
Regulation of Fibroblast Activation Protein by Transforming Growth Factor Beta-1 in Glioblastoma Microenvironment

 and
and
Abstract

1. Introduction
2. Results
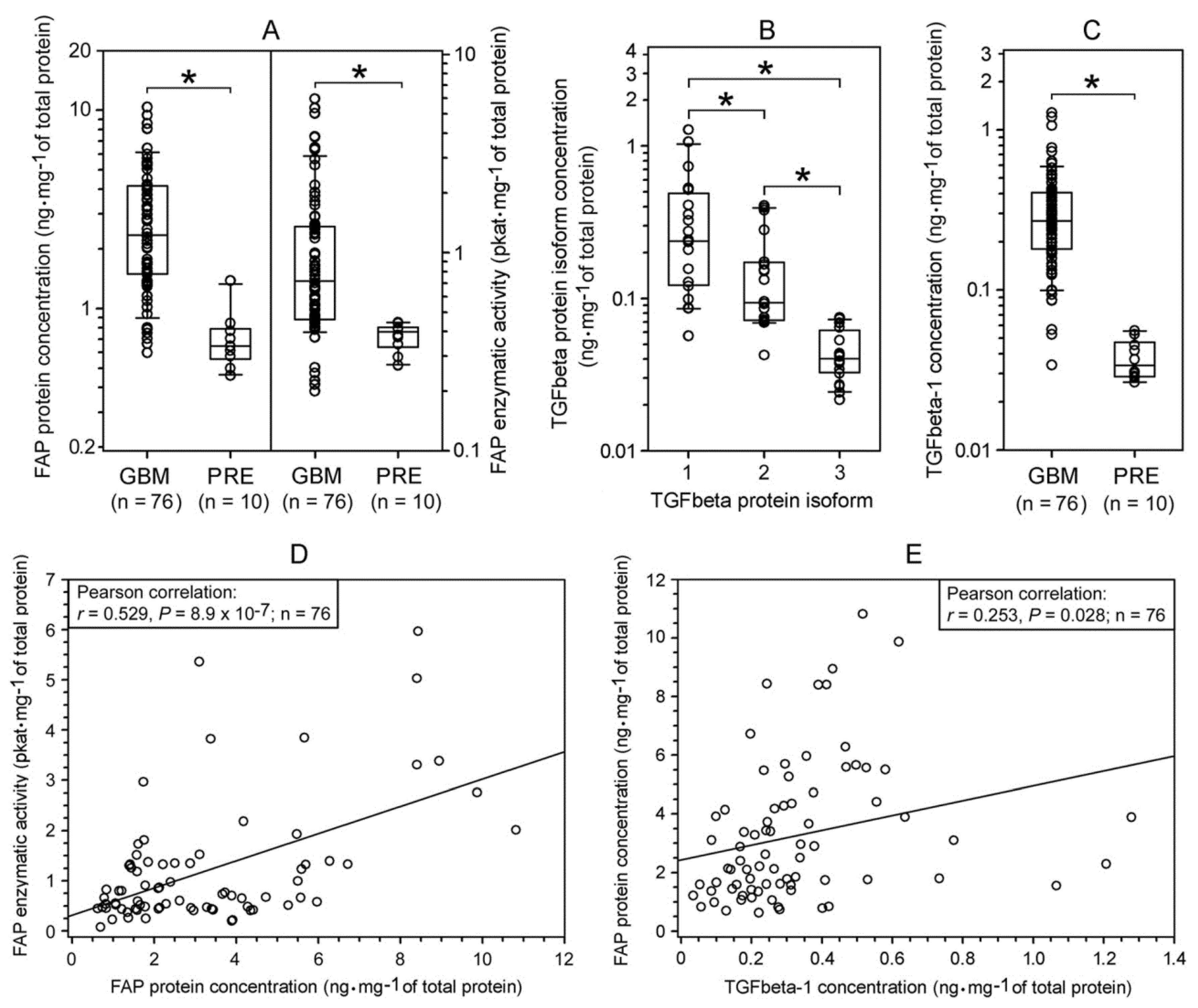
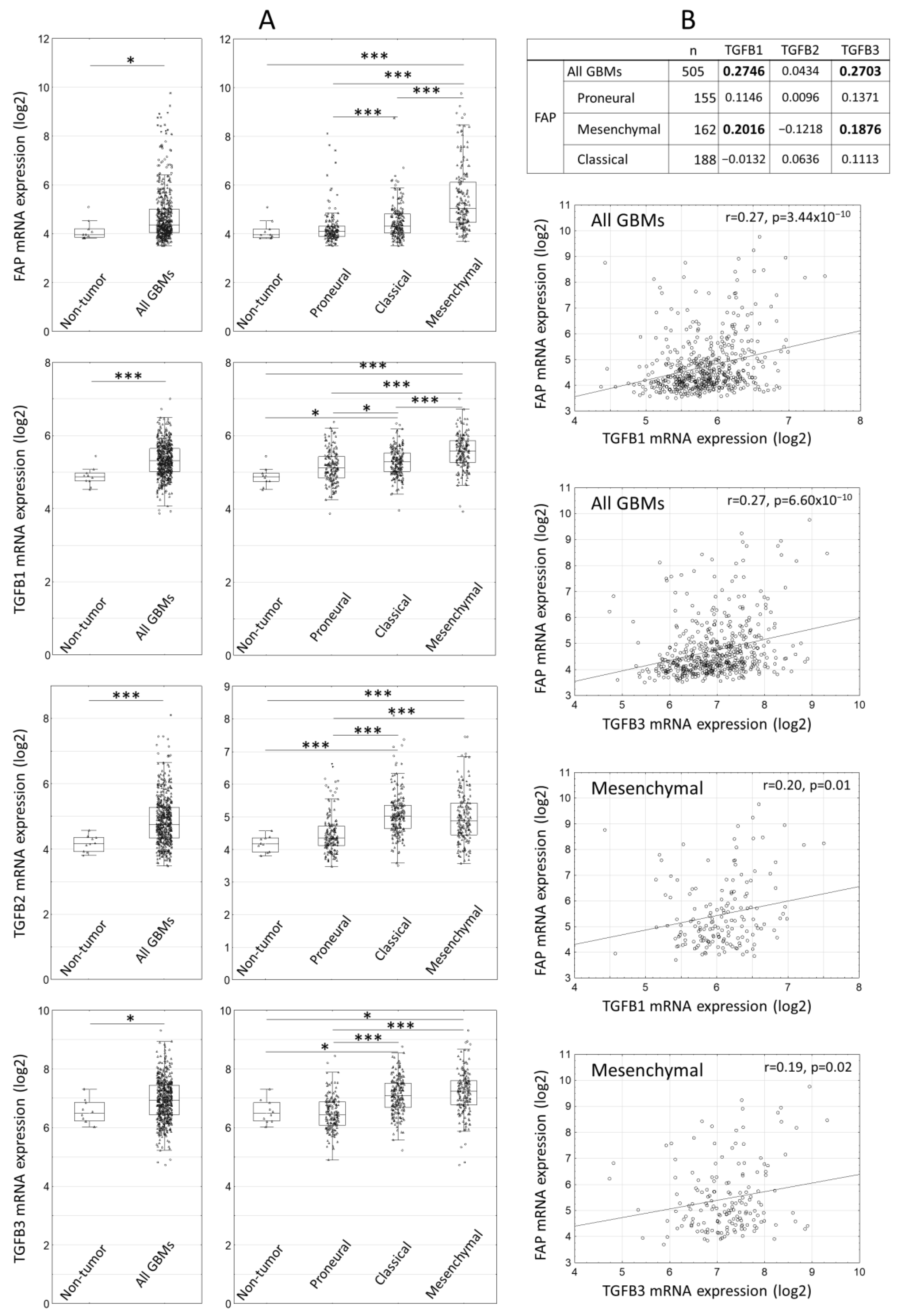
2.1. Expression of FAP and TGFbeta Isoforms in Human Glioblastomas
2.2. Immunohistochemical Localization of FAP and TGFbeta-1 in Human Glioblastomas
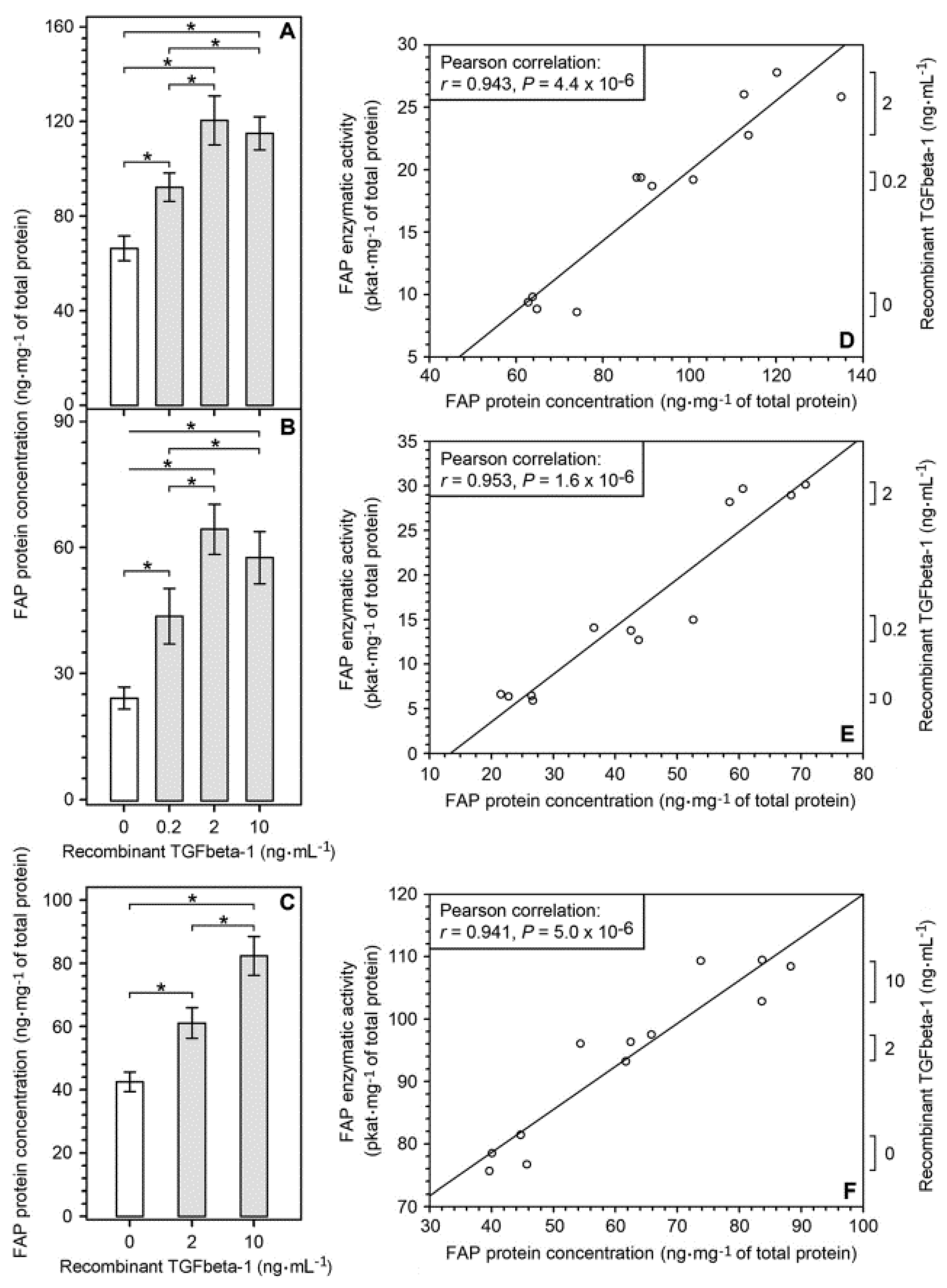
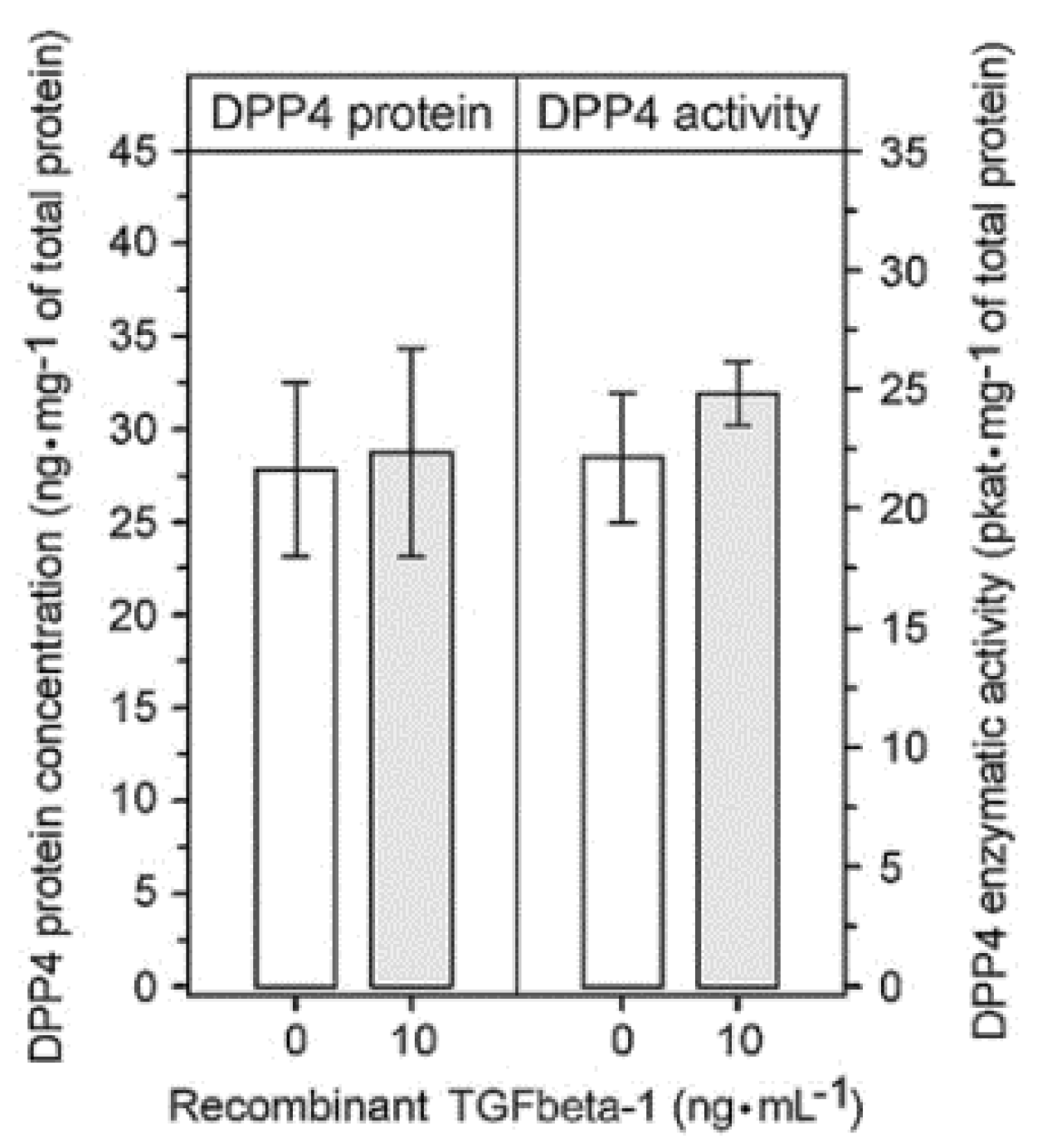
2.3. Upregulation of FAP Enzymatic Activity and FAP Protein Induced by Recombinant TGFbeta-1 in Different Cell Types Present in the Glioblastoma Microenvironment
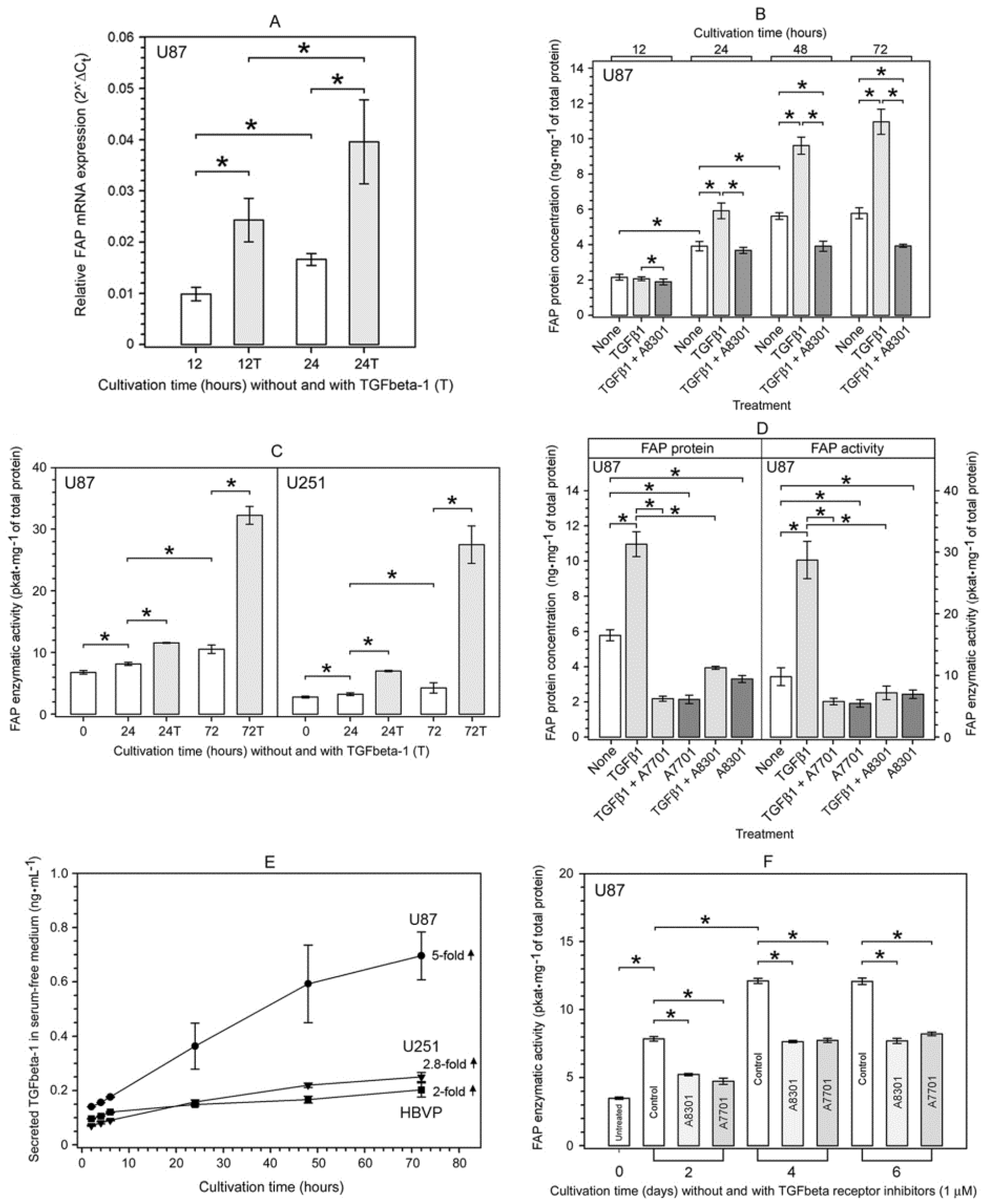
2.4. Characterization of the TGFbeta-1-Mediated Upregulation of FAP Enzymatic Activity and FAP Protein in Human Glioma Cells
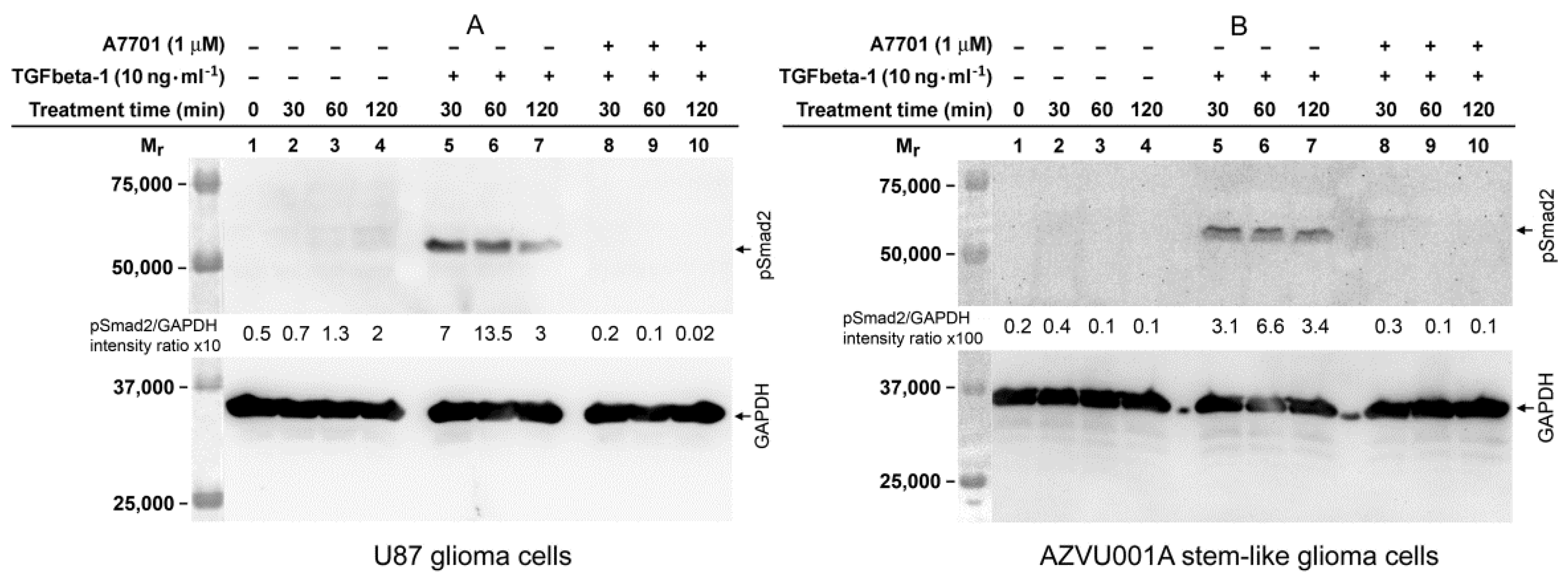
2.5. TGFbeta-1-Induced Phosphorylation of Smad2 in FAP-Upregulating and FAP-Non-Upregulating Human Glioma Cells
3. Discussion
4. Materials and Methods
4.1. Patients and Tissue Samples
4.2. The Cancer Genome Atlas (TCGA) Database Data
4.3. Immunohistochemistry and Confocal Microscopy
4.4. Cell Culture and Experimental Treatment of Cells
4.5. Preparation of Tissue and Cell Lysates and Cell Conditioned Media
4.6. Determination of Total Protein
4.7. Determination of Enzyme Activity
4.8. ELISA Assays
4.9. Western Blot Analysis
4.10. Isolation of Total RNA and Real Time RT-PCR
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GBM | Glioblastoma |
| PRE | Pharmacoresistant epilepsy |
| TGFbeta | Transforming growth factor beta |
| FAP | Fibroblast activation protein |
| DPP4 | Dipeptidyl peptidase-4 |
| Smad | Small mothers against decapentaplegic |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| ACTB | β-Actin |
References
- Perrin, S.L.; Samuel, M.S.; Koszyca, B.; Brown, M.P.; Ebert, L.M.; Oksdath, M.; Gomez, G.A. Glioblastoma heterogeneity and the tumour microenvironment: Implications for preclinical research and development of new treatments. Biochem. Soc. Trans. 2019, 47, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Broekman, M.L.; Maas, S.L.N.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional communication in the microenvirons of glioblastoma. Nat. Rev. Neurol. 2018, 14, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Busek, P.; Prevorovsky, M.; Krepela, E.; Sedo, A. Glioma associated proteases. In Glioma Cell Biology; Sedo, A., Mentlein, R., Eds.; Springer: Vienna, Austria, 2014; pp. 317–395. [Google Scholar]
- Bušek, P.; Balaziova, E.; Matrasova, I.; Hilser, M.; Tomas, R.; Syrucek, M.; Zemanova, Z.; Krepela, E.; Belacek, J.; Šedo, A. Fibroblast activation protein alpha is expressed by transformed and stromal cells and is associated with mesenchymal features in glioblastoma. Tumor Biol. 2016, 37, 13961–13971. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.; Huang, Y.; Simms, A.E.; Mazur, A. Fibroblast activation protein-alpha: A key modulator of the microenvironment in multiple pathologies. Int. Rev. Cell. Mol. Biol. 2012, 297, 83–116. [Google Scholar] [PubMed]
- Koczorowska, M.M.; Tholen, S.; Bucher, F.; Lutz, L.; Kizhakkedathu, J.N.; De Wever, O.; Wellner, U.F.; Biniossek, M.L.; Stahl, A.; Lassmann, S.; et al. Fibroblast activation protein-alpha, a stromal cell alterations. Mol. Oncol. 2016, 10, 40–58. [Google Scholar] [CrossRef]
- Zhang, H.E.; Hamson, E.J.; Koczorowska, M.M.; Tholen, S.; Chowdhury, S.; Bailey, C.G.; Lay, A.J.; Twigg, S.M.; Lee, Q.; Roediger, B.; et al. Identification of Novel Natural Substrates of Fibroblast Activation Protein-alpha by Differential Degradomics and Proteomics. Mol. Cell. Proteom. 2019, 18, 65–85. [Google Scholar] [CrossRef]
- Bae, S.; Park, C.W.; Son, H.K.; Ju, H.K.; Paik, D.; Koh, G.Y.; Kim, J.; Kim, H.; Jeon, C.J. Fibroblast activation protein α identifies mesenchymal stromal cells from human bone marrow. Br. J. Haematol. 2008, 142, 827–830. [Google Scholar] [CrossRef]
- Bušek, P.; Hrabal, P.; Fric, P.; Šedo, A. Co-expression of the homologous proteases proteases fibroblast activation and dipeptidyl peptidase-IV in the adult human Langerhans islets. Histochem. Cell Biol. 2015, 143, 497–504. [Google Scholar] [CrossRef]
- Huber, M.A.; Kraut, N.; Schweifer, N.; Dolznig, H.; Peter, R.U.; Schubert, R.D.; Scharffetter-Kochanek, K.; Pehamberger, H.; Garin-Chesa, P. Expression of stromal cell markers in distinct compartments of human skin cancers. J. Cutan. Pathol. 2006, 33, 145–155. [Google Scholar] [CrossRef]
- Wäster, P.; Orfanidis, K.; Eriksson, I.; Rosdahl, I.; Seifert, O.; Ollinger, K. UV radiation promotes melanoma dissemination mediated by the sequential reaction axis of cathepsins-TGF-beta1-FAP-alpha. Br. J. Cancer 2017, 117, 535–544. [Google Scholar] [CrossRef]
- Jacob, M.; Chang, L.; Pure, E. Fibroblast Activation Protein in Remodeling Tissues. Curr. Mol. Med. 2012, 12, 1220–1243. [Google Scholar] [CrossRef] [PubMed]
- Juillerat-Jeanneret, L.; Tafelmeyer, P.; Golshayan, D. Fibroblast activation protein-alpha in fibrogenic disorders and cancer: More than a prolyl-specific peptidase? Expert Opin. Ther. Targets 2017, 21, 977–991. [Google Scholar] [CrossRef] [PubMed]
- Puré, E.; Blomberg, R. Pro-tumorigenic roles of fibroblast activation protein in cancer: Back to the basics. Oncogene 2018, 37, 4343–4357. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Qi, L.; Liu, B.; Liu, J.; Zhang, H.; Che, D.; Cao, J.; Shen, J.; Geng, J.; Bi, Y.; et al. Fibroblast Activation Protein Overexpression and Clinical Implications in Solid Tumors: A Meta-Analysis. PLoS ONE 2015, 10, e0116683. [Google Scholar] [CrossRef] [PubMed]
- Goscinski, M.A.; Suo, Z.H.; Nesland, J.M.; Chen, W.-T.; Zakrzewska, M.; Wang, J.; Zhang, S.; Flørenes, V.A.; Giercksky, K.E. Seprase, dipeptidyl peptidase IV and urokinase-type plasminogen activator expression in dysplasia and invasive squamous cell carcinoma of the esophagus. A study of 229 cases from Anyang Tumor Hospital, Henan Province, China. Oncology 2008, 75, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Goscinski, M.A.; Suo, Z.; Flørenes, V.A.; Vlatkovic, L.; Nesland, J.M.; Giercksky, K.E. FAP-α and uPA Show Different Expression Patterns in Premalignant and Malignant Esophageal Lesions. Ultrastruct. Pathol. 2008, 32, 89–96. [Google Scholar] [CrossRef]
- Mori, Y.; Kono, K.; Matsumoto, Y.; Fujii, H.; Yamane, T.; Mitsumata, M.; Chen, W.-T. The Expression of a Type II Transmembrane Serine Protease (Seprase) in Human Gastric Carcinoma. Oncology 2004, 67, 411–419. [Google Scholar] [CrossRef]
- Busek, P.; Mateu, R.; Zubal, M.; Kotackova, L.; Šedo, A. Targeting fibroblast activation protein in cancer—Prospects and caveats. Front. Biosci. 2018, 23, 1933–1968. [Google Scholar]
- Röhrich, M.; Loktev, A.; Wefers, A.K.; Altmann, A.; Paech, D.; Adeberg, S.; Windisch, P.; Hielscher, T.; Flechsig, P.; Floca, R.; et al. IDH-wildtype glioblastomas and grade III/IV IDH-mutant gliomas show elevated tracer uptake in fibroblast activation protein–specific PET/CT. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 2569–2580. [Google Scholar] [CrossRef]
- Šimková, A.; Bušek, P.; Šedo, A.; Konvalinka, J. Molecular recognition of fibroblast activation protein for diagnostic and therapeutic applications. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140409. [Google Scholar] [CrossRef]
- Fitzgerald, A.A.; Weiner, L.M. The role of fibroblast activation protein in health and malignancy. Cancer Metastasis Rev. 2020, 39, 783–803. [Google Scholar] [CrossRef] [PubMed]
- Tulley, S.; Chen, W.T. Transcriptional regulation of seprase in invasive melanoma cells by transforming growth factor-beta signaling. J. Biol. Chem. 2014, 289, 15280–15296. [Google Scholar] [CrossRef] [PubMed]
- Knopf, J.D.; Tholen, S.; Koczorowska, M.M.; De Wever, O.; Biniossek, M.L.; Schilling, O. The stromal cell-surface protease fibroblast activation protein-α localizes to lipid rafts and is recruited to invadopodia. Biochim. Biophys. Acta 2015, 1853, 2515–2525. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.Q.; Zhao, M.; Huang, G.Z.; Zheng, Z.N.; Chen, Y.; Zeng, W.S.; Lv, X.Z. Fibroblast Activation Protein (FAP) Overexpression Induces Epithelial-Mesenchymal Transition (EMT) in Oral Squamous Cell Carcinoma by Down-Regulating Dipeptidyl Peptidase 9 (DPP9). Onco Targets Ther. 2020, 13, 2599–2611. [Google Scholar] [CrossRef] [PubMed]
- Behnan, J.; Finocchiaro, G.; Hanna, G. The landscape of the mesenchymal signature in brain tumours. Brain 2019, 142, 847–866. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.V.; Balasubramaniyan, V.; Walenkamp, A.; Kruyt, F.A. TGF-beta as a therapeutic target in high grade gliomas —Promises and challenges. Biochem. Pharmacol. 2013, 85, 478–485. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 2017, 32, 42–56. [Google Scholar] [CrossRef]
- Heldin, C.H.; Moustakas, A. Signaling receptors for TGF-beta family members. Cold Spring Harb. Perspect. Biol. 2016, 8, a022053. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad signaling pathways of the TGF-beta family. Cold Spring Harb. Perspect. Biol. 2017, 9, a022129. [Google Scholar] [CrossRef]
- Budi, E.H.; Duan, D.; Derynck, R. Transforming growth factor-beta receptors and smads: Regulatory complexity and functional versatility. Trends Cell Biol. 2017, 27, 658–672. [Google Scholar] [CrossRef]
- Yeh, H.W.; Lee, S.S.; Chang, C.Y.; Lang, Y.D.; Jou, Y.S. A new switch for TGFbeta in cancer. Cancer Res. 2019, 79, 3797–3805. [Google Scholar] [CrossRef] [PubMed]
- Bainbridge, T.W.; Dunshee, D.R.; Kljavin, N.M.; Skelton, N.J.; Sonoda, J.; Ernst, J.A. Selective Homogeneous Assay for Circulating Endopeptidase Fibroblast Activation Protein (FAP). Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Roy, L.O.; Poirier, M.B.; Fortin, D. Differential Expression and Clinical Significance of Transforming Growth Factor-Beta Isoforms in GBM Tumors. Int. J. Mol. Sci. 2018, 19, 1113. [Google Scholar] [CrossRef]
- Tojo, M.; Hamashima, Y.; Hanyu, A.; Kajimoto, T.; Saitoh, M.; Miyazono, K.; Node, M.; Imamura, T. The ALK-5 inhibitor A-83-01 inhibits Smad signaling and epithelial-to-mesenchymal transition by transforming growth factor-beta. Cancer Sci. 2005, 96, 791–800. [Google Scholar] [CrossRef]
- Goldstein, L.A.; Ghersi, G.; Piñeiro-Sánchez, M.L.; Salamone, M.; Yeh, Y.; Flessate, D.; Chen, W.-T. Molecular cloning of seprase: A serine integral membrane protease from human melanoma. Biochim. Biophys. Acta 1997, 1361, 11–19. [Google Scholar] [CrossRef]
- Balaziova, E.; Bušek, P.; Stremenová, J.; Sromova, L.; Křepela, E.; Lizcova, L.; Šedo, A. Coupled expression of dipeptidyl peptidase-IV and fibroblast activation protein-α in transformed astrocytic cells. Mol. Cell. Biochem. 2011, 354, 283–289. [Google Scholar] [CrossRef]
- Constam, D.B.; Philipp, J.; Malipiero, U.V.; Ten Dijke, P.; Schachner, M.; Fontana, A. Differential expression of transforming growth factor-beta 1, -beta 2, and -beta 3 by glioblastoma cells, astrocytes, and microglia. J. Immunol. 1992, 148, 1404–1410. [Google Scholar]
- Sasaki, A.; Naganuma, H.; Satoh, E.; Nagasaka, M.; Isoe, S.; Nakano, S.; Nukui, H. Secretion of transforming growth factor-beta 1 and -beta 2 by malignant glioma cells. Neurol. Med. Chir. (Tokyo) 1995, 35, 423–430. [Google Scholar] [CrossRef]
- Leitlein, J.; Aulwurm, S.; Waltereit, R.; Naumann, U.; Wagenknecht, B.; Garten, W.; Weller, M.; Platten, M. Processing of immunosuppressive pro-TGF-beta 1,2 by human glioblastoma cells involves cytoplasmic and secreted furin-like proteases. J. Immunol. 2001, 166, 7238–7243. [Google Scholar] [CrossRef]
- Frei, K.; Gramatzki, D.; Tritschler, I.; Schroeder, J.J.; Espinoza, L.; Rushing, E.J.; Weller, M. Transforming growth factor-beta pathway activity in glioblastoma. Oncotarget 2015, 6, 5963–5977. [Google Scholar] [CrossRef]
- Herbertz, S.; Sawyer, J.S.; Stauber, A.J.; Gueorguieva, I.; Driscoll, K.E.; Estrem, S.T.; Cleverly, A.L.; Desaiah, D.; Guba, S.C.; Benhadji, K.A.; et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Dev. Ther. 2015, 9, 4479–4499. [Google Scholar] [CrossRef]
- Akhurst, R.J. Targeting TGF-beta signaling for therapeutic gain. Cold Spring Harb. Perspect. Biol. 2017, 9, a022301. [Google Scholar] [CrossRef] [PubMed]
- Imamichi, Y.; Waidmann, O.; Hein, R.; Eleftheriou, P.; Giehl, K.; Menke, A. TGF beta-induced focal complex formation in epithelial cells is mediated by activated ERK and JNK MAP kinases and is independent of Smad4. Biol. Chem. 2005, 386, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Ikushima, H.; Todo, T.; Ino, Y.; Takahashi, M.; Miyazawa, K.; Miyazono, K. Autocrine TGF-beta signaling maintains tumorigenicity of glioma-initiating cells through Sry-related HMG-box factors. Cell Stem Cell 2009, 5, 504–514. [Google Scholar] [CrossRef]
- Bandyopadhyay, B.; Han, A.; Dai, J.; Fan, J.; Li, Y.; Chen, M.; Woodley, D.T.; Li, W. TbetaRI/ Alk5-independent TbetaRII signaling to ERK1/2 in human skin cells according to distinct levels of TbetaRII expression. J. Cell Sci. 2011, 124, 19–24. [Google Scholar] [CrossRef]
- Hamidi, A.; Song, J.; Thakur, N.; Itoh, S.; Marcusson, A.; Bergh, A.; Heldin, C.H.; Landstrom, M. TGF-beta promotes PI3K-AKT signaling and prostate cancer cell migration through the TRAF6-mediated ubiquitylation of p85alpha. Sci. Signal. 2017, 10, 486. [Google Scholar] [CrossRef]
- Zhang, J.; Valianou, M.; Cheng, J.D. Identification and characterization of the promoter of fibroblast activation protein. Front. Biosci. (Elite Ed.) 2010, 2, 1154–1163. [Google Scholar] [CrossRef]
- Mikheeva, S.A.; Mikheev, A.M.; Petit, A.; Beyer, R.; Oxford, R.G.; Khorasani, L.; Maxwell, J.-P.; Glackin, C.A.; Wakimoto, H.; González-Herrero, I.; et al. TWIST1 promotes invasion through mesenchymal change in human glioblastoma. Mol. Cancer 2010, 9, 194. [Google Scholar] [CrossRef]
- Yi, Y.; Wang, Z.; Sun, Y.; Chen, J.; Zhang, B.; Wu, M.; Li, T.; Hu, L.; Zeng, J. The EMT-related transcription factor snail up-regulates FAPalpha in malignant melanoma cells. Exp. Cell Res. 2018, 364, 160–167. [Google Scholar] [CrossRef]
- Chen, S.J.; Ning, H.; Ishida, W.; Sodin-Semrl, S.; Takagawa, S.; Mori, Y.; Varga, J. The early-immediate gene EGR-1 is induced by transforming growth factor-beta and mediates stimulation of collagen gene expression. J. Biol. Chem. 2006, 281, 21183–21197. [Google Scholar] [CrossRef]
- Cho, H.J.; Baek, K.E.; Saika, S.; Jeong, M.J.; Yoo, J. Snail is required for transforming growth factor-beta-induced epithelial-mesenchymal transition by activating PI3 kinase/Akt signal pathway. Biochem. Biophys. Res. Commun. 2007, 353, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.J.; Thuault, S.; Caja, L.; Carletti, T.; Heldin, C.H.; Moustakas, A. Regulation of Transcription Factor Twist Expression by the DNA Architectural Protein High Mobility Group A2 during Epithelial-to-Mesenchymal Transition. J. Biol. Chem. 2012, 287, 7134–7145. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.V.; Conroy, S.; Tomar, T.; Eggens-Meijer, E.; Bhat, K.; Copray, S.; Walenkamp, A.M.; Boddeke, E.; Balasubramanyian, V.; Wagemakers, M.; et al. TGF-beta is an inducer of ZEB1-dependent mesenchymal transdifferentiation in glioblastoma that is associated with tumor invasion. Cell Death Dis. 2014, 5, e1443. [Google Scholar] [CrossRef] [PubMed]
- Fuxe, J.; Vincent, T.; Garcia de Hereos, A. Transcriptional crosstalk between TGF- beta and stem cell pathways in tumor cell invasion: Role of EMT promoting Smad complexes. Cell Cycle 2010, 9, 2363–2374. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.; Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro. Oncol. 2017, 19, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Sana, J.; Bušek, P.; Fadrus, P.; Besse, A.; Radova, L.; Vecera, M.; Reguli, S.; Stollinova-Sromova, L.; Hilser, M.; Lipina, R.; et al. Identification of microRNAs differentially expressed in glioblastoma stem-like cells and their association with patient survival. Sci. Rep. 2018, 8, 2836. [Google Scholar] [CrossRef]
- Miebach, S.; Grau, S.; Hummel, V.; Rieckmann, P.; Tonn, J.-C.; Goldbrunner, R.H. Isolation and Culture of Microvascular Endothelial Cells from Gliomas of Different WHO Grades. J. Neuro-Oncol. 2006, 76, 39–48. [Google Scholar] [CrossRef]
- Charalambous, C.; Hofman, F.M.; Chen, T.C. Functional and phenotypic differences between glioblastoma multiforme—derived and normal human brain endothelial cells. J. Neurosurg. 2005, 102, 699–705. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Keane, F.M.; Yao, T.-W.; Seelk, S.; Gall, M.G.; Chowdhury, S.; Poplawski, S.E.; Lai, J.H.; Li, Y.; Wu, W.; Farrell, P.; et al. Quantitation of fibroblast activation protein (FAP)-specific protease activity in mouse, baboon and human fluids and organs. FEBS Open Bio 2013, 4, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Dvořáková, P.; Bušek, P.; Knedlík, T.; Schimer, J.; Etrych, T.; Kostka, L.; Stollinova-Sromova, L.; Šubrt, V.; Šácha, P.; Šedo, A.; et al. Inhibitor-Decorated Polymer Conjugates Targeting Fibroblast Activation Protein. J. Med. Chem. 2017, 60, 8385–8393. [Google Scholar] [CrossRef] [PubMed]
- Bušek, P.; Vanickova, Z.; Hrabal, P.; Brabec, M.; Frič, P.; Zavoral, M.; Skrha, J.; Kmochová, K.; Laclav, M.; Bunganič, B.; et al. Increased tissue and circulating levels of dipeptidyl peptidase-IV enzymatic activity in patients with pancreatic ductal adenocarcinoma. Pancreatology 2016, 16, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Matrasova, I.; Bušek, P.; Balaziova, E.; Šedo, A. Heterogeneity of molecular forms of dipeptidyl peptidase-IV and fibroblast activation protein in human glioblastomas. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 2017, 161, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Krepela, E.; Bušek, P.; Hilser, M.; Vanickova, Z.; Šedo, A. Species-specific real-time RT-PCR analysis of expression of stromal cell genes in a tumor xenotransplantation model in mice. Biochem. Biophys. Res. Commun. 2017, 491, 126–133. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diagnosis | n | Age at Surgery a (Years) | Sex (Male/Female) | IDH b Status (Wild/Mutated) |
|---|---|---|---|---|
| Glioblastoma multiforme | 76 | 62 (41–80) | 49/27 | 76/0 |
| Pharmacoresistant epilepsy | 10 | 32 (22–62) | 3/7 | N.D. c |
| Antibody, Source | Dilution | Temperature, Incubation Time | Secondary Antibody (1:500, Room Temperature, 1 h) |
|---|---|---|---|
| Anti-FAP mouse IgG1 kappa isolated from F19 mouse hybridoma (ATCC CRL-2733) | 56 μg·mL−1 | 4 °C, overnight | Anti-mouse Alexa Fluor 488 (Invitrogen, A21202) |
| Anti-TE-7 (CBL271, Millipore) | 1:100 | room temperature, 1 h | Anti-mouse Alexa Fluor 488 (Invitrogen, A21202) |
| Anti-PDGFRbeta (LS-C11443, LSBio) | 1:50 | 4 °C, overnight | Anti-mouse Alexa Fluor 488 (Invitrogen, A21202) |
| Anti-vWF (A0083, Dako) | 1:200 | room temperature, 1 h | Anti-rabbit Alexa Fluor 488 (Invitrogen, A11010) |
| Anti-GFAP (11-255-M001, Exbio) | 1:200 | 4 °C, overnight | Anti-mouse Alexa Fluor 488 (Invitrogen, A21202) |
| Anti-beta III Tubulin (TU-20, ab7751, Abcam) | 1:250 | 4 °C, overnight | Anti-mouse Alexa Fluor 488 (Invitrogen, A21202) |
| Human Transcript | GenBank Accession No. | Primer Names | Sequences of Primers and TaqMan Probes |
|---|---|---|---|
| Fibroblast activation protein (FAP) | NM_004460.4 | Forward primer: | 5′-TCTGCTGTGCTTGCCTTATTG-3′ |
| Reverse primer: | 5′-ATGAAGATATTCTTGTCCTGAAATCC-3′ | ||
| TaqMan probe: | 5′-(6-FAM)TGCATTGTCTTACGCCCTTCAAGAGTTCA(TAMRA)-3′ | ||
| β-actin (ACTB) | NM_001101.2 | Forward primer: | 5′-ATGGCCACGGCTGCTT-3′ |
| Reverse primer: | 5′-CCATGCCCAGGAAGGAA-3′ | ||
| TaqMan probe: | 5′-(6-FAM)CCCTGGAGAAGAGCTACGAGCTGCCT(TAMRA)-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krepela, E.; Vanickova, Z.; Hrabal, P.; Zubal, M.; Chmielova, B.; Balaziova, E.; Vymola, P.; Matrasova, I.; Busek, P.; Sedo, A. Regulation of Fibroblast Activation Protein by Transforming Growth Factor Beta-1 in Glioblastoma Microenvironment. Int. J. Mol. Sci. 2021, 22, 1046. https://doi.org/10.3390/ijms22031046
Krepela E, Vanickova Z, Hrabal P, Zubal M, Chmielova B, Balaziova E, Vymola P, Matrasova I, Busek P, Sedo A. Regulation of Fibroblast Activation Protein by Transforming Growth Factor Beta-1 in Glioblastoma Microenvironment. International Journal of Molecular Sciences. 2021; 22(3):1046. https://doi.org/10.3390/ijms22031046
Chicago/Turabian StyleKrepela, Evzen, Zdislava Vanickova, Petr Hrabal, Michal Zubal, Barbora Chmielova, Eva Balaziova, Petr Vymola, Ivana Matrasova, Petr Busek, and Aleksi Sedo. 2021. "Regulation of Fibroblast Activation Protein by Transforming Growth Factor Beta-1 in Glioblastoma Microenvironment" International Journal of Molecular Sciences 22, no. 3: 1046. https://doi.org/10.3390/ijms22031046
APA StyleKrepela, E., Vanickova, Z., Hrabal, P., Zubal, M., Chmielova, B., Balaziova, E., Vymola, P., Matrasova, I., Busek, P., & Sedo, A. (2021). Regulation of Fibroblast Activation Protein by Transforming Growth Factor Beta-1 in Glioblastoma Microenvironment. International Journal of Molecular Sciences, 22(3), 1046. https://doi.org/10.3390/ijms22031046