
Sex Differences in the Incidence of Obesity-Related Gastrointestinal Cancer


{kind=link}
{kind=link}
Abstract
1. Introduction
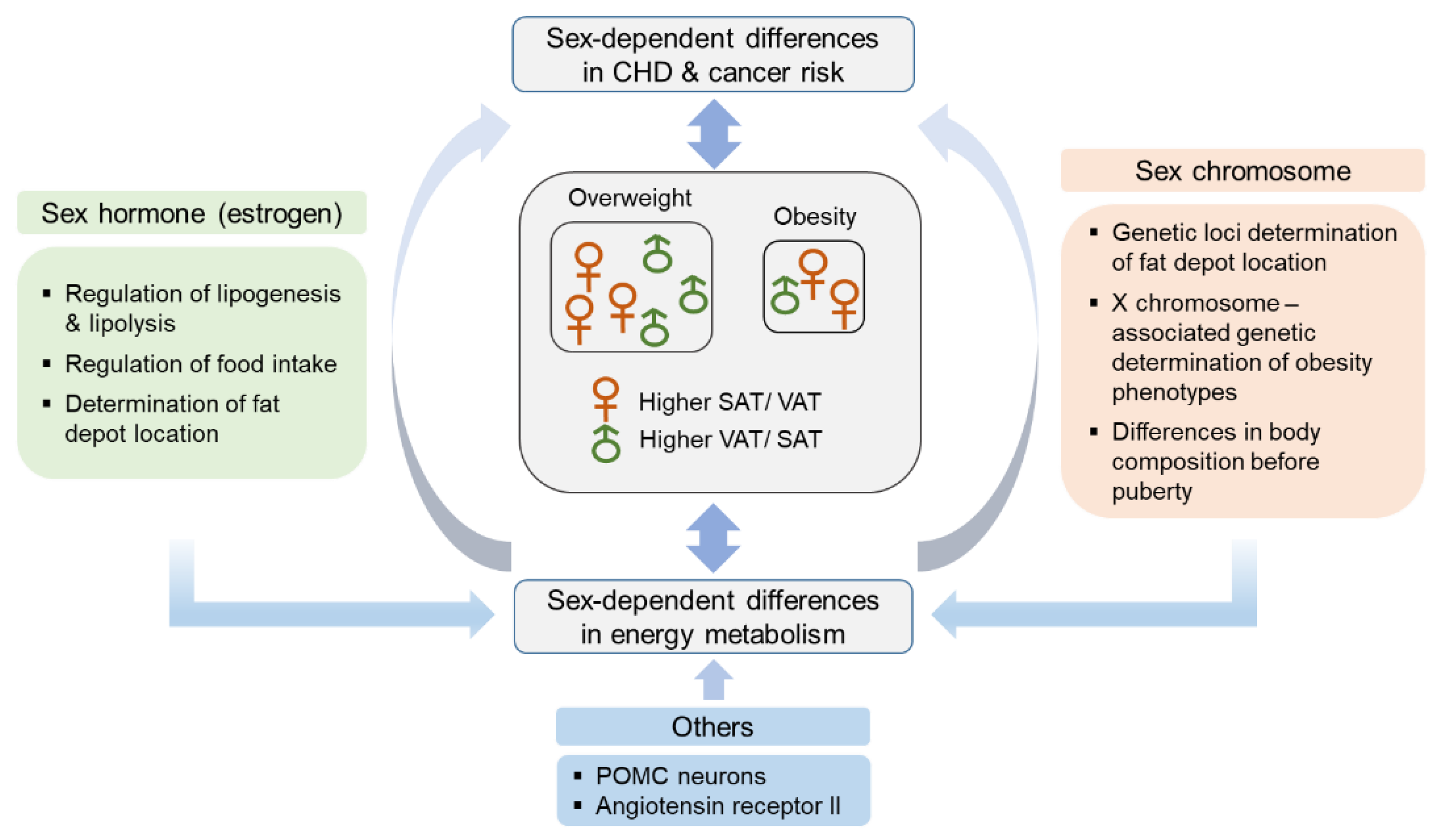
2. Sex Differences in Adipose Tissue Distribution and Energy Metabolism
2.1. Hormonal Factors
2.2. Genetic Factors
3. Sex Differences in the Incidence of Major Gastrointestinal Cancers in Association with Obesity
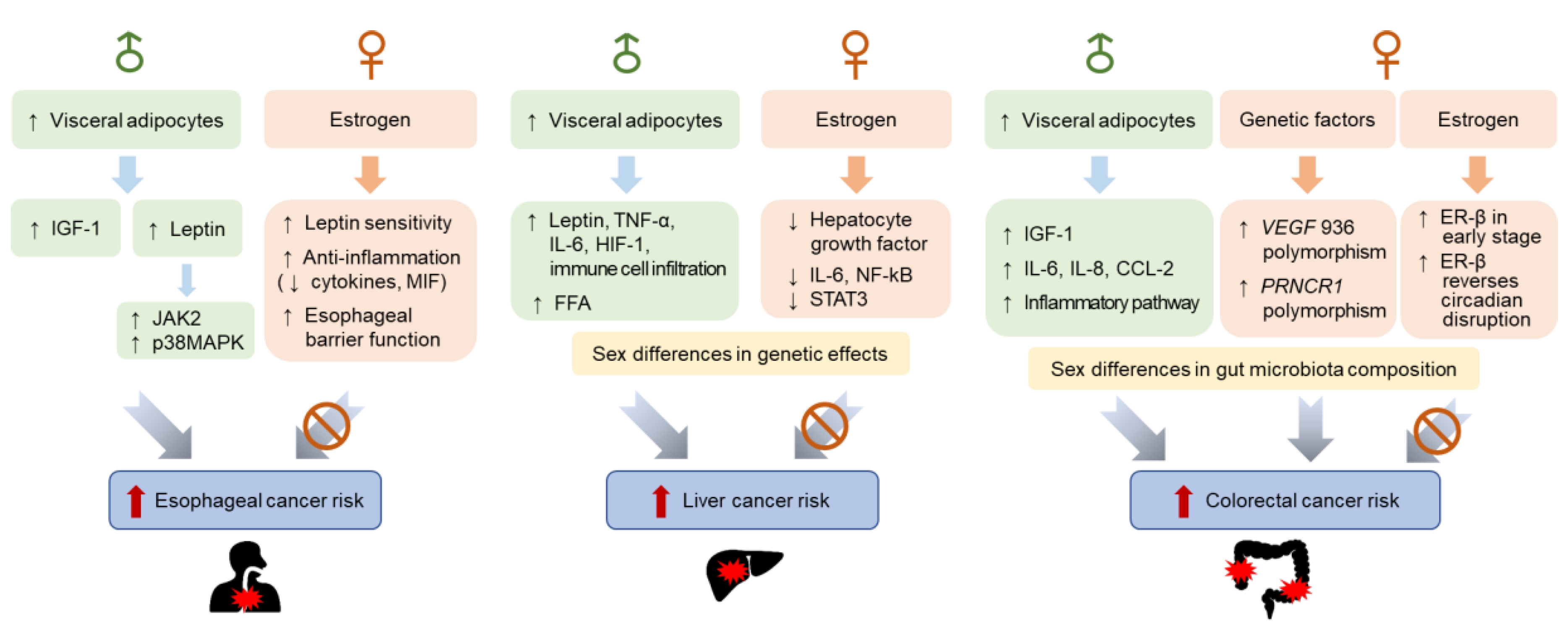
3.1. Sex Differences in Esophageal Cancer Incidence
3.1.1. Hormonal Factors
3.1.2. Genetic Factors
3.2. Sex Differences in Liver Cancer Incidence
3.2.1. Hormonal Factors
3.2.2. Genetic Factors
3.3. Sex Differences in Colorectal Cancer Incidence
3.3.1. Hormonal Factors
3.3.2. Genetic Factors
3.3.3. Gut Microbiota
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AR | androgen receptor |
| BMI | body mass index |
| CI | confidence interval |
| CRC | colorectal cancer |
| EC | esophageal cancer |
| EGFR | epidermal growth factor receptor |
| ER | estrogen receptor |
| FFAs | free fatty acids |
| GWAS | genome-wide association studies |
| HCC | hepatocellular carcinoma |
| IGF-1 | insulin-like growth factor 1 |
| IL | interleukin |
| LC | liver cancer |
| MAPK | mitogen-activated protein kinase |
| MIF | migration inhibitory factor |
| NAFLD | non-alcoholic fatty liver disease |
| NASH | non-alcoholic steatohepatitis |
| OS | overall survival |
| MAPK | mitogen-activated protein kinases |
| POMC | pro-opiomelanocortin |
| PPAR | peroxisome proliferator-activated receptor |
| RR | relative risk |
| SAT | subcutaneous adipose tissue |
| STAT | signal transducers and activators of transcription |
| TG | triacylglyceride |
| VAT | visceral adipose tissue |
| WC | waist circumference |
| WHR | waist–hip ratio |
References
- Wild, C.P.; Weiderpass, E.; Stewart, B.W. (Eds.) World Cancer Report: Cancer Research for Cancer Prevention; International Agency for Research on Cancer: Lyon, France, 2020. [Google Scholar]
- An, R.; Xiang, X. Age–period–cohort analyses of obesity prevalence in US adults. Public Health 2016, 141, 163–169. [Google Scholar] [CrossRef] [PubMed]
- World Cancer Research Fund/American Institute for Cancer Research. Diet, Nutrition, Physical Activity and Cancer: A Global Perspective; Continuous Update Project Expert Report; WCRF International: London, UK, 2018. [Google Scholar]
- Deng, T.; Lyon, C.J.; Bergin, S.; Caligiuri, M.A.; Hsueh, W. Obesity, Inflammation, and Cancer. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 421–449. [Google Scholar] [CrossRef] [PubMed]
- Calle, E.E.; Kaaks, R. Overweight, obesity and cancer: Epidemiological evidence and proposed mechanisms. Nat. Rev. Cancer 2004, 4, 579–591. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Obesity and Overweight. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 29 July 2020).
- Null, N. Trends in adult body-mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet 2016, 387, 1377–1396. [Google Scholar] [CrossRef]
- D’Eon, T.M.; Souza, S.C.; Aronovitz, M.; Obin, M.S.; Fried, S.K.; Greenberg, A.S. Estrogen Regulation of Adiposity and Fuel Partitioning. J. Biol. Chem. 2005, 280, 35983–35991. [Google Scholar] [CrossRef]
- Fried, S.K.; Lee, M.-J.; Karastergiou, K. Shaping fat distribution: New insights into the molecular determinants of depot and sex-dependent adipose biology. Obesity 2015, 23, 1345–1352. [Google Scholar] [CrossRef]
- Smith, S.R.; Lovejoy, J.C.; Greenway, F.; Ryan, D.; Dejonge, L.; De La Bretonne, J.; Volafova, J.; Bray, G.A. Contributions of total body fat, abdominal subcutaneous adipose tissue compartments, and visceral adipose tissue to the metabolic complications of obesity. Metabolism 2001, 50, 425–435. [Google Scholar] [CrossRef]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- De Vries, G.J.; Rissman, E.F.; Simerly, R.B.; Yang, L.-Y.; Scordalakes, E.M.; Auger, C.J.; Swain, A.; Lovell-Badge, R.; Burgoyne, P.S.; Arnold, A.P. A Model System for Study of Sex Chromosome Effects on Sexually Dimorphic Neural and Behavioral Traits. J. Neurosci. 2002, 22, 9005–9014. [Google Scholar] [CrossRef]
- Medrikova, D.; Jilkova, Z.M.; Bardova, K.; Janovska, P.; Rossmeisl, M.; Kopecky, J. Sex differences during the course of diet-induced obesity in mice: Adipose tissue expandability and glycemic control. Int. J. Obes. 2012, 36, 262–272. [Google Scholar] [CrossRef]
- Au-Yong, I.T.; Thorn, N.; Ganatra, R.H.; Perkins, A.C.; Symonds, M. Brown Adipose Tissue and Seasonal Variation in Humans. Diabetes 2009, 58, 2583–2587. [Google Scholar] [CrossRef] [PubMed]
- Lobo, R.A. Metabolic syndrome after menopause and the role of hormones. Maturitas 2008, 60, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.F.; Shah, N.M. Representing Sex in the Brain, One Module at a Time. Neuron 2014, 82, 261–278. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Nedungadi, T.P.; Zhu, L.; Sobhani, N.; Irani, B.G.; Davis, K.E.; Zhang, X.; Zou, F.; Gent, L.M.; Hahner, L.D.; et al. Distinct Hypothalamic Neurons Mediate Estrogenic Effects on Energy Homeostasis and Reproduction. Cell Metab. 2011, 14, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Patisaul, H.B. Sexually dimorphic expression of hypothalamic estrogen receptors α and β and kiss1 in neonatal male and female rats. J. Comp. Neurol. 2011, 519, 2954–2977. [Google Scholar] [CrossRef] [PubMed]
- Kyi-Tha-Thu, C.; Okoshi, K.; Ito, H.; Matsuda, K.-I.; Kawata, M.; Tsukahara, S. Sex differences in cells expressing green fluorescent protein under the control of the estrogen receptor-α promoter in the hypothalamus of mice. Neurosci. Res. 2015, 101, 44–52. [Google Scholar] [CrossRef]
- Dumesic, D.A.; Akopians, A.L.; Madrigal, V.K.; Ramirez, E.; Margolis, D.J.; Sarma, M.K.; Thomas, A.M.; Grogan, T.R.; Haykal, R.; Schooler, T.A.; et al. Hyperandrogenism Accompanies Increased Intra-Abdominal Fat Storage in Normal Weight Polycystic Ovary Syndrome Women. J. Clin. Endocrinol. Metab. 2016, 101, 4178–4188. [Google Scholar] [CrossRef]
- Jørgensen, J.O.L.; Vahl, N.; Hansen, T.B.; Fisker, S.; Hagen, C.; Christiansen, J.S. Influence of Growth Hormone and Androgens on Body Composition in Adults. Horm. Res. 1996, 45, 94–98. [Google Scholar] [CrossRef]
- Asarian, L.; Geary, N. Modulation of appetite by gonadal steroid hormones. Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 1251–1263. [Google Scholar] [CrossRef]
- Clegg, D.; Brown, L.M.; Woods, S.C.; Benoit, S.C. Gonadal Hormones Determine Sensitivity to Central Leptin and Insulin. Diabetes 2006, 55, 978–987. [Google Scholar] [CrossRef]
- Lu, S.-F.; McKenna, S.E.; Cologer-Clifford, A.; Nau, E.A.; Simon, N.G. Androgen Receptor in Mouse Brain: Sex Differences and Similarities in Autoregulation 1. Endocrinology 1998, 139, 1594–1601. [Google Scholar] [CrossRef] [PubMed]
- Klaver, M.; De Blok, C.J.M.; Wiepjes, C.M.; Nota, N.M.; Dekker, M.J.H.J.; De Mutsert, R.; Schreiner, T.; Fisher, A.D.; T’Sjoen, G.; Heijer, M.D. Changes in regional body fat, lean body mass and body shape in trans persons using cross-sex hormonal therapy: Results from a multicenter prospective study. Eur. J. Endocrinol. 2018, 178, 163–171. [Google Scholar] [CrossRef]
- Papadakis, G.; Hans, D.; Rodriguez, E.G.; Vollenweider, P.; Waeber, G.; Marques-Vidal, P.; Lamy, O. Menopausal Hormone Therapy Is Associated With Reduced Total and Visceral Adiposity: The OsteoLaus Cohort. J. Clin. Endocrinol. Metab. 2018, 103, 1948–1957. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.D. Gender differences in regional fatty acid metabolism before and after meal ingestion. J. Clin. Investig. 1995, 96, 2297–2303. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.; Guo, Z.; Johnson, C.M.; Hensrud, D.D.; Jensen, M.D. Splanchnic lipolysis in human obesity. J. Clin. Investig. 2004, 113, 1582–1588. [Google Scholar] [CrossRef]
- Burguera, B.; Proctor, D.; Dietz, N.; Guo, Z.; Joyner, M.; Jensen, M.D. Leg free fatty acid kinetics during exercise in men and women. Am. J. Physiol. Metab. 2000, 278, E113–E117. [Google Scholar] [CrossRef]
- Marin, P.; Lönn, L.B.; Andersson, B.; Odén, B.; Olbe, L.; A Bengtsson, B.; Björntorp, P. Assimilation of triglycerides in subcutaneous and intraabdominal adipose tissues In Vivo in men: Effects of testosterone. J. Clin. Endocrinol. Metab. 1996, 81, 1018–1022. [Google Scholar] [CrossRef]
- Votruba, S.B.; Jensen, M.D. Short-term regional meal fat storage in nonobese humans is not a predictor of long-term regional fat gain. Am. J. Physiol. Metab. 2012, 302, E1078–E1083. [Google Scholar] [CrossRef]
- Santosa, S.; Jensen, M.D. Adipocyte Fatty Acid Storage Factors Enhance Subcutaneous Fat Storage in Postmenopausal Women. Diabetes 2012, 62, 775–782. [Google Scholar] [CrossRef]
- Pulit, S.L.; Karaderi, T.; Lindgren, C.M. Sexual dimorphisms in genetic loci linked to body fat distribution. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef]
- Van Dongen, J.; Willemsen, G.; Chen, W.-M.; De Geus, E.J.C.; Boomsma, D.I. Heritability of metabolic syndrome traits in a large population-based sample. J. Lipid Res. 2013, 54, 2914–2923. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, C.; Heid, I.; Randall, J.; Lamina, C.; Steinthorsdottir, V.; Qi, L.; Speliotes, E.; Thorleifsson, G.; Willer, C.; Herrera, B.; et al. Genome-Wide Association Scan Meta-Analysis Identifies Three Loci Influencing Adiposity and Fat Distribution. PLoS Genet. 2009, 5, e1000508. [Google Scholar] [CrossRef]
- Locke, A.E.; Kahali, B.; Berndt, S.I.; Justice, A.E.; Pers, T.H.; Day, F.R.; Powell, C.; Vedantam, S.; Buchkovich, M.L.; Yang, J.; et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015, 518, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Shungin, D.; Winkler, T.W.; Croteau-Chonka, D.C.; Ferreira, T.; Locke, A.E.; Mägi, R.; Strawbridge, R.J.; Pers, T.H.; Fischer, K.; Justice, A.E.; et al. New genetic loci link adipose and insulin biology to body fat distribution. Nat. Cell Biol. 2015, 518, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Heid, I.M.; Jackson, A.U.; Randall, J.C.; Winkler, T.W.; Qi, L.; Steinthorsdottir, V.; Thorleifsson, G.; Zillikens, M.C.; Speliotes, E.K.; Magi, R. Meta-analysis identifies 13 new loci associated with waist-hip ratio and reveals sexual dimorphism in the genetic basis of fat distribution. Nat. Genet. 2010, 42, 949–960. [Google Scholar] [CrossRef]
- Grove, K.L.; Fried, S.K.; Greenberg, A.S.; Xiao, X.Q.; Clegg, D.J. A microarray analysis of sexual dimorphism of adipose tissues in high-fat-diet-induced obese mice. Int. J. Obes. 2010, 34, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Schadt, E.E.; Wang, S.; Wang, H.; Arnold, A.P.; Ingram-Drake, L.; Drake, T.A.; Lusis, A.J. Tissue-specific expression and regulation of sexually dimorphic genes in mice. Genome Res. 2006, 16, 995–1004. [Google Scholar] [CrossRef]
- Koopman, P. The delicate balance between male and female sex determining pathways: Potential for disruption of early steps in sexual development. Int. J. Androl. 2010, 33, 252–258. [Google Scholar] [CrossRef]
- Wells, J.C. Sexual dimorphism of body composition. Best Pr. Res. Clin. Endocrinol. Metab. 2007, 21, 415–430. [Google Scholar] [CrossRef]
- Taylor, B.J.; Grant, A.M.; Williams, S.M.; Goulding, A. Sex Differences in Regional Body Fat Distribution from Pre to Postpuberty. Obesity 2010, 18, 1410–1416. [Google Scholar] [CrossRef]
- Chen, X.; McClusky, R.; Chen, J.; Beaven, S.W.; Tontonoz, P.; Arnold, A.P.; Reue, K. The Number of X Chromosomes Causes Sex Differences in Adiposity in Mice. PLoS Genet. 2012, 8, e1002709. [Google Scholar] [CrossRef] [PubMed]
- Lagerlöf, O.; Slocomb, J.E.; Hong, I.; Aponte, Y.; Blackshaw, S.; Hart, G.W.; Huganir, R.L. The nutrient sensor OGT in PVN neurons regulates feeding. Science 2016, 351, 1293–1296. [Google Scholar] [CrossRef] [PubMed]
- Nonogaki, K.; Strack, A.M.; Dallman, M.F.; Tecott, L.H. Leptin-independent hyperphagia and type 2 diabetes in mice with a mutated serotonin 5-HT2C receptor gene. Nat. Med. 1998, 4, 1152–1156. [Google Scholar] [CrossRef] [PubMed]
- Harno, E.; Ramamoorthy, T.G.; Coll, A.P.; White, A. POMC: The Physiological Power of Hormone Processing. Physiol. Rev. 2018, 98, 2381–2430. [Google Scholar] [CrossRef] [PubMed]
- Millington, G.W. The role of proopiomelanocortin (POMC) neurones in feeding behaviour. Nutr. Metab. 2007, 4, 1–16. [Google Scholar] [CrossRef]
- Nohara, K.; Zhang, Y.; Waraich, R.S.; Laque, A.; Tiano, J.P.; Tong, J.; Münzberg, H.; Mauvais-Jarvis, F. Early-Life Exposure to Testosterone Programs the Hypothalamic Melanocortin System. Endocrinology 2011, 152, 1661–1669. [Google Scholar] [CrossRef]
- Su, X.; Gi, Y.J.; Chakravarti, D.; Chan, I.L.; Zhang, A.; Xia, X.; Tsai, K.Y.; Flores, E.R. TAp63 Is a Master Transcriptional Regulator of Lipid and Glucose Metabolism. Cell Metab. 2012, 16, 511–525. [Google Scholar] [CrossRef]
- Ramadori, G.; Fujikawa, T.; Fukuda, M.; Anderson, J.; Morgan, D.A.; Mostoslavsky, R.; Stuart, R.C.; Perello, M.; Vianna, C.R.; Nillni, E.A.; et al. SIRT1 Deacetylase in POMC Neurons Is Required for Homeostatic Defenses against Diet-Induced Obesity. Cell Metab. 2010, 12, 78–87. [Google Scholar] [CrossRef]
- Samuel, P.; Khan, M.A.; Nag, S.; Inagami, T.; Hussain, T. Angiotensin AT(2) Receptor Contributes towards Gender Bias in Weight Gain. PLoS ONE 2013, 8, e48425. [Google Scholar] [CrossRef]
- Li, L.; Hossain, M.A.; Sadat, S.; Hager, L.; Liu, L.; Tam, L.; Schroer, S.; Huogen, L.; Fantus, I.G.; Connelly, P.W.; et al. Lecithin Cholesterol Acyltransferase Null Mice Are Protected from Diet-induced Obesity and Insulin Resistance in a Gender-specific Manner through Multiple Pathways. J. Biol. Chem. 2011, 286, 17809–17820. [Google Scholar] [CrossRef]
- Kamangar, F.; Nasrollahzadeh, D.; Safiri, S.; Sepanlou, S.G.; Fitzmaurice, C.; Ikuta, K.S.; Bisignano, C.; Islami, F.; Roshandel, G.; Lim, S.S.; et al. The global, regional, and national burden of oesophageal cancer and its attributable risk factors in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 582–597. [Google Scholar] [CrossRef]
- Arnold, M.; Laversanne, M.; Brown, L.M.; Devesa, S.S.; Bray, F. Predicting the Future Burden of Esophageal Cancer by Histological Subtype: International Trends in Incidence up to 2030. Am. J. Gastroenterol. 2017, 112, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.C.; Yang, Y.C.; Shaheen, N.J.; Hofstetter, W.L.; Sandler, R.S. An age-period-cohort analysis of obesity and incident esophageal adenocarcinoma among white males. Dis. Esophagus 2017, 30, 1–8. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Chen, N.; Hou, Y.; Wang, Z.; Zhang, Y.; Zhang, G.; Fu, J. Trends in the incidence and survival of patients with esophageal cancer: A SEER database analysis. Thorac. Cancer 2020, 11, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- Lovegrove, C. Obesity is linked with increased risk of gastroesophageal reflux disease. Nat. Clin. Pr. Gastroenterol. Hepatol. 2005, 2, 501. [Google Scholar] [CrossRef]
- Tian, J.; Zuo, C.; Liu, G.; Che, P.; Li, G.; Li, X.; Chen, H. Cumulative evidence for the relationship between body mass index and the risk of esophageal cancer: An updated meta-analysis with evidence from 25 observational studies. J. Gastroenterol. Hepatol. 2020, 35, 730–743. [Google Scholar] [CrossRef]
- Xie, S.-H.; Lagergren, J. Risk factors for oesophageal cancer. Best Pr. Res. Clin. Gastroenterol. 2018, 36–37, 3–8. [Google Scholar] [CrossRef]
- Turati, F.; Tramacere, I.; La Vecchia, C.; Negri, E. A meta-analysis of body mass index and esophageal and gastric cardia adenocarcinoma. Ann. Oncol. 2012, 24, 609–617. [Google Scholar] [CrossRef]
- Kubo, A.; Cook, M.B.; Shaheen, N.J.; Vaughan, T.L.; Whiteman, D.C.; Murray, L.; A Corley, D. Sex-specific associations between body mass index, waist circumference and the risk of Barrett’s oesophagus: A pooled analysis from the international BEACON consortium. Gut 2013, 62, 1684–1691. [Google Scholar] [CrossRef]
- Corley, D.A.; Kubo, A.; Zhao, W. Abdominal Obesity and the Risk of Esophageal and Gastric Cardia Carcinomas. Cancer Epidemiol. Biomark. Prev. 2008, 17, 352–358. [Google Scholar] [CrossRef]
- Arnold, M.; Soerjomataram, I.; Ferlay, J.; Forman, D. Global incidence of oesophageal cancer by histological subtype in 2012. Gut 2015, 64, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Doyle, S.L.; Donohoe, C.L.; Finn, S.P.; Howard, J.M.; Lithander, F.E.; Reynolds, J.V.; Pidgeon, G.P.; Lysaght, J. IGF-1 and Its Receptor in Esophageal Cancer: Association with Adenocarcinoma and Visceral Obesity. Am. J. Gastroenterol. 2012, 107, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Fowler, A.J.; Richer, A.L.; Bremner, R.M.; Inge, L.J. A high-fat diet is associated with altered adipokine production and a more aggressive esophageal adenocarcinoma phenotype In Vivo. J. Thorac. Cardiovasc. Surg. 2015, 149, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chua, S. Leptin Function and Regulation. Compr. Physiol. 2017, 8, 351–369. [Google Scholar] [CrossRef] [PubMed]
- Ogunwobi, O.; Mutungi, G.; Beales, I.L.P. Leptin Stimulates Proliferation and Inhibits Apoptosis in Barrett’s Esophageal Adenocarcinoma Cells by Cyclooxygenase-2-Dependent, Prostaglandin-E2-Mediated Transactivation of the Epidermal Growth Factor Receptor and c-Jun NH2-Terminal Kinase Activation. Endocrinology 2006, 147, 4505–4516. [Google Scholar] [CrossRef]
- Kendall, B.J.; Macdonald, G.A.; Hayward, N.K.; Prins, J.B.; Brown, I.; Walker, N.; Pandeya, N.; Green, A.C.; Webb, P.M.; Whiteman, D.C.; et al. Leptin and the risk of Barrett’s oesophagus. Gut 2007, 57, 448–454. [Google Scholar] [CrossRef]
- Montague, C.T.; Prins, J.B.; Sanders, L.; Digby, J.E.; O’Rahilly, S. Depot and Sex-Specific Differences in Human Leptin mRNA Expression: Implications for the Control of Regional Fat Distribution. Diabetes 1997, 46, 342–347. [Google Scholar] [CrossRef]
- Petrick, J.L.; Hyland, P.L.; Caron, P.; Falk, R.T.; Pfeiffer, R.M.; Dawsey, S.M.; Abnet, C.C.; Taylor, P.R.; Weinstein, S.J.; Albanes, D.; et al. Associations Between Prediagnostic Concentrations of Circulating Sex Steroid Hormones and Esophageal/Gastric Cardia Adenocarcinoma Among Men. J. Natl. Cancer Inst. 2018, 111, 34–41. [Google Scholar] [CrossRef]
- Petrick, J.L.; Falk, R.T.; Hyland, P.L.; Caron, P.; Pfeiffer, R.M.; Wood, S.N.; Dawsey, S.M.; Abnet, C.C.; Taylor, P.R.; Guillemette, C.; et al. Association between circulating levels of sex steroid hormones and esophageal adenocarcinoma in the FINBAR Study. PLoS ONE 2018, 13, e0190325. [Google Scholar] [CrossRef]
- Lagergren, K.; Lagergren, J.; Brusselaers, N. Hormone replacement therapy and oral contraceptives and risk of oesophageal adenocarcinoma: A systematic review and meta-analysis. Int. J. Cancer 2014, 135, 2183–2190. [Google Scholar] [CrossRef]
- Brusselaers, N.; Maret-Ouda, J.; Konings, P.; El-Serag, H.B.; Lagergren, P. Menopausal hormone therapy and the risk of esophageal and gastric cancer. Int. J. Cancer 2017, 140, 1693–1699. [Google Scholar] [CrossRef] [PubMed]
- Coleman, H.G.; Xie, S.-H.; Lagergren, J. The Epidemiology of Esophageal Adenocarcinoma. Gastroenterology 2018, 154, 390–405. [Google Scholar] [CrossRef]
- Jenks, M.Z.; Fairfield, H.E.; Johnson, E.C.; Morrison, R.F.; Muday, G.K. Sex Steroid Hormones Regulate Leptin Transcript Accumulation and Protein Secretion in 3T3-L1 Cells. Sci. Rep. 2017, 7, 8232. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-S.; Chae, H.-J.; Shin, T.-Y.; Kim, H.-M.; Kim, H.-R. Estrogen Regulates Cytokine Release in Human Mast Cells. Immunopharmacol. Immunotoxicol. 2001, 23, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Masaka, T.; Iijima, K.; Endo, H.; Asanuma, K.; Ara, N.; Ishiyama, F.; Asano, N.; Koike, T.; Imatani, A.; Shimosegawa, T. Gender differences in oesophageal mucosal injury in a reflux oesophagitis model of rats. Gut 2013, 62, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, G.S.; Mills, S.J.; Lei, K.; Gibbons, L.; Jeong, M.-J.; Taniguchi, M.; Burow, M.; Horan, M.A.; Wahl, S.M.; Nakayama, T. Estrogen modulates cutaneous wound healing by downregulating macrophage migration inhibitory factor. J. Clin. Investig. 2003, 111, 1309–1318. [Google Scholar] [CrossRef]
- Nishihira, J. Macrophage Migration Inhibitory Factor (MIF): Its Essential Role in the Immune System and Cell Growth. J. Interf. Cytokine Res. 2000, 20, 751–762. [Google Scholar] [CrossRef]
- Honda, J.; Iijima, K.; Asanuma, K.; Ara, N.; Shiroki, T.; Kondo, Y.; Hatta, W.; Uno, K.; Asano, N.; Koike, T.; et al. Estrogen Enhances Esophageal Barrier Function by Potentiating Occludin Expression. Dig. Dis. Sci. 2015, 61, 1028–1038. [Google Scholar] [CrossRef]
- Lin, Y.; Yngve, A.; Lagergren, P.; Lu, Y. A dietary pattern rich in lignans, quercetin and resveratrol decreases the risk of oesophageal cancer. Br. J. Nutr. 2014, 112, 1–8. [Google Scholar] [CrossRef]
- Böhmer, A.C.; Hecker, J.; Schröder, J.; Gharahkhani, P.; May, A.; Gerges, C.; Anders, M.; Becker, J.; Hess, T.; Kreuser, N.; et al. Shared Genetic Etiology of Obesity-Related Traits and Barrett’s Esophagus/Adenocarcinoma: Insights from Genome-Wide Association Studies. Cancer Epidemiol. Biomark. Prev. 2019, 29, 427–433. [Google Scholar] [CrossRef]
- Dong, J.; Maj, C.; Tsavachidis, S.; Ostrom, Q.T.; Gharahkhani, P.; Anderson, L.A.; Wu, A.H.; Ye, W.; Bernstein, L.; Borisov, O.; et al. Sex-Specific Genetic Associations for Barrett’s Esophagus and Esophageal Adenocarcinoma. Gastroenterology 2020, 159, 2065–2076. [Google Scholar] [CrossRef] [PubMed]
- Petrick, J.L.; Braunlin, M.; Laversanne, M.; Valery, P.C.; Bray, F.; McGlynn, K.A. International trends in liver cancer incidence, overall and by histologic subtype, 1978–2007. Int. J. Cancer 2016, 139, 1534–1545. [Google Scholar] [CrossRef] [PubMed]
- Jelic, S.; Sotiropoulos, G.C. On behalf of the ESMO Guidelines Working Group Hepatocellular carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2010, 21 (Suppl. 5), v59–v64. [Google Scholar] [CrossRef] [PubMed]
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E. Nonalcoholic Fatty Liver Disease: A systematic review. JAMA 2015, 313, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Baecker, A.; Liu, X.; La Vecchia, C.; Zhang, Z.-F. Worldwide incidence of hepatocellular carcinoma cases attributable to major risk factors. Eur. J. Cancer Prev. 2018, 27, 205–212. [Google Scholar] [CrossRef]
- Larsson, S.C.; Wolk, A. Overweight, obesity and risk of liver cancer: A meta-analysis of cohort studies. Br. J. Cancer 2007, 97, 1005–1008. [Google Scholar] [CrossRef]
- Schlesinger, S.; Aleksandrova, K.; Pischon, T.; Fedirko, V.; Jenab, M.; Trepo, E.; Boffetta, P.; Dahm, C.C.; Overvad, K.; Tjønneland, A.; et al. Abdominal obesity, weight gain during adulthood and risk of liver and biliary tract cancer in a European cohort. Int. J. Cancer 2013, 132, 645–657. [Google Scholar] [CrossRef]
- Saunders, D.; Seidel, D.; Allison, M.; Lyratzopoulos, G. Systematic review: The association between obesity and hepatocellular carcinoma—Epidemiologic evidence. Aliment. Pharmacol. Ther. 2010, 31, 1051–1063. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, X.; Wang, J.; Yan, Z.-P.; Luo, J. Excess body weight and the risk of primary liver cancer: An updated meta-analysis of prospective studies. Eur. J. Cancer 2012, 48, 2137–2145. [Google Scholar] [CrossRef]
- Renehan, A.G.; Tyson, M.; Egger, M.; Heller, R.F.; Zwahlen, M. Body-mass index and incidence of cancer: A systematic review and meta-analysis of prospective observational studies. Lancet 2008, 371, 569–578. [Google Scholar] [CrossRef]
- Setiawan, V.W.; Lim, U.; Lipworth, L.; Lu, S.C.; Shepherd, J.; Ernst, T.; Wilkens, L.R.; Henderson, B.E.; Le Marchand, L. Sex and Ethnic Differences in the Association of Obesity With Risk of Hepatocellular Carcinoma. Clin. Gastroenterol. Hepatol. 2016, 14, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Ohki, T.; Tateishi, R.; Shiina, S.; Goto, E.; Sato, T.; Nakagawa, H.; Masuzaki, R.; Goto, T.; Hamamura, K.; Kanai, F.; et al. Visceral fat accumulation is an independent risk factor for hepatocellular carcinoma recurrence after curative treatment in patients with suspected NASH. Gut 2009, 58, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Cheung, O.K.-W.; Cheng, A.S. Gender Differences in Adipocyte Metabolism and Liver Cancer Progression. Front. Genet. 2016, 7, 168. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, E.; Tokushige, K. Prevalence, gender, ethnic variations, and prognosis of NASH. J. Gastroenterol. 2010, 46, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Wu, E.M.; Wong, L.L.; Hernandez, B.Y.; Ji, J.-F.; Jia, W.; Kwee, S.A.; Kalathil, S. Gender differences in hepatocellular cancer: Disparities in nonalcoholic fatty liver disease/steatohepatitis and liver transplantation. Hepatoma Res. 2018, 4, 66. [Google Scholar] [CrossRef] [PubMed]
- Rich, N.E.; Murphy, C.C.; Yopp, A.C.; Tiro, J.; Marrero, J.A.; Singal, A.G. Sex disparities in presentation and prognosis of 1110 patients with hepatocellular carcinoma. Aliment. Pharmacol. Ther. 2020, 52, 701–709. [Google Scholar] [CrossRef]
- Yang, D.; Hanna, D.L.; Usher, J.; Lococo, J.; Chaudhari, P.; Lenz, H.-J.; Setiawan, V.W.; El-Khoueiry, A. Impact of sex on the survival of patients with hepatocellular carcinoma: A Surveillance, Epidemiology, and End Results analysis. Cancer 2014, 120, 3707–3716. [Google Scholar] [CrossRef]
- Li, Y.; Li, H.; Spitsbergen, J.M.; Gong, Z. Males develop faster and more severe hepatocellular carcinoma than females in krasV12 transgenic zebrafish. Sci. Rep. 2017, 7, srep41280. [Google Scholar] [CrossRef]
- Nemoto, Y.; Toda, K.; Ono, M.; Fujikawa-Adachi, K.; Saibara, T.; Onishi, S.; Enzan, H.; Okada, T.; Shizuta, Y. Altered expression of fatty acid–metabolizing enzymes in aromatase-deficient mice. J. Clin. Investig. 2000, 105, 1819–1825. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Xu, G.; Jia, W.-D.; Han, S.-J.; Ren, W.-H.; Wang, W.; Liu, W.-B.; Zhang, C.-H.; Chen, H. Estrogen Suppresses Metastasis in Rat Hepatocellular Carcinoma through Decreasing Interleukin-6 and Hepatocyte Growth Factor Expression. Inflammation 2012, 35, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wei, Y.; Zhang, Y.; Xu, Y.; Li, F.; Liu, J.; Zhang, W.; Han, X.; Tan, R.; Shen, P. Oestrogen attenuates tumour progression in hepatocellular carcinoma. J. Pathol. 2012, 228, 216–229. [Google Scholar] [CrossRef]
- Hou, J.; Xu, J.; Jiang, R.; Wang, Y.; Chen, C.; Deng, L.; Huang, X.; Wang, X.; Sun, B. Estrogen-sensitive PTPRO expression represses hepatocellular carcinoma progression by control of STAT3. Hepatolgy 2013, 57, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.-W.; Yang, Y.-C.; Yang, S.-Y.; Cheng, S.-W.; Liaw, Y.-F.; Lin, S.-M.; Chen, C.-J. Hormonal Markers and Hepatitis B Virus-Related Hepatocellular Carcinoma Risk: A Nested Case-Control Study among Men. J. Natl. Cancer Inst. 2001, 93, 1644–1651. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.-L.; Hsu, C.; Wu, M.; Wu, C.; Wu, C.; Lai, J.; Jou, Y.; Cheng-Lung, H.; Yeh, S.; Chang, C. Androgen Receptor Is a New Potential Therapeutic Target for the Treatment of Hepatocellular Carcinoma. Gastroenterology 2008, 135, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, Y.; Lu, J.-W.; Huo, X.; Gong, Z. Liver-specific androgen receptor knockout attenuates early liver tumor development in zebrafish. Sci. Rep. 2019, 9, 10645. [Google Scholar] [CrossRef]
- Chen, Y.-M.A.; Shiu, J.-Y.A.; Tzeng, S.J.; Shih, L.-S.; Lui, W.-Y.; Chen, P.-H. Characterization of glycine-N-methyltransferase-gene expression in human hepatocellular carcinoma. Int. J. Cancer 1998, 75, 787–793. [Google Scholar] [CrossRef]
- Liao, Y.-J.; Liu, S.-P.; Lee, C.-M.; Yen, C.-H.; Chuang, P.-C.; Chen, C.-Y.; Tsai, T.-F.; Huang, S.-F.; Lee, Y.-H.W.; Chen, Y.-M.A. Characterization of a glycine N-methyltransferase gene knockout mouse model for hepatocellular carcinoma: Implications of the gender disparity in liver cancer susceptibility. Int. J. Cancer 2009, 124, 816–826. [Google Scholar] [CrossRef]
- Natri, H.M.; Wilson, M.A.; Buetow, K.H. Distinct molecular etiologies of male and female hepatocellular carcinoma. BMC Cancer 2019, 19, 951. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Ye, P.; Xi, Y.; Huang, Z.; Xu, P. Linking Obesity with Colorectal Cancer: Epidemiology and Mechanistic Insights. Cancers 2020, 12, 1408. [Google Scholar] [CrossRef] [PubMed]
- Bardou, M.; Barkun, A.N.; Martel, M. Obesity and colorectal cancer. Gut 2013, 62, 933–947. [Google Scholar] [CrossRef] [PubMed]
- Vazzana, N.; Riondino, S.; Toto, V.; Guadagni, F.; Roselli, M.; Davì, G.; Ferroni, P. Obesity-Driven Inflammation and Colorectal Cancer. Curr. Med. Chem. 2012, 19, 5837–5853. [Google Scholar] [CrossRef] [PubMed]
- Riondino, S.; Roselli, M.; Palmirotta, R.; Della-Morte, D.; Ferroni, P.; Guadagni, F. Obesity and colorectal cancer: Role of adipokines in tumor initiation and progression. World J. Gastroenterol. 2014, 20, 5177–5190. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Giovannucci, E.L. Sex differences in the association of obesity and colorectal cancer risk. Cancer Causes Control 2017, 28, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Colditz, G.A.; Peterson, L.L. Obesity and Cancer: Evidence, Impact, and Future Directions. Clin. Chem. 2018, 64, 154–162. [Google Scholar] [CrossRef]
- Garcia, H.; Song, M. Early-life obesity and adulthood colorectal cancer risk: A meta-analysis. Rev. Panam. Salud Pública 2019, 43, e3. [Google Scholar] [CrossRef]
- Jensen, B.W.; Bjerregaard, L.G.; Ängquist, L.; Gögenur, I.; Renehan, A.G.; Osler, M.; Sørensen, T.I.; Baker, J.L. Change in weight status from childhood to early adulthood and late adulthood risk of colon cancer in men: A population-based cohort study. Int. J. Obes. 2018, 42, 1797–1803. [Google Scholar] [CrossRef]
- Bull, C.J.; Bell, J.A.; Murphy, N.; Sanderson, E.; Smith, G.D.; Timpson, N.J.; Banbury, B.L.; Albanes, D.; Berndt, S.I.; Bézieau, S.; et al. Adiposity, metabolites, and colorectal cancer risk: Mendelian randomization study. BMC Med. 2020, 18, 396. [Google Scholar] [CrossRef]
- Kabat, G.C.; Kim, M.Y.; Stefanick, M.; Ho, G.Y.; Lane, D.S.; Odegaard, A.O.; Simon, M.S.; Bea, J.W.; Luo, J.; Wassertheil-Smoller, S.; et al. Metabolic obesity phenotypes and risk of colorectal cancer in postmenopausal women. Int. J. Cancer 2018, 143, 543–551. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Lee, H.-S.; Lee, D.-C.; Chu, S.-H.; Jeon, J.Y.; Kim, N.-K.; Lee, J.-W. Visceral Fat Accumulation Is Associated with Colorectal Cancer in Postmenopausal Women. PLoS ONE 2014, 9, e110587. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Kim, B.C.; Nam, S.Y.; Nam, J.H.; Ryu, K.H.; Park, B.J.; Sohn, D.K.; Hong, C.W.; Han, K.S.; Kim, H.B. Visceral Adipose Tissue Volume and the Occurrence of Colorectal Adenoma in Follow-up Colonoscopy for Screening and Surveillance. Nutr. Cancer 2017, 69, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Seo, I.-K.; Kim, B.J.; Kim, B.; Choi, C.H.; Kim, J.W.; Kim, J.G.; Chang, S.K.; Kang, H. Abdominal fat distribution measured using computed tomography is associated with an increased risk of colorectal adenoma in men. Medicine 2017, 96, e8051. [Google Scholar] [CrossRef] [PubMed]
- Haffa, M.; Holowatyj, A.N.; Kratz, M.; Toth, R.; Benner, A.; Gigic, B.; Habermann, N.; Schrotz-King, P.; Böhm, J.; Brenner, H.; et al. Transcriptome Profiling of Adipose Tissue Reveals Depot-Specific Metabolic Alterations among Patients with Colorectal Cancer. J. Clin. Endocrinol. Metab. 2019, 104, 5225–5237. [Google Scholar] [CrossRef]
- Del Cornò, M.; D’Archivio, M.; Conti, L.; Scazzocchio, B.; Varì, R.; Donninelli, G.; Varano, B.; Giammarioli, S.; De Meo, S.; Silecchia, G.; et al. Visceral fat adipocytes from obese and colorectal cancer subjects exhibit distinct secretory and ω6 polyunsaturated fatty acid profiles and deliver immunosuppressive signals to innate immunity cells. Oncotarget 2016, 7, 63093–63105. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, G.; He, J.; Ren, S.; Wu, F.; Zhang, J.; Wang, F. Gender differences in colorectal cancer survival: A meta-analysis. Int. J. Cancer 2017, 141, 1942–1949. [Google Scholar] [CrossRef]
- Majek, O.; Gondos, A.; Jansen, L.; Emrich, K.; Holleczek, B.; Katalinic, A.; Nennecke, A.; Eberle, A.; Brenner, H. Sex Differences in Colorectal Cancer Survival: Population-Based Analysis of 164,996 Colorectal Cancer Patients in Germany. PLoS ONE 2013, 8, e68077. [Google Scholar] [CrossRef]
- Koo, J.H.; Jalaludin, B.; Wong, S.K.C.; Kneebone, A.; Connor, S.J.; Leong, R.W. Improved Survival in Young Women with Colorectal Cancer. Am. J. Gastroenterol. 2008, 103, 1488–1495. [Google Scholar] [CrossRef]
- Chen, J.; Iverson, D. Estrogen in obesity-associated colon cancer: Friend or foe? Protecting postmenopausal women but promoting late-stage colon cancer. Cancer Causes Control 2012, 23, 1767–1773. [Google Scholar] [CrossRef]
- Hartman, J.; Edvardsson, K.; Lindberg, K.; Zhao, C.; Williams, C.; Ström, A.; Gustafsson, J.A. Tumor Repressive Functions of Estrogen Receptor β in SW480 Colon Cancer Cells. Cancer Res. 2009, 69, 6100–6106. [Google Scholar] [CrossRef]
- Papaxoinis, K.; Triantafyllou, K.; Sasco, A.J.; Nicolopoulou-Stamati, P.; Ladas, S.D. Subsite-specific differences of estrogen receptor beta expression in the normal colonic epithelium: Implications for carcinogenesis and colorectal cancer epidemiology. Eur. J. Gastroenterol. Hepatol. 2010, 22, 614–619. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Sundquist, J.; Sundquist, K. Use of hormone replacement therapy improves the prognosis in patients with colorectal cancer: A population-based study in Sweden. Int. J. Cancer 2018, 142, 2003–2010. [Google Scholar] [CrossRef] [PubMed]
- Hases, L.; Archer, A.; Indukuri, R.; Birgersson, M.; Savva, C.; Korach-André, M.; Williams, C. High-fat diet and estrogen impacts the colon and its transcriptome in a sex-dependent manner. Sci. Rep. 2020, 10, 16160. [Google Scholar] [CrossRef] [PubMed]
- Benedix, F.; Kube, R.; Meyer, F.; Schmidt, U.; Gastinger, I.; Lippert, H. Comparison of 17,641 Patients with Right- and Left-Sided Colon Cancer: Differences in Epidemiology, Perioperative Course, Histology, and Survival. Dis. Colon Rectum 2010, 53, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-E. Sex and gender-specific disparities in colorectal cancer risk. World J. Gastroenterol. 2015, 21, 5167–5175. [Google Scholar] [CrossRef] [PubMed]
- Warren, R.S.; Atreya, C.E.; Niedzwiecki, D.; Weinberg, V.K.; Donner, D.B.; Mayer, R.J.; Goldberg, R.M.; Compton, C.C.; Zuraek, M.B.; Ye, C.; et al. Association of TP53 Mutational Status and Gender with Survival after Adjuvant Treatment for Stage III Colon Cancer: Results of CALGB 89803. Clin. Cancer Res. 2013, 19, 5777–5787. [Google Scholar] [CrossRef]
- Bae, S.J.; Kim, J.W.; Kang, H.; Hwang, S.G.; Oh, D.; Kim, N.K. Gender-specific association between polymorphism of vascular endothelial growth factor (VEGF 936 C>T) gene and colon cancer in Korea. Anticancer. Res. 2008, 28, 1271–1276. [Google Scholar]
- Almutairi, M.; Parine, N.R.; Shaik, J.P.; Aldhaian, S.; Azzam, N.A.; Aljebreen, A.M.; Alharbi, O.; Almadi, M.A.; Al-Balbeesi, A.O.; Alanazi, M. Association between polymorphisms in PRNCR1 and risk of colorectal cancer in the Saudi population. PLoS ONE 2019, 14, e0220931. [Google Scholar] [CrossRef]
- Hasakova, K.; Vician, M.; Reis, R.; Zeman, M.; Herichova, I. Sex-dependent correlation between survival and expression of genes related to the circadian oscillator in patients with colorectal cancer. Chronobiol. Int. 2018, 35, 1423–1434. [Google Scholar] [CrossRef]
- Morgan, M.N.; Dvuchbabny, S.; Martinez, C.-A.; Kerr, B.; A Cistulli, P.; Cook, K.M. The Cancer Clock Is (Not) Ticking: Links between Circadian Rhythms and Cancer. Clocks Sleep 2019, 1, 34. [Google Scholar] [CrossRef]
- Alhinai, E.A.; Walton, G.E.; Commane, D.M. The Role of the Gut Microbiota in Colorectal Cancer Causation. Int. J. Mol. Sci. 2019, 20, 5295. [Google Scholar] [CrossRef] [PubMed]
- Sobhani, I.; Bergsten, E.; Couffin, S.; Amiot, A.; Nebbad, B.; Barau, C.; De’Angelis, N.; Rabot, S.; Canoui-Poitrine, F.; Mestivier, D.; et al. Colorectal cancer-associated microbiota contributes to oncogenic epigenetic signatures. Proc. Natl. Acad. Sci. USA 2019, 116, 24285–24295. [Google Scholar] [CrossRef] [PubMed]
- Campisciano, G.; De Manzini, N.; Delbue, S.; Cason, C.; Cosola, D.; Basile, G.; Ferrante, P.; Comar, M.; Palmisano, S. The Obesity-Related Gut Bacterial and Viral Dysbiosis Can Impact the Risk of Colon Cancer Development. Microorganisms 2020, 8, 431. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Roberts, J.D.; Grimm, S.A.; Lih, F.B.; Deterding, L.J.; Li, R.; Chrysovergis, K.; Wade, P.A. An obesity-associated gut microbiome reprograms the intestinal epigenome and leads to altered colonic gene expression. Genome Biol. 2018, 19, 7. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Kim, N.; Yoon, H.; Nam, R.H.; Lee, D.H. Microbial Changes and Host Response in F344 Rat Colon Depending on Sex and Age Following a High-Fat Diet. Front. Microbiol. 2018, 9, 2236. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heo, J.-W.; Kim, S.-E.; Sung, M.-K. Sex Differences in the Incidence of Obesity-Related Gastrointestinal Cancer. Int. J. Mol. Sci. 2021, 22, 1253. https://doi.org/10.3390/ijms22031253
Heo J-W, Kim S-E, Sung M-K. Sex Differences in the Incidence of Obesity-Related Gastrointestinal Cancer. International Journal of Molecular Sciences. 2021; 22(3):1253. https://doi.org/10.3390/ijms22031253
Chicago/Turabian StyleHeo, Ji-Won, Sung-Eun Kim, and Mi-Kyung Sung. 2021. "Sex Differences in the Incidence of Obesity-Related Gastrointestinal Cancer" International Journal of Molecular Sciences 22, no. 3: 1253. https://doi.org/10.3390/ijms22031253
APA StyleHeo, J.-W., Kim, S.-E., & Sung, M.-K. (2021). Sex Differences in the Incidence of Obesity-Related Gastrointestinal Cancer. International Journal of Molecular Sciences, 22(3), 1253. https://doi.org/10.3390/ijms22031253