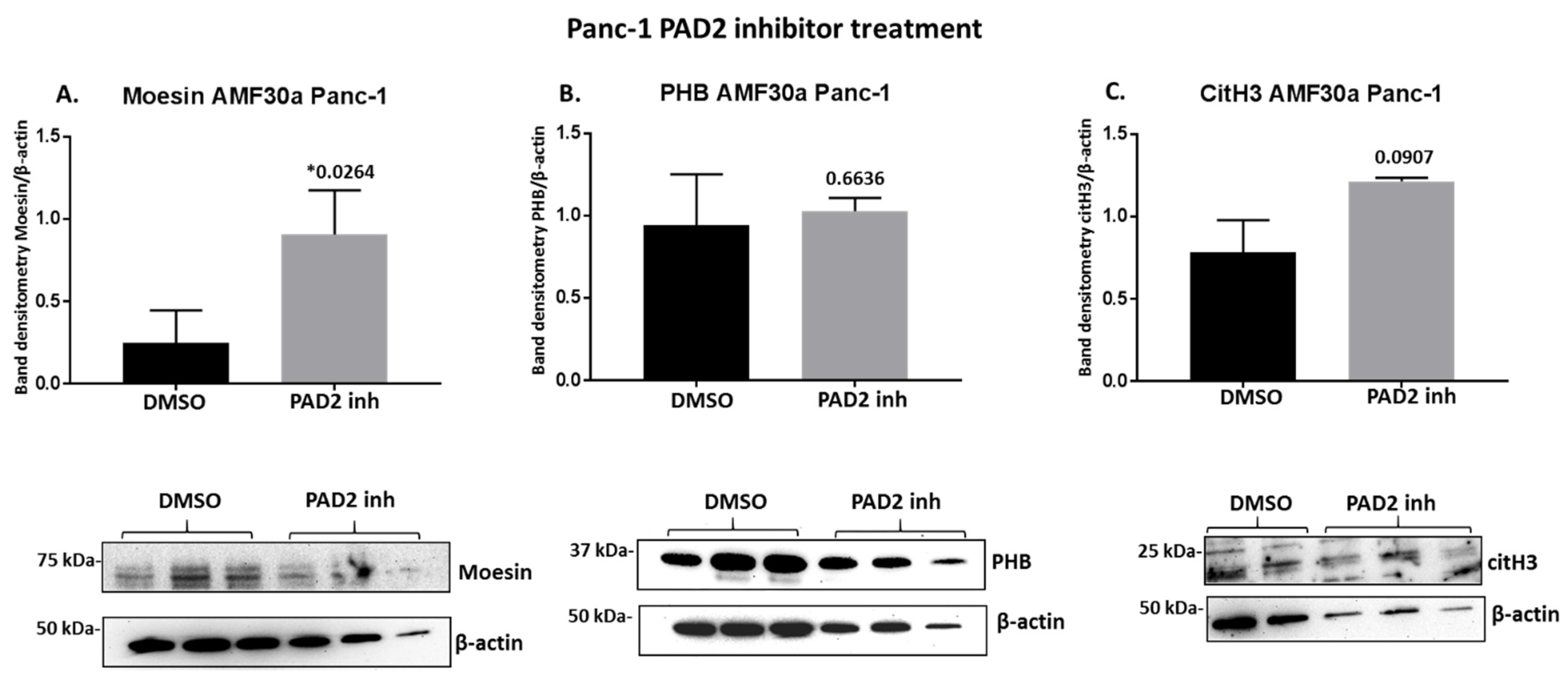
2.4. PAD Inhibitors Differently Affect Moesin, PHB, and Deiminated Histone H3 Protein Levels in Panc-1 Cells, Following 1 h Treatment
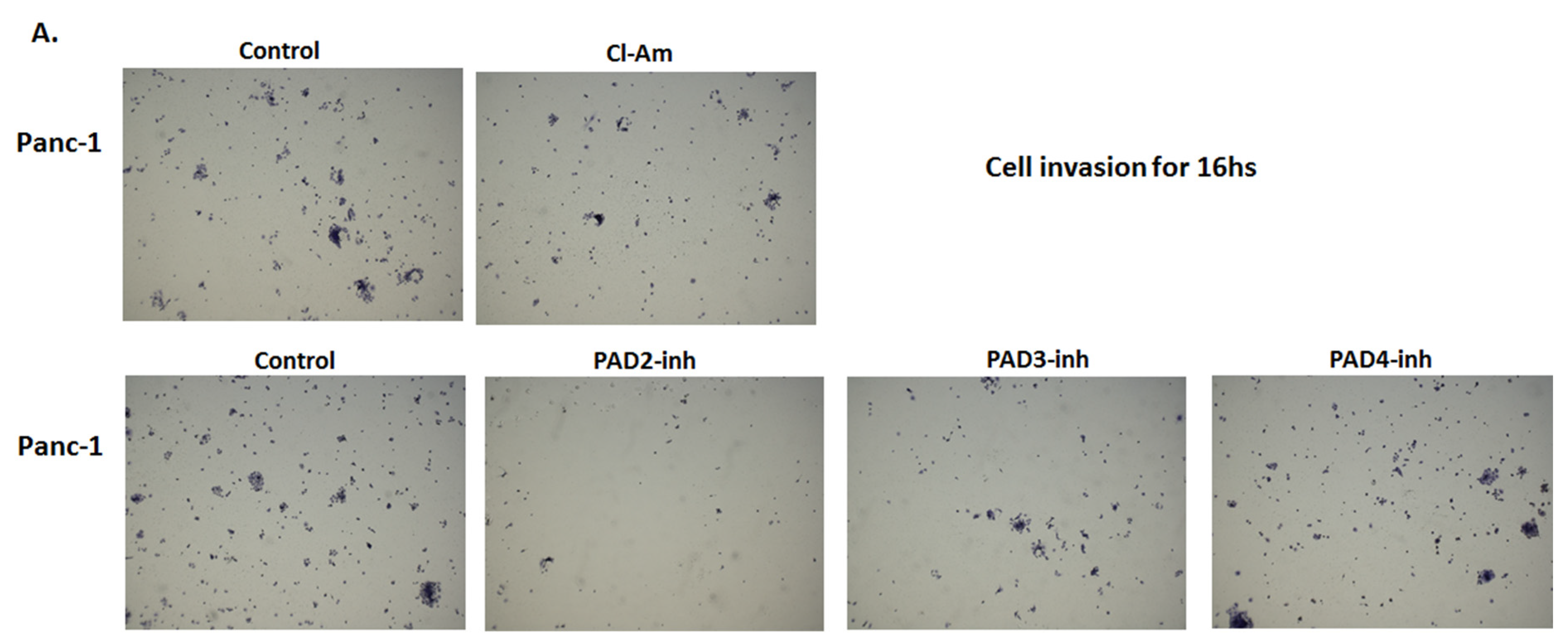
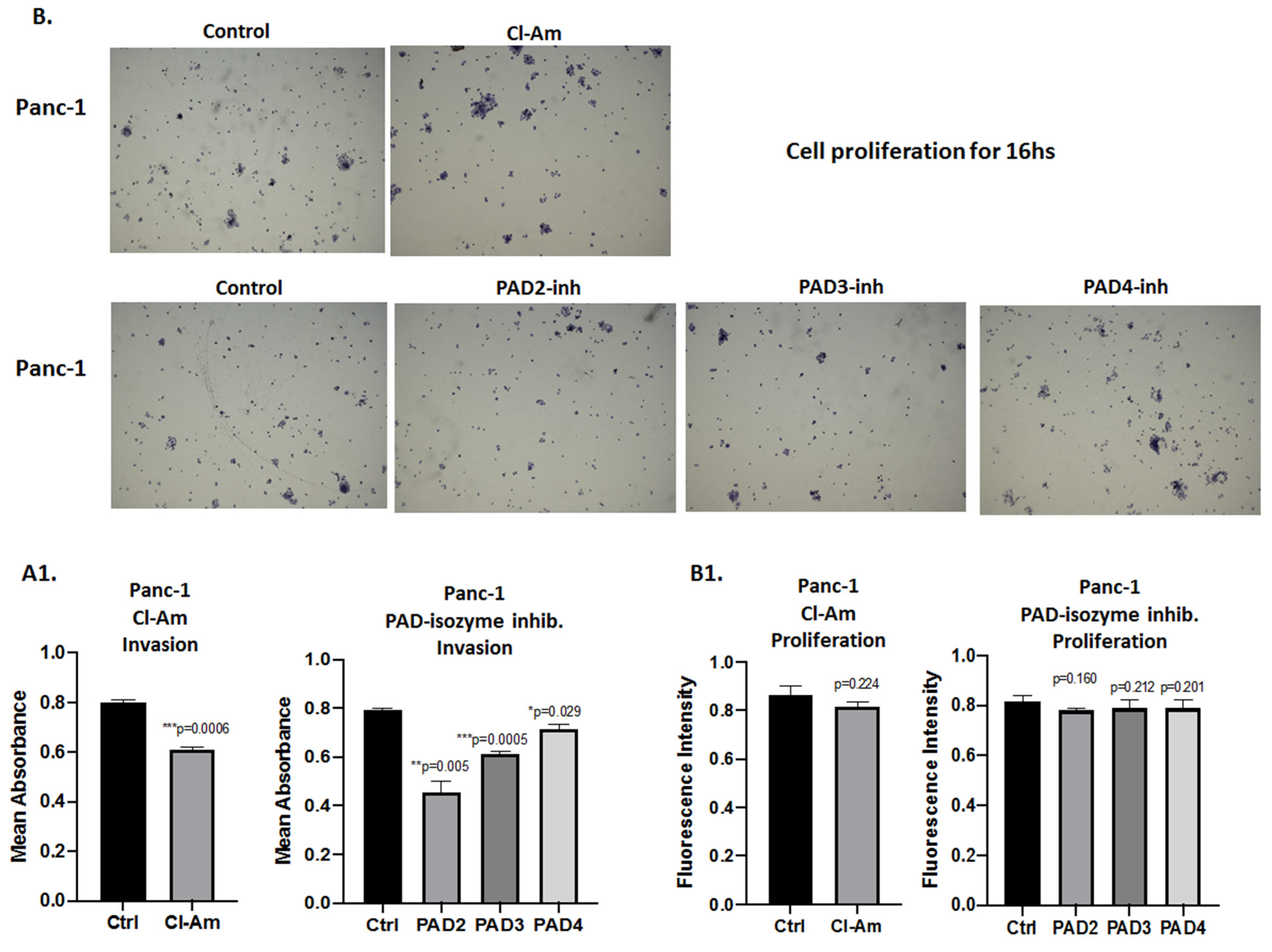
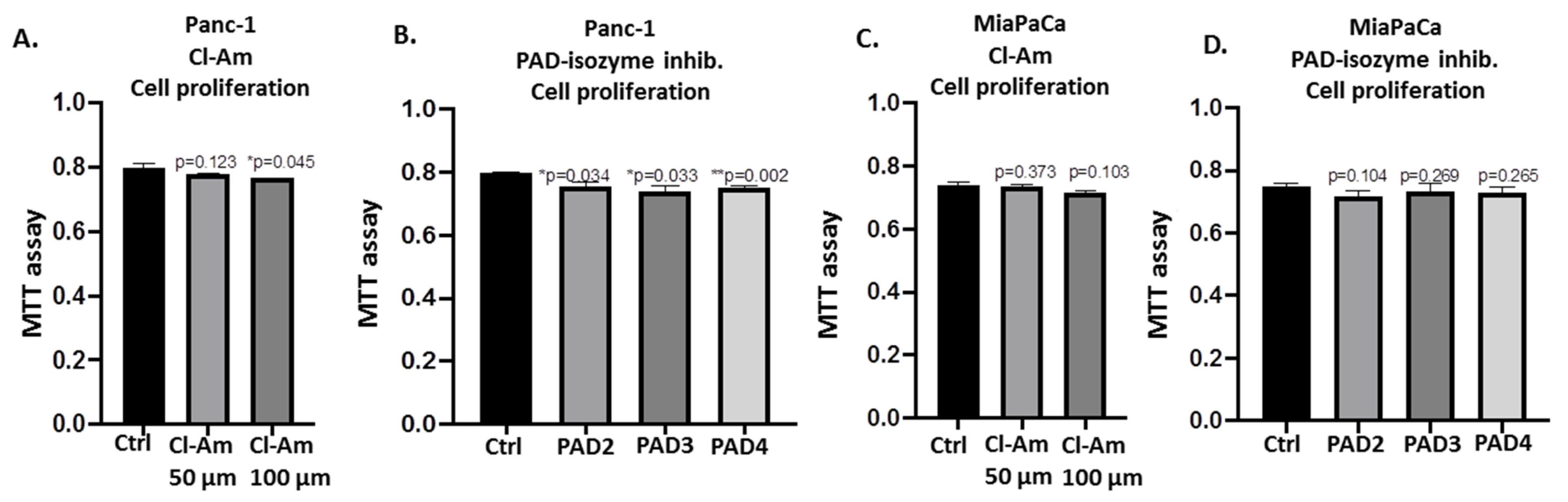
2.5. PAD Isozyme-Specific Inhibitors Differently Affect Panc-1 Cell Invasion, Independent of Cell Proliferation
3. Discussion
4. Materials and Methods
4.1. Pancreatic Cell Cultures and PAD Inhibitor Treatment
4.2. Cell Viability Assays Following PAD Inhibitor Treatment
4.3. Modulation of EV Release Using Cl-Amidine, PAD2, PAD3, and PAD4 Isozyme-Specific Inhibitors Following 1 h Treatment
4.4. EV Isolation and Quantification by Nanoparticle Tracking Analysis
4.5. EV Characterisation by Transmission Electron Microscopy
4.6. Analysis of MicroRNAs miR-21, miR-126, and miR-221 in EV Cargo Following 1 h PAD Inhibitor Treatment
4.7. Western Blotting Analysis
4.8. Cancer Cell Invasion Assay
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Von Hoff, D.D.; Korn, R.; Mousses, S. Pancreatic Cancer—Could It Be that Simple? A Different Context of Vulnerability. Cancer Cell 2009, 16, 7–8. [Google Scholar] [CrossRef][Green Version]
- Srivastava, S.K.; Arora, S.; Singh, S.; Bhardwaj, A.; Averett, C.; Singh, A.P. MicroRNAs in pancreatic malignancy: Progress and promises. Cancer Lett. 2014, 347, 167–174. [Google Scholar] [CrossRef]
- Abbruzzese, J.L. Adjuvant therapy for surgically resected pancreatic adenocarcinoma. JAMA 2008, 299, 1066–1067. [Google Scholar] [CrossRef]
- Scara, S.; Bottoni, P.; Scatena, R. CA 19-9: Biochemical and Clinical Aspects. Adv. Exp. Med. Biol. 2015, 867, 247–260. [Google Scholar] [CrossRef]
- Chan, A.; Prassas, I.; Dimitromanolakis, A.; Brand, R.E.; Serra, S.; Diamandis, E.P.; Blasutig, I.M. Validation of Biomarkers That Complement CA19.9 in Detecting Early Pancreatic Cancer. Clin. Cancer Res. 2014, 20, 5787–5795. [Google Scholar] [CrossRef]
- Vossenaar, E.R.; Zendman, A.J.; van Venrooij, W.J.; Pruijn, G.J.M. PAD, a growing family of citrullinating enzymes: Genes, features and involvement in disease. BioEssays 2003, 25, 1106–1118. [Google Scholar] [CrossRef] [PubMed]
- György, B.; Tóth, E.; Tarcsa, E.; Falus, A.; Buzás, E.I. Citrullination: A posttranslational modification in health and disease. Int. J. Biochem. Cell Biol. 2006, 38, 1662–1677. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, Y. Peptidylarginine deiminases in citrullination, gene regulation, health and pathogenesis. Biochim. Biophys. Acta Bioenerg. 2013, 1829, 1126–1135. [Google Scholar] [CrossRef] [PubMed]
- Henderson, B.; Martin, A.C.R. Protein moonlighting: A new factor in biology and medicine. Biochem. Soc. Trans. 2014, 42, 1671–1678. [Google Scholar] [CrossRef]
- Jeffrey, C.J. Protein moonlighting: What is it, and why is it important? Philos. Trans. R Soc. B Biol. Sci. 2018, 373, 20160523. [Google Scholar] [CrossRef]
- Kholia, S.; Jorfi, S.; Thompson, P.R.; Causey, C.P.; Nicholas, A.P.; Inal, J.; Lange, S. A Novel Role for Peptidylarginine Deiminases (PADs) in Microvesicle Release: A Therapeutic Potential for PAD Inhibitors to Sensitize Prostate Cancer Cells to Chemotherapy. J. Extracell. Vesicles. 2015, 4, 26192. [Google Scholar] [CrossRef] [PubMed]
- Kosgodage, U.S.; Trindade, R.P.; Thompson, P.R.; Inal, J.; Lange, S. Chloramidine/Bisindolylmaleimide-I-Mediated Inhibition of Exosome and Microvesicle Release and Enhanced Efficacy of Cancer Chemotherapy. Int. J. Mol. Sci. 2017, 18, 1007. [Google Scholar] [CrossRef] [PubMed]
- Lange, S.; Gallagher, M.; Kholia, S.; Kosgodage, U.S.; Hristova, M.; Hardy, J.J.; Inal, J. Peptidylarginine Deiminases—Roles in Cancer and Neurodegeneration and Possible Avenues for Therapeutic Intervention via Modulation of Exosome and Microvesicle (EMV) Release? Int. J. Mol. Sci. 2017, 18, 1196. [Google Scholar] [CrossRef] [PubMed]
- Witalison, P.R.; Thompson, P.R.; Hofseth, L.J. Protein Arginine Deiminases and Associated Citrullination: Physiological Functions and Diseases Associated with Dysregulation. Curr. Drug Targets 2015, 16, 700–710. [Google Scholar] [CrossRef]
- Kosgodage, U.S.; Uysal-Onganer, P.; MacLatchy, A.; Kraev, I.; Chatterton, N.P.; Nicholas, A.P.; Inal, J.M.; Lange, S. Pepti-dylarginine Deiminases Post-Translationally Deiminate Prohibitin and Modulate Extracellular Vesicle Release and MicroRNAs in Glioblastoma Multiforme. Int. J. Mol. Sci. 2018, 20, 103. [Google Scholar] [CrossRef]
- Kosgodage, U.S.; Matewele, P.; Mastroianni, G.; Kraev, I.; Brotherton, D.; Awamaria, B.; Nicholas, A.P.; Lange, S.; Inal, J. Peptidylarginine Deiminase Inhibitors Reduce Bacterial Membrane Vesicle Release and Sensitize Bacteria to Antibiotic Treatment. Front. Cell. Infect. Microbiol. 2019, 9, 227. [Google Scholar] [CrossRef]
- Uysal-Onganer, P.; MacLatchy, A.; Mahmoud, R.; Kraev, I.; Thompson, P.R.; Inal, J.; Lange, S. Peptidylarginine Deiminase Isozyme-Specific PAD2, PAD3 and PAD4 Inhibitors Differentially Modulate Extracellular Vesicle Signatures and Cell Invasion in Two Glioblastoma Multiforme Cell Lines. Int. J. Mol. Sci. 2020, 21, 1495. [Google Scholar] [CrossRef]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Tvall, J.O.L.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef]
- Fatima, F.; Nawaz, M. Vesiculated Long Non-Coding RNAs: Offshore Packages Deciphering Trans-Regulation between Cells, Cancer Progression and Resistance to Therapies. Non-Coding RNA 2017, 3, 10. [Google Scholar] [CrossRef]
- Ma, P.; Pan, Y.; Li, W.; Sun, C.; Liu, J.; Xu, T.; Shu, Y.-Q. Extracellular vesicles-mediated noncoding RNAs transfer in cancer. J. Hematol. Oncol. 2017, 10, 1–11. [Google Scholar] [CrossRef]
- Moeng, S.; Son, S.W.; Lee, J.S.; Lee, H.Y.; Kim, T.H.; Choi, S.Y.; Kuh, H.-J.; Park, J.K. Extracellular Vehicles (EVs) and Pancreatic Cancer: From the Role of EVs to the Interference with EV-Mediated Reciprocal Communication. Biomedicines 2020, 8, 267. [Google Scholar] [CrossRef]
- Qiu, J.; Yang, G.; Feng, M.; Zheng, S.; Cao, Z.; You, L.; Zheng, L.; Zhang, T.; Zhao, Y. Extracellular vesicles as mediators of the progression and chemoresistance of pancreatic cancer and their potential clinical applications. Mol. Cancer 2018, 17, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.; Thakur, B.K.; Weiss, J.M.; Kim, H.S.; Peinado, H.; Lyden, D. Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Metastasis. Cancer Cell 2016, 30, 836–848. [Google Scholar] [CrossRef]
- Clancy, J.; D’Souza-Schorey, C. Extracellular Vesicles in Cancer: Purpose and Promise. Cancer J. 2018, 24, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Bebelman, M.P.; Smit, M.J.; Pegtel, D.M.; Baglio, S.R. Biogenesis and function of extracellular vesicles in cancer. Pharmacol. Ther. 2018, 188, 1–11. [Google Scholar] [CrossRef]
- Kosgodage, U.S.; Mould, R.; Henley, A.B.; Nunn, A.V.; Guy, G.W.; Thomas, E.L.; Inal, J.M.; Bell, J.D.; Lange, S. Cannabidiol (CBD) Is a Novel Inhibitor for Exosome and Microvesicle (EMV) Release in Cancer. Front. Pharmacol. 2018, 9, 889. [Google Scholar] [CrossRef] [PubMed]
- Kosgodage, U.S.; Uysal-Onganer, P.; MacLatchy, A.; Mould, R.; Nunn, A.V.; Guy, G.W.; Kraev, I.; Chatterton, N.P.; Thomas, E.L.; Inal, J.; et al. Cannabidiol Affects Extracellular Vesicle Release, miR21 and miR126, and Reduces Prohibitin Protein in Glioblastoma Multiforme Cells. Transl. Oncol. 2019, 12, 513–522. [Google Scholar] [CrossRef]
- Federici, C.; Petrucci, F.; Caimi, S.; Cesolini, A.; Logozzi, M.; Borghi, M.; D’Ilio, S.; Lugini, L.; Violante, N.; Azzarito, T.; et al. Exosome release and low pH belong to a framework of resistance of human melanoma cells to cisplatin. PLoS ONE 2014, 9, e88193. [Google Scholar] [CrossRef]
- Jorfi, S.; Ansa-Addo, E.A.; Kholia, S.; Stratton, D.; Valley, S.; Lange, S.; Inal, J. Inhibition of microvesiculation sensitizes prostate cancer cells to chemotherapy and reduces docetaxel dose required to limit tumor growth in vivo. Sci. Rep. 2015, 5, 13006. [Google Scholar] [CrossRef]
- Koch, R.; Aung, T.; Vogel, D.; Chapuy, B.; Wenzel, D.; Becker, S.; Sinzig, U.; Venkataramani, V.; Von Mach, T.; Jacob, R.; et al. Nuclear Trapping through Inhibition of Exosomal Export by Indomethacin Increases Cytostatic Efficacy of Doxorubicin and Pixantrone. Clin. Cancer Res. 2016, 22, 395–404. [Google Scholar] [CrossRef]
- Muralidharan-Chari, V.; Kohan, H.G.; Asimakopoulos, A.G.; Sudha, T.; Sell, S.; Kannan, K.; Boroujerdi, M.; Davis, P.J.; Mousa, S.A. Microvesicle removal of anticancer drugs contributes to drug resistance in human pancreatic cancer cells. Oncotarget 2016, 7, 50365–50379. [Google Scholar] [CrossRef] [PubMed]
- Maacha, S.; Bhat, A.A.; Jimenez, L.; Raza, A.; Haris, M.; Uddin, S.; Grivel, J.-C. Extracellular vesicles-mediated intercellular communication: Roles in the tumor microenvironment and anti-cancer drug resistance. Mol. Cancer 2019, 18, 1–16. [Google Scholar] [CrossRef]
- Catalano, M.; O’Driscoll, L. Inhibiting extracellular vesicles formation and release: A review of EV inhibitors. J. Extracell. Vesicles 2020, 9, 1703244. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Gilles, M.-E.; Hao, L.; Huang, L.; Rupaimoole, R.; Lopez-Casas, P.P.; Pulver, E.; Jeong, J.C.; Muthuswamy, S.K.; Hidalgo, M.; Bhatia, S.N.; et al. Personalized RNA Medicine for Pancreatic Cancer. Clin. Cancer Res. 2018, 24, 1734–1747. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.R.; Frampton, A.E.; Jacob, J.; Pellegrino, L.; Krell, J.; Giamas, G.; Tsim, N.; Vlavianos, P.; Cohen, P.; Ahmad, R.; et al. MicroRNAs targeting oncogenes are down-regulated in pancreatic malignant transfor-mation from benign tumours. PLoS ONE 2012, 7, e32068. [Google Scholar] [CrossRef]
- Vorvis, C.; Koutsioumpa, M.; Iliopoulos, D. Developments in miRNA gene signaling pathways in pancreatic cancer. Futur. Oncol. 2016, 12, 1135–1150. [Google Scholar] [CrossRef]
- Frezzetti, D.; de Menna, M.; Zoppoli, P.; Guerra, C.; Ferraro, A.; Bello, A.M.; de Luca, P.; Calabrese, C.; Fusco, A.; Ceccarelli, M.; et al. Upregulation of miR-21 by Ras in vivo and its role in tumor growth. Oncogene 2010, 30, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.-N.; Ma, Y.-H.; Zhao, W.-G.; Jin, X.-L.; Yang, H.-Y.; Liu, P.-P.; Chen, J. KRAS-related noncoding RNAs in pancreatic ductal adenocarcinoma. Chronic Dis. Transl. Med. 2016, 2, 215–222. [Google Scholar] [CrossRef]
- Wang, S.; Lin, R.J. MicroRNA and HER2-overexpressing cancer. Microrna 2013, 2, 137–147. [Google Scholar] [CrossRef]
- Garajová, I.; le Large, T.Y.; Frampton, A.E.; Rolfo, C.; Voortman, J.; Giovannetti, E. Molecular Mechanisms Underlying the Role of MicroRNAs in the Chemoresistance of Pancreatic Cancer. BioMed Res. Int. 2014, 2014, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Komoto, M.; Nakata, B.; Amano, R.; Yamada, N.; Yashiro, M.; Ohira, M.; Wakasa, K.; Hirakawa, K. HER2 overexpression correlates with survival after curative resection of pancreatic cancer. Cancer Sci. 2009, 100, 1243–1247. [Google Scholar] [CrossRef] [PubMed]
- Mosquera, C.; Maglic, D.; Zervos, E. Molecular targeted therapy for pancreatic adenocarcinoma: A review of completed and ongoing late phase clinical trials. Cancer Genet. 2016, 209, 567–581. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A.; Lee, H.Y.; Lee, E.S.; Kim, I.; Bae, J.W. Prognostic Implications of MicroRNA-21 Overexpression in Invasive Ductal Carcinomas of the Breast. J. Breast Cancer 2011, 14, 269–275. [Google Scholar] [CrossRef]
- Collisson, E.A.; Trejo, C.L.; Silva, J.M.; Gu, S.; Korkola, J.E.; Heiser, L.M.; Charles, R.P.; Rabinovich, B.A.; Hann, B.; Dankort, D.; et al. A central role for RAF→MEK→ERK signaling in the genesis of pan-creatic ductal adenocarcinoma. Cancer Discov. 2012, 2, 685–693. [Google Scholar] [CrossRef]
- Zhao, Q.; Chen, S.; Zhu, Z.; Yu, L.; Ren, Y.; Jiang, M.; Weng, J.; Li, B. miR-21 promotes EGF-induced pancreatic cancer cell proliferation by targeting Spry2. Cell Death Dis. 2018, 9, 1157. [Google Scholar] [CrossRef]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef]
- Yachida, S.; Fu, B.; Yachida, S.; Luo, M.; Abe, H.; Henderson, C.M.; Vilardell, F.; Wang, Z.; Keller, J.W.; Banerjee, P.; et al. DPC4 Gene Status of the Primary Carcinoma Correlates with Patterns of Failure in Patients with Pancreatic Cancer. J. Clin. Oncol. 2009, 27, 1806–1813. [Google Scholar] [CrossRef]
- Truty, M.J.; Urrutia, R. Basics of TGF-beta and pancreatic cancer. Pancreatology 2007, 7, 423–435. [Google Scholar] [CrossRef]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic Adenocarcinoma. New Engl. J. Med. 2014, 371, 1039–1049. [Google Scholar] [CrossRef]
- Bloomston, M.; Frankel, W.L.; Petrocca, F.; Volinia, S.; Alder, H.; Hagan, J.P.; Liu, C.-G.; Bhatt, D.; Taccioli, C.; Croce, C.M. MicroRNA Expression Patterns to Differentiate Pancreatic Adenocarcinoma from Normal Pancreas and Chronic Pancreatitis. JAMA 2007, 297, 1901–1908. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, M.; Wang, H.; Fisher, W.E.; Lin, P.H.; Yao, Q.; Chen, C. Profiling of 95 MicroRNAs in Pancreatic Cancer Cell Lines and Surgical Specimens by Real-Time PCR Analysis. World J. Surg. 2008, 33, 698–709. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Yang, Y.; Xia, L.; Yang, Y.; Wang, F.; Song, M.; Chen, X.; Liu, J.; Song, Y.; Zhao, Y.; et al. MiR-221 Promotes Capan-2 Pancreatic Ductal Adenocarcinoma Cells Proliferation by Targeting PTEN-Akt. Cell. Physiol. Biochem. 2016, 38, 2366–2374. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Liu, J.; Wang, L.; Wu, H.; Zhou, C.; Zhu, H.; Xu, N.; Xie, Y. Association of Serum MicroRNA Expression in Hepatocellular Carcinomas Treated with Transarterial Chemoembolization and Patient Survival. PLoS ONE 2014, 9, e109347. [Google Scholar] [CrossRef] [PubMed]
- Tao, K.; Yang, J.; Guo, Z.; Hu, Y.; Sheng, H.; Gao, H.; Yu, H. Prognostic value of mir-221-3p, mir-342-3p and mir-491-5p ex-pression in colon cancer. Am. J. Transl. Res. 2014, 6, 391–401. [Google Scholar]
- Yau, T.O.; Wu, C.W.; Dong, Y.; Tang, C.-M.; Ng, S.S.M.; Chan, F.K.L.; Sung, J.J.Y.; Yu, J. microRNA-221 and microRNA-18a identification in stool as potential biomarkers for the non-invasive diagnosis of colorectal carcinoma. Br. J. Cancer 2014, 111, 1765–1771. [Google Scholar] [CrossRef]
- Zheng, Q.; Peskoe, S.B.; Ribas, J.; Rafiqi, F.; Kudrolli, T.; Meeker, A.K.; de Marzo, A.M.; Platz, E.A.; Lupold, S.E. Investigation of mir-21, mir-141, and mir-221 expression levels in prostate adenocarcinoma for associated risk of recurrence after radical prosta-tectomy. Prostate 2014, 74, 1655–1662. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Komatsu, S.; Ichikawa, D.; Morimura, R.; Tsujiura, M.; Konishi, H.; Takeshita, H.; Nagata, H.; Arita, T.; Hirajima, S.; et al. Clinical impact of circulating miR-221 in plasma of patients with pancreatic cancer. Br. J. Cancer 2013, 108, 361–369. [Google Scholar] [CrossRef]
- Su, A.; He, S.; Tian, B.; Hu, W.; Zhang, Z. MicroRNA-221 Mediates the Effects of PDGF-BB on Migration, Proliferation, and the Epithelial-Mesenchymal Transition in Pancreatic Cancer Cells. PLoS ONE 2013, 8, e71309. [Google Scholar] [CrossRef]
- Masamune, A.; Nakano, E.; Hamada, S.; Takikawa, T.; Yoshida, N.; Shimosegawa, T. Alteration of the microRNA expression profile during the activation of pancreatic stellate cells. Scand. J. Gastroenterol. 2013, 49, 323–331. [Google Scholar] [CrossRef]
- Gallop, J.L. Filopodia and their links with membrane traffic and cell adhesion. Semin. Cell Dev. Biol. 2020, 102, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Abiatari, I.; Esposito, I.; de Oliveira, T.; Felix, K.; Xin, H.; Penzel, R.; Giese, T.; Friess, H.; Kleeff, J. Moesin-dependent cytoskeleton remodelling is associated with an anaplastic phenotype of pancreatic cancer. J. Cell. Mol. Med. 2009, 14, 1166–1179. [Google Scholar] [CrossRef] [PubMed]
- Törer, N.; Kayaselçuk, F.; Nursal, T.Z.; Yildirim, S.; Tarim, A.; Noyan, T.; Karakayali, H. Adhesion molecules as prognostic markers in pancreatic adenocarcinoma. J. Surg. Oncol. 2007, 96, 419–423. [Google Scholar] [CrossRef]
- Liang, L.; Dong, M.; Cong, K.; Chen, Y.; Ma, Z. Correlations of Moesin expression with the pathological stage, nerve infiltration, tumor location and pain severity in patients with pancreatic cancer. J. BUON 2019, 24, 1225–1232. [Google Scholar] [PubMed]
- Cui, Y.; Wu, J.; Zong, M.; Song, G.; Jia, Q.; Jiang, J.; Han, J. Proteomic profiling in pancreatic cancer with and without lymph node metastasis. Int. J. Cancer 2009, 124, 1614–1621. [Google Scholar] [CrossRef]
- Peng, Y.-T.; Chen, P.; Ouyang, R.-Y.; Song, L. Multifaceted role of prohibitin in cell survival and apoptosis. Apoptosis 2015, 20, 1135–1149. [Google Scholar] [CrossRef]
- Hernando-Rodríguez, B.; Artal-Sanz, M. Mitochondrial Quality Control Mechanisms and the PHB (Prohibitin) Complex. Cells 2018, 7, 238. [Google Scholar] [CrossRef]
- Signorile, A.; Sgaramella, G.; Bellomo, F.; de Rasmo, D. Prohibitins: A Critical Role in Mitochondrial Functions and Implication in Diseases. Cells 2019, 8, 71. [Google Scholar] [CrossRef]
- Wang, W.; Xu, L.; Yang, Y.; Dong, L.; Zhao, B.; Lu, J.; Zhang, T.; Zhao, Y. A novel prognostic marker and immunogenic mem-brane antigen: Prohibitin (PHB) in pancreatic cancer. Clin. Transl. Gastroenterol. 2018, 9, 178. [Google Scholar] [CrossRef]
- Lange, S.; Gögel, S.; Leung, K.-Y.; Vernay, B.; Nicholas, A.P.; Causey, C.P.; Thompson, P.R.; Greene, N.D.; Ferretti, P. Protein deiminases: New players in the developmentally regulated loss of neural regenerative ability. Dev. Biol. 2011, 355, 205–214. [Google Scholar] [CrossRef]
- Lange, S.; Rocha-Ferreira, E.; Thei, L.; Mawjee, P.; Bennett, K.; Thompson, P.R.; Subramanian, V.; Nicholas, A.P.; Peebles, D.; Hristova, M.; et al. Peptidylarginine deiminases: Novel drug targets for prevention of neuronal damage following hypoxic ischemic insult (HI) in neonates. J. Neurochem. 2014, 130, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Lange, S. Peptidylarginine deiminases and extracellular vesicles: Prospective drug targets and biomarkers in central nervous system diseases and repair. Neural Regen. Res. 2021, 16, 934–938. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Arita, K.; Bhatia, M.; Knuckley, B.; Lee, Y.-H.; Stallcup, M.R.; Sato, M.; Thompson, P.R. Inhibitors and Inactivators of Protein Arginine Deiminase 4: Functional and Structural Characterization. Biochemistry 2006, 45, 11727–11736. [Google Scholar] [CrossRef] [PubMed]
- Knuckley, B.; Causey, C.P.; Jones, J.E.; Bhatia, M.; Dreyton, C.J.; Osborne, T.C.; Takahara, H.; Thompson, P.R. Substrate Specificity and Kinetic Studies of PADs 1, 3, and 4 Identify Potent and Selective Inhibitors of Protein Arginine Deiminase 3. Biochemistry 2010, 49, 4852–4863. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ma, L.; Sui, Y.; Chang, X.-T. Screening of citrullinated proteins in ten tumor cell lines. Zhonghua Zhong Liu Za Zhi Chin. J. Oncol. 2015, 37, 165–169. [Google Scholar]
- Hisada, Y.; Grover, S.P.; Maqsood, A.; Houston, R.; Ay, C.; Noubouossie, D.F.; Cooley, B.C.; Wallén, H.; Key, N.S.; Thålin, C.; et al. Neutrophils and neutrophil extracellular traps enhance venous thrombosis in mice bearing human pancreatic tumors. Haematologica 2019, 105, 218–225. [Google Scholar] [CrossRef]
- Hisada, Y.; Mackman, N. Update from the laboratory: Mechanistic studies of pathways of cancer-associated venous thrombosis using mouse models. Hematology 2019, 2019, 182–186. [Google Scholar] [CrossRef]
- Jin, W.; Xu, H.X.; Zhang, S.R.; Li, H.; Wang, W.Q.; Gao, H.L.; Wu, C.T.; Xu, J.Z.; Qi, Z.H.; Li, S.; et al. Tu-mor-Infiltrating NETs Predict Postsurgical Survival in Patients with Pancreatic Ductal Adenocarcinoma. Ann Surg Oncol. 2019, 26, 635–643. [Google Scholar] [CrossRef]
- Cook, K.; Daniels, I.; Symonds, P.; Pitt, T.; Gijon, M.; Xue, W.; Metheringham, R.; Durrant, L.G.; Brentville, V. Citrullinated α-enolase is an effective target for anti-cancer immunity. OncoImmunology 2018, 7, e1390642. [Google Scholar] [CrossRef]
- Kumar, S.; Sethi, S.; Irani, F.; Bode, B.Y. Anticyclic Citrullinated Peptide Antibody-Positive Paraneoplastic Polyarthritis in a Patient With Metastatic Pancreatic Cancer. Am. J. Med. Sci. 2009, 338, 511–512. [Google Scholar] [CrossRef]
- Öğrendik, M. Oral bacteria in pancreatic cancer: Mutagenesis of the p53 tumour suppressor gene. Int. J. Clin. Exp. Pathol. 2015, 8, 11835–11836. [Google Scholar] [PubMed]
- Yu, J.; Li, A.; Hong, S.-M.; Hruban, R.H.; Goggins, M. MicroRNA Alterations of Pancreatic Intraepithelial Neoplasias. Clin. Cancer Res. 2012, 18, 981–992. [Google Scholar] [CrossRef] [PubMed]
- Krichevsky, A.M.; Gabriely, G. miR-21: A small multi-faceted RNA. J. Cell. Mol. Med. 2008, 13, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-K.; Lee, E.J.; Esau, C.; Schmittgen, T.D. Antisense Inhibition of microRNA-21 or -221 Arrests Cell Cycle, Induces Apoptosis, and Sensitizes the Effects of Gemcitabine in Pancreatic Adenocarcinoma. Pancreas 2009, 38, e190–e199. [Google Scholar] [CrossRef] [PubMed]
- Chu, I.M.; Hengst, L.; Slingerland, J.M. The Cdk inhibitor p27 in human cancer: Prognostic potential and relevance to anticancer therapy. Nat. Rev. Cancer 2008, 8, 253–267. [Google Scholar] [CrossRef]
- Le Sage, C.; Nagel, R.; Egan, D.A.; Schrier, M.; Mesman, E.; Mangiola, A.; Anile, C.; Maira, G.; Mercatelli, N.; Ciafrè, S.A.; et al. Regulation of the p27Kip1 tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J. 2007, 26, 3699–3708. [Google Scholar] [CrossRef]
- Almas, I.; Afzal, S.; Idrees, M.; Ashraf, M.U.; Amin, I.; Shahid, M.; Zahid, K.; Zahid, S. Role of circulatory microRNAs in the pathogenesis of hepatitis C virus. Virus Disease 2017, 28, 360–367. [Google Scholar] [CrossRef]
- Feng, R.; Chen, X.; Yu, Y.; Su, L.; Yu, B.; Li, J.; Cai, Q.; Yan, M.; Liu, B.; Zhu, Z. miR-126 functions as a tumour suppressor in human gastric cancer. Cancer Lett. 2010, 298, 50–63. [Google Scholar] [CrossRef]
- Grutzmann, R.; Luttges, J.; Sipos, B.; Ammerpohl, O.; Dobrowolski, F.; Alldinger, I.; Kersting, S.; Ockert, D.; Koch, R.M.; Kalthoff, H.; et al. ADAM9 expression in pancreatic cancer is associated with tumour type and is a prognostic factor in ductal adenocarcinoma. Br. J. Cancer 2004, 90, 1053–1058. [Google Scholar] [CrossRef]
- Luan, Z.; He, Y.; Alattar, M.; Chen, Z.; He, F. Targeting the prohibitin scaffold-CRAF kinase interaction in RAS-ERK-driven pancreatic ductal adenocarcinoma. Mol. Cancer 2014, 13, 38. [Google Scholar] [CrossRef]
- Cherrington, B.D.; Zhang, X.; McElwee, J.L.; Morency, E.; Anguish, L.J.; Coonrod, S.A. Potential Role for PAD2 in Gene Regulation in Breast Cancer Cells. PLoS ONE 2012, 7, e41242. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, V.; Nicholas, A.P.; Thompson, P.R.; Ferretti, P. Modulation of calcium-induced cell death in human neural stem cells by the novel peptidylarginine deiminase–AIF pathway. Biochim. Biophys. Acta Bioenerg. 2014, 1843, 1162–1171. [Google Scholar] [CrossRef]
- Watanabe, K.; Akiyama, K.; Hikichi, K.; Ohtsuka, R.; Okuyama, A.; Senshu, T. Combined biochemical and immunochemical comparison of peptidylarginine deiminases present in various tissues. Biochim. Biophys. Acta Gen. Subj. 1988, 966, 375–383. [Google Scholar] [CrossRef]
- McElwee, J.L.; Mohanan, S.; Griffith, O.L.; Breuer, H.C.; Anguish, L.J.; Cherrington, B.D.; Palmer, A.M.; Howe, L.R.; Subramanian, V.; Causey, C.P.; et al. Identification of PADI2 as a potential breast cancer biomarker and therapeutic target. BMC Cancer 2012, 12, 500. [Google Scholar] [CrossRef]
- Cuthbert, G.L.; Daujat, S.; Snowden, A.W.; Erdjument-Bromage, H.; Hagiwara, T.; Yamada, M.; Schneider, R.; Gregory, P.D.; Tempst, P.; Bannister, A.J.; et al. Histone Deimination Antagonizes Arginine Methylation. Cell 2004, 118, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bolt, M.; Guertin, M.J.; Chen, W.; Zhang, S.; Cherrington, B.D.; Slade, D.J.; Dreyton, C.J.; Subramanian, V.; Bicker, K.L.; et al. Peptidylarginine deiminase 2-catalyzed histone H3 arginine 26 cit-rullination facilitates estrogen receptor α target gene activation. Proc. Natl. Acad. Sci. USA. 2012, 109, 13331–13336. [Google Scholar] [CrossRef]
- Karvar, S.; Ansa-Addo, E.A.; Suda, J.; Singh, S.; Zhu, L.; Li, Z.; Rockey, D.C. Moesin, an ERM family member, regulates hepatic fibrosis. Hepatology 2020, 7, 1073–1084. [Google Scholar] [CrossRef]
- Jacquemet, G.; Baghirov, H.; Georgiadou, M.; Sihto, H.; Peuhu, E.; Cettour-Janet, P.; He, T.; Perälä, M.; Kronqvist, P.; Joensuu, H.; et al. L-type calcium channels regulate filopodia stability and cancer cell invasion downstream of integrin signalling. Nat. Commun. 2016, 7, 13297. [Google Scholar] [CrossRef]
- Jacquemet, G.; Hamidi, H.; Ivaska, J. Filopodia in cell adhesion, 3D migration and cancer cell invasion. Curr. Opin. Cell Biol. 2015, 36, 23–31. [Google Scholar] [CrossRef]
- Jacquemet, G.; Stubb, A.; Saup, R.; Miihkinen, M.; Kremneva, E.; Hamidi, H.; Ivaska, J. Filopodome Mapping Identifies p130Cas as a Mechanosensitive Regulator of Filopodia Stability. Curr. Biol. 2019, 29, 202–216. [Google Scholar] [CrossRef]
- Yu, L.; Zhao, L.; Wu, H.; Zhao, H.; Yu, Z.; He, M.; Jin, F.; Wei, M. Moesin is an independent prognostic marker for ER‑positive breast cancer. Oncol. Lett. 2018, 17, 1921–1933. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Lu, X.; Wang, J.; Yang, Z.; Hoffman, R.M.; Wu, X. Moesin Up-regulation Is Associated with Enhanced Tumor Pro-gression Imaged Non-invasively in an Orthotopic Mouse Model of Human Glioblastoma. Anticancer Res. 2018, 38, 3267–3272. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Morales, F.C.; Agarwal, N.K.; Dogruluk, T.; Gagea, M.; Georgescu, M.-M. Moesin Is a Glioma Progression Marker That Induces Proliferation and Wnt/β-Catenin Pathway Activation via Interaction with CD44. Cancer Res. 2013, 73, 1142–1155. [Google Scholar] [CrossRef] [PubMed]
- Dart, D.A.; Arisan, D.E.; Owen, S.; Hao, C.; Jiang, W.G.; Uysal-Onganer, P. Wnt-11 Expression Promotes Invasiveness and Correlates with Survival in Human Pancreatic Ductal Adenocarcinoma. Genes 2019, 10, 921. [Google Scholar] [CrossRef]
- Guo, W.; Zheng, Y.; Xu, B.; Ma, F.; Li, C.; Zhang, X.; Wang, Y.; Chang, X. Investigating the expression, effect and tumorigenic pathway of PADI2 in tumors. OncoTargets Ther. 2017, 10, 1475–1485. [Google Scholar] [CrossRef]
- Wang, H.; Xu, B.; Zhang, X.; Zheng, Y.; Zhao, Y.; Chang, X. PADI2 gene confers susceptibility to breast cancer and plays tu-morigenic role via ACSL4, BINC3 And CA9 signaling. Cancer Cell Int. 2016, 16, 61. [Google Scholar] [CrossRef]
- Funayama, R.; Taniguchi, H.; Mizuma, M.; Fujishima, F.; Kobayashi, M.; Ohnuma, S.; Unno, M.; Nakayama, K. Protein-arginine deiminase 2 suppresses proliferation of colon cancer cells through protein citrullination. Cancer Sci. 2017, 108, 713–718. [Google Scholar] [CrossRef]
- Cantariño, N.; Musulén, E.; Valero, V.; Peinado, M.A.; Perucho, M.; Moreno, V.; Forcales, S.-V.; Douet, J.; Buschbeck, M. Downregulation of the Deiminase PADI2 Is an Early Event in Colorectal Carcinogenesis and Indicates Poor Prognosis. Mol. Cancer Res. 2016, 14, 841–848. [Google Scholar] [CrossRef]
- Stadler, S.C.; Vincent, C.T.; Fedorov, V.D.; Patsialou, A.; Cherrington, B.D.; Wakshlag, J.J.; Mohanan, S.; Zee, B.M.; Zhang, X.; Garcia, B.A.; et al. Dysregulation of PAD4-mediated citrullination of nuclear GSK3β activates TGF-β signaling and induces epithelial-to-mesenchymal transition in breast cancer cells. Proc. Natl. Acad. Sci. USA. 2013, 110, 11851–11856. [Google Scholar] [CrossRef]
- Cui, X.; Witalison, E.E.; Chumanevich, A.P.; Chumanevich, A.A.; Poudyal, D.; Subramanian, V.; Schetter, A.J.; Harris, C.C.; Thompson, P.R.; Hofseth, L.J. The Induction of microRNA-16 in Colon Cancer Cells by Protein Arginine Deiminase Inhibition Causes a p53-Dependent Cell Cycle Arrest. PLoS ONE 2013, 8, e53791. [Google Scholar] [CrossRef]
- Flies, A.; Darby, J.M.; Lennard, P.R.; Murphy, P.R.; Ong, C.E.B.; Pinfold, T.L.; de Luca, A.; Lyons, A.B.; Woods, G.M.; Patchett, A.L. A novel system to map protein interactions reveals evolutionarily conserved immune evasion pathways on transmissible cancers. Sci. Adv. 2020, 6, eaba5031. [Google Scholar] [CrossRef] [PubMed]
- Patchett, A.L.; Coorens, T.H.H.; Darby, J.; Wilson, R.; McKay, M.J.; Kamath, K.S.; Rubin, A.F.; Wakefield, M.J.; McIntosh, L.; Mangiola, S.; et al. Two of a kind: Transmissible Schwann cell cancers in the endangered Tasmanian devil (Sarcophilus harrisii). Cell. Mol. Life Sci. 2019, 77, 1847–1858. [Google Scholar] [CrossRef] [PubMed]
- Muth, A.; Subramanian, V.; Beaumont, E.; Nagar, M.; Kerry, P.; McEwan, P.; Srinath, H.; Clancy, K.; Parelkar, S.; Thompson, P.R. Development of a Selective Inhibitor of Protein Arginine Deiminase 2. J. Med. Chem. 2017, 60, 3198–3211. [Google Scholar] [CrossRef] [PubMed]
- Willis, V.C.; Banda, N.K.; Cordova, K.N.; Chandra, P.E.; Robinson, W.H.; Cooper, D.C.; Lugo, D.; Mehta, G.; Taylor, S.; Tak, P.P.; et al. Protein arginine deiminase 4 inhibition is sufficient for the amelioration of colla-gen-induced arthritis. Clin. Exp. Immunol. 2017, 188, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Uysal-Onganer, P.; Kawano, Y.; Caro, M.; Walker, M.M.; Diez, S.; Darrington, R.S.; Waxman, J.; Kypta, R. Wnt-11 promotes neuroendocrine-like differentiation, survival and migration of prostate cancer cells. Mol. Cancer 2010, 9, 55. [Google Scholar] [CrossRef]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uysal-Onganer, P.; D’Alessio, S.; Mortoglou, M.; Kraev, I.; Lange, S. Peptidylarginine Deiminase Inhibitor Application, Using Cl-Amidine, PAD2, PAD3 and PAD4 Isozyme-Specific Inhibitors in Pancreatic Cancer Cells, Reveals Roles for PAD2 and PAD3 in Cancer Invasion and Modulation of Extracellular Vesicle Signatures. Int. J. Mol. Sci. 2021, 22, 1396. https://doi.org/10.3390/ijms22031396
Uysal-Onganer P, D’Alessio S, Mortoglou M, Kraev I, Lange S. Peptidylarginine Deiminase Inhibitor Application, Using Cl-Amidine, PAD2, PAD3 and PAD4 Isozyme-Specific Inhibitors in Pancreatic Cancer Cells, Reveals Roles for PAD2 and PAD3 in Cancer Invasion and Modulation of Extracellular Vesicle Signatures. International Journal of Molecular Sciences. 2021; 22(3):1396. https://doi.org/10.3390/ijms22031396
Chicago/Turabian StyleUysal-Onganer, Pinar, Stefania D’Alessio, Maria Mortoglou, Igor Kraev, and Sigrun Lange. 2021. "Peptidylarginine Deiminase Inhibitor Application, Using Cl-Amidine, PAD2, PAD3 and PAD4 Isozyme-Specific Inhibitors in Pancreatic Cancer Cells, Reveals Roles for PAD2 and PAD3 in Cancer Invasion and Modulation of Extracellular Vesicle Signatures" International Journal of Molecular Sciences 22, no. 3: 1396. https://doi.org/10.3390/ijms22031396
APA StyleUysal-Onganer, P., D’Alessio, S., Mortoglou, M., Kraev, I., & Lange, S. (2021). Peptidylarginine Deiminase Inhibitor Application, Using Cl-Amidine, PAD2, PAD3 and PAD4 Isozyme-Specific Inhibitors in Pancreatic Cancer Cells, Reveals Roles for PAD2 and PAD3 in Cancer Invasion and Modulation of Extracellular Vesicle Signatures. International Journal of Molecular Sciences, 22(3), 1396. https://doi.org/10.3390/ijms22031396