Emerging Role of the Inflammasome and Pyroptosis in Hypertension



Abstract
1. Inflammation and Hypertension
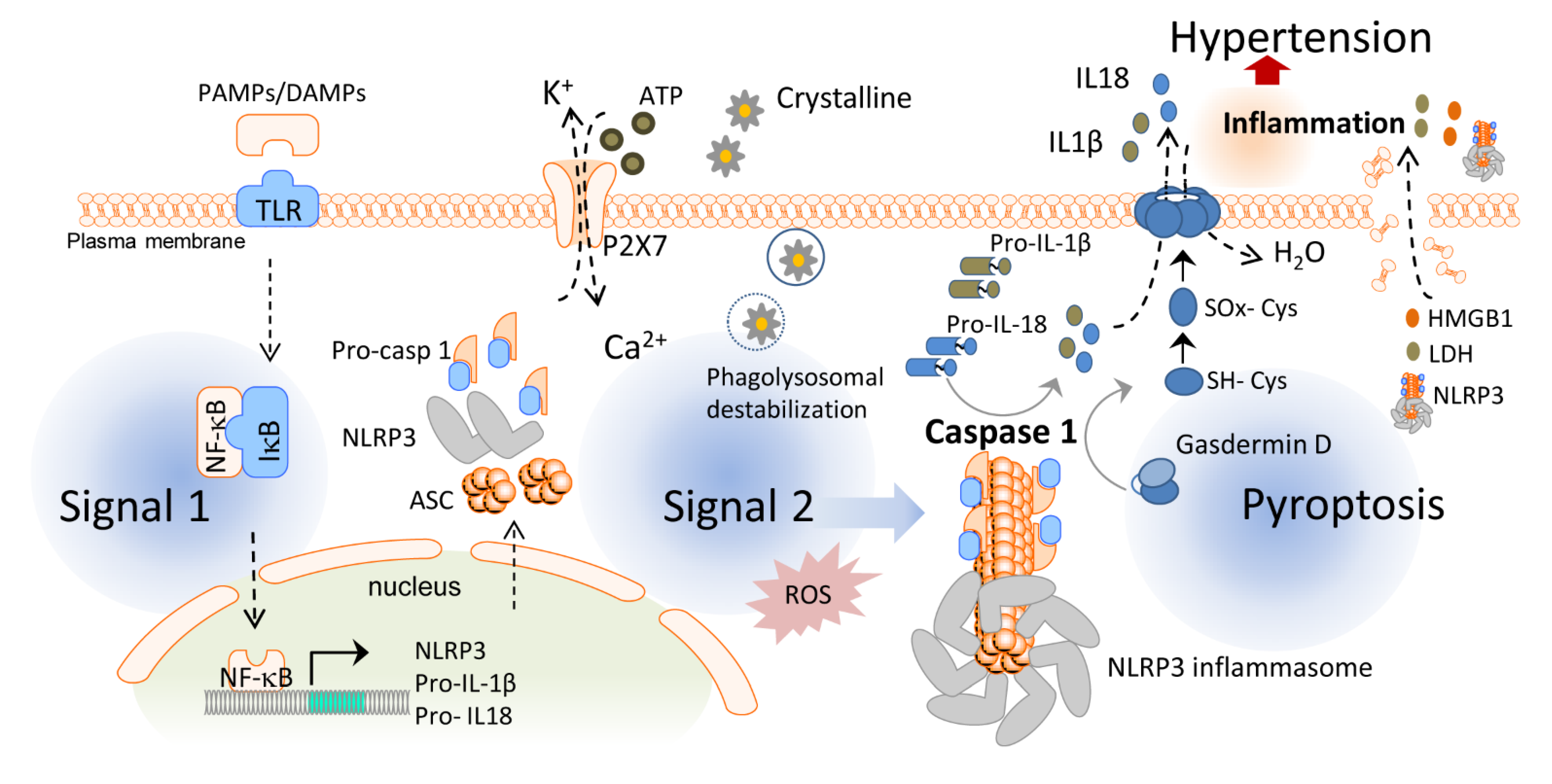
2. Inflammasomes and Pyroptotic Cell Death
3. Novel Roles of the NLRP3 Inflammasome and Pyroptosis in Blood Pressure Regulation
3.1. NLRP3 Inflammasome in Human Pulmonary Hypertension
3.2. NLRP3 Inflammasome in Pre-Eclampsia and Systemic Hypertension
3.3. Renal Inflammasome Activation and Pyroptosis in Hypertension
3.4. Vascular Inflammasome and Pyroptosis Activation in Hypertension
3.5. Hypothalamic Activation of the Inflammasome During Hypertension
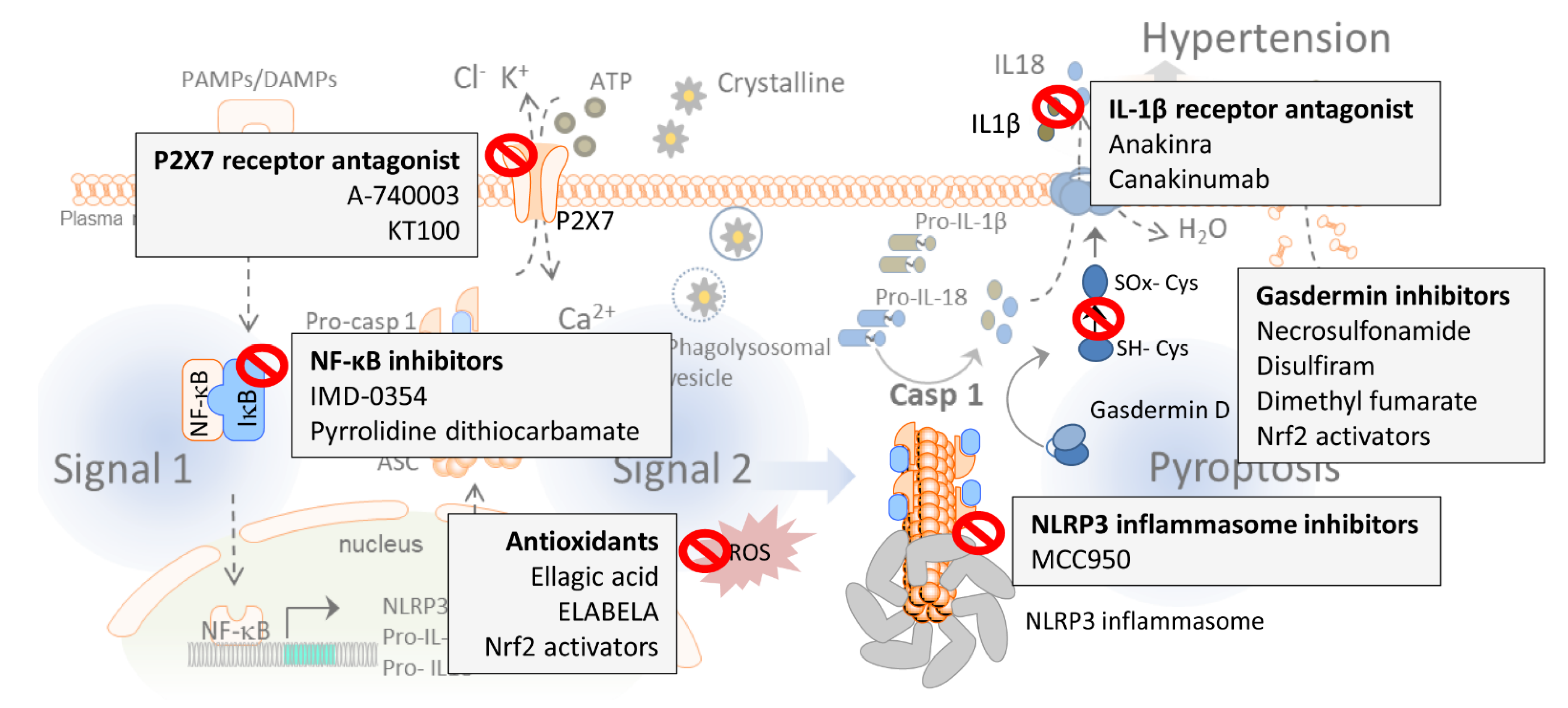
4. Targeting the NLRP3 Inflammasome and Pyroptosis in Hypertension: Emerging Pharmacological Approaches
4.1. P2X7 Receptors Antagonism
4.2. Reactive Oxygen Species (ROS) Production Inhibitors
4.3. NLRP3 Inhibitors
4.4. NF-κB Inhibitors
4.5. IL-1β Receptor Antagonism
4.6. Gasdermin D Inhibitors
5. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Whelton, P.K.; Carey, R.M.; Aronow, W.S.; Casey, D.E., Jr.; Collins, K.J.; Dennison Himmelfarb, C.; De Palma, S.M.; Gidding, S.; Jamerson, K.A.; Jones, D.W.; et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: A report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines. Circulation 2018, 138, e484–e594. [Google Scholar] [CrossRef] [PubMed]
- Global Burden of Metabolic Risk Factors for Chronic Diseases Collaboration. Cardiovascular disease, chronic kidney disease, and diabetes mortality burden of cardiometabolic risk factors from 1980 to 2010: A comparative risk assessment. Lancet Diabetes Endocrinol. 2014, 2, 634–647. [Google Scholar] [CrossRef]
- Outeda, P.; Menezes, L.; Hartung, E.A.; Bridges, S.; Zhou, F.; Zhu, X.; Xu, H.; Huang, Q.; Yao, Q.; Qian, F.; et al. A novel model of autosomal recessive polycystic kidney questions the role of the fibrocystin C-terminus in disease mechanism. Kidney Int. 2017, 92, 1130–1144. [Google Scholar] [CrossRef] [PubMed]
- Bromfield, S.; Muntner, P. High blood pressure: The leading global burden of disease risk factor and the need for worldwide prevention programs. Curr. Hypertens. Rep. 2013, 15, 134–136. [Google Scholar] [CrossRef]
- Sokabe, H.; Grollman, A. A study of hypertension in the rat induced by infarction of the kidney. Tex. Rep. Biol. Med. 1963, 21, 93–100. [Google Scholar] [PubMed]
- White, F.N.; Grollman, A. Autoimmune Factors associated with infarction of the kidney. Nephron 1964, 1, 93–102. [Google Scholar] [CrossRef]
- Okuda, T.; Grollman, A. Passive transfer of autoimmune induced hypertension in the rat by lymph node cells. Tex. Rep. Biol. Med. 1967, 25, 257–264. [Google Scholar]
- Svendsen, U.G. Evidence for an initial, thymus independent and a chronic, thymus dependent phase of DOCA and salt hypertension in mice. Acta Pathol. Microbiol. Scand. A 1976, 84, 523–528. [Google Scholar] [CrossRef]
- Svendsen, U.G. The role of thymus for the development and prognosis of hypertension and hypertensive vascular disease in mice following renal infarction. Acta Pathol. Microbiol. Scand. A 1976, 84, 235–243. [Google Scholar] [CrossRef]
- Svendsen, U.G. The importance of thymus for hypertension and hypertensive vascular disease in rats and mice. Acta Pathol. Microbiol. Scand. Suppl. 1978, 267, 1–15. [Google Scholar]
- Caillon, A.; Mian, M.O.R.; Fraulob-Aquino, J.C.; Huo, K.G.; Barhoumi, T.; Ouerd, S.; Sinnaeve, P.R.; Paradis, P.; Schiffrin, E.L. γδ T cells mediate angiotensin ii-induced hypertension and vascular injury. Circulation 2017, 135, 2155–2162. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Iturbe, B.; Vaziri, N.D.; Herrera-Acosta, J.; Johnson, R.J. Oxidative stress, renal infiltration of immune cells, and salt-sensitive hypertension: All for one and one for all. Am. J. Physiol. Renal Physiol. 2004, 286, F606–F616. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Rodriguez-Iturbe, B.; Nakagawa, T.; Kang, D.H.; Feig, D.I.; Herrera-Acosta, J. Subtle renal injury is likely a common mechanism for salt-sensitive essential hypertension. Hypertension 2005, 45, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Herrera-Acosta, J.; Schreiner, G.F.; Rodriguez-Iturbe, B. Subtle acquired renal injury as a mechanism of salt-sensitive hypertension. N. Engl. J. Med. 2002, 346, 913–923. [Google Scholar] [CrossRef]
- Guzik, T.J.; Hoch, N.E.; Brown, K.A.; McCann, L.A.; Rahman, A.; Dikalov, S.; Goronzy, J.; Weyand, C.; Harrison, D.G. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J. Exp. Med. 2007, 204, 2449–2460. [Google Scholar] [CrossRef] [PubMed]
- Barbaro, N.R.; Foss, J.D.; Kryshtal, D.O.; Tsyba, N.; Kumaresan, S.; Xiao, L.; Mernaugh, R.L.; Itani, H.A.; Loperena, R.; Chen, W.; et al. Dendritic cell amiloride-sensitive channels mediate sodium-induced inflammation and hypertension. Cell Rep. 2017, 21, 1009–1020. [Google Scholar] [CrossRef]
- Van Beusecum, J.P.; Barbaro, N.R.; McDowell, Z.; Aden, L.A.; Xiao, L.; Pandey, A.K.; Itani, H.A.; Himmel, L.E.; Harrison, D.G.; Kirabo, A.; et al. High salt activates CD11c(+) antigen-presenting cells via SGK (Serum Glucocorticoid Kinase) 1 to promote renal inflammation and salt-sensitive hypertension. Hypertension 2019, 74, 555–563. [Google Scholar] [CrossRef]
- Norlander, A.E.; Saleh, M.A.; Pandey, A.K.; Itani, H.A.; Wu, J.; Xiao, L.; Kang, J.; Dale, B.L.; Goleva, S.B.; Laroumanie, F.; et al. A salt-sensing kinase in T lymphocytes, SGK1, drives hypertension and hypertensive end-organ damage. JCI Insight. 2017, 2. [Google Scholar] [CrossRef]
- Itani, H.A.; McMaster, W.G., Jr.; Saleh, M.A.; Nazarewicz, R.R.; Mikolajczyk, T.P.; Kaszuba, A.M.; Konior, A.; Prejbisz, A.; Januszewicz, A.; Norlander, A.E.; et al. Activation of human T cells in hypertension: Studies of Humanized mice and hypertensive humans. Hypertension 2016, 68, 123–132. [Google Scholar] [CrossRef]
- Fehrenbach, D.J.; Abais-Battad, J.M.; Dasinger, J.H.; Lund, H.; Keppel, T.; Zemaj, J.; Cherian-Shaw, M.; Gundry, R.L.; Geurts, A.M.; Dwinell, M.R.; et al. Sexual dimorphic role of CD14 (Cluster of Differentiation 14) in Salt-sensitive hypertension and renal injury. Hypertension 2021, 77, 228–240. [Google Scholar] [CrossRef]
- Alsheikh, A.J.; Dasinger, J.H.; Abais-Battad, J.M.; Fehrenbach, D.J.; Yang, C.; Cowley, A.W., Jr.; Mattson, D.L. CCL2 mediates early renal leukocyte infiltration during salt-sensitive hypertension. Am. J. Physiol. Renal Physiol. 2020, 318, F982–F993. [Google Scholar] [CrossRef] [PubMed]
- Fehrenbach, D.J.; Abais-Battad, J.M.; Dasinger, J.H.; Lund, H.; Mattson, D.L. Salt-sensitive increase in macrophages in the kidneys of Dahl SS rats. Am. J. Physiol. Renal Physiol. 2019, 317, F361–F374. [Google Scholar] [CrossRef] [PubMed]
- Abais-Battad, J.M.; Lund, H.; Fehrenbach, D.J.; Dasinger, J.H.; Mattson, D.L. Rag1-null Dahl SS rats reveal that adaptive immune mechanisms exacerbate high protein-induced hypertension and renal injury. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R28–R35. [Google Scholar] [CrossRef] [PubMed]
- De Miguel, C.; Lund, H.; Mattson, D.L. High dietary protein exacerbates hypertension and renal damage in Dahl SS rats by increasing infiltrating immune cells in the kidney. Hypertension 2011, 57, 269–274. [Google Scholar] [CrossRef]
- De Miguel, C.; Das, S.; Lund, H.; Mattson, D.L. T lymphocytes mediate hypertension and kidney damage in Dahl salt-sensitive rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1136–R1142. [Google Scholar] [CrossRef]
- De Miguel, C.; Guo, C.; Lund, H.; Feng, D.; Mattson, D.L. Infiltrating T lymphocytes in the kidney increase oxidative stress and participate in the development of hypertension and renal disease. Am. J. Physiol. Renal Physiol. 2011, 300, F734–F742. [Google Scholar] [CrossRef]
- Khraibi, A.A.; Norman, R.A., Jr.; Dzielak, D.J. Chronic immunosuppression attenuates hypertension in Okamoto spontaneously hypertensive rats. Am. J. Physiol. 1984, 247, H722–H726. [Google Scholar] [CrossRef]
- Norman, R.A., Jr.; Galloway, P.G.; Dzielak, D.J.; Huang, M. Mechanisms of partial renal infarct hypertension. J. Hypertens. 1988, 6, 397–403. [Google Scholar] [CrossRef]
- Mattson, D.L.; James, L.; Berdan, E.A.; Meister, C.J. Immune suppression attenuates hypertension and renal disease in the Dahl salt-sensitive rat. Hypertension 2006, 48, 149–156. [Google Scholar] [CrossRef]
- Mattson, D.L.; Lund, H.; Guo, C.; Rudemiller, N.; Geurts, A.M.; Jacob, H. Genetic mutation of recombination activating gene 1 in Dahl salt-sensitive rats attenuates hypertension and renal damage. Am. J. Physiol. Regul Integr. Comp. Physiol. 2013, 304, R407–R414. [Google Scholar] [CrossRef]
- Rudemiller, N.P.; Lund, H.; Priestley, J.R.; Endres, B.T.; Prokop, J.W.; Jacob, H.J.; Geurts, A.M.; Cohen, E.P.; Mattson, D.L. Mutation of SH2B3 (LNK), a genome-wide association study candidate for hypertension, attenuates Dahl salt-sensitive hypertension via inflammatory modulation. Hypertension 2015, 65, 1111–1117. [Google Scholar] [CrossRef]
- Lu, X.; Rudemiller, N.P.; Privratsky, J.R.; Ren, J.; Wen, Y.; Griffiths, R.; Crowley, S.D. Classical dendritic cells mediate hypertension by promoting renal oxidative stress and fluid retention. Hypertension 2020, 75, 131–138. [Google Scholar] [CrossRef]
- Zhang, J.; Rudemiller, N.P.; Patel, M.B.; Karlovich, N.S.; Wu, M.; McDonough, A.A.; Griffiths, R.; Sparks, M.A.; Jeffs, A.D.; Crowley, S.D.; et al. Interleukin-1 receptor activation potentiates salt reabsorption in angiotensin II-induced hypertension via the NKCC2 Co-transporter in the Nephron. Cell Metab. 2016, 23, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Madhur, M.S.; Lob, H.E.; McCann, L.A.; Iwakura, Y.; Blinder, Y.; Guzik, T.J.; Harrison, D.G. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension 2010, 55, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.A.; McMaster, W.G.; Wu, J.; Norlander, A.E.; Funt, S.A.; Thabet, S.R.; Kirabo, A.; Xiao, L.; Chen, W.; Itani, H.A.; et al. Lymphocyte adaptor protein LNK deficiency exacerbates hypertension and end-organ inflammation. J. Clin. Investig. 2015, 125, 1189–1202. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.T.; Sobey, C.G.; Lieu, M.; Ferens, D.; Kett, M.M.; Diep, H.; Kim, H.A.; Krishnan, S.M.; Lewis, C.V.; Salimova, E.; et al. Obligatory role for B cells in the development of angiotensin II-dependent hypertension. Hypertension 2015, 66, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Caillon, A.; Paradis, P.; Schiffrin, E.L. Role of immune cells in hypertension. Br. J. Pharmacol. 2019, 176, 1818–1828. [Google Scholar] [CrossRef] [PubMed]
- Wade, B.; Abais-Battad, J.M.; Mattson, D.L. Role of immune cells in salt-sensitive hypertension and renal injury. Curr. Opin. Nephrol. Hypertens. 2016, 25, 22–27. [Google Scholar] [CrossRef]
- De Miguel, C.; Rudemiller, N.P.; Abais, J.M.; Mattson, D.L. Inflammation and hypertension: New understandings and potential therapeutic targets. Curr. Hypertens. Rep. 2015, 17, 507. [Google Scholar] [CrossRef]
- Xiao, L.; Harrison, D.G. Inflammation in hypertension. Can. J. Cardiol. 2020, 36, 635–647. [Google Scholar] [CrossRef]
- Bomfim, G.F.; Rodrigues, F.L.; Carneiro, F.S. Are the innate and adaptive immune systems setting hypertension on fire? Pharmacol. Res. 2017, 117, 377–393. [Google Scholar] [CrossRef]
- Mattson, D.L. Immune mechanisms of salt-sensitive hypertension and renal end-organ damage. Nat. Rev. Nephrol. 2019, 15, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Foulquier, S. Brain perivascular macrophages: Connecting inflammation to autonomic activity in hypertension. Hypertens. Res. 2020, 43, 148–150. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.J. An update on immune system activation in the pathogenesis of hypertension. Hypertension 2013, 62, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.G.; Marvar, P.J.; Titze, J.M. Vascular inflammatory cells in hypertension. Front Physiol. 2012, 3, 128. [Google Scholar] [CrossRef] [PubMed]
- Mattson, D.L. Infiltrating immune cells in the kidney in salt-sensitive hypertension and renal injury. Am. J. Physiol. Renal Physiol. 2014, 307, F499–F508. [Google Scholar] [CrossRef]
- Markó, L.; Kvakan, H.; Park, J.K.; Qadri, F.; Spallek, B.; Binger, K.J.; Bowman, E.P.; Kleinewietfeld, M.; Fokuhl, V.; Dechend, R.; et al. Interferon-γ signaling inhibition ameliorates angiotensin II-induced cardiac damage. Hypertension 2012, 60, 1430–1436. [Google Scholar] [CrossRef]
- Ramseyer, V.D.; Garvin, J.L. Tumor necrosis factor-α: Regulation of renal function and blood pressure. Am. J. Physiol. Renal Physiol. 2013, 304, F1231–F1242. [Google Scholar] [CrossRef]
- Lee, D.L.; Sturgis, L.C.; Labazi, H.; Osborne, J.B., Jr.; Fleming, C.; Pollock, J.S.; Manhiani, M.; Imig, J.D.; Brands, M.W. Angiotensin II hypertension is attenuated in interleukin-6 knockout mice. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H935–H940. [Google Scholar] [CrossRef]
- Wu, J.; Thabet, S.R.; Kirabo, A.; Trott, D.W.; Saleh, M.A.; Xiao, L.; Madhur, M.S.; Chen, W.; Harrison, D.G. Inflammation and mechanical stretch promote aortic stiffening in hypertension through activation of p38 mitogen-activated protein kinase. Circ. Res. 2014, 114, 616–625. [Google Scholar] [CrossRef]
- Satou, R.; Miyata, K.; Gonzalez-Villalobos, R.A.; Ingelfinger, J.R.; Navar, L.G.; Kobori, H. Interferon-γ biphasically regulates angiotensinogen expression via a JAK-STAT pathway and suppressor of cytokine signaling 1 (SOCS1) in renal proximal tubular cells. FASEB J. 2012, 26, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Costerousse, O.; Allegrini, J.; Lopez, M.; Alhenc-Gelas, F. Angiotensin I-converting enzyme in human circulating mononuclear cells: Genetic polymorphism of expression in T-lymphocytes. Biochem. J. 1993, 290, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Jurewicz, M.; McDermott, D.H.; Sechler, J.M.; Tinckam, K.; Takakura, A.; Carpenter, C.B.; Milford, E.; Abdi, R. Human T and natural killer cells possess a functional renin-angiotensin system: Further mechanisms of angiotensin II-induced inflammation. J. Am. Soc. Nephrol. 2007, 18, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Okamura, A.; Rakugi, H.; Ohishi, M.; Yanagitani, Y.; Takiuchi, S.; Moriguchi, K.; Fennessy, P.A.; Higaki, J.; Ogihara, T. Upregulation of renin-angiotensin system during differentiation of monocytes to macrophages. J. Hypertens. 1999, 17, 537–545. [Google Scholar] [CrossRef]
- Elisa, T.; Antonio, P.; Giuseppe, P.; Alessandro, B.; Giuseppe, A.; Federico, C.; Marzia, D.; Ruggero, B.; Giacomo, M.; Andrea, O.; et al. Endothelin receptors expressed by immune cells are involved in modulation of inflammation and in fibrosis: Relevance to the pathogenesis of systemic sclerosis. J. Immunol. Res. 2015, 2015, 147616. [Google Scholar] [CrossRef]
- Soldano, S.; Pizzorni, C.; Paolino, S.; Trombetta, A.C.; Montagna, P.; Brizzolara, R.; Ruaro, B.; Sulli, A.; Cutolo, M. Alternatively activated (M2) macrophage phenotype is inducible by endothelin-1 in cultured human macrophages. PLoS ONE 2016, 11, e0166433. [Google Scholar] [CrossRef]
- Bray, M.A.; Gordon, D. Prostaglandin production by macrophages and the effect of anti-inflammatory drugs. Br. J. Pharmacol. 1978, 63, 635–642. [Google Scholar] [CrossRef]
- Lone, A.; Taskén, K. Proinflammatory and immunoregulatory roles of eicosanoids in T Cells. Front. Immunol. 2013, 4. [Google Scholar] [CrossRef]
- Cardinale, J.P.; Sriramula, S.; Mariappan, N.; Agarwal, D.; Francis, J. Angiotensin II-induced hypertension is modulated by nuclear factor-κBin the paraventricular nucleus. Hypertension 2012, 59, 113–121. [Google Scholar] [CrossRef]
- Masson, G.S.; Costa, T.S.; Yshii, L.; Fernandes, D.C.; Soares, P.P.; Laurindo, F.R.; Scavone, C.; Michelini, L.C. Time-dependent effects of training on cardiovascular control in spontaneously hypertensive rats: Role for brain oxidative stress and inflammation and baroreflex sensitivity. PLoS ONE 2014, 9, e94927. [Google Scholar] [CrossRef]
- Rodríguez-Iturbe, B.; Franco, M.; Tapia, E.; Quiroz, Y.; Johnson, R.J. Renal inflammation, autoimmunity and salt-sensitive hypertension. Clin. Exp. Pharmacol. Physiol. 2012, 39, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Kamat, N.V.; Thabet, S.R.; Xiao, L.; Saleh, M.A.; Kirabo, A.; Madhur, M.S.; Delpire, E.; Harrison, D.G.; McDonough, A.A. Renal transporter activation during angiotensin-II hypertension is blunted in interferon-γ-/- and interleukin-17A-/- mice. Hypertension 2015, 65, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Caruso, R.; Warner, N.; Inohara, N.; Nunez, G. NOD1 and NOD2: Signaling, host defense, and inflammatory disease. Immunity 2014, 41, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Enosi Tuipulotu, D.; Tan, W.H.; Kay, C.; Man, S.M. Emerging activators and regulators of inflammasomes and pyroptosis. Trends Immunol. 2019, 40, 1035–1052. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.I.; Lu, A.; Chen, J.W.; Ruan, J.; Tang, C.; Wu, H.; Ploegh, H.L. A single domain antibody fragment that recognizes the adaptor ASC defines the role of ASC domains in inflammasome assembly. J. Exp. Med. 2016, 213, 771–790. [Google Scholar] [CrossRef]
- Chan, A.H.; Schroder, K. Inflammasome signaling and regulation of interleukin-1 family cytokines. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Van Opdenbosch, N.; Gurung, P.; Vande Walle, L.; Fossoul, A.; Kanneganti, T.D.; Lamkanfi, M. Activation of the NLRP1b inflammasome independently of ASC-mediated caspase-1 autoproteolysis and speck formation. Nat. Commun. 2014, 5, 3209. [Google Scholar] [CrossRef]
- Broz, P.; Pelegrin, P.; Shao, F. The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 2020, 20, 143–157. [Google Scholar] [CrossRef]
- Miao, E.A.; Rajan, J.V.; Aderem, A. Caspase-1-induced pyroptotic cell death. Immunol. Rev. 2011, 243, 206–214. [Google Scholar] [CrossRef]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, C.; Yang, J.; Zhou, B.; Yang, R.; Ramachandran, R.; Abbott, D.W.; Xiao, T.S. Crystal structures of the full-length murine and human gasdermin d reveal mechanisms of autoinhibition, lipid binding, and oligomerization. Immunity 2019, 51, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Xia, S.; Liu, X.; Lieberman, J.; Wu, H. Cryo-EM structure of the gasdermin A3 membrane pore. Nature 2018, 557, 62–67. [Google Scholar] [CrossRef]
- Vince, J.E.; Silke, J. The intersection of cell death and inflammasome activation. Cell Mol. Life Sci. 2016, 73, 2349–2367. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.B.; Nakashima, A.; Huber, W.J.; Davis, S.; Banerjee, S.; Huang, Z.; Saito, S.; Sadovsky, Y.; Sharma, S. Pyroptosis is a critical inflammatory pathway in the placenta from early onset preeclampsia and in human trophoblasts exposed to hypoxia and endoplasmic reticulum stressors. Cell Death Dis. 2019, 10, 927. [Google Scholar] [CrossRef]
- Man, S.M.; Karki, R.; Kanneganti, T.D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Dombrowski, Y.; Peric, M.; Koglin, S.; Kammerbauer, C.; Goss, C.; Anz, D.; Simanski, M.; Glaser, R.; Harder, J.; Hornung, V.; et al. Cytosolic DNA triggers inflammasome activation in keratinocytes in psoriatic lesions. Sci. Transl. Med. 2011, 3, 82ra38. [Google Scholar] [CrossRef] [PubMed]
- Raupach, B.; Peuschel, S.K.; Monack, D.M.; Zychlinsky, A. Caspase-1-mediated activation of interleukin-1beta (IL-1beta) and IL-18 contributes to innate immune defenses against Salmonella enterica serovar Typhimurium infection. Infect. Immun. 2006, 74, 4922–4926. [Google Scholar] [CrossRef]
- Monack, D.M.; Raupach, B.; Hromockyj, A.E.; Falkow, S. Salmonella typhimurium invasion induces apoptosis in infected macrophages. Proc. Natl. Acad. Sci. USA 1996, 93, 9833–9838. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef]
- Gong, W.; Shi, Y.; Ren, J. Research progresses of molecular mechanism of pyroptosis and its related diseases. Immunobiology 2020, 225, 151884. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Kanneganti, T.D. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nature Rev. Immunol. 2016, 16, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Xia, S. Biological mechanisms and therapeutic relevance of the gasdermin family. Mol. Aspects Med. 2020. [Google Scholar] [CrossRef]
- Tartey, S.; Kanneganti, T.D. Inflammasomes in the pathophysiology of autoinflammatory syndromes. J. Leukoc. Biol. 2020, 107, 379–391. [Google Scholar] [CrossRef]
- de Torre-Minguela, C.; Mesa del Castillo, P.; Pelegrín, P. The NLRP3 and Pyrin Inflammasomes: Implications in the Pathophysiology of Autoinflammatory Diseases. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Jo, E.K.; Kim, J.K.; Shin, D.M.; Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell. Mol. Immunol. 2016, 13, 148–159. [Google Scholar] [CrossRef]
- Martín-Sánchez, F.; Martínez-García, J.J.; Muñoz-García, M.; Martínez-Villanueva, M.; Noguera-Velasco, J.A.; Andreu, D.; Rivas, L.; Pelegrín, P. Lytic cell death induced by melittin bypasses pyroptosis but induces NLRP3 inflammasome activation and IL-1β release. Cell Death Dis. 2017, 8, e2984. [Google Scholar] [CrossRef]
- Baroja-Mazo, A.; Martin-Sanchez, F.; Gomez, A.I.; Martinez, C.M.; Amores-Iniesta, J.; Compan, V.; Barbera-Cremades, M.; Yague, J.; Ruiz-Ortiz, E.; Anton, J.; et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat. Immunol. 2014, 15, 738–748. [Google Scholar] [CrossRef]
- Compan, V.; Baroja-Mazo, A.; Lopez-Castejon, G.; Gomez, A.I.; Martinez, C.M.; Angosto, D.; Montero, M.T.; Herranz, A.S.; Bazan, E.; Reimers, D.; et al. Cell volume regulation modulates NLRP3 inflammasome activation. Immunity 2012, 37, 487–500. [Google Scholar] [CrossRef]
- Pelegrin, P. P2X7 receptor and the NLRP3 inflammasome: Partners in crime. Biochem. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Weber, A.N.R.; Bittner, Z.A.; Shankar, S.; Liu, X.; Chang, T.H.; Jin, T.; Tapia-Abellan, A. Recent insights into the regulatory networks of NLRP3 inflammasome activation. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef] [PubMed]
- Prochnicki, T.; Mangan, M.S.; Latz, E. Recent insights into the molecular mechanisms of the NLRP3 inflammasome activation. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Planillo, R.; Kuffa, P.; Martinez-Colon, G.; Smith, B.L.; Rajendiran, T.M.; Nunez, G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Akira, S. TLR signaling pathways. Semin. Immunol. 2004, 16, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.S. Functional crosstalk between non-canonical caspase-11 and canonical NLRP3 inflammasomes during infection-mediated inflammation. Immunology 2020, 159, 142–155. [Google Scholar] [CrossRef]
- Evavold, C.L.; Ruan, J.; Tan, Y.; Xia, S.; Wu, H.; Kagan, J.C. The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity 2018, 48, 35–44. [Google Scholar] [CrossRef]
- Ruhl, S.; Shkarina, K.; Demarco, B.; Heilig, R.; Santos, J.C.; Broz, P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 2018, 362, 956–960. [Google Scholar] [CrossRef]
- Baroja-Mazo, A.; Compan, V.; Martin-Sanchez, F.; Tapia-Abellan, A.; Couillin, I.; Pelegrin, P. Early endosome autoantigen 1 regulates IL-1beta release upon caspase-1 activation independently of gasdermin D membrane permeabilization. Sci. Rep. 2019, 9, 5788. [Google Scholar] [CrossRef]
- Briard, B.; Fontaine, T.; Samir, P.; Place, D.E.; Muszkieta, L.; Malireddi, R.K.S.; Karki, R.; Christgen, S.; Bomme, P.; Vogel, P.; et al. Galactosaminogalactan activates the inflammasome to provide host protection. Nature 2020, 588, 688–692. [Google Scholar] [CrossRef]
- Chen, J.; Chen, Z.J. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 2018, 564, 71–76. [Google Scholar] [CrossRef]
- Magupalli, V.G.; Negro, R.; Tian, Y.; Hauenstein, A.V.; di Caprio, G.; Skillern, W.; Deng, Q.; Orning, P.; Alam, H.B.; Maliga, Z.; et al. HDAC6 mediates an aggresome-like mechanism for NLRP3 and pyrin inflammasome activation. Science 2020, 369. [Google Scholar] [CrossRef]
- Zhu, J.; Yang, Y.; Hu, S.G.; Zhang, Q.B.; Yu, J.; Zhang, Y.M. T-lymphocyte K(v)1.3 channel activation triggers the NLRP3 inflammasome signaling pathway in hypertensive patients. Exp. Ther. Med. 2017, 14, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ortega, M.; Lorenzo, O.; Rupérez, M.; Blanco, J.; Egido, J. Systemic infusion of angiotensin II into normal rats activates nuclear factor-kappaB and AP-1 in the kidney: Role of AT(1) and AT(2) receptors. Am. J. Pathol. 2001, 158, 1743–1756. [Google Scholar] [CrossRef]
- Dalekos, G.N.; Elisaf, M.; Bairaktari, E.; Tsolas, O.; Siamopoulos, K.C. Increased serum levels of interleukin-1beta in the systemic circulation of patients with essential hypertension: Additional risk factor for atherogenesis in hypertensive patients? J. Lab. Clin. Med. 1997, 129, 300–308. [Google Scholar] [CrossRef]
- Rabkin, S.W. The role of interleukin 18 in the pathogenesis of hypertension-induced vascular disease. Nature Clinical Practice. Cardiovasc. Med. 2009, 6, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Kunnas, T.; Maatta, K.; Nikkari, S.T. NLR family pyrin domain containing 3 (NLRP3) inflammasome gene polymorphism rs7512998 (C>T) predicts aging-related increase of blood pressure, the TAMRISK study. Immun. Ageing 2015, 12, 19. [Google Scholar] [CrossRef] [PubMed]
- Omi, T.; Kumada, M.; Kamesaki, T.; Okuda, H.; Munkhtulga, L.; Yanagisawa, Y.; Utsumi, N.; Gotoh, T.; Hata, A.; Soma, M.; et al. An intronic variable number of tandem repeat polymorphisms of the cold-induced autoinflammatory syndrome 1 (CIAS1) gene modifies gene expression and is associated with essential hypertension. Eur. J. Hum. Genet. 2006, 14, 1295–1305. [Google Scholar] [CrossRef]
- Scott, T.E.; Kemp-Harper, B.K.; Hobbs, A.J. Inflammasomes: A novel therapeutic target in pulmonary hypertension? Br. J. Pharmacol. 2019, 176, 1880–1896. [Google Scholar] [CrossRef]
- Heng, T.S.; Painter, M.W. The immunological genome project: Networks of gene expression in immune cells. Nat. Immunol. 2008, 9, 1091–1094. [Google Scholar] [CrossRef]
- Xiang, M.; Shi, X.; Li, Y.; Xu, J.; Yin, L.; Xiao, G.; Scott, M.J.; Billiar, T.R.; Wilson, M.A.; Fan, J. Hemorrhagic shock activation of NLRP3 inflammasome in lung endothelial cells. J. Immunol. 2011, 187, 4809–4817. [Google Scholar] [CrossRef]
- Xia, M.; Boini, K.M.; Abais, J.M.; Xu, M.; Zhang, Y.; Li, P.L. Endothelial NLRP3 inflammasome activation and enhanced neointima formation in mice by adipokine visfatin. Am. J. Pathol. 2014, 184, 1617–1628. [Google Scholar] [CrossRef] [PubMed]
- De Nardo, D.; de Nardo, C.M.; Latz, E. New insights into mechanisms controlling the NLRP3 inflammasome and its role in lung disease. Am. J. Pathol. 2014, 184, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, T.; Kato, S.; Oka, N.; Imaoka, H.; Kinoshita, T.; Takei, S.; Kitasato, Y.; Kawayama, T.; Imaizumi, T.; Yamada, K.; et al. Pulmonary inflammation and emphysema: Role of the cytokines IL-18 and IL-13. Am. J. Respir. Crit. Care Med. 2007, 176, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Morisawa, D.; Hirotani, S.; Oboshi, M.; Nishimura, K.; Sawada, H.; Eguchi, A.; Okuhara, Y.; Iwasaku, T.; Naito, Y.; Mano, T.; et al. Interleukin-18 disruption suppresses hypoxia-induced pulmonary artery hypertension in mice. Int. J. Cardiol. 2016, 202, 522–524. [Google Scholar] [CrossRef]
- Kang, M.J.; Homer, R.J.; Gallo, A.; Lee, C.G.; Crothers, K.A.; Cho, S.J.; Rochester, C.; Cain, H.; Chupp, G.; Yoon, H.J.; et al. IL-18 is induced and IL-18 receptor alpha plays a critical role in the pathogenesis of cigarette smoke-induced pulmonary emphysema and inflammation. J. Immunol. 2007, 178, 1948–1959. [Google Scholar] [CrossRef]
- Cero, F.T.; Hillestad, V.; Sjaastad, I.; Yndestad, A.; Aukrust, P.; Ranheim, T.; Lunde, I.G.; Olsen, M.B.; Lien, E.; Zhang, L.; et al. Absence of the inflammasome adaptor ASC reduces hypoxia-induced pulmonary hypertension in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L378–L387. [Google Scholar] [CrossRef]
- Soon, E.; Holmes, A.M.; Treacy, C.M.; Doughty, N.J.; Southgate, L.; Machado, R.D.; Trembath, R.C.; Jennings, S.; Barker, L.; Nicklin, P.; et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 2010, 122, 920–927. [Google Scholar] [CrossRef]
- Ross, D.J.; Strieter, R.M.; Fishbein, M.C.; Ardehali, A.; Belperio, J.A. Type I immune response cytokine-chemokine cascade is associated with pulmonary arterial hypertension. J. Heart Lung Transplant. 2012, 31, 865–873. [Google Scholar] [CrossRef]
- Villegas, L.R.; Kluck, D.; Field, C.; Oberley-Deegan, R.E.; Woods, C.; Yeager, M.E.; El Kasmi, K.C.; Savani, R.C.; Bowler, R.P.; Nozik-Grayck, E. Superoxide dismutase mimetic, MnTE-2-PyP, attenuates chronic hypoxia-induced pulmonary hypertension, pulmonary vascular remodeling, and activation of the NALP3 inflammasome. Antioxid. Redox Signal. 2013, 18, 1753–1764. [Google Scholar] [CrossRef]
- Groth, A.; Vrugt, B.; Brock, M.; Speich, R.; Ulrich, S.; Huber, L.C. Inflammatory cytokines in pulmonary hypertension. Respir. Res. 2014, 15, 47. [Google Scholar] [CrossRef]
- Tang, B.; Chen, G.X.; Liang, M.Y.; Yao, J.P.; Wu, Z.K. Ellagic acid prevents monocrotaline-induced pulmonary artery hypertension via inhibiting NLRP3 inflammasome activation in rats. Int. J. Cardiol. 2015, 180, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Parpaleix, A.; Amsellem, V.; Houssaini, A.; Abid, S.; Breau, M.; Marcos, E.; Sawaki, D.; Delcroix, M.; Quarck, R.; Maillard, A.; et al. Role of interleukin-1 receptor 1/MyD88 signalling in the development and progression of pulmonary hypertension. Eur. Respir. J. 2016, 48, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; You, S.; Liu, H.; Chen, L.; Zhang, C.; Hu, H.; Xue, M.; Cheng, W.; Wang, Y.; Li, X.; et al. Role of P2X(7)R in the development and progression of pulmonary hypertension. Respir. Res. 2017, 18, 127. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Guo, S.L.; Wei, B.; Gao, X.C.; Zhou, Y.C.; Li, J.Q. Activation of nicotinic acetylcholine α7 receptor attenuates progression of monocrotaline-induced pulmonary hypertension in rats by downregulating the NLRP3 Inflammasome. Front. Pharmacol. 2019, 10, 128. [Google Scholar] [CrossRef]
- Zhang, M.; Xin, W.; Yu, Y.; Yang, X.; Ma, C.; Zhang, H.; Liu, Y.; Zhao, X.; Guan, X.; Wang, X.; et al. Programmed death-ligand 1 triggers PASMCs pyroptosis and pulmonary vascular fibrosis in pulmonary hypertension. J. Mol. Cell. Cardiol. 2020, 138, 23–33. [Google Scholar] [CrossRef]
- Matias, M.L.; Romão, M.; Weel, I.C.; Ribeiro, V.R.; Nunes, P.R.; Borges, V.T.; Araújo, J.P., Jr.; Peraçoli, J.C.; de Oliveira, L.; Peraçoli, M.T. Endogenous and Uric acid-induced activation of NLRP3 inflammasome in pregnant women with Preeclampsia. PLoS ONE 2015, 10, e0129095. [Google Scholar] [CrossRef]
- Dörffel, Y.; Franz, S.; Pruss, A.; Neumann, G.; Rohde, W.; Burmester, G.R.; Scholze, J. Preactivated monocytes from hypertensive patients as a factor for atherosclerosis? Atherosclerosis 2001, 157, 151–160. [Google Scholar] [CrossRef]
- Alexander, M.R.; Norlander, A.E.; Elijovich, F.; Atreya, R.V.; Gaye, A.; Gnecco, J.S.; Laffer, C.L.; Galindo, C.L.; Madhur, M.S. Human monocyte transcriptional profiling identifies IL-18 receptor accessory protein and lactoferrin as novel immune targets in hypertension. Br. J. Pharmacol. 2019, 176, 2015–2027. [Google Scholar] [CrossRef]
- Krishnan, S.M.; Dowling, J.K.; Ling, Y.H.; Diep, H.; Chan, C.T.; Ferens, D.; Kett, M.M.; Pinar, A.; Samuel, C.S.; Vinh, A.; et al. Inflammasome activity is essential for one kidney/deoxycorticosterone acetate/salt-induced hypertension in mice. Br. J. Pharmacol. 2016, 173, 752–765. [Google Scholar] [CrossRef]
- Krishnan, S.M.; Ling, Y.H.; Huuskes, B.M.; Ferens, D.M.; Saini, N.; Chan, C.T.; Diep, H.; Kett, M.M.; Samuel, C.S.; Kemp-Harper, B.K.; et al. Pharmacological inhibition of the NLRP3 inflammasome reduces blood pressure, renal damage, and dysfunction in salt-sensitive hypertension. Cardiovasc. Res. 2019, 115, 776–787. [Google Scholar] [CrossRef]
- Wang, Q.; So, A.; Nussberger, J.; Tschopp, J.; Burnier, M. Impact of nlrp3 inflammasome on the development of hypertension and renal and cardiac hypertrophy in 2k1c and doca/salt mice. Kidney Res. Clin. Pract. 2012, 31, A83. [Google Scholar] [CrossRef][Green Version]
- Sogawa, Y.; Nagasu, H.; Itano, S.; Kidokoro, K.; Taniguchi, S.; Takahashi, M.; Kadoya, H.; Satoh, M.; Sasaki, T.; Kashihara, N. The eNOS-NO pathway attenuates kidney dysfunction via suppression of inflammasome activation in aldosterone-induced renal injury model mice. PLoS ONE 2018, 13, e0203823. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Youn, J.-Y.; Cai, H. Mechanisms and consequences of endothelial nitric oxide synthase dysfunction in hypertension. J. Hypertens. 2015, 33, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Shesely, E.G.; Maeda, N.; Kim, H.-S.; Desai, K.M.; Krege, J.H.; Laubach, V.E.; Sherman, P.A.; Sessa, W.C.; Smithies, O. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 1996, 93, 13176–13181. [Google Scholar] [CrossRef] [PubMed]
- Klinger, J.R.; Abman, S.H.; Gladwin, M.T. Nitric oxide deficiency and endothelial dysfunction in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2013, 188, 639–646. [Google Scholar] [CrossRef]
- Zambom, F.F.F.; Oliveira, K.C.; Foresto-Neto, O.; Faustino, V.D.; Ávila, V.F.; Albino, A.H.; Arias, S.C.A.; Volpini, R.A.; Malheiros, D.; Saraiva Camara, N.O.; et al. Pathogenic role of innate immunity in a model of chronic NO inhibition associated with salt overload. Am. J. Physiol. Renal Physiol. 2019, 317, F1058–F1067. [Google Scholar] [CrossRef]
- Furman, D.; Chang, J.; Lartigue, L.; Bolen, C.R.; Haddad, F.; Gaudilliere, B.; Ganio, E.A.; Fragiadakis, G.K.; Spitzer, M.H.; Douchet, I.; et al. Expression of specific inflammasome gene modules stratifies older individuals into two extreme clinical and immunological states. Nat. Med. 2017, 23, 174–184. [Google Scholar] [CrossRef]
- Ulrich, C.; Wildgrube, S.; Fiedler, R.; Seibert, E.; Kneser, L.; Fick, S.; Schäfer, C.; Markau, S.; Trojanowicz, B.; Girndt, M. NLRP3 inflammasome activation in hemodialysis and hypertensive patients with intact kidney function. Toxins 2020, 12, 675. [Google Scholar] [CrossRef]
- Shahzad, K.; Bock, F.; Dong, W.; Wang, H.; Kopf, S.; Kohli, S.; Al-Dabet, M.M.; Ranjan, S.; Wolter, J.; Wacker, C.; et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 2015, 87, 74–84. [Google Scholar] [CrossRef]
- Haque, S.; Lan, X.; Wen, H.; Lederman, R.; Chawla, A.; Attia, M.; Bongu, R.P.; Husain, M.; Mikulak, J.; Saleem, M.A.; et al. HIV promotes NLRP3 inflammasome complex activation in murine HIV-associated nephropathy. Am. J. Pathol. 2016, 186, 347–358. [Google Scholar] [CrossRef]
- Gu, J.; Huang, W.; Zhang, W.; Zhao, T.; Gao, C.; Gan, W.; Rao, M.; Chen, Q.; Guo, M.; Xu, Y.; et al. Sodium butyrate alleviates high-glucose-induced renal glomerular endothelial cells damage via inhibiting pyroptosis. Int. Immunopharmacol. 2019, 75, 105832. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.J.; Suarez-Alvarez, B.; Grigorescu, M.; Foresto-Neto, O.; Steiger, S.; Desai, J.; Marschner, J.A.; Honarpisheh, M.; Shi, C.; Jordan, J.; et al. The macrophage phenotype and inflammasome component NLRP3 contributes to nephrocalcinosis-related chronic kidney disease independent from IL-1-mediated tissue injury. Kidney Int. 2018, 93, 656–669. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Boini, K.M.; Xia, M.; Abais, J.M.; Li, X.; Liu, Q.; Li, P.L. Activation of Nod-like receptor protein 3 inflammasomes turns on podocyte injury and glomerular sclerosis in hyperhomocysteinemia. Hypertension 2012, 60, 154–162. [Google Scholar] [CrossRef]
- Abais, J.M.; Xia, M.; Li, G.; Gehr, T.W.; Boini, K.M.; Li, P.L. Contribution of endogenously produced reactive oxygen species to the activation of podocyte NLRP3 inflammasomes in hyperhomocysteinemia. Free Radical Biol. Med. 2014, 67, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Abais, J.M.; Zhang, C.; Xia, M.; Liu, Q.; Gehr, T.W.; Boini, K.M.; Li, P.L. NADPH oxidase-mediated triggering of inflammasome activation in mouse podocytes and glomeruli during hyperhomocysteinemia. Antioxid. Redox Signal. 2013, 18, 1537–1548. [Google Scholar] [CrossRef]
- Araujo, M.; Wilcox, C.S. Oxidative stress in hypertension: Role of the kidney. Antioxid. Redox Signal. 2014, 20, 74–101. [Google Scholar] [CrossRef]
- Wilcox, C.S. Reactive oxygen species: Roles in blood pressure and kidney function. Curr. Hypertens. Rep. 2002, 4, 160–166. [Google Scholar] [CrossRef]
- Solini, A.; Menini, S.; Rossi, C.; Ricci, C.; Santini, E.; Blasetti Fantauzzi, C.; Iacobini, C.; Pugliese, G. The purinergic 2X7 receptor participates in renal inflammation and injury induced by high-fat diet: Possible role of NLRP3 inflammasome activation. J. Pathol. 2013, 231, 342–353. [Google Scholar] [CrossRef]
- Chi, K.; Geng, X.; Liu, C.; Cai, G.; Hong, Q. Research progress on the role of inflammasomes in kidney disease. Mediat. Inflamm. 2020, 2020, 8032797. [Google Scholar] [CrossRef]
- Sun, H.J.; Ren, X.S.; Xiong, X.Q.; Chen, Y.Z.; Zhao, M.X.; Wang, J.J.; Zhou, Y.B.; Han, Y.; Chen, Q.; Li, Y.H.; et al. NLRP3 inflammasome activation contributes to VSMC phenotypic transformation and proliferation in hypertension. Cell Death Dis. 2017, 8, e3074. [Google Scholar] [CrossRef]
- Zhang, X.; Hong, S.; Qi, S.; Liu, W.; Zhang, X.; Shi, Z.; Chen, W.; Zhao, M.; Yin, X. NLRP3 Inflammasome is involved in calcium-sensing receptor-induced aortic remodeling in SHRs. Mediat. Inflamm. 2019, 2019, 6847087. [Google Scholar] [CrossRef] [PubMed]
- Xi, H.; Zhang, Y.; Xu, Y.; Yang, W.Y.; Jiang, X.; Sha, X.; Cheng, X.; Wang, J.; Qin, X.; Yu, J.; et al. Caspase-1 Inflammasome activation mediates homocysteine-induced pyrop-apoptosis in endothelial cells. Circ. Res. 2016, 118, 1525–1539. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lu, Y.; Cao, Z.; Ma, Q.; Pi, H.; Fang, Y.; Yu, Z.; Hu, H.; Zhou, Z. Cadmium induces NLRP3 inflammasome-dependent pyroptosis in vascular endothelial cells. Toxicol. Lett. 2016, 246, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Jiang, L.; Li, Q.; Liu, X.; Zhang, T.; Dong, L.; Liu, T.; Liu, L.; Hu, G.; Sun, X. Acrolein induces NLRP3 inflammasome-mediated pyroptosis and suppresses migration via ROS-dependent autophagy in vascular endothelial cells. Toxicology 2018, 410, 26–40. [Google Scholar] [CrossRef]
- Hafeez, N.; Chan, S.Y. A New “TYK” tok era for the study of long noncoding RNAs in pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2020, 202, 1339–1341. [Google Scholar] [CrossRef]
- Bai, B.; Yang, Y.; Wang, Q.; Li, M.; Tian, C.; Liu, Y.; Aung, L.H.H.; Li, P.-F.; Yu, T.; Chu, X.-M. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis. 2020, 11, 776. [Google Scholar] [CrossRef]
- Wu, L.M.; Wu, S.G.; Chen, F.; Wu, Q.; Wu, C.M.; Kang, C.M.; He, X.; Zhang, R.Y.; Lu, Z.F.; Li, X.H.; et al. Atorvastatin inhibits pyroptosis through the lncRNA NEXN-AS1/NEXN pathway in human vascular endothelial cells. Atherosclerosis 2020, 293, 26–34. [Google Scholar] [CrossRef]
- Qi, Y.; Du, X.; Yao, X.; Zhao, Y. Vildagliptin inhibits high free fatty acid (FFA)-induced NLRP3 inflammasome activation in endothelial cells. Artif Cells Nanomed. Biotechnol. 2019, 47, 1067–1074. [Google Scholar] [CrossRef]
- Zhou, X.; Wu, Y.; Ye, L.; Wang, Y.; Zhang, K.; Wang, L.; Huang, Y.; Xian, S.; Zhang, Y.; Chen, Y. Aspirin alleviates endothelial gap junction dysfunction through inhibition of NLRP3 inflammasome activation in LPS-induced vascular injury. Acta Pharm. Sin. B 2019, 9, 711–723. [Google Scholar] [CrossRef]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Munoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef]
- Ren, X.S.; Tong, Y.; Ling, L.; Chen, D.; Sun, H.J.; Zhou, H.; Qi, X.H.; Chen, Q.; Li, Y.H.; Kang, Y.M.; et al. NLRP3 gene deletion attenuates angiotensin II-induced phenotypic transformation of vascular smooth muscle cells and vascular remodeling. Cell Physiol. Biochem. 2017, 44, 2269–2280. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Chen, J.K.; Lu, W.J.; Jiang, Y.J.; Wang, Y.Y.; Li, D.J.; Shen, F.M. Inflammasome-independent NALP3 contributes to high-salt induced endothelial dysfunction. Front. Pharmacol. 2018, 9, 968. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Yu, X.J.; Shi, X.L.; Gao, H.L.; Yi, Q.Y.; Tan, H.; Fan, X.Y.; Zhang, Y.; Song, X.A.; Cui, W.; et al. NF-kappaB blockade in hypothalamic paraventricular nucleus inhibits high-salt-induced hypertension through NLRP3 and caspase-1. Cardiovasc. Toxicol. 2016, 16, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-L.; Kang, Y.-M.; Li, X.-G.; Su, Q.; Li, H.-B.; Liu, K.-L.; Fu, L.-Y.; Saahene, R.O.; Li, Y.; Tan, H.; et al. Central blockade of NLRP3 reduces blood pressure via regulating inflammation microenvironment and neurohormonal excitation in salt-induced prehypertensive rats. J. Neuroinflamm. 2018, 15, 95. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.C.; Morris, B.J. Association analysis of polymorphisms at the interleukin-1 locus in essential hypertension. Am. J. Med. Gen. 2002, 107, 311–316. [Google Scholar] [CrossRef]
- Rubio-Guerra, A.F.; Rodriguez-Lopez, L.; Vargas-Ayala, G.; Huerta-Ramirez, S.; Serna, D.C.; Lozano-Nuevo, J.J. Depression increases the risk for uncontrolled hypertension. Exp. Clin. Cardiol. 2013, 18, 10–12. [Google Scholar]
- Li, R.; Wang, X.; Qin, T.; Qu, R.; Ma, S. Apigenin ameliorates chronic mild stress-induced depressive behavior by inhibiting interleukin-1beta production and NLRP3 inflammasome activation in the rat brain. Behav. Brain Res. 2016, 296, 318–325. [Google Scholar] [CrossRef]
- Huang, Q.; Ye, X.; Wang, L.; Pan, J. Salvianolic acid B abolished chronic mild stress-induced depression through suppressing oxidative stress and neuro-inflammation via regulating NLRP3 inflammasome activation. J. Food Biochem. 2018, e12742. [Google Scholar] [CrossRef]
- Hermann, M.; Ruschitzka, F. Novel anti-inflammatory drugs in hypertension. Nephrol. Dial. Transplant. 2006, 21, 859–864. [Google Scholar] [CrossRef]
- Aljadhey, H.; Tu, W.; Hansen, R.A.; Blalock, S.J.; Brater, D.C.; Murray, M.D. Comparative effects of non-steroidal anti-inflammatory drugs (NSAIDs) on blood pressure in patients with hypertension. BMC Cardiovasc. Dis. 2012, 12, 93. [Google Scholar] [CrossRef]
- Snowden, S.; Nelson, R. The effects of nonsteroidal anti-inflammatory drugs on blood pressure in hypertensive patients. Cardiol. Rev. 2011, 19, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.; Karimi Galougahi, K.; Besnier, M.; Genetzakis, E.; Tsang, M.; Finemore, M.; O’Brien-Brown, J.; Di Bartolo, B.A.; Kassiou, M.; Bubb, K.J.; et al. The novel P2X7 receptor antagonist PKT100 improves cardiac function and survival in pulmonary hypertension by direct targeting of the right ventricle. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H183–H191. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, J.; Canete, J.D. Anakinra for the treatment of rheumatoid arthritis: A safety evaluation. Expert Opin. Drug Saf. 2018, 17, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.H.; Krishnan, S.M.; Chan, C.T.; Diep, H.; Ferens, D.; Chin-Dusting, J.; Kemp-Harper, B.K.; Samuel, C.S.; Hewitson, T.D.; Latz, E.; et al. Anakinra reduces blood pressure and renal fibrosis in one kidney/DOCA/salt-induced hypertension. Pharmacol. Res. 2017, 116, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Trankle, C.R.; Canada, J.M.; Kadariya, D.; Markley, R.; De Chazal, H.M.; Pinson, J.; Fox, A.; Van Tassell, B.W.; Abbate, A.; Grinnan, D. IL-1 blockade reduces inflammation in pulmonary arterial hypertension and right ventricular failure: A single-arm, open-label, phase IB/II pilot study. Am. J. Respir. Crit. Care Med. 2019, 199, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Rothman, A.M.; MacFadyen, J.; Thuren, T.; Webb, A.; Harrison, D.G.; Guzik, T.J.; Libby, P.; Glynn, R.J.; Ridker, P.M. Effects of Interleukin-1β inhibition on blood pressure, incident hypertension, and residual inflammatory risk: A secondary analysis of CANTOS. Hypertension 2020, 75, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Rathkey, J.K.; Zhao, J.; Liu, Z.; Chen, Y.; Yang, J.; Kondolf, H.C.; Benson, B.L.; Chirieleison, S.M.; Huang, A.Y.; Dubyak, G.R.; et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef]
- Hu, J.J.; Liu, X.; Xia, S.; Zhang, Z.; Zhang, Y.; Zhao, J.; Ruan, J.; Luo, X.; Lou, X.; Bai, Y.; et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol. 2020, 21, 736–745. [Google Scholar] [CrossRef]
- Humphries, F.; Shmuel-Galia, L.; Ketelut-Carneiro, N.; Li, S.; Wang, B.; Nemmara, V.V.; Wilson, R.; Jiang, Z.; Khalighinejad, F.; Muneeruddin, K.; et al. Succination inactivates gasdermin D and blocks pyroptosis. Science 2020, 369, 1633–1637. [Google Scholar] [CrossRef]
- Pang, Y.; Zhang, P.C.; Lu, R.R.; Li, H.L.; Li, J.C.; Fu, H.X.; Cao, Y.W.; Fang, G.X.; Liu, B.H.; Wu, J.B.; et al. Andrade-oliveira salvianolic acid B modulates caspase-1-mediated pyroptosis in renal ischemia-reperfusion injury via Nrf2 pathway. Front. Pharmacol. 2020, 11, 541426. [Google Scholar] [CrossRef]
- Diao, C.; Chen, Z.; Qiu, T.; Liu, H.; Yang, Y.; Liu, X.; Wu, J.; Wang, L. Inhibition of PRMT5 attenuates oxidative stress-induced pyroptosis via activation of the Nrf2/HO-1 signal pathway in a mouse model of renal ischemia-reperfusion injury. Oxid. Med. Cell. Longev. 2019, 2019, 2345658. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Zhang, T.; Yi, L.; Zhou, X.; Mi, M. Dihydromyricetin inhibits NLRP3 inflammasome-dependent pyroptosis by activating the Nrf2 signaling pathway in vascular endothelial cells. BioFactors 2018, 44, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.D.; Lai, T.Y.; Chien, C.T.; Yu, H.J. Activating Nrf-2 signaling depresses unilateral ureteral obstruction-evoked mitochondrial stress-related autophagy, apoptosis and pyroptosis in kidney. PLoS ONE 2012, 7, e47299. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhu, X.; Yuan, S.; Wen, S.; Liu, X.; Wang, C.; Qu, Z.; Li, J.; Liu, H.; Sun, L.; et al. TLR4/NF-kappaB signaling induces GSDMD-related pyroptosis in tubular cells in diabetic kidney disease. Front. Endocrinol. 2019, 10, 603. [Google Scholar] [CrossRef]
- Tang, Y.S.; Zhao, Y.H.; Zhong, Y.; Li, X.Z.; Pu, J.X.; Luo, Y.C.; Zhou, Q.L. Neferine inhibits LPS-ATP-induced endothelial cell pyroptosis via regulation of ROS/NLRP3/Caspase-1 signaling pathway. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. 2019, 68, 727–738. [Google Scholar] [CrossRef]
- Ling, W.C.; Liu, J.; Lau, C.W.; Murugan, D.D.; Mustafa, M.R.; Huang, Y. Treatment with salvianolic acid B restores endothelial function in angiotensin II-induced hypertensive mice. Biochem. Pharmacol. 2017, 136, 76–85. [Google Scholar] [CrossRef]
- Li, Q.; Wang, J.; Zhu, X.; Zeng, Z.; Wu, X.; Xu, Y.; Xie, J.; Yu, J. Dihydromyricetin prevents monocrotaline-induced pulmonary arterial hypertension in rats. Biomed. Pharmacother. 2017, 96, 825–833. [Google Scholar] [CrossRef]
- Senanayake, G.V.; Banigesh, A.; Wu, L.; Lee, P.; Juurlink, B.H. The dietary phase 2 protein inducer sulforaphane can normalize the kidney epigenome and improve blood pressure in hypertensive rats. Am. J. Hypertens. 2012, 25, 229–235. [Google Scholar] [CrossRef]
- Wicha, P.; Onsa-Ard, A.; Chaichompoo, W.; Suksamrarn, A.; Tocharus, C. Vasorelaxant and antihypertensive effects of neferine in rats: An in vitro and in vivo study. Planta Med. 2020, 86, 496–504. [Google Scholar] [CrossRef]
- Wang, L.; Zhu, Q.; Lu, A.; Liu, X.; Zhang, L.; Xu, C.; Liu, X.; Li, H.; Yang, T. Sodium butyrate suppresses angiotensin II-induced hypertension by inhibition of renal (pro)renin receptor and intrarenal renin-angiotensin system. J. Hypertens. 2017, 35, 1899–1908. [Google Scholar] [CrossRef]
- Ferreira, N.S.; Bruder-Nascimento, T.; Pereira, C.A.; Zanotto, C.Z.; Prado, D.S.; Silva, J.F.; Rassi, D.M.; Foss-Freitas, M.C.; Alves-Filho, J.C.; Carlos, D.; et al. NLRP3 Inflammasome and mineralocorticoid receptors are associated with vascular dysfunction in type 2 diabetes mellitus. Cells 2019, 8, 1595. [Google Scholar] [CrossRef] [PubMed]
- Ren, P.; Wu, D.; Appel, R.; Zhang, L.; Zhang, C.; Luo, W.; Robertson, A.A.B.; Cooper, M.A.; Coselli, J.S.; Milewicz, D.M.; et al. Targeting the NLRP3 inflammasome with inhibitor MCC950 prevents aortic aneurysms and dissections in mice. J. Am. Heart Assoc. 2020, 9, e014044. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wu, C.; Liu, Y.; Li, H.; Zhu, Y.; Huang, C.; Lin, H.; Qiao, Q.; Huang, M.; Zhu, Q.; et al. ELABELA attenuates deoxycorticosterone acetate/salt-induced hypertension and renal injury by inhibition of NADPH oxidase/ROS/NLRP3 inflammasome pathway. Cell Death Dis. 2020, 11, 698. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, S.; Haraguchi, G.; Sasaki, A.; Arai, H.; Muto, S.; Itai, A.; Doi, S.; Mizutani, S.; Isobe, M. Pathophysiological roles of nuclear factor kappaB (NF-kB) in pulmonary arterial hypertension: Effects of synthetic selective NF-kB inhibitor IMD-0354. Cardiovasc. Res. 2013, 99, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Volonte, C.; Apolloni, S.; Skaper, S.D.; Burnstock, G. P2X7 receptors: Channels, pores and more. CNS Neurol. Dis. Drug Targets 2012, 11, 705–721. [Google Scholar] [CrossRef]
- Wang, D.; Wang, H.; Gao, H.; Zhang, H.; Zhang, H.; Wang, Q.; Sun, Z. P2X7 receptor mediates NLRP3 inflammasome activation in depression and diabetes. Cell Biosci. 2020, 10, 28. [Google Scholar] [CrossRef]
- Coll, R.C.; Hill, J.R.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.L.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A.B.; et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 2019, 15, 556–559. [Google Scholar] [CrossRef]
- Tapia-Abellan, A.; Angosto-Bazarra, D.; Martinez-Banaclocha, H.; de Torre-Minguela, C.; Ceron-Carrasco, J.P.; Perez-Sanchez, H.; Arostegui, J.I.; Pelegrin, P. MCC950 closes the active conformation of NLRP3 to an inactive state. Nat. Chem. Biol. 2019, 15, 560–564. [Google Scholar] [CrossRef]
- Welsh, P.; Grassia, G.; Botha, S.; Sattar, N.; Maffia, P. Targeting inflammation to reduce cardiovascular disease risk: A realistic clinical prospect? Br. J. Pharmacol. 2017, 174, 3898–3913. [Google Scholar] [CrossRef]
- Andrzejczak, D.; Gorska, D.; Czarnecka, E. Influence of enalapril, quinapril and losartan on lipopolysaccharide (LPS)-induced serum concentrations of TNF-α, IL-1 β, IL-6 in spontaneously hypertensive rats (SHR). Pharmacol. Rep. 2007, 59, 437–446. [Google Scholar]
- Huang, G.; Niu, T.; Peng, S.; Ling, D.; Liu, J.; Zhang, X.; Xu, X. Association between the interleukin-1beta C(-511)T polymorphism and blood pressure in a Chinese hypertensive population. Immunol. Lett. 2004, 91, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.A. Exonic polymorphism (rs315952, Ser133Ser) of interleukin 1 receptor antagonist (IL1RN) is related to overweigh/obese with hypertension. J. Exerc. Rehabilit. 2014, 10, 332–336. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Krishnan, S.M.; Sobey, C.G.; Latz, E.; Mansell, A.; Drummond, G.R. IL-1β and IL-18: Inflammatory markers or mediators of hypertension? Br. J. Pharmacol. 2014, 171, 5589–5602. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, M.; Ferrucci, L.; Corsi, A.M.; Macchi, C.; Lauretani, F.; Bonafe, M.; Olivieri, F.; Giovagnetti, S.; Franceschi, C.; Paolisso, G. Is chronic inflammation a determinant of blood pressure in the elderly? Am. J. Hypertens. 2003, 16, 537–543. [Google Scholar] [CrossRef][Green Version]
- Liu, Y.; Liu, T.; McCarron, R.M.; Spatz, M.; Feuerstein, G.; Hallenbeck, J.M.; Siren, A.L. Evidence for activation of endothelium and monocytes in hypertensive rats. Am. J. Physiol. 1996, 270, H2125–H2131. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Nishimura, M.; Sakamoto, M.; Ikegaki, I.; Nakanishi, T.; Yoshimura, M. Effects of interleukin-1 beta on blood pressure, sympathetic nerve activity, and pituitary endocrine functions in anesthetized rats. Am. J. Hypertens. 1992, 5, 224–229. [Google Scholar] [CrossRef]
- Fox, E.; Jayaprakash, N.; Pham, T.H.; Rowley, A.; McCully, C.L.; Pucino, F.; Goldbach-Mansky, R. The serum and cerebrospinal fluid pharmacokinetics of anakinra after intravenous administration to non-human primates. J. Neuroimmunol. 2010, 223, 138–140. [Google Scholar] [CrossRef]
- Rodriguez-Smith, J.; Lin, Y.C.; Tsai, W.L.; Kim, H.; Montealegre-Sanchez, G.; Chapelle, D.; Huang, Y.; Sibley, C.H.; Gadina, M.; Wesley, R.; et al. Cerebrospinal fluid cytokines correlate with aseptic meningitis and blood-brain barrier function in neonatal-onset multisystem inflammatory disease: Central nervous system biomarkers in neonatal-onset multisystem inflammatory disease correlate with central nervous system inflammation. Arthritis Rheumatol. 2017, 69, 1325–1336. [Google Scholar] [CrossRef]
- Jeong, J.; Jung, Y.; Na, S.; Jeong, J.; Lee, E.; Kim, M.S.; Choi, S.; Shin, D.H.; Paek, E.; Lee, H.Y.; et al. Novel oxidative modifications in redox-active cysteine residues. Mol. Cell. Proteom. MCP 2011, 10, M110-000513. [Google Scholar] [CrossRef]
- Sevier, C.S.; Kaiser, C.A. Formation and transfer of disulphide bonds in living cells. Nat. Rev. Mol. Cell Biol. 2002, 3, 836–847. [Google Scholar] [CrossRef]
- Blatnik, M.; Thorpe, S.R.; Baynes, J.W. Succination of proteins by fumarate: Mechanism of inactivation of glyceraldehyde-3-phosphate dehydrogenase in diabetes. Ann. N. Y. Acad. Sci. 2008, 1126, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, P.; Chen, Q.; Huang, Z.; Zou, D.; Zhang, J.; Gao, X.; Lin, Z. Mitochondrial ROS promote macrophage pyroptosis by inducing GSDMD oxidation. J. Mol. Cell Biol. 2019, 11, 1069–1082. [Google Scholar] [CrossRef] [PubMed]
- Bharathi Priya, L.; Baskaran, R.; Huang, C.Y.; Vijaya Padma, V. Neferine modulates IGF-1R/Nrf2 signaling in doxorubicin treated H9c2 cardiomyoblasts. J. Cell. Biochem. 2018, 119, 1441–1452. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Jia, Y.; Liu, X.; Zhang, H.; Li, T.; Huang, W.; Chen, X.; Wang, F.; Sun, W.; Wu, H. Sodium butyrate activates NRF2 to ameliorate diabetic nephropathy possibly via inhibition of HDAC. J. Endocrinol. 2017, 232, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.Y.; Kang, B.; Suh, H.J.; Choi, H.S. Parthenolide, a feverfew-derived phytochemical, ameliorates obesity and obesity-induced inflammatory responses via the Nrf2/Keap1 pathway. Pharmacol. Res. 2019, 145, 104259. [Google Scholar] [CrossRef]
- Hennig, P.; Garstkiewicz, M.; Grossi, S.; Di Filippo, M.; French, L.E.; Beer, H.D. The Crosstalk between Nrf2 and Inflammasomes. Int. J. Mol. Sci. 2018, 19, 562. [Google Scholar] [CrossRef]
- Cuevas, S.; Yang, Y.; Konkalmatt, P.; Asico, L.D.; Feranil, J.; Jones, J.; Villar, V.A.; Armando, I.; Jose, P.A. Role of nuclear factor erythroid 2-related factor 2 in the oxidative stress-dependent hypertension associated with the depletion of DJ-1. Hypertension 2015, 65, 1251–1257. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Drug | Mechanism of Action | Effect on Blood Pressure Regulation | Organism | Publications |
|---|---|---|---|---|
| A-740003 | P2X7 receptor antagonist | Decreases mean RV pressure associated with pulmonary hypertension | Rat | [123] |
| PKT100 | P2X7 receptor antagonist | Improves survival in the pulmonary hypertension model | Mouse | [172] |
| Anakinra | IL-1 receptor antagonist | Reverses salt-induced hypertension/reduces pulmonary blood pressure | Mouse/human | [172,173,174,175] |
| Canakinumab | IL-1β blocker | No effect on blood pressure regulation | Human | [176] |
| NSA | Gasdermin inhibitor | No evidence | Human/mouse | [177] |
| Disulfiram | Gasdermin inhibitor | No evidence | Human/mouse | [178] |
| DMF | Gasdermin inhibitor | No evidence | Mouse | [179] |
| Nrf2 activators | Gasdermin inhibitors, antioxidants | Reduces systolic blood pressure | Mouse | [180,181,182,183,184,185,186,187,188,189,190] |
| MCC950 | NLRP3 inflammasome inhibitor | Reverses salt-induced hypertension/reduces blood pressure | Mouse/rat | [129,130,160,191,192] |
| Ellagic acid | Antioxidant | Reduces pulmonary artery hypertension | Rat | [121] |
| ELABELA | Antioxidant | Mitigates hypertension | Human renal tubular cells | [193] |
| IMD-0354 | NF-κB inhibitor | Reduces RV pressure | Rat | [194] |
| PDTC | NF-κB inhibitor | Reverses salt-induced hypertension | Rat | [163] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Miguel, C.; Pelegrín, P.; Baroja-Mazo, A.; Cuevas, S. Emerging Role of the Inflammasome and Pyroptosis in Hypertension. Int. J. Mol. Sci. 2021, 22, 1064. https://doi.org/10.3390/ijms22031064
De Miguel C, Pelegrín P, Baroja-Mazo A, Cuevas S. Emerging Role of the Inflammasome and Pyroptosis in Hypertension. International Journal of Molecular Sciences. 2021; 22(3):1064. https://doi.org/10.3390/ijms22031064
Chicago/Turabian StyleDe Miguel, Carmen, Pablo Pelegrín, Alberto Baroja-Mazo, and Santiago Cuevas. 2021. "Emerging Role of the Inflammasome and Pyroptosis in Hypertension" International Journal of Molecular Sciences 22, no. 3: 1064. https://doi.org/10.3390/ijms22031064
APA StyleDe Miguel, C., Pelegrín, P., Baroja-Mazo, A., & Cuevas, S. (2021). Emerging Role of the Inflammasome and Pyroptosis in Hypertension. International Journal of Molecular Sciences, 22(3), 1064. https://doi.org/10.3390/ijms22031064