
The Carbohydrate Metabolism of Lactiplantibacillus plantarum


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. The Sugar Fermentation Capacity of L. plantarum Strains
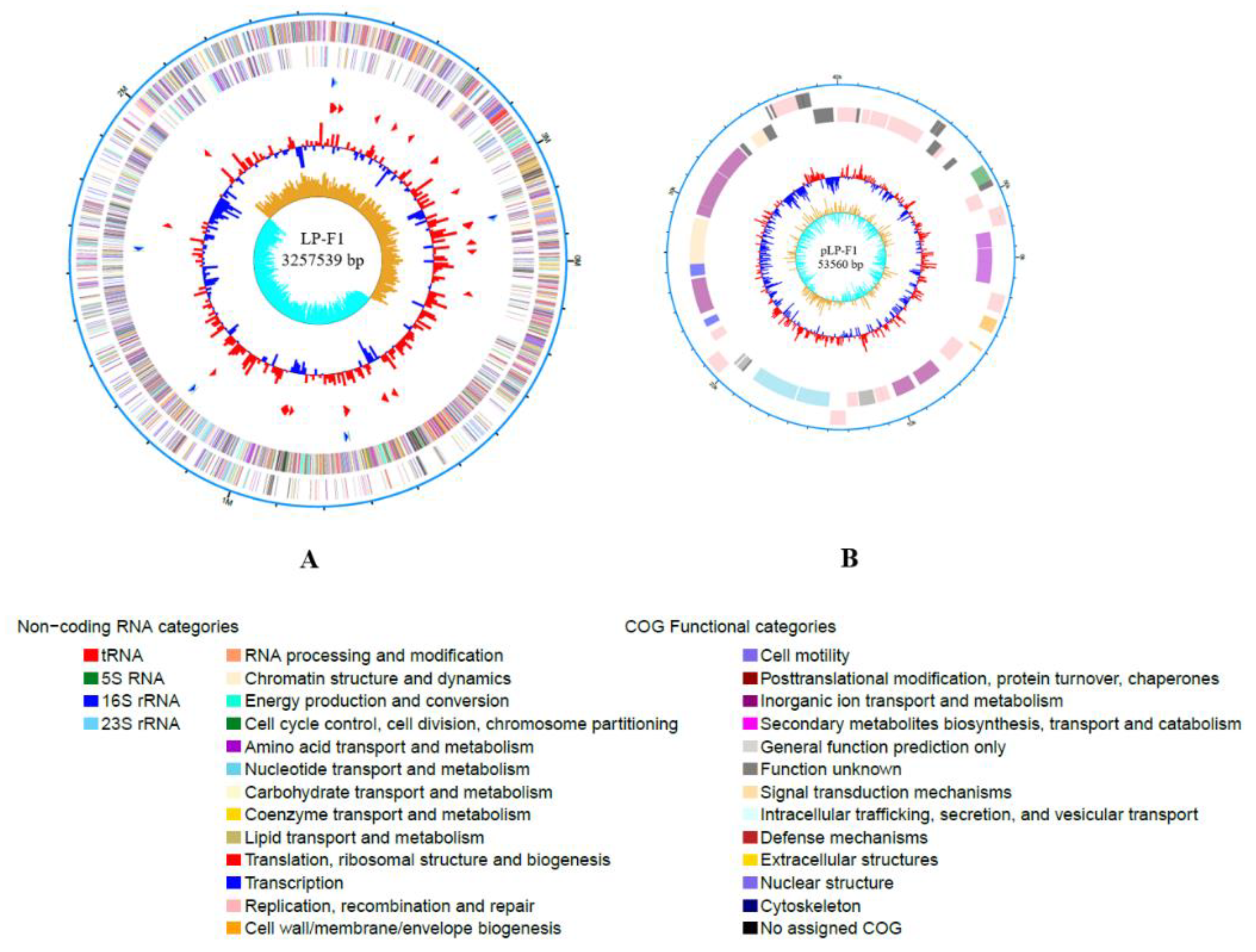
2.2. Genomic Analysis of L. plantarum Strain LP-F1
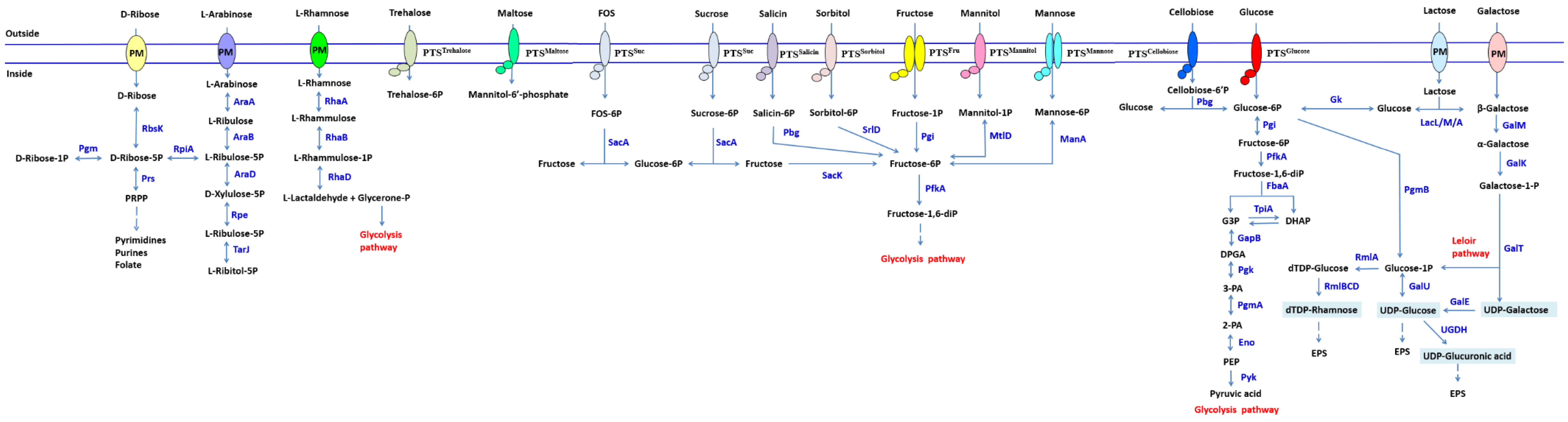
2.3. Carbohydrate Metabolism Systems Analysis of LP-F1 Based on the Genome
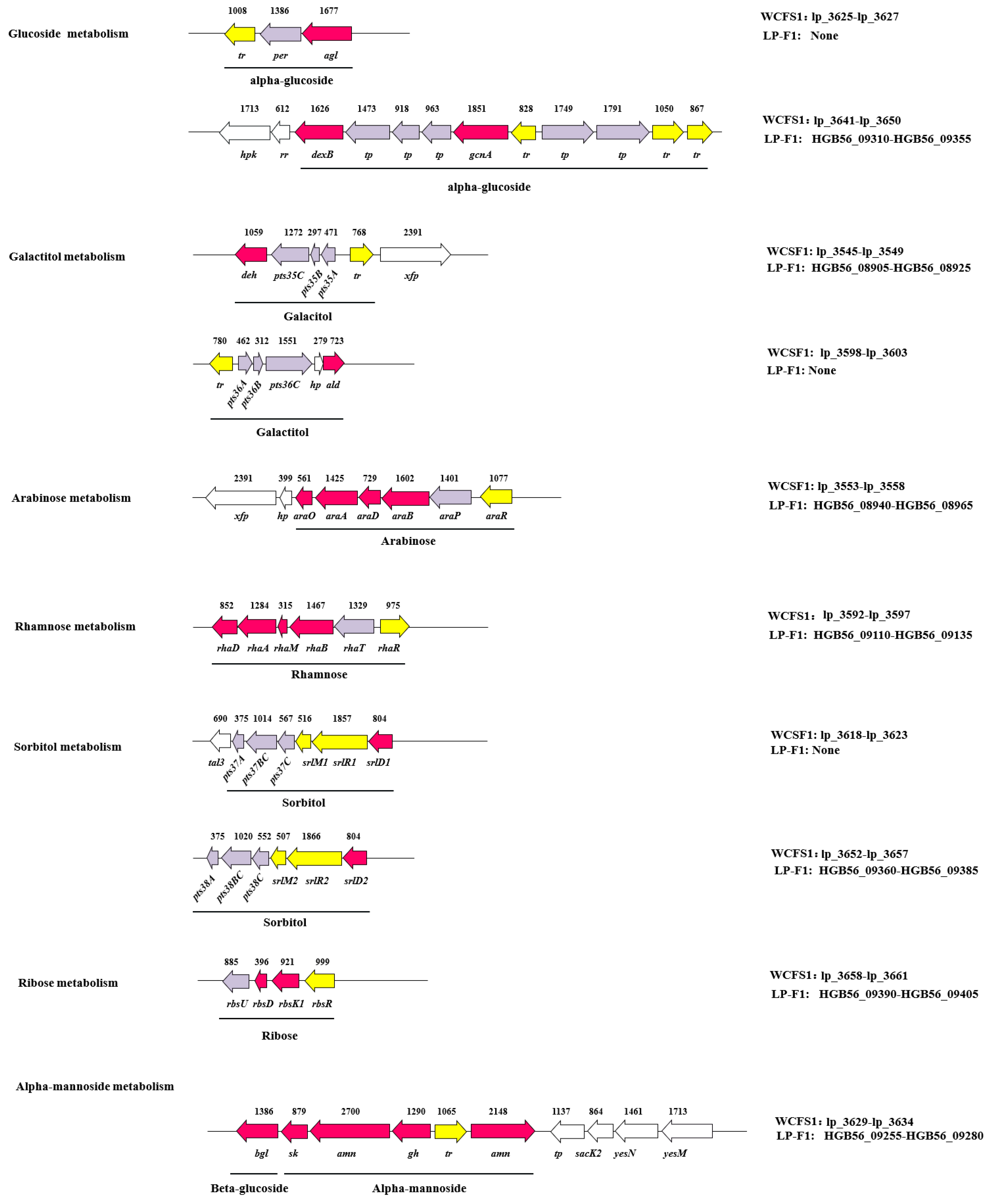
2.3.1. Monosaccharide and Disaccharide
Glucose
Lactose and Galactose
Fructose and Sucrose
Arabinose
Rhamnose
2.3.2. Sugar Alcohol
Galactitol
Sorbitol
2.3.3. Oligosaccharides and Polysaccharides
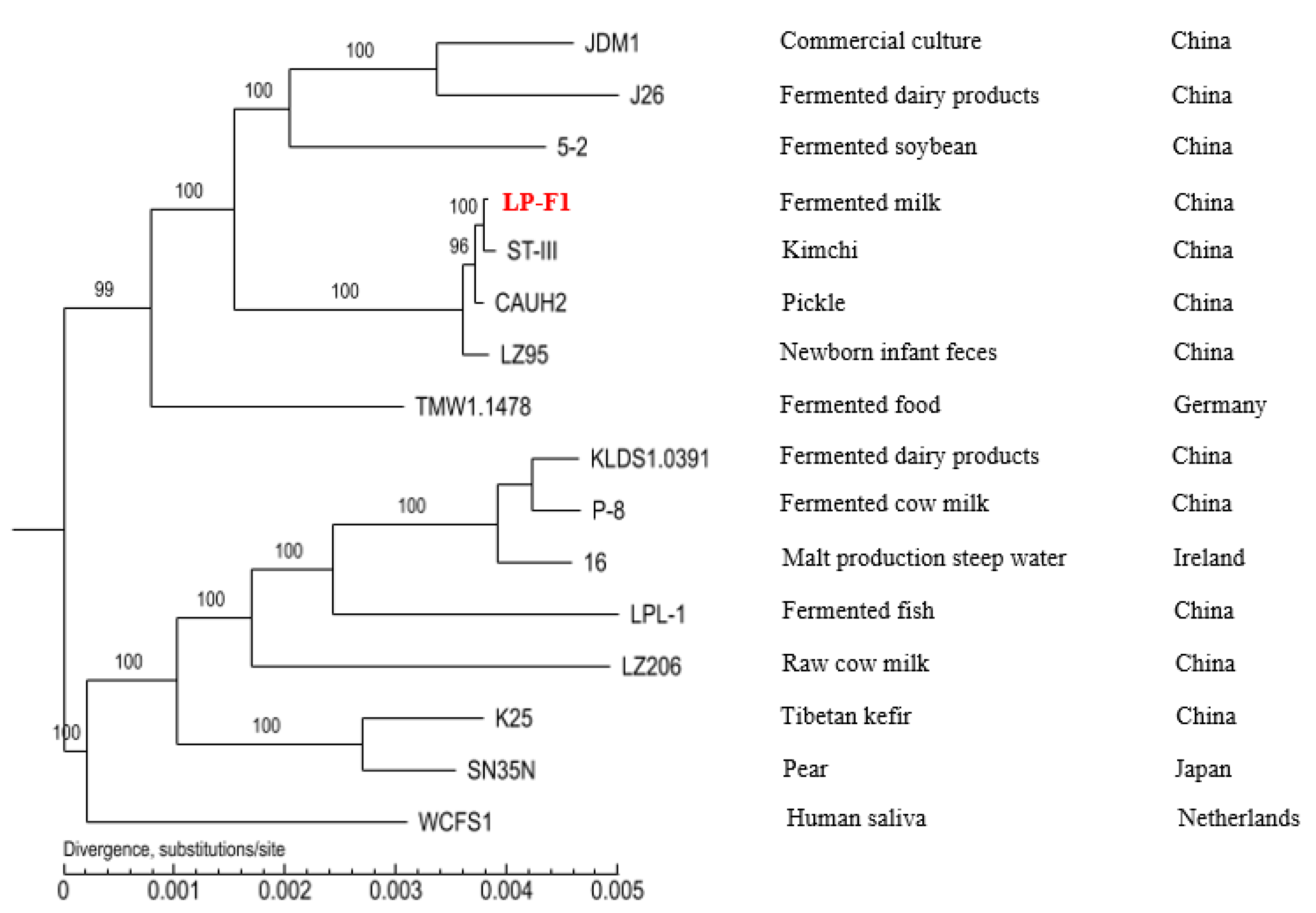
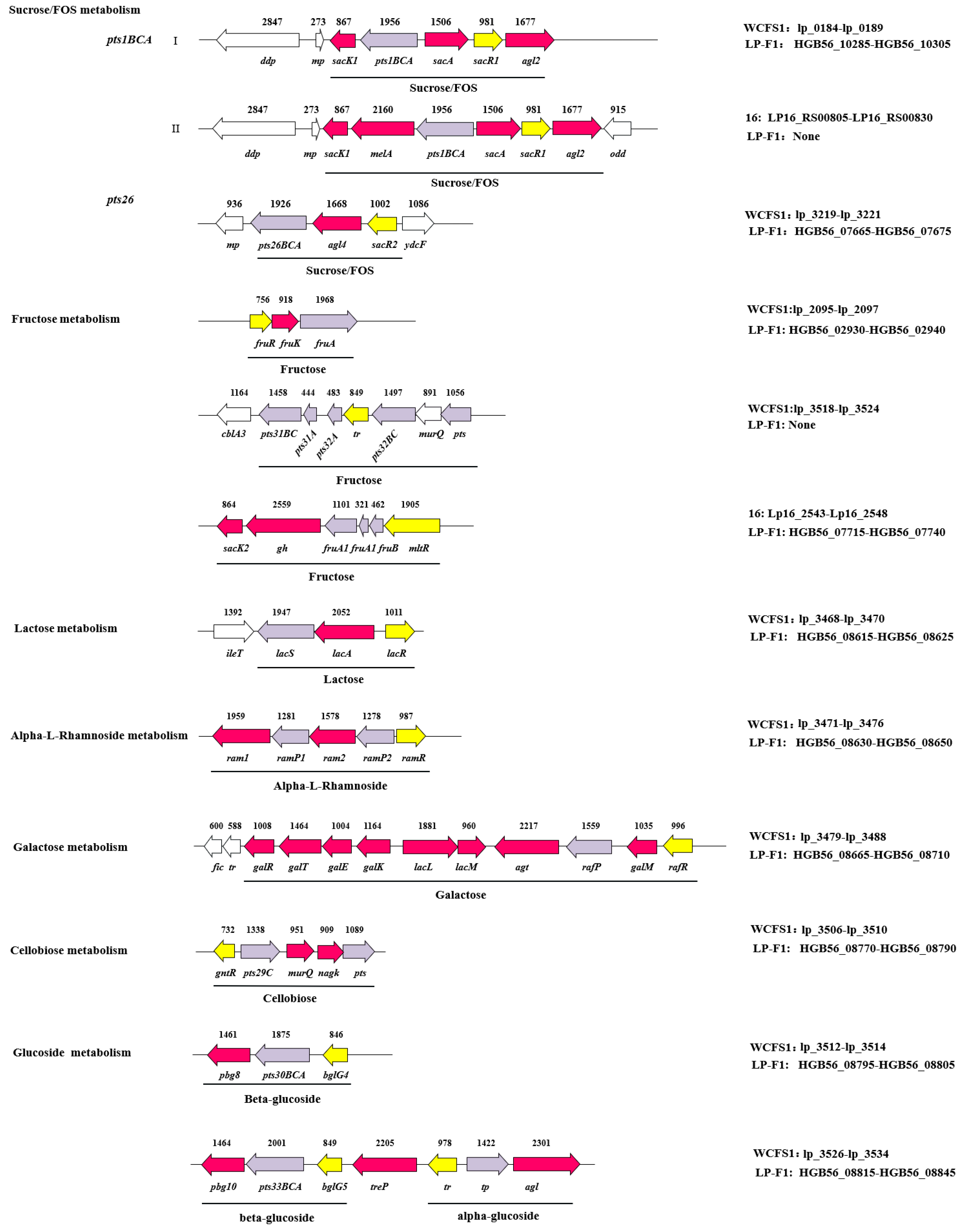
2.3.4. The Difference of Different L. plantarum Strains in Carbohydrates Utilization Genes
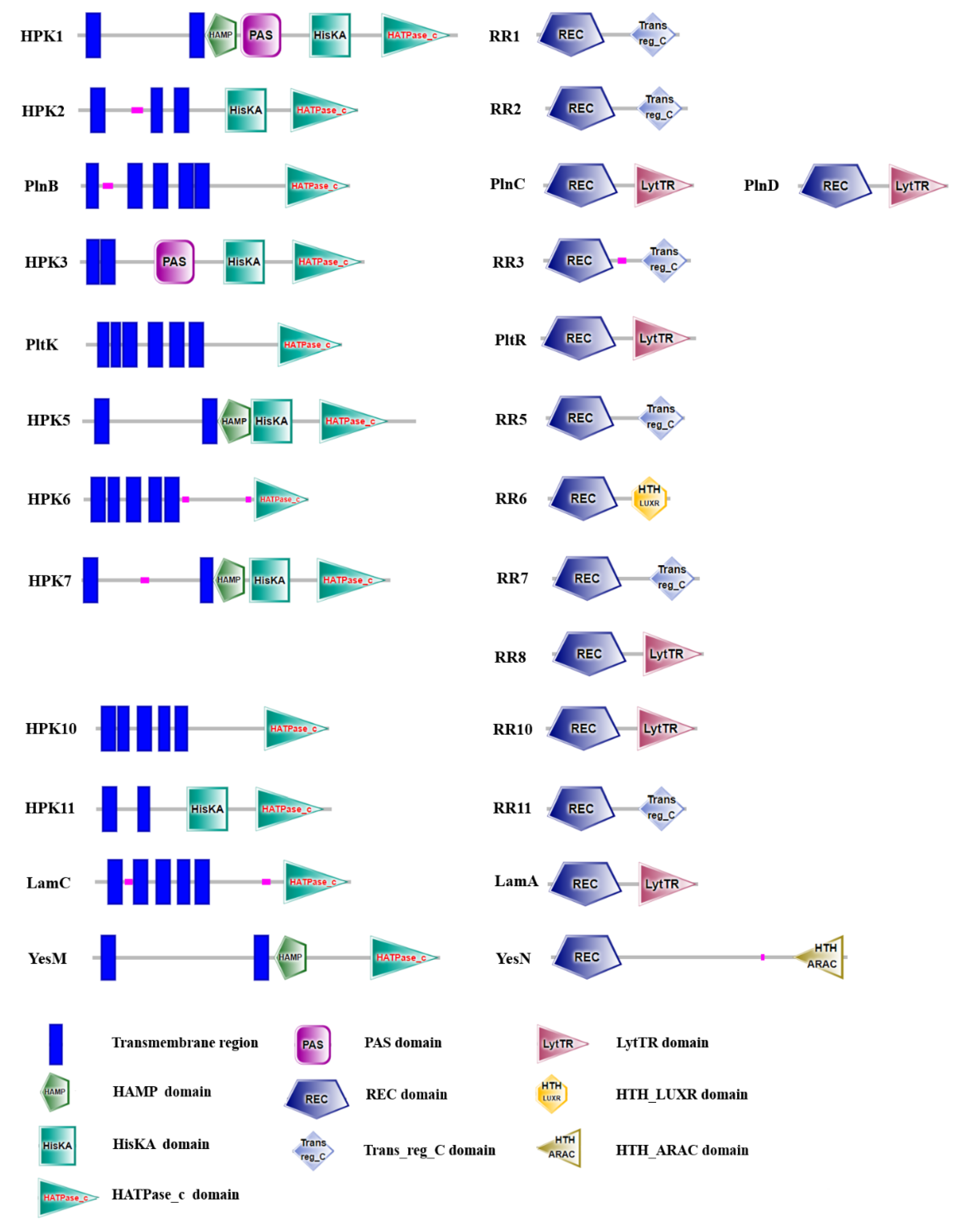
2.4. Two-Component Systems Analysis of LP-F1 Based on the Genome
2.5. Carbohydrate Metabolism Ability Analysis of LP-F1
2.6. Quantification of the Expressions of Sugar Metabolism Related Genes in LP-F1
2.6.1. The Different Fermentation Times
2.6.2. The Effect of Different Sugars on the Expressions of Sugar Metabolism Related Genes under 4 h
2.6.3. The Effect of Different Sugars on the Expressions of Sugar Metabolism Related Genes under 12 h
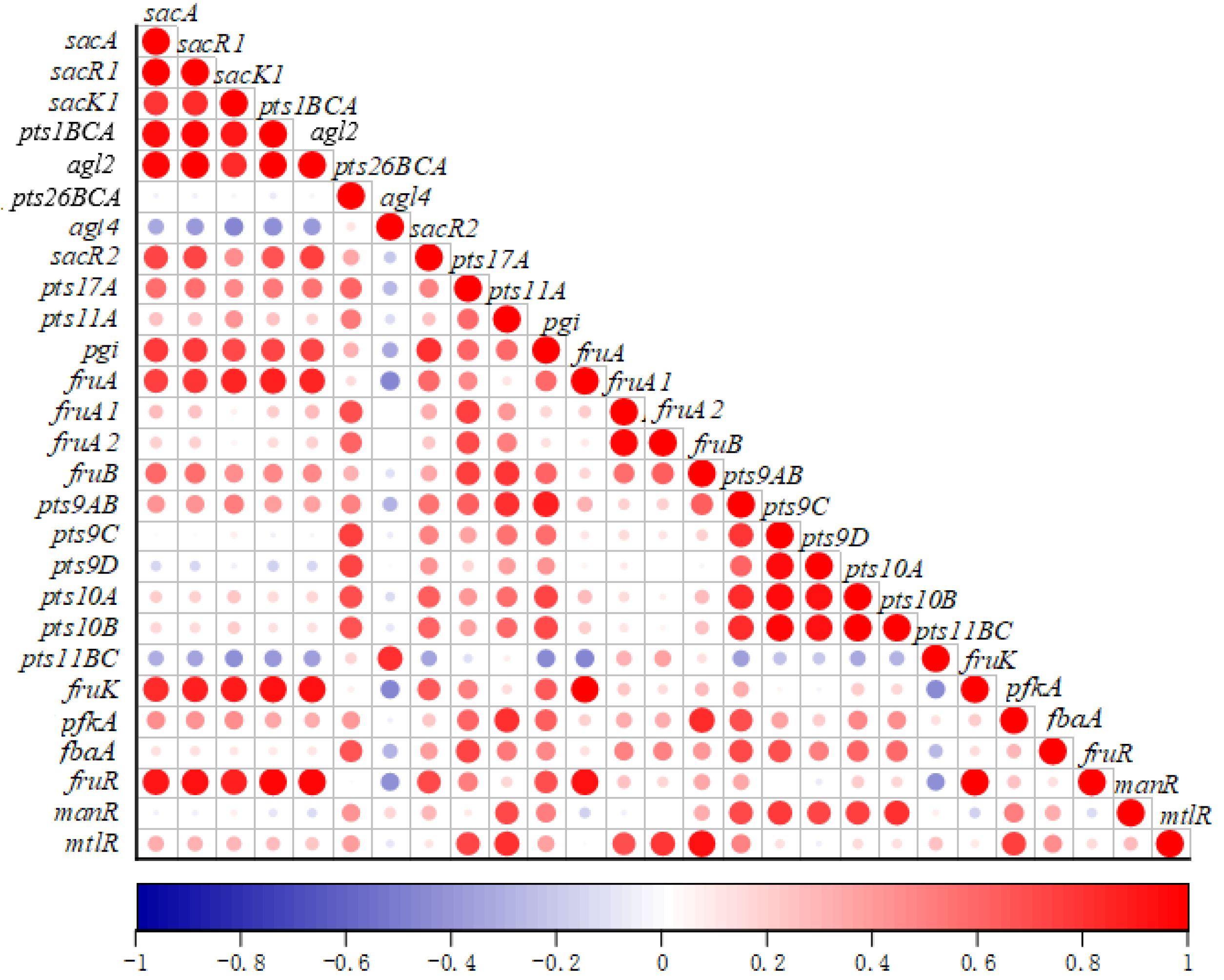
2.6.4. The Correlation of the Sugar Metabolism Related Genes
2.7. Quantification of the Expressions of TCSs Genes in LP-F1
2.7.1. The Fermentation Stages
2.7.2. The Effect of Different Sugars on the Expressions of Genes under 4 h
2.7.3. The Effect of Different Sugars on the Expressions of Genes under 12 h
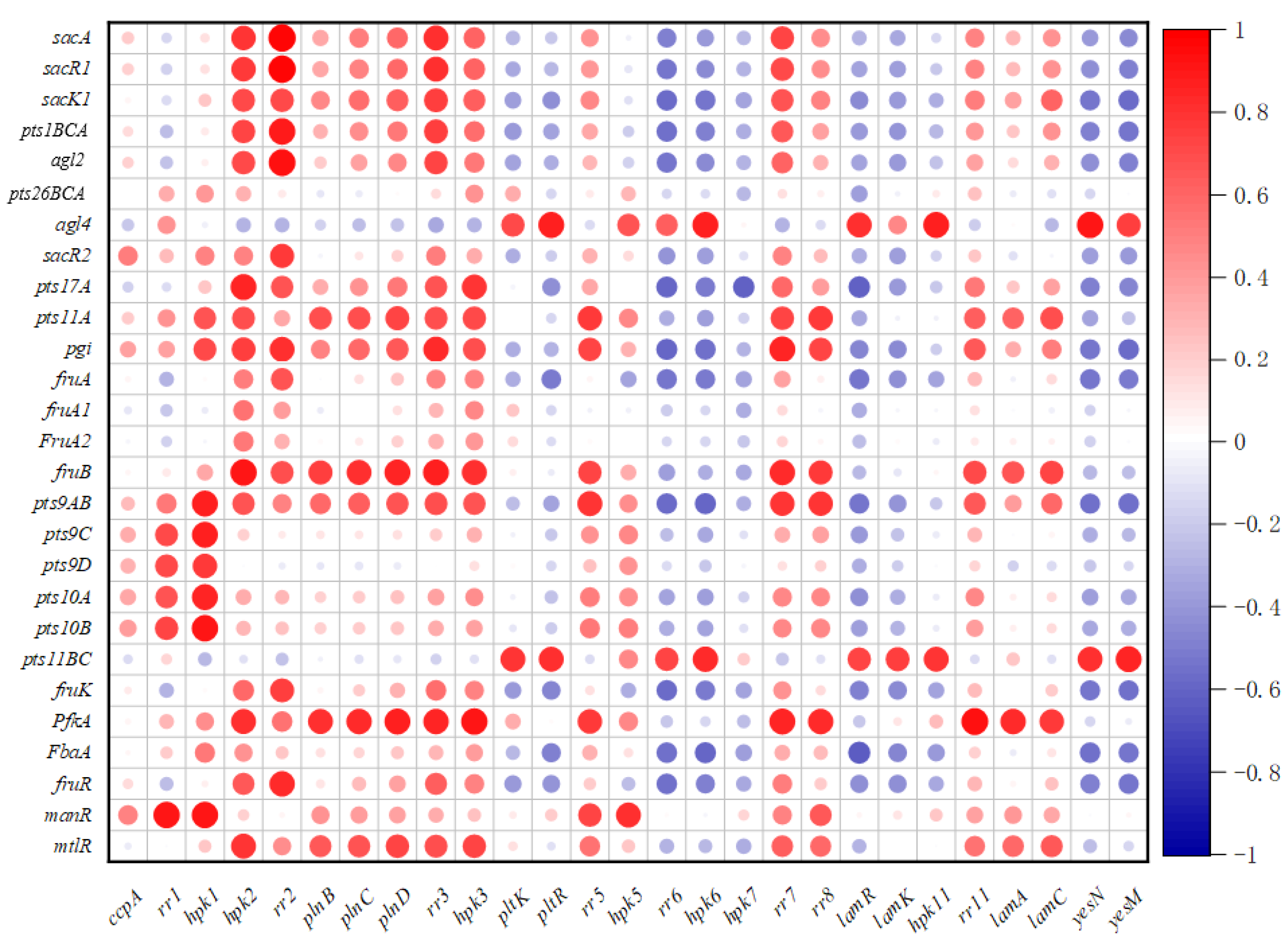
2.8. Correlation between the Sugar Metabolism Related Genes and Regulatory Genes
3. Discussion
4. Materials and Methods
4.1. Bacterial Strain and Growth Conditions
4.2. Genome Sequencing of the L. plantarum LP-F1
4.3. In Silico Analysis of the Carbohydrate Metabolism Genes of L. plantarum
4.4. Carbohydrate Fermentation Tests
4.5. Quantitative Real-Time -PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Turpin, W.; Weiman, M.; Guyot, J.P.; Lajus, A.; Cruveiller, S.; Humblot, C. The genomic and transcriptomic basis of the potential of Lactobacillus plantarum A6 to improve the nutritional quality of a cereal based fermented food. Int. J. Food Microbiol. 2018, 266, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Aquilanti, L.; Santarelli, S.; Silvestri, G.; Osimani, A.; Petruzzelli, A.; Clementi, F. The microbial ecology of a typical Italian salami during its natural fermentation. Int. J. Food Microbiol. 2007, 120, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Axelsson, L.; Rud, I.; Naterstad, K.; Blom, H.; Renckens, B.; Boekhorst, J.; Kleerebezem, M.; van Hijum, S.; Siezen, R.J. Genome sequence of the naturally plasmid-free Lactobacillus plantarum strain NC8 (CCUG 61730). J. Bacteriol. 2012, 194, 2391–2392. [Google Scholar] [CrossRef] [PubMed]
- Kleerebezem, M.; Boekhorst, J.; van Kranenburg, R.; Molenaar, D.; Kuipers, O.P.; Leer, R.; Tarchini, R.; Peters, S.A.; Sandbrink, H.M.; Fiers, M.W.; et al. Complete genome sequence of Lactobacillus plantarum WCFS1. Proc. Natl. Acad. Sci. USA 2003, 100, 1990–1995. [Google Scholar] [CrossRef]
- Li, X.; Gu, Q.; Lou, X.; Zhang, X.; Song, D.; Shen, L.; Zhao, Y. Complete genome sequence of the probiotic Lactobacillus plantarum strain ZJ316. Genome Announc. 2013, 1, e0009413. [Google Scholar] [CrossRef]
- Seddik, H.A.; Bendali, F.; Gancel, F.; Fliss, I.; Spano, G.; Drider, D. Lactobacillus plantarum and its probiotic and food potentialities. Probiotics Antimicrob. Proteins 2017, 9, 111–122. [Google Scholar] [CrossRef]
- Zhou, Y.; Cui, Y.; Qu, X. Exopolysaccharides of lactic acid bacteria: Structure, bioactivity and associations. Carbohydr. Polym. 2019, 207, 317–332. [Google Scholar] [CrossRef]
- Cui, Y.; Miao, K.; Niyaphorn, S.; Qu, X. Production of gamma-aminobutyric acid from lactic acid bacteria: A systematic review. Int. J. Mol. Sci. 2020, 21, 995. [Google Scholar] [CrossRef]
- Wegkamp, A.; Mars, A.E.; Faijes, M.; Molenaar, D.; de Vos, R.C.; Klaus, S.M.; Hanson, A.D.; de Vos, W.M.; Smid, E.J. Physiological responses to folate overproduction in Lactobacillus plantarum WCFS1. Microb. Cell Factories 2010, 9, 100. [Google Scholar] [CrossRef]
- Buntin, N.; Hongpattarakere, T.; Ritari, J.; Douillard, F.P.; Paulin, L.; Boeren, S.; Shetty, S.A.; de Vos, W.M. An inducible operon is involved in inulin utilization in Lactobacillus plantarum strains, as revealed by comparative proteogenomics and metabolic profiling. Appl. Environ. Microb. 2017, 83, e02402-16. [Google Scholar] [CrossRef]
- Fuhren, J.; Rösch, C.; Napel, M.T.; Schols, H.A.; Kleerebezem, M. Synbiotic matchmaking in Lactobacillus plantarum: Substrate screening and gene-trait matching to characterize strain-specific carbohydrate utilization. Appl. Environ. Microbiol. 2020, 86, e01081-20. [Google Scholar] [CrossRef]
- Siezen, R.J.; Tzeneva, V.A.; Castioni, A.; Wels, M.; Phan, H.T.; Rademaker, J.L.; Starrenburg, M.J.; Kleerebezem, M.; Molenaar, D.; van Hylckama Vlieg, J.E. Phenotypic and genomic diversity of Lactobacillus plantarum strains isolated from various environmental niches. Environ. Microbiol. 2010, 12, 758–773. [Google Scholar] [CrossRef]
- Mao, B.; Yin, R.; Li, X.; Cui, S.; Zhang, H.; Zhao, J.; Chen, W. Comparative genomic analysis of Lactiplantibacillus plantarum isolated from different niches. Genes 2021, 12, 241. [Google Scholar] [CrossRef]
- Cui, Y.; Qu, X. Genetic mechanisms of prebiotic carbohydrate metabolism in lactic acid bacteria: Emphasis on Lacticaseibacillus casei and Lacticaseibacillus paracasei as flexible, diverse and outstanding prebiotic carbohydrate starters. Trends Food Sci. Technol. 2021, 115, 486–499. [Google Scholar] [CrossRef]
- Bringel, F.; Quenee, P.; Tailliez, P. Polyphasic investigation of the diversity within Lactobacillus plantarum related strains revealed two L. plantarum subgroups. Syst. Appl. Microbiol. 2001, 24, 561–571. [Google Scholar] [CrossRef]
- Molenaar, D.; Bringel, F.; Schuren, F.H.; de Vos, W.M.; Siezen, R.J.; Kleerebezem, M. Exploring Lactobacillus plantarum genome diversity by using microarrays. J. Bacteriol. 2005, 187, 6119–6127. [Google Scholar] [CrossRef]
- Siezen, R.; van Hylckama Vlieg, J. Genomic diversity and versatility of Lactobacillus plantarum, a natural metabolic engineer. Microb. Cell Factories 2011, 10, S3. [Google Scholar] [CrossRef]
- Choi, S.; Jin, G.D.; Park, J.; You, I.; Kim, E.B. Pan-genomics of Lactobacillus plantarum revealed group-specific genomic profiles without habitat association. J. Microbiol. Biotechnol. 2018, 28, 1352–1359. [Google Scholar] [CrossRef]
- Alcantara, C.; Revilla-Guarinos, A.; Zuniga, M. Influence of two-component signal transduction systems of Lactobacillus casei BL23 on tolerance to stress conditions. Appl. Environ. Microb. 2011, 77, 1516–1519. [Google Scholar] [CrossRef]
- Azcarate-Peril, M.; McAuliffe, O.; Altermann, E.; Lick, S.; Russell, W.M.; Klaenhammer, T.R. Microarray analysis of a two-component regulatory system involved in acid resistance and proteolytic activity in Lactobacillus acidophilus. Appl. Environ. Microbiol. 2005, 71, 5794–5804. [Google Scholar] [CrossRef]
- Bourret, R.B.; Silversmith, R.E. Two-component signal transduction. Curr. Opin. Microbiol. 2010, 13, 113–115. [Google Scholar] [CrossRef]
- Thevenard, B.; Besset, C.; Choinard, S.; Fourcassié, P.; Boyaval, P.; Monnet, V. Response of S. thermophilus LMD-9 to bacitracin: Involvement of a BceRS/AB-like module and of the rhamnose-glucose polysaccharide synthesis pathway. Int. J. Food Microbiol. 2014, 177, 89–97. [Google Scholar] [CrossRef]
- Cui, Y.; Liu, W.; Qu, X.; Chen, Z.; Zhang, X.; Liu, T. A two component system is involved in acid adaptation of Lactobacillus delbrueckii subsp. bulgaricus. Microbiol. Res. 2012, 167, 253–261. [Google Scholar] [CrossRef]
- Hu, T.; Zhang, Y.; Cui, Y.; Zhao, C.; Jiang, X.; Zhu, X.; Wang, Y.; Qu, X. Technological properties assessment and two component systems distribution of Streptococcus thermophilus strains isolated from fermented milk. Arch. Microbiol. 2018, 200, 567–580. [Google Scholar] [CrossRef]
- Sturme, M.H.J.; Francke, C.; Siezen, R.J.; de Vos, W.M.; Kleerebezem, M. Making sense of quorum sensing in lactobacilli: A special focus on Lactobacillus plantarum WCFS1. Microbiology 2007, 153, 3939–3947. [Google Scholar] [CrossRef]
- Diep, D.B.; Johnsborg, O.; Risøen, P.A.; Nes, I.F. Evidence for dual functionality of the operon plnABCD in the regulation of bacteriocin production in Lactobacillus plantarum. Mol. Microbiol. 2001, 41, 633–644. [Google Scholar] [CrossRef]
- Sturme, M.H.J.; Nakayama, J.; Molenaar, D.; Murakami, Y.; Kunugi, R.; Fujii, T.; Vaughan, E.E.; Kleerebezem, M.; de Vos, W.M. An agr-like two-component regulatory system in Lactobacillus plantarum is involved in production of a novel cyclic peptide and regulation of adherence. J. Bacteriol. 2005, 187, 5224–5235. [Google Scholar] [CrossRef]
- Fujii, T.; Ingham, C.; Nakayama, J.; Beerthuyzen, M.; Kunuki, R.; Molenaar, D.; Sturme, M.; Vaughan, E.; Kleerebezem, M.; de Vos, W. Two homologous Agr-like quorum-sensing systems cooperatively control adherence, cell morphology, and cell viability properties in Lactobacillus plantarum WCFS1. J. Bacteriol. 2008, 190, 7655–7665. [Google Scholar] [CrossRef]
- Miao, K. Optimization of Fermentation Conditions of γ-Aminobutytic Acid Produced by Lactobacillus plantrum and Genome Analysis. Master’s Thesis, Harbin Institute of Technology, Harbin, China, 2020. [Google Scholar]
- Crowley, S.; Bottacini, F.; Mahony, J.; van Sinderen, D. Complete genome sequence of Lactobacillus plantarum strain 16, a broad-spectrum antifungal-producing lactic acid bacterium. Genome Announc. 2013, 1, e00533-13. [Google Scholar] [CrossRef]
- Jia, F.F.; Zhang, L.J.; Pang, X.H.; Gu, X.X.; Abdelazez, A.; Liang, Y.; Sun, S.R.; Meng, X.C. Complete genome sequence of bacteriocin-producing Lactobacillus plantarum KLDS1.0391, a probiotic strain with gastrointestinal tract resistance and adhesion to the intestinal epithelial cells. Genomics 2017, 109, 432–437. [Google Scholar] [CrossRef]
- Jiang, Y.Y.; Zhang, J.; Zhao, X.; Zhao, W.; Yu, Z.J.; Chen, C.; Yang, Z.N. Complete genome sequencing of exopolysaccharide-producing Lactobacillus plantarum K25 provides genetic evidence for the probiotic functionality and cold endurance capacity of the strain. Biosci. Biotechnol. Biochem. 2018, 82, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wang, R.; Gong, F.; Liu, X.; Zheng, H.; Luo, Y.; Li, X. Complete genome sequences and comparative genome analysis of Lactobacillus plantarum strain 5-2 isolated from fermented soybean. Genomics 2015, 106, 404–411. [Google Scholar] [CrossRef]
- Noda, M.; Shiraga, M.; Kumagai, T.; Danshiitsoodol, N.; Sugiyama, M. Characterization of the SN35N strain-specific exopolysaccharide encoded in the whole circular genome of a plant-derived Lactobacillus plantarum. Biol. Pharm. Bull. 2018, 41, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Sun, Z.; Bilige, M.; Zhang, H. Complete genome sequence of probiotic Lactobacillus plantarum P-8 with antibacterial activity. J. Biotechnol. 2015, 193, 41–42. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Z.Y.; Yang, Y.; Wang, J.J.; Wang, G.H.; Ren, F.Z.; Hao, Y.L. Complete genome sequencing of Lactobacillus plantarum CAUH2 reveals a novel plasmid pCAUH203 associated with oxidative stress tolerance. 3 Biotech 2019, 9, 116. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Chen, C.; Ai, L.Z.; Zhou, F.F.; Zhou, Z.M.; Wang, L.; Zhang, H.; Chen, W.; Guo, B.H. Complete genome sequence of the probiotic Lactobacillus plantarum ST-III. J. Bacteriol. 2011, 193, 313–314. [Google Scholar] [CrossRef]
- Martino, M.E.; Bayjanov, J.R.; Caffrey, B.E.; Wels, M.; Joncour, P.; Hughes, S.; Gillet, B.; Kleerebezem, M.; van Hijum, S.A.F.T.; Leulier, F. Nomadic lifestyle of Lactobacillus plantarum revealed by comparative genomics of 54 strains isolated from different habitats. Environ. Microbiol. 2016, 18, 4974–4989. [Google Scholar] [CrossRef]
- Saulnier, D.M.A.; Molenaar, D.; de Vos, W.M.; Gibson, G.R.; Kolida, S. Identification of prebiotic fructooligosaccharide metabolism in Lactobacillus plantarum WCFS1 through microarrays. Appl. Environ. Microb. 2007, 73, 1753–1765. [Google Scholar] [CrossRef]
- Chen, C.; Zhao, G.; Chen, W.; Guo, B. Metabolism of fructooligosaccharides in Lactobacillus plantarum ST-III via differential gene transcription and alteration of cell membrane fluidity. Appl. Environ. Microb. 2015, 81, 7697–7707. [Google Scholar] [CrossRef]
- Beck, B.R.; Park, G.S.; Lee, Y.H.; Im, S.; Jeong, D.Y.; Kang, J. Whole genome analysis of Lactobacillus plantarum strains isolated from Kimchi and determination of probiotic properties to treat mucosal infections by Candida albicans and Gardnerella vaginalis. Front Microbiol. 2019, 10, 433. [Google Scholar] [CrossRef]
- Wang, Y.; Shang, N.; Qin, Y.; Zhang, Y.; Zhang, J.; Li, P. The complete genome sequence of Lactobacillus plantarum LPL-1, a novel antibacterial probiotic producing class IIa bacteriocin. J. Biotechnol. 2018, 266, 84–88. [Google Scholar] [CrossRef]
- Taylor, B.L.; Zhulin, I.B. PAS domains: Internal sensors of oxygen, redox potential, and light. Microbiol. Mol. Biol. Rev. 1999, 63, 479–506. [Google Scholar] [CrossRef]
- Jackman, S.D.; Vandervalk, B.P.; Mohamadi, H.; Chu, J.; Yeo, S.; Hammond, S.A.; Jahesh, G.; Khan, H.; Coombe, L.; Warren, R.L.; et al. ABySS 2.0: Resource-efficient assembly of large genomes using a bloom filter. Genome Res. 2017, 27, 768–777. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Yin, Y.; Mao, X.; Yang, J.; Chen, X.; Mao, F.; Xu, Y. dbCAN: A web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012, 40, 445–451. [Google Scholar] [CrossRef]
- Lombard, V.; Ramulu, H.G.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef]
- Andreas, U.; Ioana, C.; Triinu, K.; Jian, Y.; Faircloth, B.C.; Maido, R. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔC(T) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, Y.; Wang, M.; Zheng, Y.; Miao, K.; Qu, X. The Carbohydrate Metabolism of Lactiplantibacillus plantarum. Int. J. Mol. Sci. 2021, 22, 13452. https://doi.org/10.3390/ijms222413452
Cui Y, Wang M, Zheng Y, Miao K, Qu X. The Carbohydrate Metabolism of Lactiplantibacillus plantarum. International Journal of Molecular Sciences. 2021; 22(24):13452. https://doi.org/10.3390/ijms222413452
Chicago/Turabian StyleCui, Yanhua, Meihong Wang, Yankun Zheng, Kai Miao, and Xiaojun Qu. 2021. "The Carbohydrate Metabolism of Lactiplantibacillus plantarum" International Journal of Molecular Sciences 22, no. 24: 13452. https://doi.org/10.3390/ijms222413452
APA StyleCui, Y., Wang, M., Zheng, Y., Miao, K., & Qu, X. (2021). The Carbohydrate Metabolism of Lactiplantibacillus plantarum. International Journal of Molecular Sciences, 22(24), 13452. https://doi.org/10.3390/ijms222413452