
Conformational Ensembles by NMR and MD Simulations in Model Heptapeptides with Select Tri-Peptide Motifs


Abstract
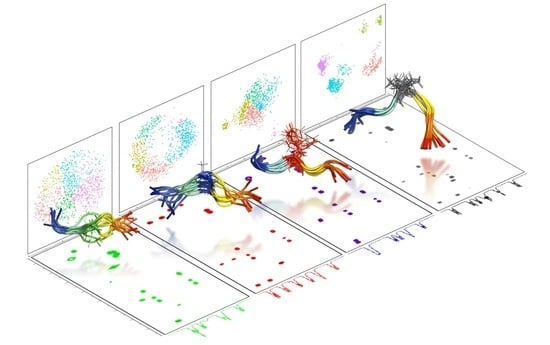
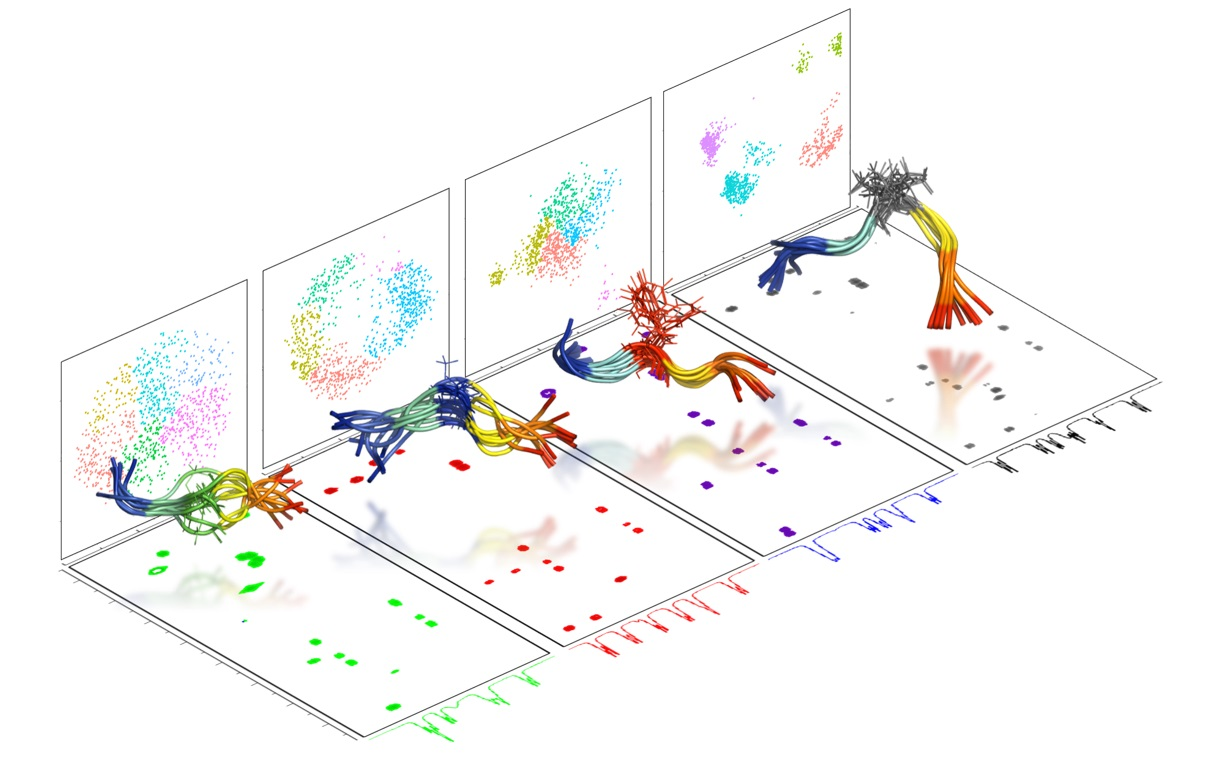{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
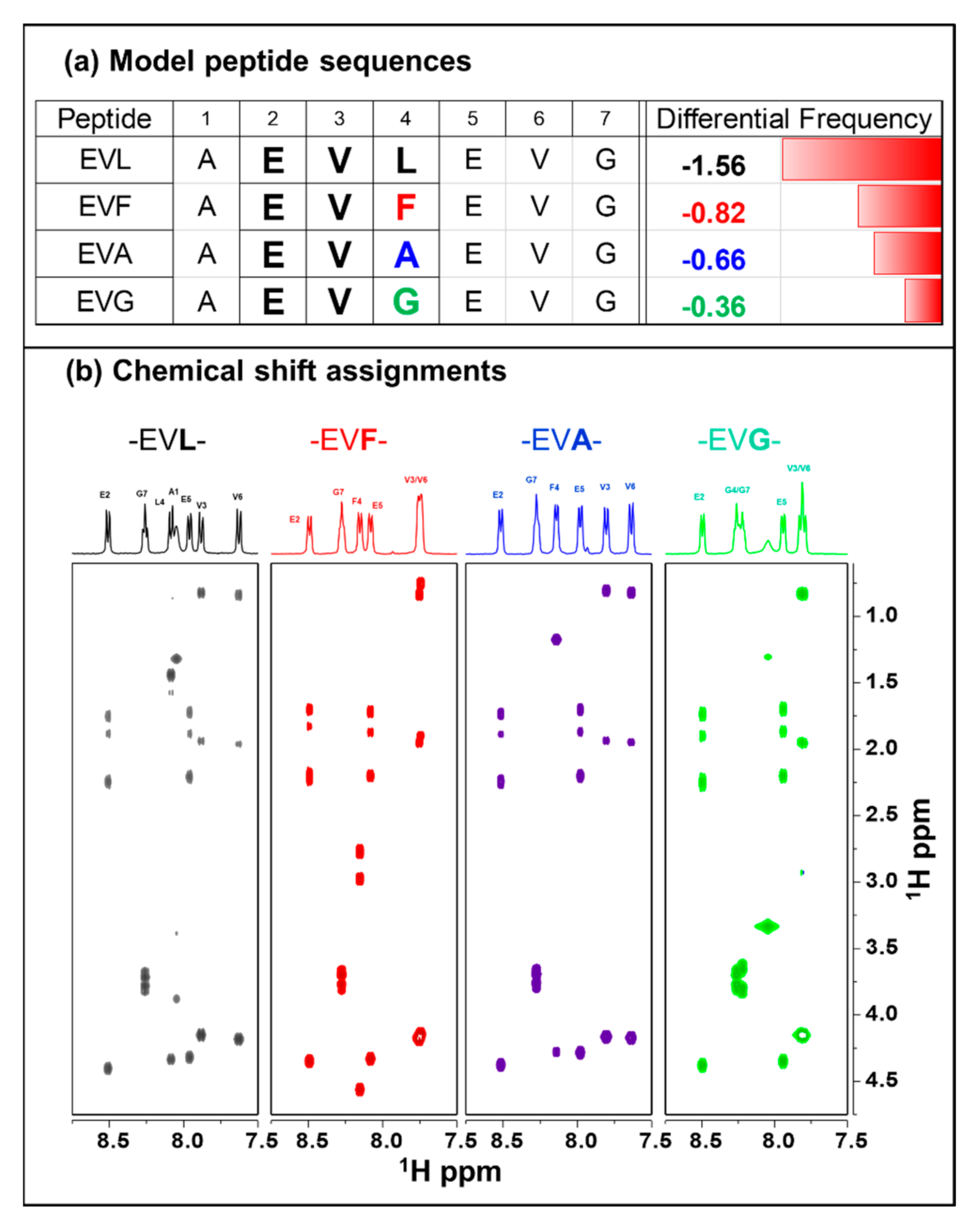
2.1. Chemical Shift Assignments
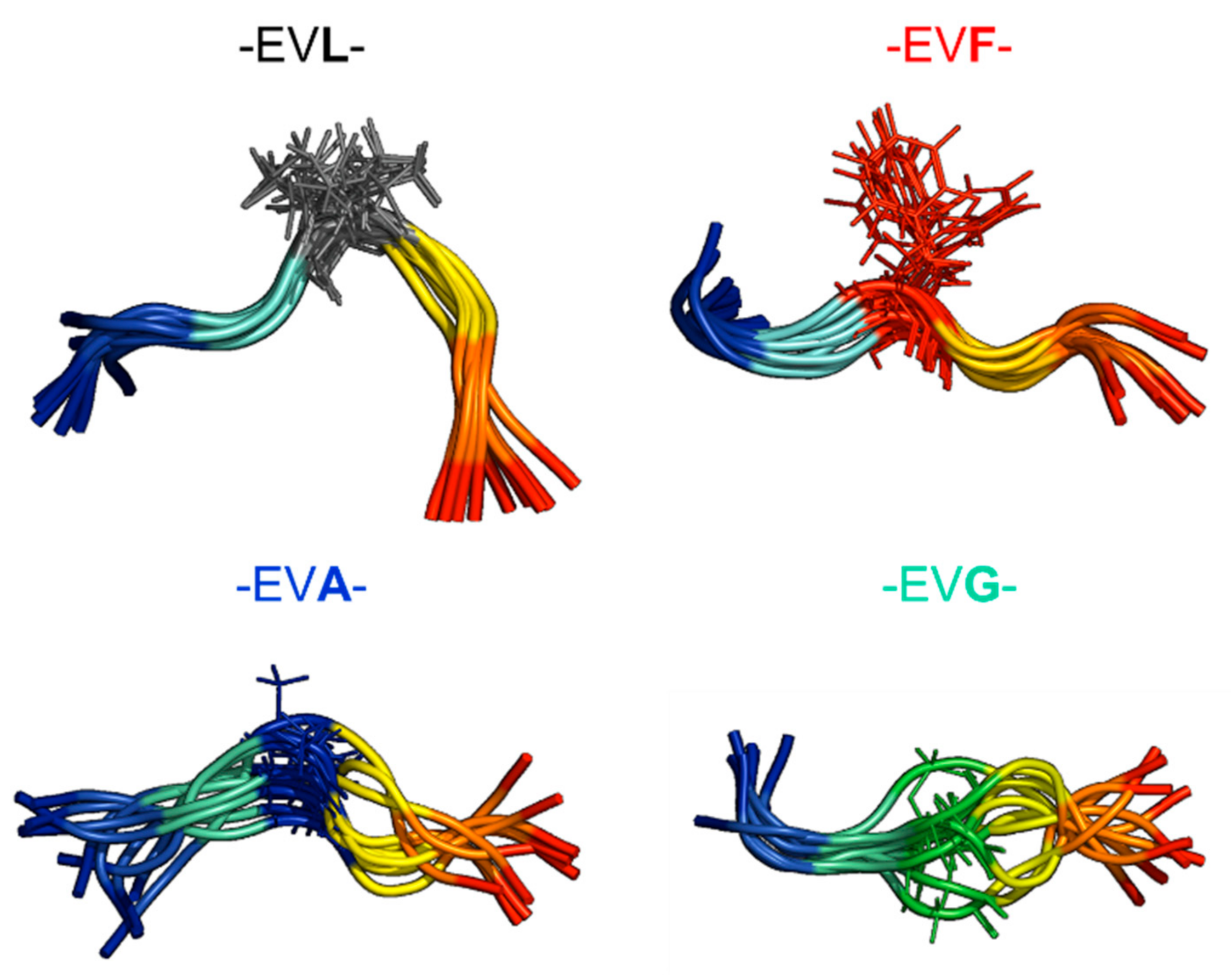
2.2. Experimental Ensemble of Structures
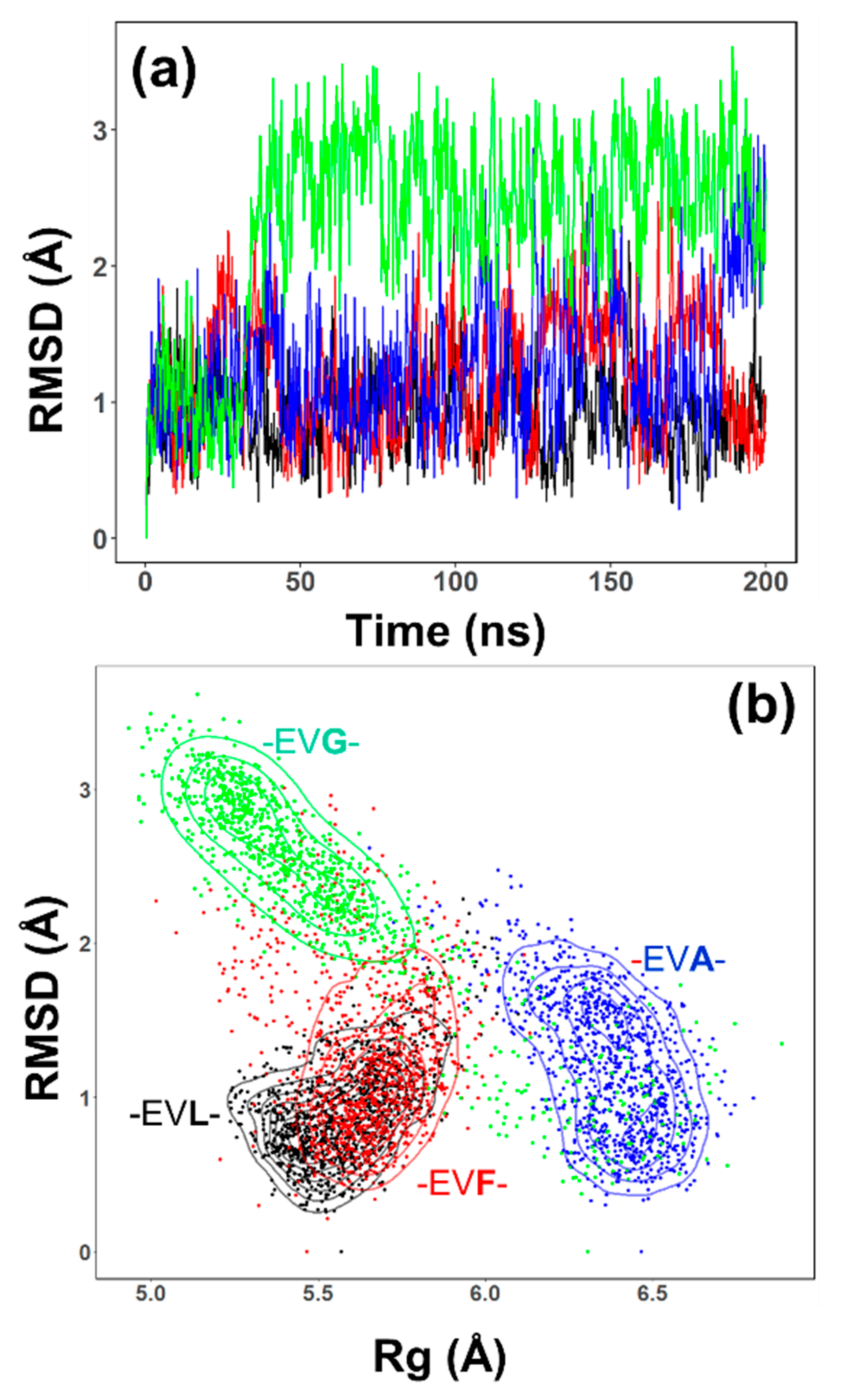
2.3. Conformational Sampling by MD Simulations
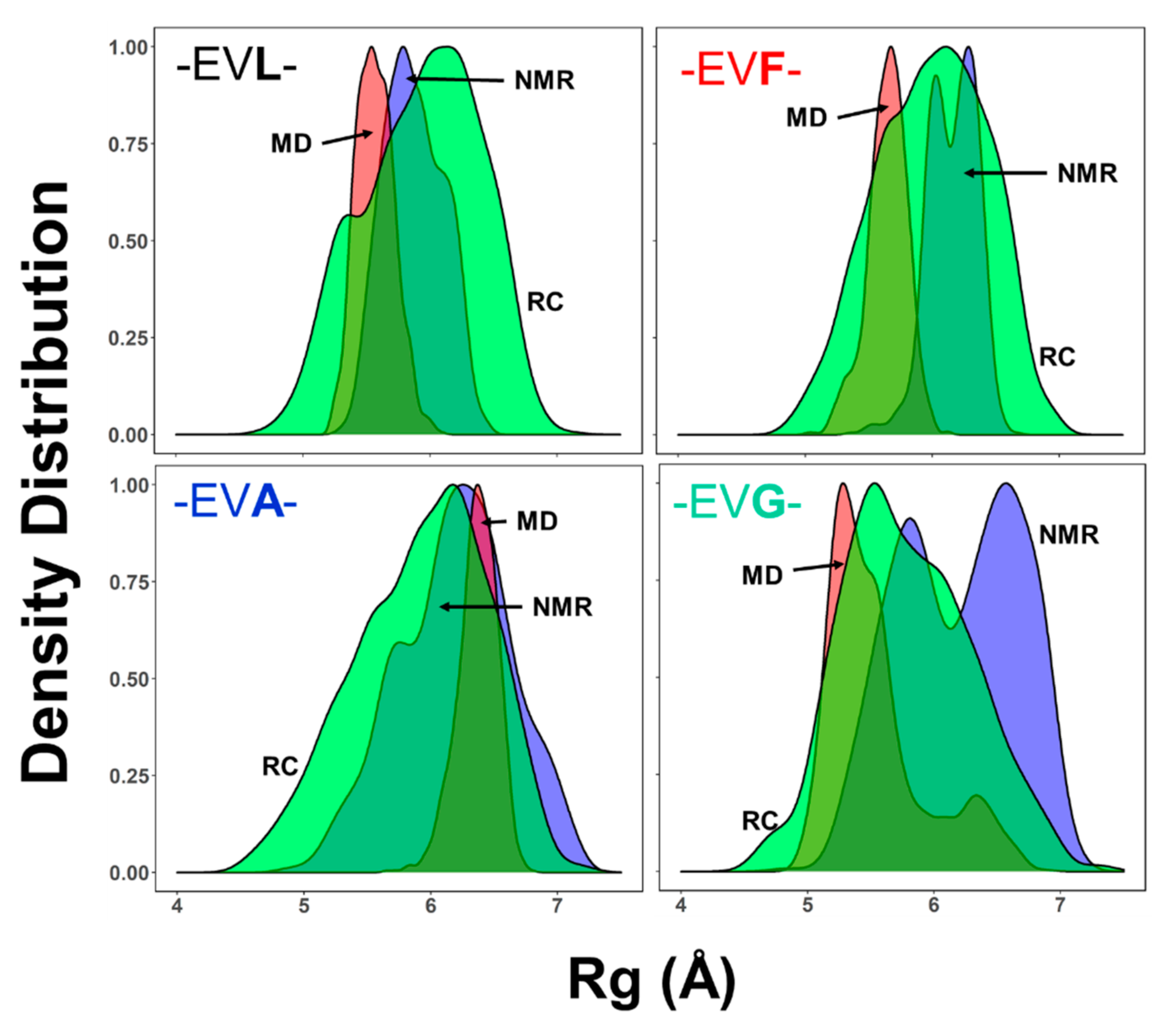
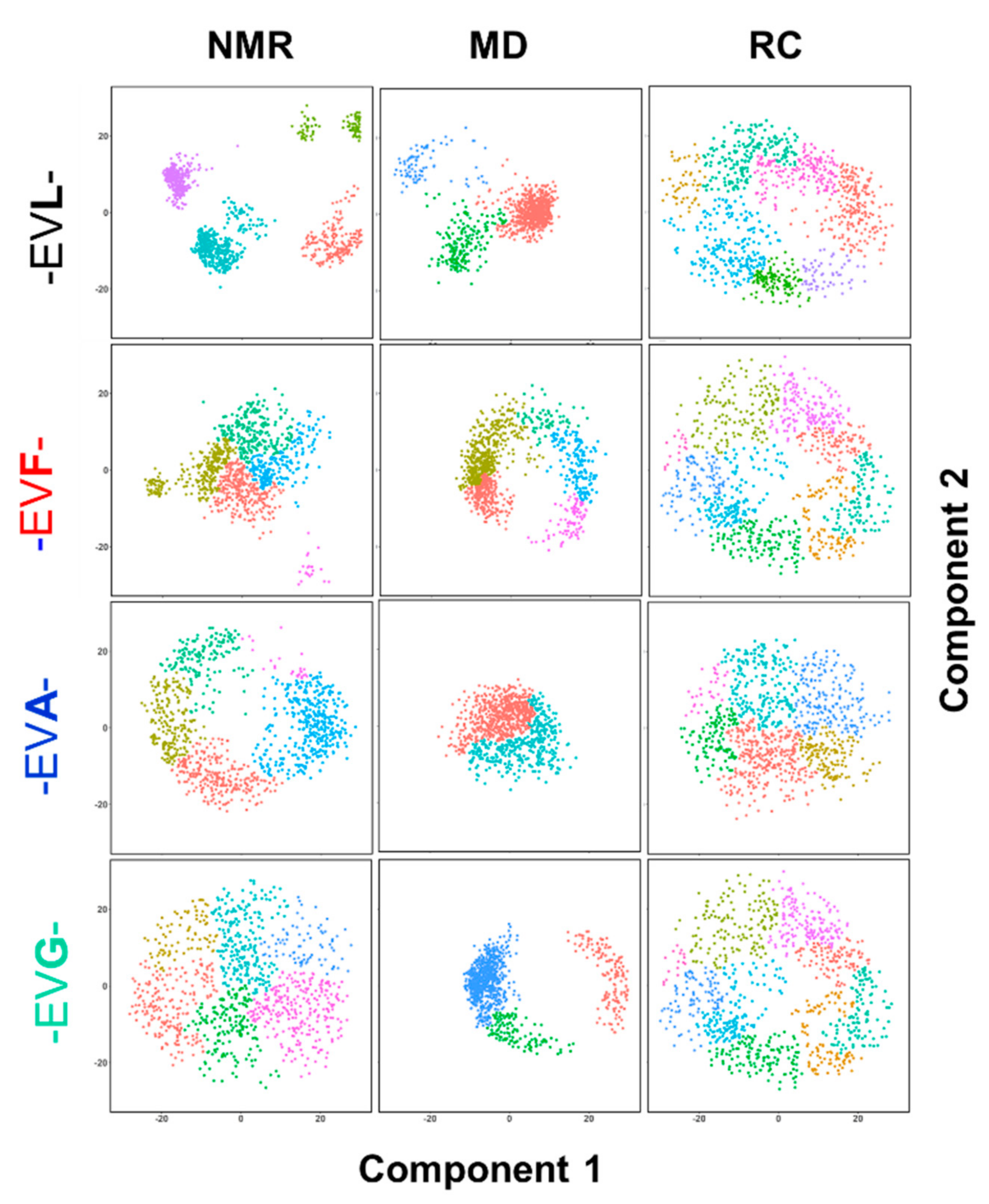
2.4. Ensemble of Structures NMR, MD, and the Random Coil Distributions
3. Materials and Methods
3.1. Peptide Synthesis
3.2. NMR Spectroscopy
3.3. Ensemble of NMR Structures
3.4. Generation of Random Structures
3.5. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koepnick, B.; Flatten, J.; Husain, T.; Ford, A.; Silva, D.-A.; Bick, M.J.; Bauer, A.; Liu, G.; Ishida, Y.; Boykov, A.; et al. De novo protein design by citizen scientists. Nature 2019, 570, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Koutsopoulos, S. Peptide Applications in Biomedicine, Biotechnology and Bioengineering; Woodhead Publishing: Cambridge, UK, 2017. [Google Scholar]
- Fairman, J.W.; Noinaj, N.; Buchanan, S.K. The structural biology of β-barrel membrane proteins: A summary of recent reports. Curr. Opin. Struct. Biol. 2011, 21, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Gromiha, M.M. Chapter 2—Protein Sequence Analysis. In Protein Bioinformatics; Gromiha, M.M., Ed.; Academic Press: Singapore, 2010; pp. 29–62. [Google Scholar] [CrossRef]
- Gromiha, M.M.; Ahmad, S.; Suwa, M. Application of residue distribution along the sequence for discriminating outer membrane proteins. Comput. Biol. Chem. 2005, 29, 135–142. [Google Scholar] [CrossRef]
- Wüthrich, K. NMR studies of structure and function of biological macromolecules (Nobel Lecture). J. Biomol. NMR 2003, 27, 13–39. [Google Scholar] [CrossRef]
- Her, C.; Yeh, Y.; Krishnan, V.V. The Ensemble of Conformations of Antifreeze Glycoproteins (AFGP8): A Study Using Nuclear Magnetic Resonance Spectroscopy. Biomolecules 2019, 9, 235. [Google Scholar] [CrossRef] [PubMed]
- Billeter, M.; Wagner, G.; Wüthrich, K. Solution NMR structure determination of proteins revisited. J. Biomol. NMR 2008, 42, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Rucker, S.P.; Shaka, A. Broadband homonuclear cross polarization in 2D NMR using DIPSI-2. Mol. Phys. 1989, 68, 509–517. [Google Scholar] [CrossRef]
- Bothner-By, A.A.; Stephens, R.; Lee, J.; Warren, C.D.; Jeanloz, R. Structure determination of a tetrasaccharide: Transient nuclear Overhauser effects in the rotating frame. J. Am. Chem. Soc. 1984, 106, 811–813. [Google Scholar] [CrossRef]
- Bax, A.; Davis, D.G. Practical aspects of two-dimensional transverse NOE spectroscopy. J. Magn. Reson. 1985, 63, 207–213. [Google Scholar] [CrossRef]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Goddard, T.D.; Kneller, D.G. SPARKY 3; University of California: San Francisco, CA, USA, 2008. [Google Scholar]
- Lee, W.; Tonelli, M.; Markley, J.L. NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 2015, 31, 1325–1327. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Delaglio, F.; Cornilescu, G.; Bax, A. TALOS+: A hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 2009, 44, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Bax, A. Protein structural information derived from NMR chemical shift with the neural network program TALOS-N. Methods Mol. Biol. 2015, 1260, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Güntert, P.; Mumenthaler, C.; Wüthrich, K. Torsion angle dynamics for NMR structure calculation with the new program Dyana11Edited by P. E. Wright. J. Mol. Biol. 1997, 273, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Guntert, P. Automated NMR structure calculation with CYANA. Methods Mol. Biol. 2004, 278, 353–378. [Google Scholar] [CrossRef] [PubMed]
- Feldman, H.J.; Hogue, C.W. A fast method to sample real protein conformational space. Proteins 2000, 39, 112–131. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 84. [Google Scholar]
- Maestro-Release, D. Desmond Molecular Dynamics System, Maestro-Desmond Interoperability Tools; D. E. Shaw Research: New York, NY, USA; Schrödinger: New York, NY, USA, 2017. [Google Scholar]
- PyMOL. The PyMOL Molecular Graphics System; Version 2.0.; Schrödinger, LLC: New York, NY, USA, 2017. [Google Scholar]
- Grant, B.J.; Skjaerven, L.; Yao, X.Q. The Bio3D packages for structural bioinformatics. Protein Sci. 2021, 30, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.J.; Rodrigues, A.P.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2011. [Google Scholar]
- Chi, P.B.; Liberles, D.A. Selection on protein structure, interaction, and sequence. Protein Sci. 2016, 25, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Siltberg-Liberles, J.; Grahnen, J.A.; Liberles, D.A. The Evolution of Protein Structures and Structural Ensembles Under Functional Constraint. Genes 2011, 2, 748–762. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krishnan, V.V.; Bentley, T.; Xiong, A.; Maitra, K. Conformational Ensembles by NMR and MD Simulations in Model Heptapeptides with Select Tri-Peptide Motifs. Int. J. Mol. Sci. 2021, 22, 1364. https://doi.org/10.3390/ijms22031364
Krishnan VV, Bentley T, Xiong A, Maitra K. Conformational Ensembles by NMR and MD Simulations in Model Heptapeptides with Select Tri-Peptide Motifs. International Journal of Molecular Sciences. 2021; 22(3):1364. https://doi.org/10.3390/ijms22031364
Chicago/Turabian StyleKrishnan, V. V., Timothy Bentley, Alina Xiong, and Kalyani Maitra. 2021. "Conformational Ensembles by NMR and MD Simulations in Model Heptapeptides with Select Tri-Peptide Motifs" International Journal of Molecular Sciences 22, no. 3: 1364. https://doi.org/10.3390/ijms22031364
APA StyleKrishnan, V. V., Bentley, T., Xiong, A., & Maitra, K. (2021). Conformational Ensembles by NMR and MD Simulations in Model Heptapeptides with Select Tri-Peptide Motifs. International Journal of Molecular Sciences, 22(3), 1364. https://doi.org/10.3390/ijms22031364