Neurodegenerative Proteinopathies in the Proteoform Spectrum—Tools and Challenges



Abstract
1. Introduction
2. Neurodegenerative Proteinopathies
- Genetic variants (based on mutations).
- Isoforms (based on differences in post-transcriptional modifications).
- Proteoforms (based on differences in post-translation processing and three-dimensional conformation).
- Strains (based on differences in infectivity and incubation periods).
3. Utilizing Proteomic Platforms to Understand Neurodegenerative Proteinopathies
3.1. Two-Dimensional Gel Electrophoresis (2D-GE)
3.2. ESI and MALDI Based Top-Down Mass Spectrometry
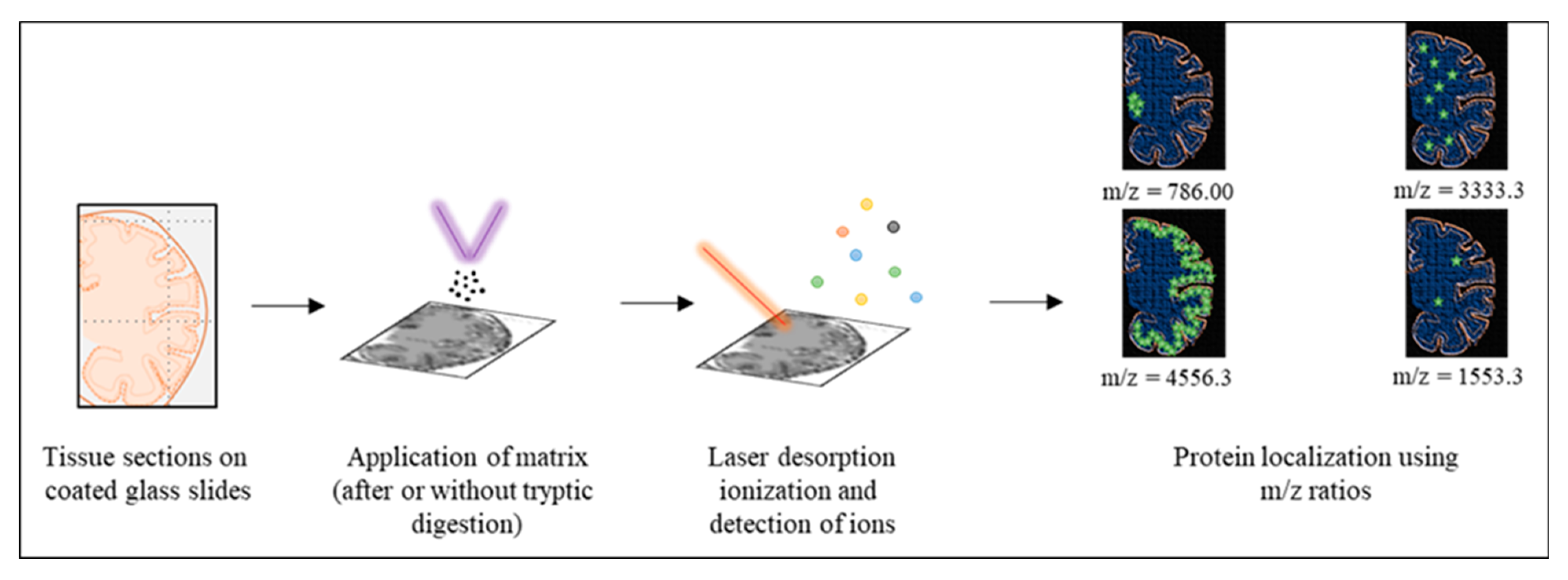
3.3. MALDI Imaging
3.4. Hydrogen/Deuterium Exchange Mass Spectrometry
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carrell, R.W.; Lomas, D.A. Conformational disease. Lancet 1997, 350, 134–138. [Google Scholar] [CrossRef]
- Serpell, L.C.; Sunde, M.; Benson, M.D.; A Tennent, G.; Pepys, M.B.; Fraser, P.E. The protofilament substructure of amyloid fibrils11Edited by F. E. Cohen. J. Mol. Biol. 2000, 300, 1033–1039. [Google Scholar] [CrossRef]
- Knowles, T.P.J.; Vendruscolo, M.; Dobson, C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef]
- Fraser, P.E. Prions and Prion-like Proteins. J. Biol. Chem. 2014, 289, 19839–19840. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Johansson, J.; Stromberg, R.; Nilsson, L. Unfolding of the Amyloid β-Peptide Central Helix: Mechanistic Insights from Molecular Dynamics Simulations. PLoS ONE 2011, 6, e17587. [Google Scholar] [CrossRef]
- Spagnolli, G.; Rigoli, M.; Inverardi, G.N.; Codeseira, Y.B.; Biasini, E.; Requena, J.R. Modeling PrPSc Generation Through Deformed Templating. Front. Bioeng. Biotechnol. 2020, 8, 590501. [Google Scholar] [CrossRef] [PubMed]
- Gillam, J.E.; Macphee, C.E. Modelling amyloid fibril formation kinetics: Mechanisms of nucleation and growth. J. Phys. Condens. Matter 2013, 25, 373101. [Google Scholar] [CrossRef] [PubMed]
- Khurana, R.; Ionescu-Zanetti, C.; Pope, M.; Li, J.; Nielson, L.; Ramírez-Alvarado, M.; Regan, L.; Fink, A.L.; Carter, S.A. A General Model for Amyloid Fibril Assembly Based on Morphological Studies Using Atomic Force Microscopy. Biophys. J. 2003, 85, 1135–1144. [Google Scholar] [CrossRef]
- Scheidt, T.; Łapińska, U.; Kumita, J.R.; Whiten, D.R.; Klenerman, D.; Wilson, M.R.; Cohen, S.I.A.; Linse, S.; Vendruscolo, M.; Dobson, C.M.; et al. Secondary nucleation and elongation occur at different sites on Alzheimer’s amyloid-β aggregates. Sci. Adv. 2019, 5, eaau3112. [Google Scholar] [CrossRef] [PubMed]
- Palmqvist, S.; Schöll, M.; Strandberg, O.; Mattsson, N.; Stomrud, E.; Zetterberg, H.; Blennow, K.; Landau, S.; Jagust, W.; Hansson, O. Earliest accumulation of β-amyloid occurs within the default-mode network and concurrently affects brain connectivity. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef]
- Sinha, M.S.; Ansell-Schultz, A.; Civitelli, L.; Hildesjö, C.; Larsson, M.; Lannfelt, L.; Ingelsson, M.; Hallbeck, M. Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol. 2018, 136, 41–56. [Google Scholar] [CrossRef]
- Theint, T.; Nadaud, P.S.; Aucoin, D.; Helmus, J.J.; Pondaven, S.P.; Surewicz, K.; Surewicz, W.K.; Jaroniec, C.P. Species-dependent structural polymorphism of Y145Stop prion protein amyloid revealed by solid-state NMR spectroscopy. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef]
- Ma, T.; Deng, J.; Ma, S.; Zhao, W.; Chang, Z.; Yu, K.; Yang, J. Structural Mechanism of Barriers to Interspecies Seeding Transmissibility of Full-Length Prion Protein Amyloid. ChemBioChem 2019, 20, 2757–2766. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2019 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Prince, M.; Guerchet, M.; Prina, M. The global impact of dementia 2013–2050. Alzheimer’s Dis. Int. 2013, 8, 23. [Google Scholar]
- Johnson, S.C.; Schmitz, T.; Moritz, C.; Meyerand, M.; Rowley, H.; Alexander, A.; Hansen, K.; Gleason, C.; Carlsson, C.; Ries, M.; et al. Activation of brain regions vulnerable to Alzheimer’s disease: The effect of mild cognitive impairment. Neurobiol. Aging 2006, 27, 1604–1612. [Google Scholar] [CrossRef]
- Sun, Q.; Xie, N.; Tang, B.; Li, R.; Shen, Y. Alzheimer’s Disease: From Genetic Variants to the Distinct Pathological Mechanisms. Front. Mol. Neurosci. 2017, 10, 319. [Google Scholar] [CrossRef]
- Cacabelos, R. Parkinson’s Disease: From Pathogenesis to Pharmacogenomics. Int. J. Mol. Sci. 2017, 18, 551. [Google Scholar] [CrossRef]
- Manix, M.; Kalakoti, P.; Henry, M.; Thakur, J.D.; Menger, R.; Guthikonda, B.; Nanda, A. Creutzfeldt-Jakob disease: Updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy. Neurosurg. Focus 2015, 39, E2. [Google Scholar] [CrossRef]
- Levin, J.; Kurz, A.; Arzberger, T.; Giese, A.; Höglinger, G.U. The Differential Diagnosis and Treatment of Atypical Parkinsonism. Dtsch. Aerzteblatt Online 2016, 113, 61–69. [Google Scholar] [CrossRef]
- Myers, R.H. Huntington’s disease genetics. NeuroRX 2004, 1, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Wijesekera, L.L.; Leigh, P.N. Amyotrophic lateral sclerosis. Orphanet J. Rare Dis. 2009, 4, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Govoni, V.; Della-Coletta, E.; Cesnik, E.; Casetta, I.; Granieri, E. Can the age at onset give a clue to the pathogenesis of ALS? Acta Neurol. Belg. 2016, 117, 221–227. [Google Scholar] [CrossRef]
- Knauer, M.F.; Soreghan, B.; Burdick, D.; Kosmoski, J.; Glabe, C.G. Intracellular accumulation and resistance to degradation of the Alzheimer amyloid A4/beta protein. Proc. Natl. Acad. Sci. USA 1992, 89, 7437–7441. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.; Wallon, D.; Goupil, C.; Richard, A.-C.; Pottier, C.; Dorval, V.; Sarov-Rivière, M.; Riant, F.; Hervé, D.; Amouyel, P.; et al. Mutation in the 3′untranslated region of APP as a genetic determinant of cerebral amyloid angiopathy. Eur. J. Hum. Genet. 2016, 24, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Wildburger, N.C.; Esparza, T.J.; LeDuc, R.D.; Fellers, R.T.; Thomas, P.M.; Cairns, N.J.; Kelleher, N.L.; Bateman, R.J.; Brody, D.L. Diversity of Amyloid-beta Proteoforms in the Alzheimer’s Disease Brain. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Trojanowski, J.Q.; Goedert, M.; Iwatsubo, T.; Lee, V.M.-Y. Fatal attractions: Abnormal protein aggregation and neuron death in Parkinson’s disease and Lewy body dementia. Cell Death Differ. 1998, 5, 832–837. [Google Scholar] [CrossRef]
- Kellie, J.F.; Higgs, R.E.; Ryder, J.W.; Major, A.; Beach, T.G.; Adler, C.H.; Merchant, K.; Knierman, M.D. Quantitative Measurement of Intact Alpha-Synuclein Proteoforms from Post-Mortem Control and Parkinson’s Disease Brain Tissue by Intact Protein Mass Spectrometry. Sci. Rep. 2014, 4, 5797. [Google Scholar] [CrossRef]
- Prusiner, S.B.; Westaway, D. Current ReviewInfectious and Genetic Manifestations of Prion Diseases. Mol. Plant. Microbe Interact. 1991, 4, 226–233. [Google Scholar] [CrossRef]
- Zafar, S.; Younas, N.; Shafiq, M.; Zerr, I. Prion Protein Strain Diversity and Disease Pathology. In Prions—Some Physiological and Pathophysiological Aspects; IntechOpen: London, UK, 2019. [Google Scholar]
- Mompeán, M.; Baralle, M.; Buratti, E.; Laurents, D.V. An Amyloid-Like Pathological Conformation of TDP-43 Is Stabilized by Hypercooperative Hydrogen Bonds. Front. Mol. Neurosci. 2016, 9, 125. [Google Scholar] [CrossRef]
- di Donato, M.; Craig, L.; Huff, M.E.; Thayer, M.M.; Cardoso, R.M.; Kassmann, C.J.; Lo, T.P.; Bruns, C.K.; Powers, E.T.; Kelly, J.W.; et al. ALS Mutants of Human Superoxide Dismutase Form Fibrous Aggregates Via Framework Destabilization. J. Mol. Biol. 2003, 332, 601–615. [Google Scholar] [CrossRef]
- Molnar, K.S.; Karabacak, N.M.; Johnson, J.L.; Wang, Q.; Tiwari, A.; Hayward, L.J.; Coales, S.J.; Hamuro, Y.; Agar, J.N. A Common Property of Amyotrophic Lateral Sclerosis-associated Variants. J. Biol. Chem. 2009, 284, 30965–30973. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Jakes, R. Mutations causing neurodegenerative tauopathies. Biochim. Biophys. Acta Mol. Basis Dis. 2005, 1739, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Lee, J.H.; Jeon, J.H.; Lee, M.J. Degradation or aggregation: The ramifications of post-translational modifications on tau. BMB Rep. 2018, 51, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, M.J. Transthyretin mutations in hyperthyroxinemia and amyloid diseases. Hum. Mutat. 2001, 17, 493–503. [Google Scholar] [CrossRef]
- Pont, L.; Poturcu, K.; Benavente, F.; Barbosa, J.; Sanz-Nebot, V. Comparison of capillary electrophoresis and capillary liquid chromatography coupled to mass spectrometry for the analysis of transthyretin in human serum. J. Chromatogr. A 2016, 1444, 145–153. [Google Scholar] [CrossRef]
- di Figlia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of Huntingtin in Neuronal Intranuclear Inclusions and Dystrophic Neurites in Brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef]
- Ehrnhoefer, D.E.; Sutton, L.; Hayden, M.R. Small changes, big impact: Posttranslational modifications and function of hun-tingtin in Huntington disease. Neuroscientist 2011, 17, 475–492. [Google Scholar] [CrossRef]
- Morales, R. Prion strains in mammals: Different conformations leading to disease. PLoS Pathog. 2017, 13, e1006323. [Google Scholar] [CrossRef]
- Lewis, P.A.; Tattum, M.H.; Jones, S.; Bhelt, D.; Batchelor, M.; Clarke, A.R.; Collinge, J.; Jackson, G.S. Codon 129 polymorphism of the human prion protein influences the kinetics of amyloid formation. J. Gen. Virol. 2006, 87, 2443–2449. [Google Scholar] [CrossRef]
- Jaunmuktane, Z.; Brandner, S. Transmissible human proteopathies: An expanding field. Diagn. Histopathol. 2019, 25, 16–22. [Google Scholar] [CrossRef]
- Petersen, C.; Nolan, A.L.; Resende, E.D.P.F.; Miller, Z.; Ehrenberg, A.J.; Gorno-Tempini, M.L.; Rosen, H.J.; Kramer, J.H.; Spina, S.; Rabinovici, G.D.; et al. Alzheimer’s disease clinical variants show distinct regional patterns of neurofibrillary tangle accumulation. Acta Neuropathol. 2019, 138, 597–612. [Google Scholar] [CrossRef]
- Tolosa, E.; Campdelacreu, J. Clinical overview of the synucleinopathies. Mov. Disord. 2003, 18, 21–27. [Google Scholar] [CrossRef]
- Irwin, D. Tauopathies as clinicopathological entities. Park. Relat. Disord. 2016, 22, S29–S33. [Google Scholar] [CrossRef]
- Burdick, D.; Soreghan, B.; Kwon, M.; Kosmoski, J.; Knauer, M.; Henschen, A.; Yates, J.; Cotman, C.; Glabe, C. Assembly and aggregation properties of synthetic Alzheimer’s A4/beta amyloid peptide analogs. J. Biol. Chem. 1992, 1, 546–554. [Google Scholar] [CrossRef]
- Martel, C.L.; Mackic, J.B.; McComb, J.; Ghiso, J.; Zlokovic, B.V. Blood-brain barrier uptake of the 40 and 42 amino acid sequences of circulating Alzheimer’s amyloid β in guinea pigs. Neurosci. Lett. 1996, 206, 157–160. [Google Scholar] [CrossRef]
- Chakraborty, S.; Das, P. Emergence of Alternative Structures in Amyloid Beta 1-42 Monomeric Landscape by N-terminal Hexapeptide Amyloid Inhibitors. Sci. Rep. 2017, 7, 9941. [Google Scholar] [CrossRef]
- Rasmussen, J.; Jucker, M.; Walker, L.C. Aβ seeds and prions: How close the fit? Prion 2017, 11, 215–225. [Google Scholar] [CrossRef]
- Schmidt, C.; Redyk, K.; Meissner, B.; Krack, L.; von Ahsen, N.; Roeber, S.; Kretzschmar, H.A.; Zerr, I. Clinical Features of Rapidly Progressive Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. 2010, 29, 371–378. [Google Scholar] [CrossRef]
- Garcia-Esparcia, P.; Sideris-Lampretsas, G.; Hernandez-Ortega, K.; Grau-Rivera, O.; Sklaviadis, T.; Gelpi, E.; & Ferrer, I. Al-tered mechanisms of protein synthesis in frontal cortex in Alzheimer disease and a mouse model. Am. J. Neurodegener. Dis. 2017, 6, 15. [Google Scholar]
- Espinoza, M.; de Silva, R.; Dickson, D.W.; Davies, P. Differential Incorporation of Tau Isoforms in Alzheimer’s Disease. J. Alzheimer’s Dis. 2008, 14, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Boban, M.; Šarac, H.; Mimica, N.; Mladinov, M.; Süßmair, C.; Ackl, N.; Šimić, G. CSF tau proteins in differential di-agnosis of dementia. Transl. Neurosci. 2010, 1, 43–48. [Google Scholar] [CrossRef][Green Version]
- Galante, D.; Corsaro, A.; Florio, T.; Vella, S.; Pagano, A.; Sbrana, F.; Vassalli, M.; Perico, A.; D’Arrigo, C. Differential toxicity, conformation and morphology of typical initial aggregation states of Aβ1-42 and Aβpy3-42 beta-amyloids. Int. J. Biochem. Cell Biol. 2012, 44, 2085–2093. [Google Scholar] [CrossRef]
- Stein, K.C.; True, H.L. Prion Strains and Amyloid Polymorphism Influence Phenotypic Variation. PLoS Pathog. 2014, 10, e1004328. [Google Scholar] [CrossRef]
- Peters, C.; Bascuñán, D.; Opazo, C.; Aguayo, L.G. Differential Membrane Toxicity of Amyloid-β Fragments by Pore Forming Mechanisms. J. Alzheimer’s Dis. 2016, 51, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Qiang, W.; Yau, W.-M.; Lu, J.-X.; Collinge, J.; Tycko, R. Structural variation in amyloid-β fibrils from Alzheimer’s disease clinical subtypes. Nat. Cell Biol. 2017, 541, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Grassi, D.; Howard, S.; Zhou, M.; Diaz-Perez, N.; Urban, N.T.; Guerrero-Given, D.; Kamasawa, N.; Volpicelli-Daley, L.A.; Lograsso, P.; Lasmézas, C.I. Identification of a highly neurotoxic α-synuclein species inducing mitochondrial damage and mitophagy in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, E2634–E2643. [Google Scholar] [CrossRef]
- Zhan, X.; Zhou, T. Application of Two-Dimensional Gel Electrophoresis in Combination with Mass Spectrometry in the Study of Hormone Proteoforms. In Mass Spectrometry—Future Perceptions and Applications; IntechOpen: London, UK, 2019. [Google Scholar]
- Silva, C.J. Applying the tools of chemistry (mass spectrometry and covalent modification by small molecule reagents) to the detection of prions and the study of their structure. Prion 2014, 8, 42–50. [Google Scholar] [CrossRef]
- Patrie, S.M. Top-Down Mass Spectrometry: Proteomics to Proteoforms. In Taurine 6; Springer Nature: Berlin/Heidelberg, Germany, 2016; Volume 919, pp. 171–200. [Google Scholar]
- Stoeckli, M.; Chaurand, P.; Hallahan, D.E.; Caprioli, R.M. Imaging mass spectrometry: A new technology for the analysis of protein expression in mammalian tissues. Nat. Med. 2001, 7, 493–496. [Google Scholar] [CrossRef]
- di Lillo, M.; Ait-Belkacem, R.; Esteve, C.; Pellegrini, D.; Nicolardi, S.; Costa, M.; Vannini, E.; de Graaf, E.L.; Caleo, M.; McDonnell, L.A. Ultra-High Mass Resolution MALDI Imaging Mass Spectrometry of Proteins and Metabolites in a Mouse Model of Glioblastoma. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Huang, R.Y.-C.; Iacob, R.E.; Sankaranarayanan, S.; Yang, L.; Ahlijanian, M.; Tao, L.; Tymiak, A.A.; Chen, G. Probing Conformational Dynamics of Tau Protein by Hydrogen/Deuterium Exchange Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2017, 29, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, F. Highlights on the capacities of “Gel-based” proteomics. Proteome Sci. 2010, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, P.H. High resolution two-dimensional electrophoresis of proteins. J. Biol. Chem. 1975, 250, 4007–4021. [Google Scholar] [CrossRef]
- Zong, C.; Young, G.W.; Wang, Y.; Lu, H.; Deng, N.; Drews, O.; Ping, P. Two-dimensional electrophoresis-based characterization of post-translational modifications of mammalian 20S proteasome complexes. Proteomics 2008, 8, 5025–5037. [Google Scholar] [CrossRef]
- Nizhnikov, A.A.; Alexandrov, A.I.; Ryzhova, T.A.; Mitkevich, O.V.; Dergalev, A.A.; Ter-Avanesyan, M.D.; Galkin, A.P. Proteomic Screening for Amyloid Proteins. PLoS ONE 2014, 9, e116003. [Google Scholar] [CrossRef]
- Kaplan, B.; Shtrasburg, S.; Pras, M. Micropurification techniques in the analysis of amyloid proteins. J. Clin. Pathol. 2003, 56, 86–90. [Google Scholar] [CrossRef]
- Esparza, T.J.; Wildburger, N.C.; Jiang, H.; Gangolli, M.; Cairns, N.J.; Bateman, R.J.; Brody, D.L. Soluble Amyloid-beta Aggregates from Human Alzheimer’s Disease Brains. Sci. Rep. 2016, 6, 38187. [Google Scholar] [CrossRef]
- Kryndushkin, D.S.; Alexandrov, I.M.; Ter-Avanesyan, M.D.; Kushnirov, V.V. Yeast [PSI+] Prion Aggregates Are Formed by Small Sup35 Polymers Fragmented by Hsp104. J. Biol. Chem. 2003, 278, 49636–49643. [Google Scholar] [CrossRef]
- Schmittschmitt, J.P.; Scholtz, J.M. The role of protein stability, solubility, and net charge in amyloid fibril formation. Protein Sci. 2009, 12, 2374–2378. [Google Scholar] [CrossRef]
- Proc, J.L.; Kuzyk, M.A.; Hardie, D.B.; Yang, J.; Smith, D.S.; Jackson, A.M.; Parker, C.E.; Borchers, C.H. A Quantitative Study of the Effects of Chaotropic Agents, Surfactants, and Solvents on the Digestion Efficiency of Human Plasma Proteins by Trypsin. J. Proteome Res. 2010, 9, 5422–5437. [Google Scholar] [CrossRef]
- Rabilloud, T.; Lelong, C. Two-dimensional gel electrophoresis in proteomics: A tutorial. J. Proteom. 2011, 74, 1829–1841. [Google Scholar] [CrossRef]
- Kang, S.-C.; Li, R.; Wang, C.; Pan, T.; Liu, T.; Rubenstein, R.; Barnard, G.; Wong, B.-S.; Sy, M.-S. Guanidine hydrochloride extraction and detection of prion proteins in mouse and hamster prion diseases by ELISA. J. Pathol. 2003, 199, 534–541. [Google Scholar] [CrossRef]
- Kim, J.R.; Muresan, A.; Lee, K.Y.C.; Murphy, R.M. Urea modulation of β-amyloid fibril growth: Experimental studies and kinetic models. Protein Sci. 2008, 13, 2888–2898. [Google Scholar] [CrossRef]
- Vernaglia, B.A.; Huang, J.; Clark, E.D. Guanidine Hydrochloride Can Induce Amyloid Fibril Formation from Hen Egg-White Lysozyme. Biomacromolecules 2004, 5, 1362–1370. [Google Scholar] [CrossRef]
- Maler, J.M.; Klafki, H.-W.; Paul, S.; Spitzer, P.; Groemer, T.W.; Henkel, A.W.; Esselmann, H.; Lewczuk, P.; Kornhuber, J.; Wiltfang, J. Urea-based two-dimensional electrophoresis of beta-amyloid peptides in human plasma: Evidence for novel Aβ species. Proteomics 2007, 7, 3815–3820. [Google Scholar] [CrossRef]
- Kawooya, J.K.; Emmons, T.L.; A Gonzalez-DeWhitt, P.; Camp, M.C.; D’Andrea, S.C. Electrophoretic mobility of Alzheimer’s amyloid-β peptides in urea–sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Anal. Biochem. 2003, 323, 103–113. [Google Scholar] [CrossRef]
- Wiltfang, J.; Esselmann, H.; Bibl, M.; Smirnov, A.; Otto, M.; Paul, S.; Schmidt, B.; Klafki, H.-W.; Maler, M.; Dyrks, T.; et al. Highly conserved and disease-specific patterns of carboxyterminally truncated Aβ peptides 1-37/38/39 in addition to 1-40/42 in Alzheimer’s disease and in patients with chronic neuroinflammation. J. Neurochem. 2002, 81, 481–496. [Google Scholar] [CrossRef]
- Katorcha, E.; Baskakov, I.V. Analysis of Covalent Modifications of Amyloidogenic Proteins Using Two-Dimensional Electrophoresis: Prion Protein and Its Sialylation. Toxic. Assess. 2018, 1779, 241–255. [Google Scholar] [CrossRef]
- Fiorini, M.; Bongianni, M.; Monaco, S.; Zanusso, G. Biochemical Characterization of Prions. Prog. Mol. Biol. Transl. Sci. 2017, 150, 389–407. [Google Scholar] [CrossRef]
- Shen, P.; Dang, J.; Wang, Z.; Zhang, W.; Yuan, J.; Lang, Y.; Ding, M.; Mitchell, M.; Kong, Q.; Feng, J.; et al. Characterization of Anchorless Human PrP With Q227X Stop Mutation Linked to Gerstmann-Sträussler-Scheinker Syndrome In Vivo and In Vitro. Mol. Neurobiol. 2021, 58, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.; Shelanski, M.L. Microheterogeneity of Micro tubule-Associated ? Proteins Is Due to Differences in Phosphorylation. J. Neurochem. 1986, 47, 1517–1522. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, A.; Nishizawa, Y.; Matsuura, I.; Hayashi, F.; Usukura, J.; Bondarenko, V.A. Microtubule-associated protein tau in bovine retinal photoreceptor rod outer segments: Comparison with brain tau. Biochim. Biophys. Acta Bioenerg. 2013, 1832, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Sergeant, N.; Fernández-Gómez, F.J.; Obriot, H.; Eddarkaoui, S.; Buée-Scherrer, V.; Buée, L. Two-Dimensional Electrophoresis Protocols to Analyze the Microtubule-Associated Tau Proteins from Several Biological Sources. Adv. Struct. Saf. Stud. 2016, 1523, 251–261. [Google Scholar] [CrossRef]
- Aebersold, R.; Agar, J.N.; Amster, I.J.; Baker, M.S.; Bertozzi, C.R.; Boja, E.S.; Costello, C.E.; Cravatt, B.F.; Fenselau, C.; A Garcia, B.; et al. How many human proteoforms are there? Nat. Chem. Biol. 2018, 14, 206–214. [Google Scholar] [CrossRef]
- Kelleher, N.L.; Costello, C.A.; Begley, T.P.; McLafferty, F.W. Thiaminase I (42 kDa) heterogeneity, sequence refinement, and active site location from high-resolution tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 1995, 6, 981–984. [Google Scholar] [CrossRef][Green Version]
- Mortz, E.; O’Connor, P.B.; Roepstorff, P.; Kelleher, N.L.; Wood, T.D.; McLafferty, F.W.; Mann, M. Sequence tag identification of intact proteins by matching tanden mass spectral data against sequence data bases. Proc. Natl. Acad. Sci. USA 1996, 93, 8264–8267. [Google Scholar] [CrossRef]
- Toby, T.K.; Fornelli, L.; Kelleher, N.L. Progress in Top-Down Proteomics and the Analysis of Proteoforms. Annu. Rev. Anal. Chem. 2016, 9, 499–519. [Google Scholar] [CrossRef]
- Wu, S.; Brown, J.N.; Tolić, N.; Meng, D.; Liu, X.; Zhang, H.; Zhao, R.; Moore, R.J.; Pevzner, P.; Smith, R.D.; et al. Quantitative analysis of human salivary gland-derived intact proteome using top-down mass spectrometry. Proteom. 2014, 14, 1211–1222. [Google Scholar] [CrossRef]
- Brodie, N.I.; Huguet, R.; Zhang, T.; Viner, R.; Zabrouskov, V.; Pan, J.; Borchers, C.H. Top-down hydrogen–deuterium ex-change analysis of protein structures using ultraviolet photodissociation. Anal. Chem. 2018, 90, 3079–3082. [Google Scholar] [CrossRef]
- Davis, R.G.; Park, H.M.; Kim, K.; Greer, J.B.; Fellers, R.T.; LeDuc, R.D.; Yau, P.M. Top-Down Proteomics Enables Com-parative Analysis of Brain Proteoforms Between Mouse Strains. Anal. Chem. 2018, 90, 3802–3810. [Google Scholar] [CrossRef] [PubMed]
- Wongkongkathep, P.; Han, J.Y.; Choi, T.S.; Yin, S.; Kim, H.I.; Loo, J.A. Native Top-Down Mass Spectrometry and Ion Mobility MS for Characterizing the Cobalt and Manganese Metal Binding of α-Synuclein Protein. J. Am. Soc. Mass Spectrom. 2018, 29, 1870–1880. [Google Scholar] [CrossRef] [PubMed]
- Nshanian, M.; Lantz, C.; Wongkongkathep, P.; Schrader, T.; Klärner, F.-G.; Blümke, A.; Despres, C.; Ehrmann, M.; Smet-Nocca, C.; Bitan, G.; et al. Native Top-Down Mass Spectrometry and Ion Mobility Spectrometry of the Interaction of Tau Protein with a Molecular Tweezer Assembly Modulator. J. Am. Soc. Mass Spectrom. 2018, 30, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Mori, H.; Cardiff, R.D. Methods of Immunohistochemistry and Immunofluorescence: Converting Invisible to Visible. Toxic. Assess. 2016, 1458, 1–12. [Google Scholar] [CrossRef]
- Ucal, Y.; Durer, Z.A.; Atak, H.; Kadioglu, E.; Sahin, B.; Coskun, A.; Baykal, A.T.; Ozpinar, A. Clinical applications of MALDI imaging technologies in cancer and neurodegenerative diseases. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 795–816. [Google Scholar] [CrossRef]
- Winter, M.; Tholey, A.; Kristen, A.; Röcken, C. MALDI Mass Spectrometry Imaging: A Novel Tool for the Identification and Classification of Amyloidosis. Proteomics 2017, 17, 1700236. [Google Scholar] [CrossRef]
- Angel, P.M.; Norris-Caneda, K.; Drake, R.R. In Situ Imaging of Tryptic Peptides by MALDI Imaging Mass Spectrometry Using Fresh-Frozen or Formalin-Fixed, Paraffin-Embedded Tissue. Curr. Protoc. Protein Sci. 2018, 94, e65. [Google Scholar] [CrossRef]
- Ikegawa, M.; Nirasawa, T.; Kakuda, N.; Miyasaka, T.; Kuzuhara, Y.; Murayama, S.; Ihara, Y. Visualization of Amyloid β Deposits in the Human Brain with Matrix-assisted Laser Desorption/Ionization Imaging Mass Spectrometry. J. Vis. Exp. 2019, 145, e57645. [Google Scholar] [CrossRef]
- Michno, W.; Wehrli, P.M.; Blennow, K.; Zetterberg, H.; Hanrieder, J. Molecular imaging mass spectrometry for probing protein dynamics in neurodegenerative disease pathology. J. Neurochem. 2019, 151, 488–506. [Google Scholar] [CrossRef]
- Ryan, D.J.; Spraggins, J.M.; Caprioli, R.M. Protein identification strategies in MALDI imaging mass spectrometry: A brief review. Curr. Opin. Chem. Biol. 2019, 48, 64–72. [Google Scholar] [CrossRef]
- Carlred, L.; Michno, W.; Kaya, I.; Sjövall, P.; Syvänen, S.; Hanrieder, J. Probing amyloid-β pathology in transgenic Alzheimer’s disease (tgArcSwe) mice using MALDI imaging mass spectrometry. J. Neurochem. 2016, 138, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Kakuda, N.; Miyasaka, T.; Iwasaki, N.; Nirasawa, T.; Wada-Kakuda, S.; Takahashi-Fujigasaki, J.; Murayama, S.; Ihara, Y.; Ikegawa, M. Distinct deposition of amyloid-β species in brains with Alzheimer’s disease pathology visualized with MALDI imaging mass spectrometry. Acta Neuropathol. Commun. 2017, 5, 73. [Google Scholar] [CrossRef] [PubMed]
- Arribat, Y.; Talmat-Amar, Y.; Paucard, A.; Lesport, P.; Bonneaud, N.; Bauer, C.; Bec, N.; Parmentier, M.-L.; Benigno, L.; Larroque, C.; et al. Systemic delivery of P42 peptide: A new weapon to fight Huntington’s disease. Acta Neuropathol. Commun. 2014, 2, 86. [Google Scholar] [CrossRef] [PubMed]
- Hunter, M.; de Marais, N.J.; Faull, R.L.; Grey, A.C.; Curtis, M.A. Subventricular zone lipidomic architecture loss in Huntington’s disease. J. Neurochem. 2018, 146, 613–630. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Calvo, P.; Xiao, X.; Bett, C.; Eraña, H.; Soldau, K.; Castilla, J.; Nilsson, K.P.R.; Surewicz, W.K.; Sigurdson, C.J. Post-translational modifications in PrP expand the conformational diversity of prions in vivo. Sci. Rep. 2017, 7, srep43295. [Google Scholar] [CrossRef]
- Masson, G.R.; Burke, J.E.; Ahn, N.G.; Anand, G.S.; Borchers, C.; Brier, S.; Bou-Assaf, G.M.; Engen, J.R.; Englander, S.W.; Faber, J.; et al. Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (HDX-MS) experiments. Nat. Methods 2019, 16, 595–602. [Google Scholar] [CrossRef]
- Smirnovas, V.; Baron, G.S.; Offerdahl, D.K.; Raymond, G.J.; Caughey, B.; Surewicz, W.K. Structural organization of brain-derived mammalian prions examined by hydrogen-deuterium exchange. Nat. Struct. Mol. Biol. 2011, 18, 504–506. [Google Scholar] [CrossRef]
- Zhang, Y.; Rempel, D.L.; Zhang, J.; Sharma, A.K.; Mirica, L.M.; Gross, M.L. Pulsed hydrogen–deuterium exchange mass spectrometry probes conformational changes in amyloid beta (Aβ) peptide aggregation. Proc. Natl. Acad. Sci. USA 2013, 110, 14604–14609. [Google Scholar] [CrossRef]
- Moulick, R.; Das, R.; Udgaonkar, J.B. Partially Unfolded Forms of the Prion Protein Populated under Misfolding-promoting Conditions. J. Biol. Chem. 2015, 290, 25227–25240. [Google Scholar] [CrossRef]
- Li, Q.; Wang, F.; Xiao, X.; Kim, C.; Bohon, J.; Kiselar, J.; Safar, J.G.; Ma, J.; Surewicz, W.K. Structural attributes of mammalian prion infectivity: Insights from studies with synthetic prions. J. Biol. Chem. 2018, 293, 18494–18503. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proteinopathy | Age at Onset (Years) | Primary Region | Common Symptoms |
|---|---|---|---|
| AD | 40–65 (early and late-onset variants) | Hippocampus and entorhinal cortex. | Memory and language impairment and visuospatial deficits. [16,17] |
| PD | 40–50 | Substantia nigra (midbrain). | Rigidity, resting tremor and bradykinesia. [18] |
| sCJD * | 44–70 (depends on subtype) | Cerebral cortex and cerebellum. | Cognitive impairment and myoclonus. [19] |
| DLB | 50–80 | Midbrain and neocortex. | Parkinsonian syndrome, autonomic and sleep fluctuations and hallucinations. [20] |
| HD | 20–49 | Caudate nucleus and putamen (basal ganglia). | Choreiform movements, emotional and behavioral alterations, bradykinesia. [21] |
| ALS | 45–55 | Motor neurons. | Focal muscle wasting, spasticity and flexor spasms. [22,23] |
| Amyloids | Precursor Protein | Associated Diseases | Proteoforms or Other Variants |
|---|---|---|---|
| Aβ | Amyloid beta A4 protein: Intrinsically disordered protein with 770 residues | AD, Cerebral amyloid angiopathy (CAA) [24,25]. | 26 differentially truncated and post translationally modified proteoforms [26] |
| α-Synuclein | Alpha Synuclein: Intrinsically disordered protein with 140 residues | PD and DLB [27] | 11 differentially truncated and post translationally modified proteoforms [28] |
| PrPSc | Major prion protein: Intrinsically disordered protein with 253 amino acids | CJD, Fatal Familial Insomnia (FFI), Gerstmann-Straussler-Scheinker disease (GSS), Huntington disease-like type 1 (HDL1), Kuru and Spongiform encephalopathy [29] | 2 Proteoforms based on Proteinase-K resistance Genetic variants (codon 129 polymorphism). [30] |
| ASOD | Superoxide dismutase: Intrinsically disordered protein with 154 amino acids | ALS—TDP-43 amyloids also involved. [31,32] | Genetic variants. No proteoforms reported yet. [33] |
| ATau | Microtubule-associated protein tau: Intrinsically disordered protein with 758 amino acids | Frontotemporal dementia (FTD), AD, Progressive Supranuclear Palsy (PSP), Corticobasal degeneration (CBD), Pick’s disease, Argyrophilic grain disease, Dementia with Lewy bodies and Parkinsonism linked to chromosome 17. [34] | Six isoforms. Differentially post translationally modified proteoforms. [35] |
| ATTR | Transthyretin: Mostly β-sheet with 147 amino acids | Familial Amyloid polyneuropathy, Leptomeningeal amyloidosis. [36] | Differentially oxidized proteoforms. [37] |
| AHtt | Huntington: Intrinsically disordered protein with 3142 residues | Huntington disease. [38] | Differentially post translationally modified proteoforms. [39] |
| Technique | Utility for Amyloids | Samples | Previously Targetted Amyloids |
|---|---|---|---|
| 2D-GE | Resolves minor biochemical variations among proteoforms by targeting isoelectric points and molecular weights. | Solubilized proteins with native charges preferably monomeric species (multimeric species may provide misleading results if more than one proteoform is involved). Buffers selected must not induce aggregation under experimental conditions. | PrP, Aβ, Tau |
| Top-Down MS | Identifies proteoforms and their post-translational modifications in their native forms. | Undigested proteins in their native conformations. Buffers that prevent aggregation but do not affect the spectrum of target. In case of MALDI, matices have to be tested for their capability to ionize the target. | Aβ, tau, α-Synuclein |
| MALDI IMS | Locates proteins via in situ identification of proteoforms. | Paraffin-embedded or frozen tissue sections. Matices have to be tested for their capability to ionize the target. | Aβ, tau |
| HDX-MS | Depicts 3D structures of proteins. | Undenatured, undigested proteins in their native conformations. Experimental conditions have to be carefully controlled to prevent uneven deutrium labelling among replicates. | PrP, Aβ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noor, A.; Zafar, S.; Zerr, I. Neurodegenerative Proteinopathies in the Proteoform Spectrum—Tools and Challenges. Int. J. Mol. Sci. 2021, 22, 1085. https://doi.org/10.3390/ijms22031085
Noor A, Zafar S, Zerr I. Neurodegenerative Proteinopathies in the Proteoform Spectrum—Tools and Challenges. International Journal of Molecular Sciences. 2021; 22(3):1085. https://doi.org/10.3390/ijms22031085
Chicago/Turabian StyleNoor, Aneeqa, Saima Zafar, and Inga Zerr. 2021. "Neurodegenerative Proteinopathies in the Proteoform Spectrum—Tools and Challenges" International Journal of Molecular Sciences 22, no. 3: 1085. https://doi.org/10.3390/ijms22031085
APA StyleNoor, A., Zafar, S., & Zerr, I. (2021). Neurodegenerative Proteinopathies in the Proteoform Spectrum—Tools and Challenges. International Journal of Molecular Sciences, 22(3), 1085. https://doi.org/10.3390/ijms22031085