1. Introduction
The emerging antibiotic resistance crisis represents a serious healthcare problem and was declared by the WHO as one of the biggest threats to global health, food security, and development. For the implementation of efficient disease control measures, cost-effective and fast diagnostic systems are essential tools to identify local outbreaks and to contain the further spread of multi- (MDR) and pandrug-resistant (XDR) pathogens into the community. The current gold standard in clinical microbiology for the characterization of antibiotic resistance is based on cultivation-based techniques that are very cost-effective but require several days until the results become available. In contrast, DNA-based technologies such as real-time PCR are fast (<4 h) but still too expensive for mass screenings in low-resource settings. In particular, the application of fluorescent labels and light cyclers are major cost-factors in such assays. Thus, novel technologies that are significantly more cost-effective and able to detect hundreds of antibiotic resistance genes in parallel have still to be developed.
Padlock probes are long, single-stranded oligonucleotides with two terminal recognition regions complementary to a target, a primer attachment site, and usually a DNA barcode sequence (
Figure 1) [
1]. Upon hybridization of the two ends to a target, the probe circularizes without a gap between the 3′ and 5′ ends. The probe is then ligated by a highly specific ligase.
This ligated circular probe can then be specifically replicated using rolling circle amplification (RCA) to a very high copy number. The resulting RCA products are usually detected via real-time amplification or microarrays [
2,
3]. High specificity is guaranteed due to the higher specificity of the ligase (compared to a polymerase) and the double recognition by two target recognition arms allowing to distinguish single nucleotide polymorphisms (SNPs) [
4]. Multiplexing can be done to a very high degree of up to 10,000 probes [
5,
6]. However, the degree of multiplexing is limited by the downstream analysis techniques. While microarrays allow analyzing 10,000 targets in parallel, this is not possible using quantitative PCR. The padlock probe technique was already used for the detection of pathogens and their antibiotic resistance genes [
7,
8].
Here, we describe a novel detection system independent of a costly downstream analysis platform. We integrated a complementary G-quadruplex-hemin DNAzyme sequence into the padlock probe. This sequence is amplified during an RCA and finally monomerized using a restriction site integrated into the primer attachment site. The sequence is then detected using the DNAzyme-catalyzed peroxidation of the 2,2′-azino-bis(3-ethylbenzothiozoline-6-sulfonic acid) diammonium salt (ABTS2-), which is visible by the naked eye. The scheme of the assay is shown in
Figure 2. Although this assay relies on amplification techniques, the assay is considered label-free since no fluorescent, chemiluminescent, radioactive, or electrochemical readout methods were used.
4. Materials and Methods
Chemicals, enzymes, and buffers. The ampligase thermostable ligase was obtained from Lucigen (Middleton, WI, USA), dNTPs from Invitrogen (Thermo Fisher Scientific, Waltham, MA, USA), phi29 DNA polymerase, AvaI restriction enzyme, and bovine serum albumin (BSA) from New England Biolabs (Ipswich, MA, USA), hemin from Merck KGaA (Darmstadt, Germany), 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) tablets and hydrogen peroxide solution (H2O2) from Sigma–Aldrich (St. Louis, MO, USA), and Hot MolTaq 16S/18S polymerase from Molzym (Bremen, Germany). The EnSpire multimode plate reader was from PerkinElmer (Waltham, MA, USA). The Ion Torrent sequencing chemistries were obtained from Thermo Fisher Scientific (Waltham, MA, USA). HEPES buffer A consisted of 25 mM of HEPES, 20 mM of KCl, 200 mM of NaCl, 0.05% Triton X-100, and 1% DMSO (pH 7.4 @ 25 °C). HEPES buffer B consisted of 25 mM of HEPES, 20 mM of KCl, and 200 mM of NaCl (pH 5.5 @ 25 °C).
Oligonucleotides. All oligonucleotides (
Supplementary Tables S1–S4) were obtained from Integrated DNA Technologies (Coralville, IA, USA). Each padlock probe consists of a sequence 1, insert 1, filler, insert 2, and sequence 2 as indicated in
Supplementary Table S1; the insert 1 (primer attachment site) corresponds to ACTACTTACCTCGGGC, and the insert 2 (G-quadruplex sequence) corresponds to TTTTCCCCTTTTCCCCTTTT-CCCCTTTTCCCC.
Design of the G-quadruplex padlock probes. The padlock probes were designed using a publicly available Python script. The script takes the sequences from the products of the 45-plex primer set (
Supplementary Table S2) as input, then complementary sequences are designed starting at either strand with an offset to the 5’ end of 2–25 bp (Tm = 62 °C). Thermodynamic data were calculated using SantaLucia’s model with salt correction. The complementary G-quadruplex sequences and a primer attachment sequence were inserted into the padlock probe. If the target length was shorter than 80 nt, a random filler sequence was inserted. The probes are listed in
Supplementary Table S1. Initially, 45 probes were designed to cover the full 45-plex PCR assay, but due to their false-positive and interfering signals, 17 probes were removed from the assay, resulting in a functional 27-plex detection assay.
Bacterial culture and DNA extraction. A volume of 10 µL of bacterial suspension from clinical isolates (Enterococcus faecium VRE, Klebsiella pneumoniae 08.22, Enterobacter cloacae 30676, Acinetobacter baumannii 44921) was inoculated in 5 mL of TSB. The incubation took place at 37 °C in a 15-mL conical tube on an orbital shaker set to 300 rpm for 20 h. DNA of all strains was extracted and purified using the QIAamp DNA Mini Kit (QIAGEN, Hilden, Germany). The first steps of the manufacturer’s protocol comprising cell lysis and disruption: A total of 2 mL of cell suspension was taken from liquid culture. The total volume was centrifuged for 5 min at 5000× g, then the supernatant was discarded, and the cell pellet was resuspended in 200 µL of TSB. The resuspended pellet was then added to a 2-mL vial with a screw cap containing sterile acid-washed glass beads with a diameter of 150–212 µm (Sigma–Aldrich, St. Louis, MO, USA) filled to the 100-µL mark. The suspension was subjected to 2 runs at 6500 rpm for 30 s in a MagNA Lyser instrument (Roche, Basel, Switzerland), separated by a 5 min incubation at room temperature. The vial was centrifuged for 3 min at 16,000× g, and 180 µL of the supernatant was transferred to a clean 1.5-mL Eppendorf tube. The protocol was continued with the addition of buffer AL and proteinase K according to the manufacturer’s instructions. The DNA concentration was measured on a NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
Sequencing. Whole-genome sequencing was performed on the Ion Torrent PGM platform (Thermo Fisher Scientific, Waltham, MA, USA) using reads with a length of 400 nt. Library preparation was done using the Ion Xpress Plus Fragment Library kit, 100 ng of genomic DNA input, and Ion Xpress Barcode Adapters, following the manufacturer’s instructions. The sheared DNA was size-selected on E-Gel SizeSelect II agarose gels (Thermo Fisher Scientific, Waltham, MA, USA). The total volume was recovered. Quantification was done using the Ion Library TaqMan quantitation kit following the manufacturer’s instructions on a LightCycler 480 System (Roche, Basel, Switzerland). The library was quantified and qualified on a 5300 Fragment Analyzer System (Agilent Technologies, Santa Clara, CA, USA) using the HS Small Fragment Kit (Agilent Technologies, Santa Clara, CA, USA). All samples were diluted to 100 pM, and 5 µL of each sample were pooled together. The sample for sequencing was prepared using the Ion PGM Hi-Q View OT2 kit following the manufacturer’s instructions. Clonal amplification and enrichment were carried out with the Ion OneTouch 2 instrument. Template-positive ion sphere particles (ISPs) were enriched on the Ion OneTouch ES instrument. The quality of unenriched, template-positive ISPs was assessed using the Ion Sphere Quality Control kit on the Qubit 2.0 fluorometer (Thermo Fisher Scientific, Waltham, MA, USA) following the manufacturer’s instructions. The sequencing was performed using the Ion PGM Hi-Q View Sequencing kit following the manufacturer’s instructions, except that control ISPs were not added to the sample. The chip loading was done per the manufacturer’s instructions on an Ion 318 chip. The reads were analyzed using FastQC. If necessary, additional trimming and filtering were done using PRINSEQ [
20]. The reads were assembled into contigs and scaffolds using SPAdes [
21] 3.11.1 (parameters: iontorrent, k: [
21], repeat resolution enabled, mismatch careful mode turned off, MismatchCorrector skipped, coverage cutoff turned off). The quality of the assemblies was assessed using QUAST 4.6.3 [
22]. The scaffolds were uploaded to the European Nucleotide Archive (ENA). The antibiotic resistances were identified using ResFinder 3.2 [
23] (parameters: acquired antimicrobial resistance genes: checked, threshold for %ID = 60%, minimum length = 60%). The specificity of primers and padlock probes was checked using PRIMEval [
9].
Optimization of PCR, RCA, restriction, and detection buffer. A PCR (50 µL) comprised 1 × PCR buffer (3.0 mM of MgCl2), 200 µM of each dNTP, 0.02 or 0.05 units/µL Hot MolTaq 16S/18S polymerase (as indicated), and two corresponding primers (
Supplementary Table S2) at 400 nM each. If asymmetric PCR was carried out, the forward primer was used at a final concentration of 1200 nM and the reverse primer at 40 nM. The target DNA was added to a final concentration of 0.4 ng/µL. The thermal cycling was performed as follows: 94 °C for 5 min; 40 cycles at 94 °C for 30 s, 49 °C for 30 s, 72 °C for 30 s; and a final elongation at 72 °C for 7 min. The ligation reaction (45 µL) comprised 1 × ampligase reaction buffer, 50 nM of the padlock probe targeting the gene of interest, 0.2 µg/µL of BSA, 0.20 units/µL of ampligase thermostable DNA ligase, and 10 µL of PCR products. The reaction was incubated for 5 min at 95 °C and 15 min at 55 °C. The rolling circle amplification (RCA) (40 µL) comprised 30 µL of the ligation products, 1 × phi29 DNA polymerase reaction buffer, 0.2 µg/µL BSA, 125 µM of each dNTP, 100 nM of padlock probe primer (
Supplementary Table S4), and 0.17 units/µL phi29 DNA polymerase. The RCA was incubated for either 15 min or 60 min at 37 °C, then 2 min at 65 °C. If an enzymatic restriction was done, 10 µL of the restriction mix was added to 40 µL of the RCA product. The restriction mix comprised 1 × phi29 DNA polymerase reaction buffer, 0.2 µg/µL BSA, 1 µM of the reverse complementary padlock probe primer (
Supplementary Table S4), and 2 units/µL AvaI restriction enzyme. The restriction reaction was incubated for 30 min at 37 °C and 10 min at 80 °C. The detection of the formed G-quadruplex DNAzymes was done by mixing 5 µL of the (digested) RCA products with 70 µL of the detection mix, consisting of 1 × HEPES buffer A or 1 × HEPES buffer B and 10 µM hemin. The colorimetric reaction was started by the addition of 5 mM of ABTS and 1 mM of H
2O
2. All indicated concentrations are final concentrations in the detection volume of 100 µL. The absorbance at 421 nm was measured for up to 40 min on an EnSpire multimode plate reader.
Specificity evaluation of padlock probes targeting antibiotic resistances with synthetic DNA. The 27 padlock probes (
Supplementary Table S1) and 3 controls without probes (H
2O) were put into one half of a 96-well plate (final concentration in the reaction: 50 nM). Then a master mix containing 1 × ampligase reaction buffer, 0.34 units/µL of ampligase thermostable DNA ligase, and 500 nM of synthetic DNA target (
Supplementary Table S3) was added in a final volume of 5 µL. For the ligation, the plate was incubated for 5 min at 95 °C and 15 min at 55 °C. Then, 5 µL of an RCA reaction mix was added to each well. This reaction mix consisted of 1 × phi29 DNA polymerase reaction buffer, 0.2 µg/µL of BSA, 125 µM of each dNTP, 100 nM of padlock probe primer (
Supplementary Table S4), and 0.17 units/µL of phi29 DNA polymerase. All indicated concentrations are final concentrations in the reaction volume of 10 µL. The RCA was incubated for 15 min at 37 °C and 10 min at 80 °C; then, 5 µL of the RCA products were transferred to a flat-bottom plate. The formed G-quadruplex DNAzymes were detected by the addition of the detection mix, consisting of 1 × HEPES buffer B and 10 µM of hemin. The reaction was started by the addition of 5 mM of ABTS and 1 mM of H
2O
2. All indicated concentrations are final concentrations in the detection volume of 100 µL. The absorbance at 421 nm was measured for 40 min on an EnSpire multimode plate reader. The data were normalized to the first time point and to the “no probe control”. Individual graphs were generated using the seaborn library. All end points were summarized in a heatmap generated by the seaborn library after normalization using StandardScaler of scikit-learn. In
Figure S1, the data are also normalized using Standard Scaler as represented in the heatmap.
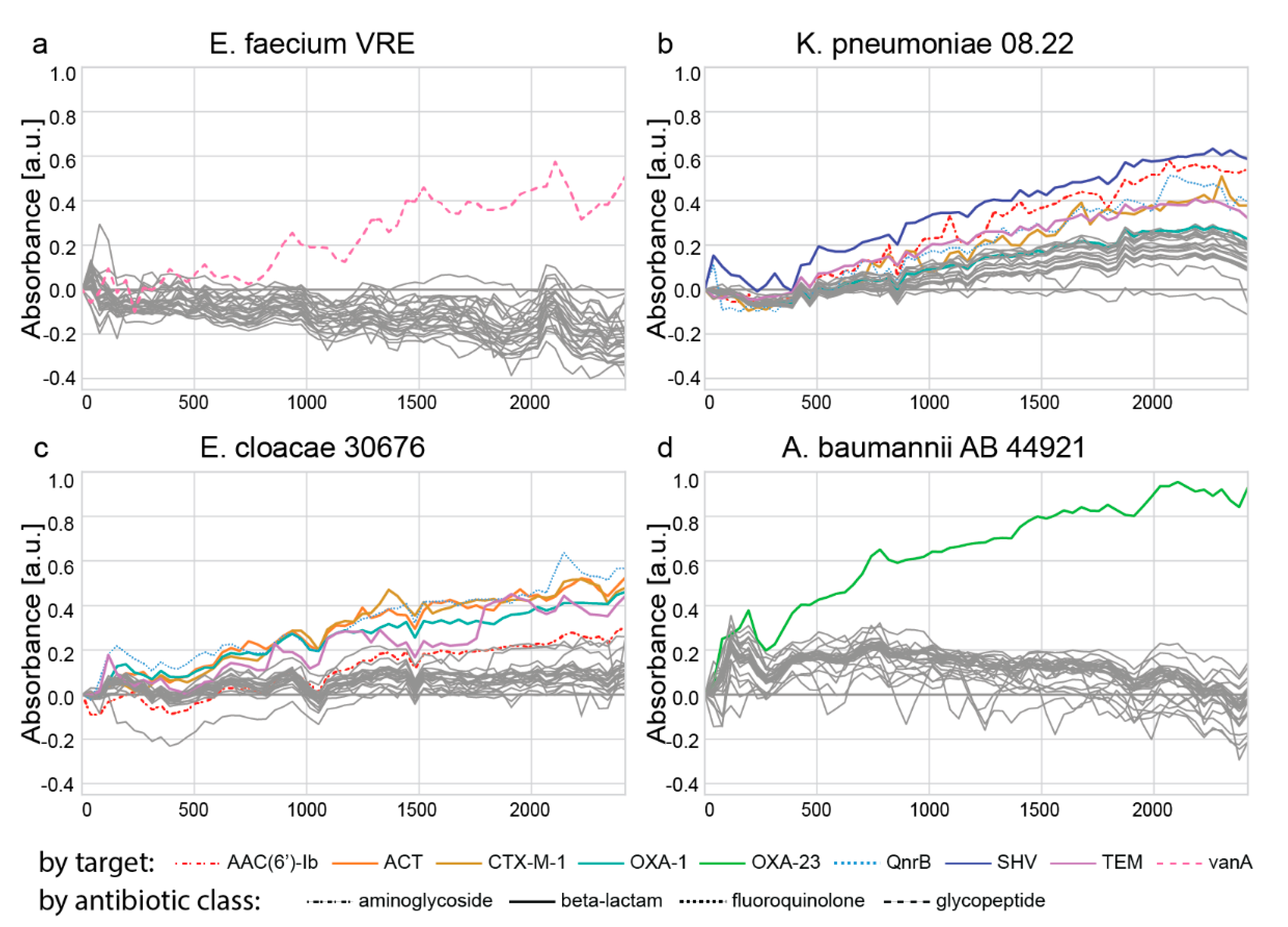
Evaluation of padlock probes targeting antibiotic resistances with genomic DNA. The 45-plex PCR (55 µL) comprised 1 × PCR buffer (3.0 mM of MgCl2), 200 µM of each dNTP, 0.05 units/µL Hot MolTaq 16S/18S polymerase, and primers targeting antibiotic resistance genes (
Supplementary Table S2) at 111 nM each (10-µM final concentration). The target DNA was added to a final concentration of 0.4 ng/µL. The thermal cycling was performed as follows: 94 °C for 5 min; 30 cycles at 94 °C for 30 s, 49 °C for 30 s, 72 °C for 30 s; and a final elongation at 72 °C for 7 min. The 27 padlock probes (
Supplementary Table S1) and 3 controls without probes (H
2O) were put twice into a 96-well plate (final concentration in the reaction: 50 nM), then a master mix containing 1 × ampligase reaction buffer, 0.34 units/µL of ampligase thermostable DNA ligase, and 1 µL of PCR product or H
2O per reaction was added. The final reaction volume was 5 µL. The plate was incubated for 5 min at 95 °C and 15 min at 55 °C. Then, 5 µL of an RCA mix was added to each well. This reaction mix consisted of 1 × phi29 DNA polymerase reaction buffer, 0.2 µg/µL of BSA, 125 µM of each dNTP, 100 nM of padlock probe primer (
Supplementary Table S4), and 0.17 units/µL phi29 DNA polymerase. All indicated concentrations are final concentrations in the reaction volume of 10 µL. The RCA was incubated for 15 min at 37 °C and 10 min at 80 °C; then, 5 µL of the RCA products was transferred to a flat-bottom plate. The formed G-quadruplex DNAzymes were detected by the addition of the detection mix, consisting of 1 × HEPES buffer B and 10 µM of hemin. The reaction was started by the addition of 5 mM of ABTS and 1 mM of H
2O
2. All indicated concentrations are final concentrations in the detection volume of 100 µL. The absorbance at 421 nm was measured for 40 min on an EnSpire multimode plate reader. For the data analysis, the negative controls were subtracted from the positive controls, then the first time point was subtracted from all other time points, and finally, the ‘no probe control’ was subtracted from all values at every time point. Individual graphs were generated using the seaborn library.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




