
Biochemical Mapping of Pyrodinium bahamense Unveils Molecular Underpinnings behind Organismal Processes

 ,
, 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
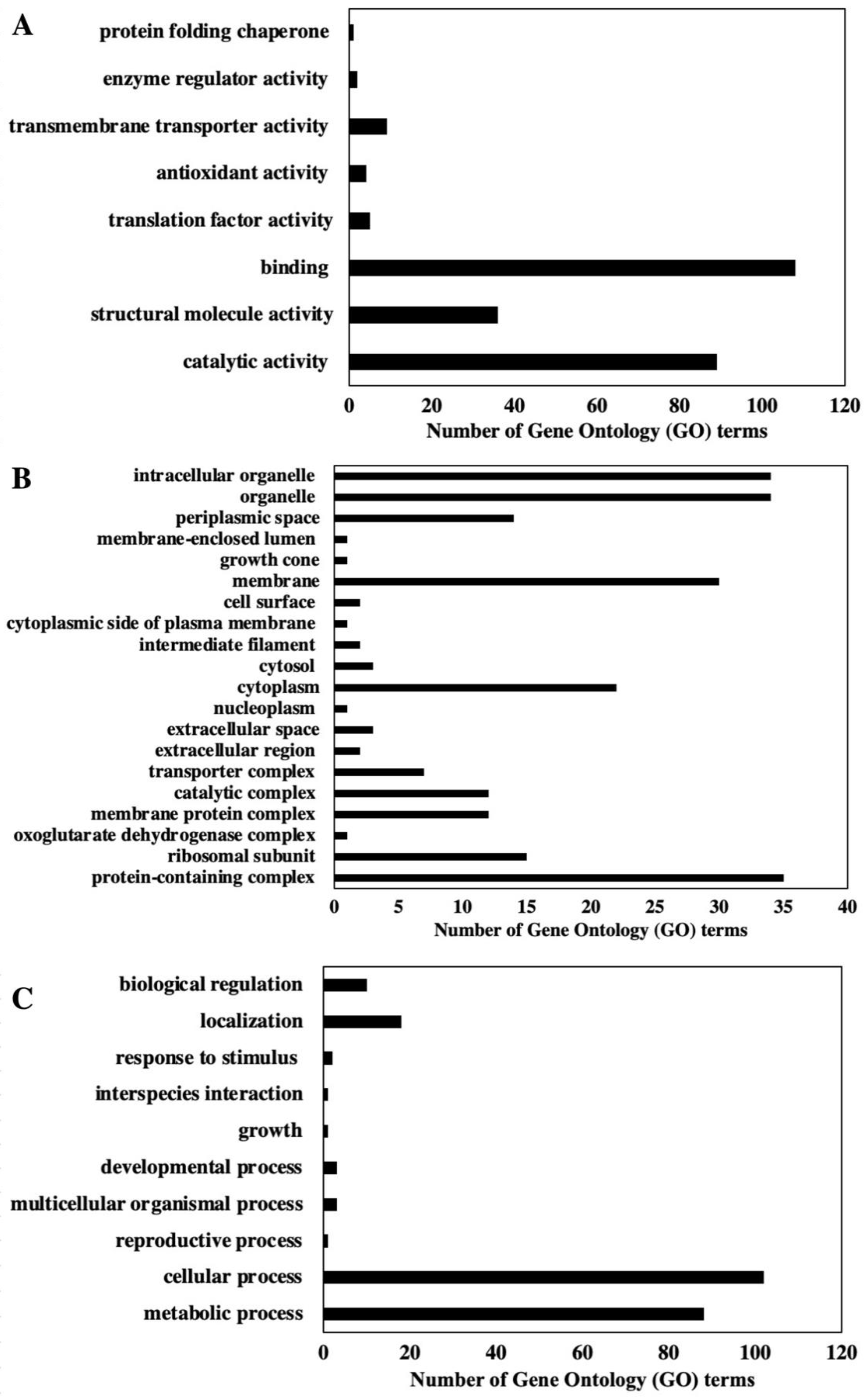
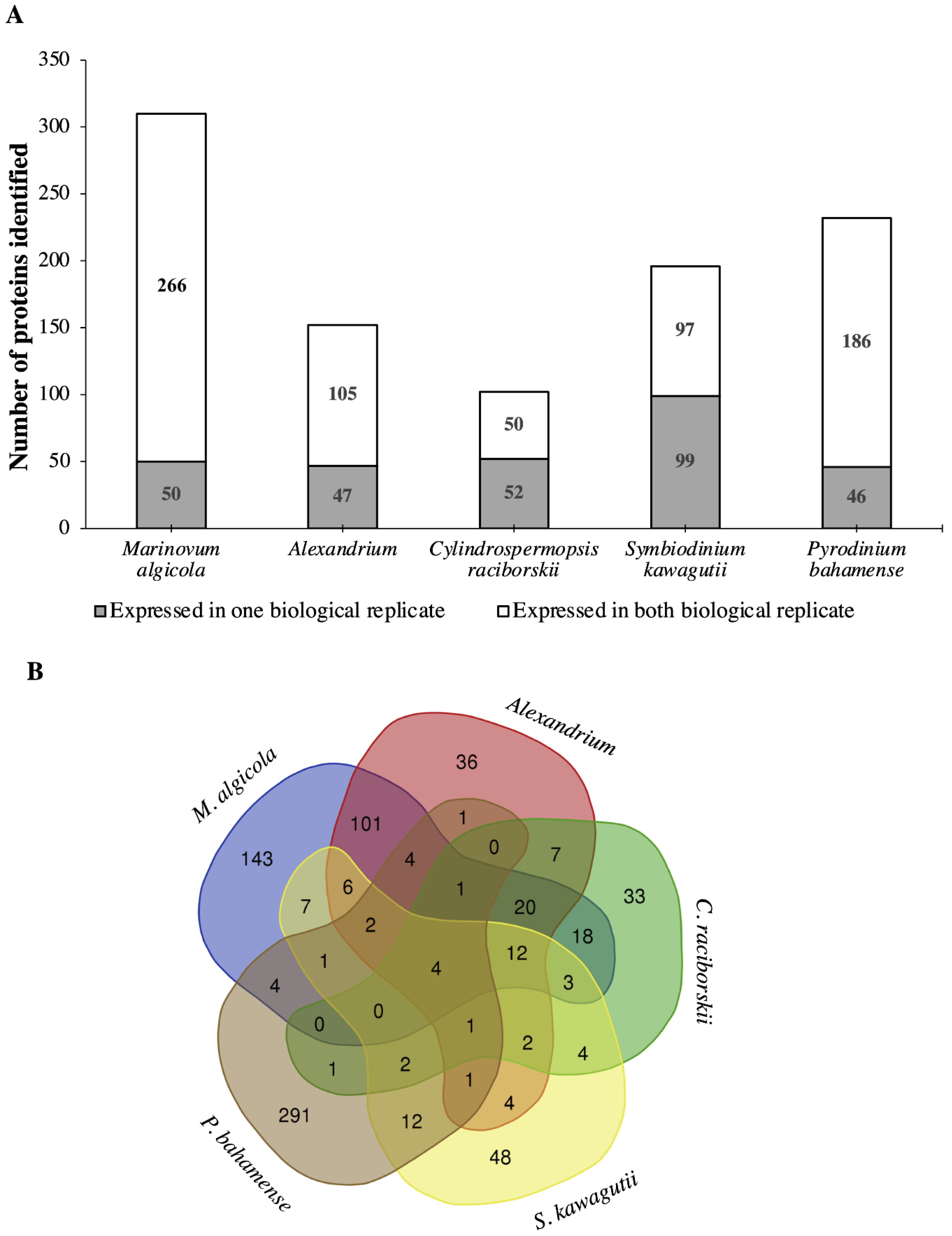
2.1. Proteome Analysis of P. bahamense
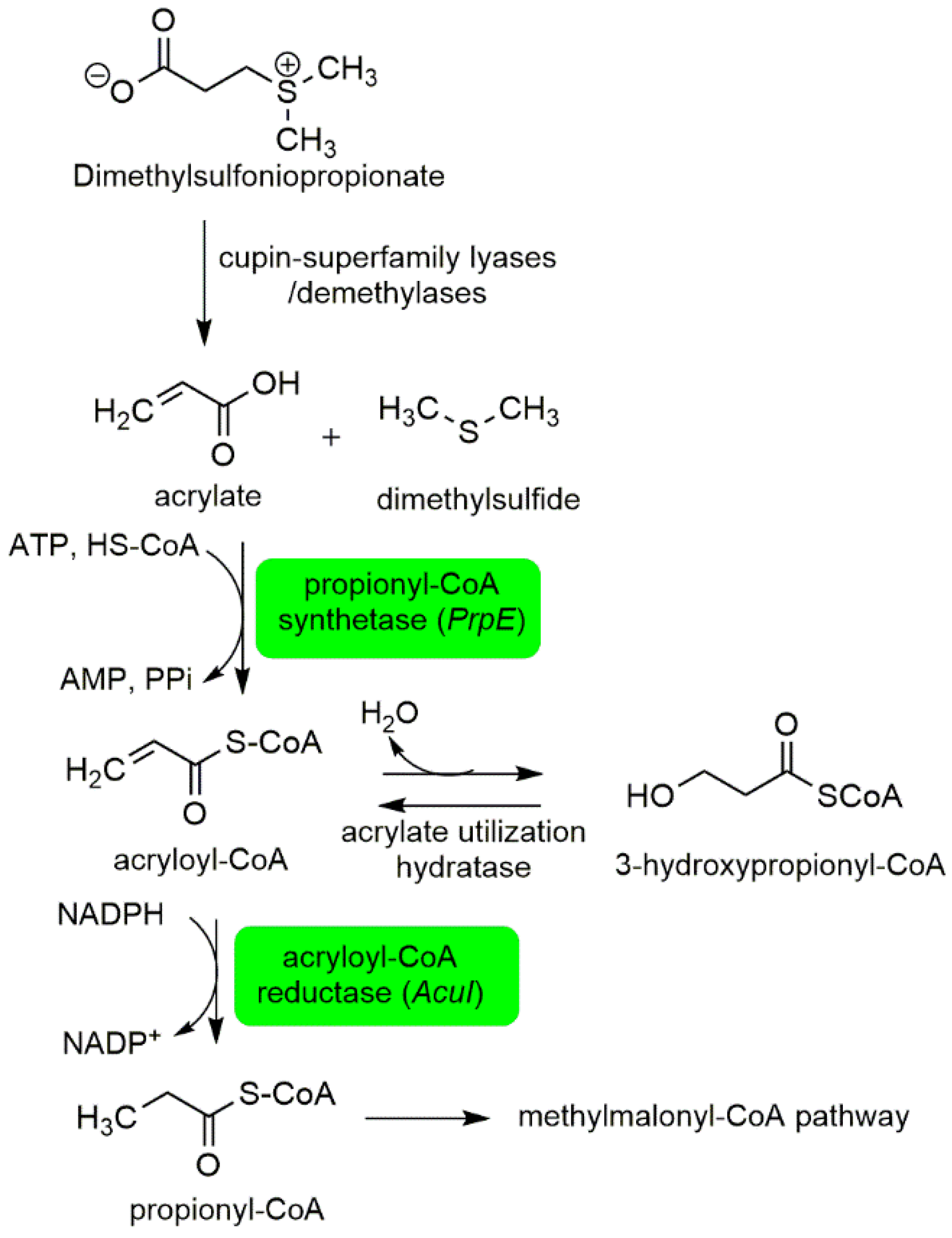
2.1.1. Interactions between P. bahamense and Associated Microorganisms
2.1.2. Toxin Production

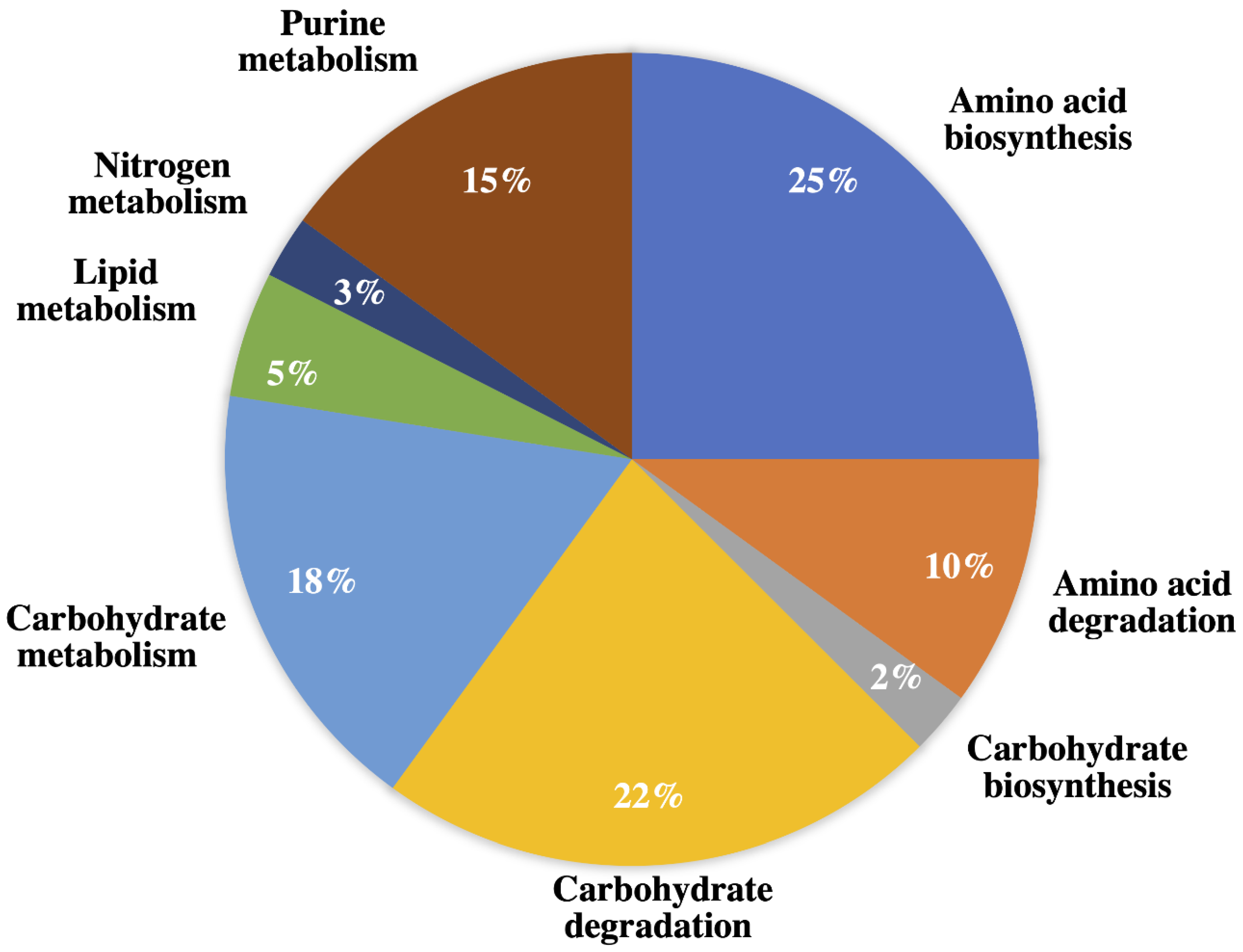
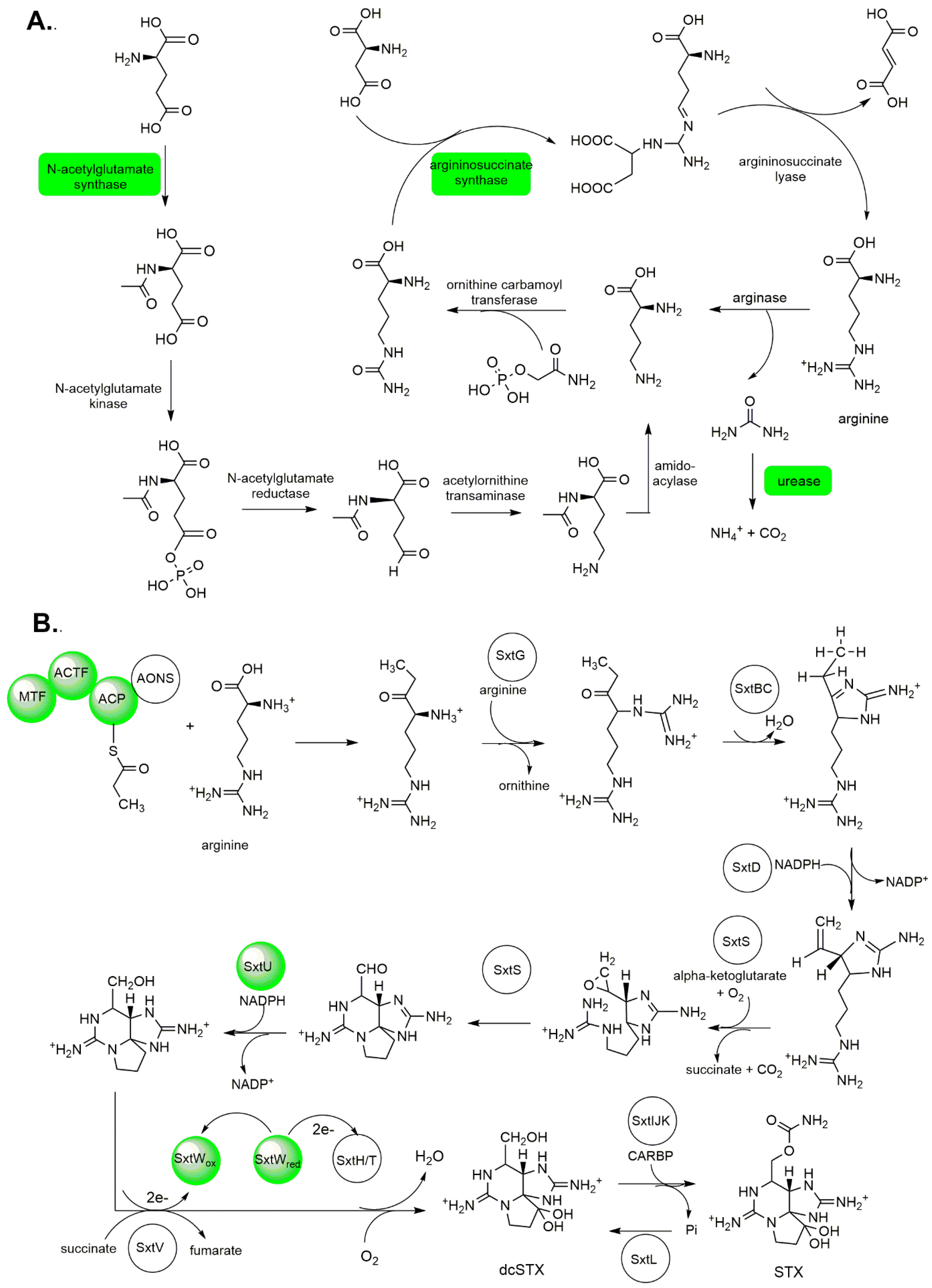
2.1.3. Amino Acid Biosynthesis, Degradation, and Nitrogen Metabolism
2.1.4. Photosynthesis and Bioluminescence
2.1.5. Circadian Rhythm and Growth
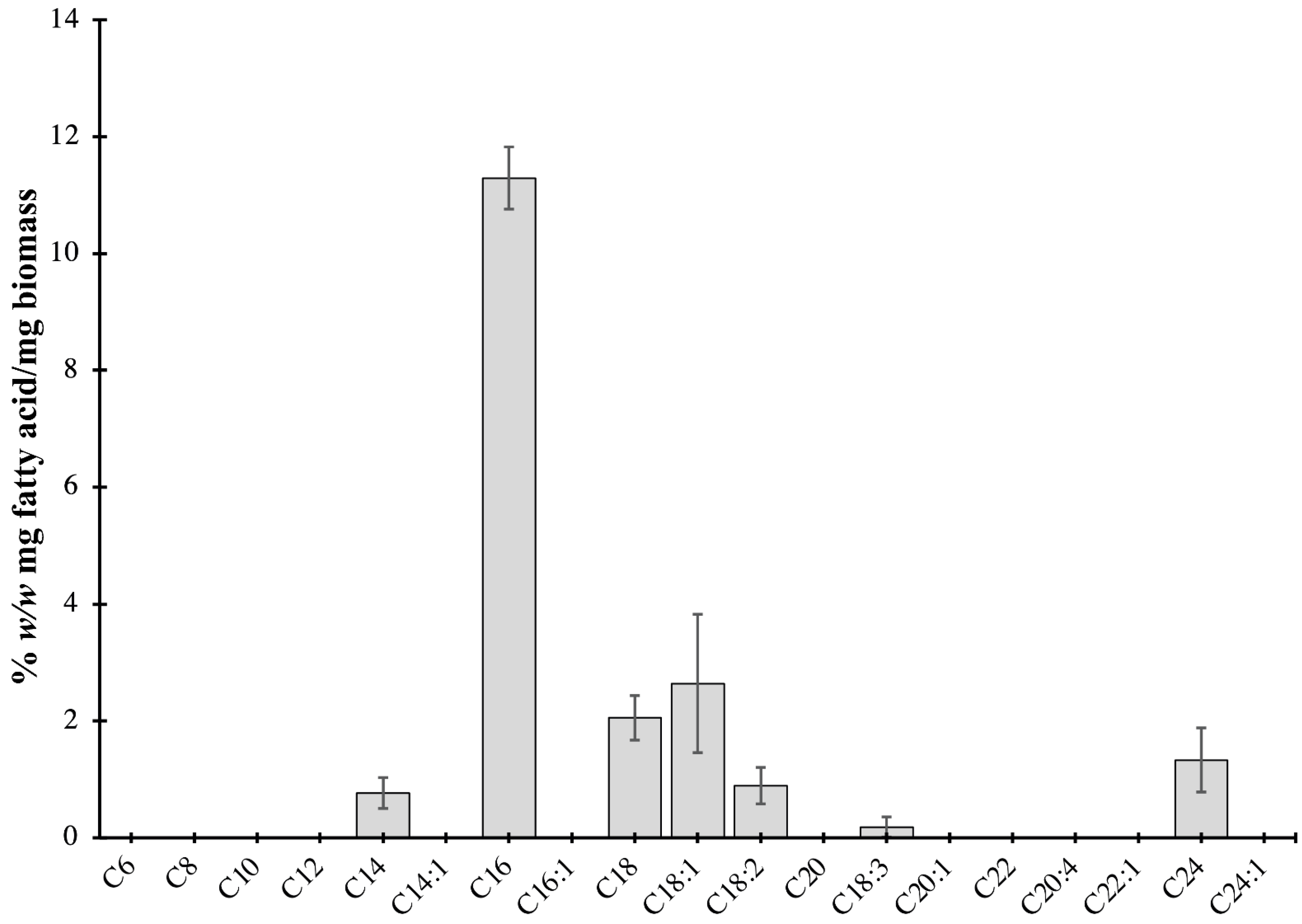
2.2. Characterization of Lipids of P. bahamense
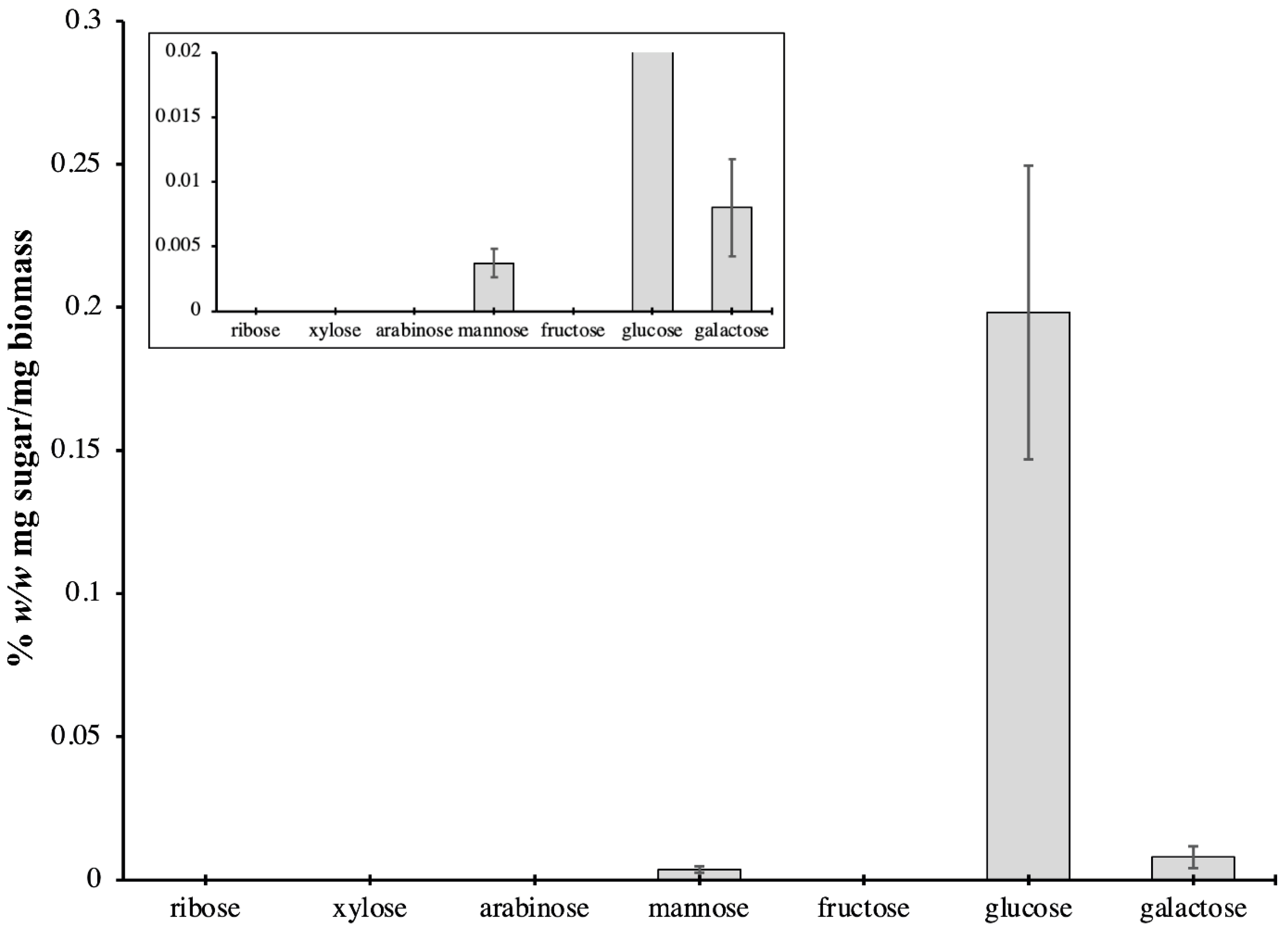
2.3. Carbohydrate Content of P. bahamense
3. Materials and Methods
3.1. Cultivation of P. bahamense
3.2. Moisture and Ash Content
3.3. Lipid Extraction and Analysis
3.4. Carbohydrate Extraction and Analysis
3.5. Proteome Analysis
3.5.1. Protein Extraction and Quantitation
3.5.2. Mass Spectrometry Analysis
3.5.3. Protein Identification and Annotation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Azanza, R.V.; Max Taylor, F.J.R. Are Pyrodinium blooms in the Southeast Asian region recurring and spreading? A view at the end of the millennium. Ambio 2001, 30, 356–364. [Google Scholar] [CrossRef]
- Yñiguez, A.T.; Lim, P.T.; Leaw, C.P.; Jipanin, S.J.; Iwataki, M.; Benico, G.; Azanza, R.V. Over 30 years of HABs in the Philippines and Malaysia: What have we learned? Harmful Algae 2020, 102, 1–15. [Google Scholar] [CrossRef]
- Balech, E. The Genus Alexandrium Halim (Dinoflagellata); Sherkin Island Marine Station: Cork, Ireland, 1995. [Google Scholar]
- Usup, G.; Leaw, C.P.; Ahmad, A.; Lim, P.T. Alexandrium (Dinophyceae) species in Malaysian waters. Harmful Algae 2002, 1, 265–275. [Google Scholar] [CrossRef]
- Leaw, C.P.; Lim, P.T.; Ng, B.K.; Cheah, M.Y.; Ahmad, A.; Usup, G. Phylogenetic analysis of Alexandrium species and Pyrodinium bahamense (Dinophyceae) based on theca morphology and nuclear ribosomal gene sequence. Phycologia 2005, 44, 550–565. [Google Scholar] [CrossRef]
- Morquecho, L. Pyrodinium bahamense one the most significant harmful dinoflagellate in Mexico. Front. Mar. Sci. 2019, 6, 1. [Google Scholar] [CrossRef]
- Gedaria, A.I.; Luckas, B.; Reinhardt, K.; Azanza, R.V. Growth response and toxin concentration of cultured Pyrodinium bahamense var. compressum to varying salinity and temperature conditions. Toxicon 2007, 50, 518–529. [Google Scholar] [CrossRef]
- Usup, G.; Kulis, D.M.; Anderson, D.M. Growth and toxin production of the toxic dinoflagellate Pyrodinium bahamense var. compressum in laboratory cultures. Nat. Toxins 1994, 2, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Rosales-Loessener, F. The Guatemalan experience with red tides and paralytic shellfish poisoning. In Biology, Epidemiology and Management of Pyrodinium Red Tides; Hallegraeff, G.M., McLean, J.L., Eds.; ICLARM Conference Proceedings; Fisheries Department, Ministry of Development, Brunei Darussalam and International Center for Living Aquatic Resources Management: Manila, Philippines, 1989; Volume 21, pp. 49–51. [Google Scholar]
- Landsberg, J.H.; Hall, S.; Johannessen, J.N.; White, K.D.; Conrad, S.M.; Abbott, J.P.; Flewelling, L.J.; Richardson, R.W.; Dickey, R.W.; Jester, E.L.E.; et al. Saxitoxin puffer fish poisoning in the United States, with the first report of Pyrodinium bahamense as the putative toxin source. Environ. Health Perspect. 2006, 114, 1502–1507. [Google Scholar] [CrossRef]
- Siringan, F.P.; Azanza, R.V.; Macalalad, N.J.H.; Zamora, P.B.; Maria, M.Y.Y.S. Temporal changes in the cyst densities of Pyrodinium bahamense var. compressum and other dinoflagellates in Manila Bay, Philippines. Harmful Algae 2008, 7, 523–531. [Google Scholar] [CrossRef]
- Sombrito, E.Z.; Bulos, A.D.M.; Sta Maria, E.J.; Honrado, M.C.V.; Azanza, R.V.; Furio, E.F. Application of 210Pb-derived sedimentation rates and dinoflagellate cyst analyses in understanding Pyrodinium bahamense harmful algal blooms in Manila Bay and Malampaya Sound, Philippines. J. Environ. Radioact. 2004, 76, 177–194. [Google Scholar] [CrossRef]
- Likumahua, S.; Sangiorgi, F.; Boer, M.K.; De Tatipatta, W.M.; Pelasula, D.D.; Polnaya, D.; Hehuwat, J.; Siahaya, D.M.; Anita, G.; Buma, J. Dinoflagellate cyst distribution in surface sediments of Ambon Bay (eastern Indonesia): Environmental conditions and harmful blooms. Mar. Pollut. Bull. 2021, 166, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Carrera, M.; Garet, E.; Barreiro, A.; Garcés, E.; Pérez, D.; Guisande, C.; González-Fernández, Á. Generation of monoclonal antibodies for the specific immunodetection of the toxic dinoflagellate Alexandrium minutum Halim from Spanish waters. Harmful Algae 2010, 9, 272–280. [Google Scholar] [CrossRef]
- Round, F.E. Biology, epidemiology and management of Pyrodinium red tides. Aquat. Bot. 1991, 40, 393–394. [Google Scholar] [CrossRef]
- Azanza, R.V.; Brosnahan, M.L.; Anderson, D.M.; Hense, I.; Montresor, M. The Role of Life Cycle Characteristics in Harmful Algal Bloom Dynamics. In Global Ecology and Oceanography of Harmful Algal Blooms; Glibert, P.M., Berdalet, E., Burford, M.A., Pitcher, G.C., Zhou, M., Eds.; Springer: Cham, Switzerland, 2018; pp. 133–161. [Google Scholar]
- Morquecho, L.; Alonso-Rodríguez, R.; Martínez-Tecuapacho, G.A. Cyst morphology, germination characteristics, and potential toxicity of Pyrodinium bahamense in the Gulf of California. Bot. Mar. 2014, 57, 303–314. [Google Scholar] [CrossRef]
- Onda, D.F.L.; Lluisma, A.O.; Azanza, R.V. Development, morphological characteristics and viability of temporary cysts of Pyrodinium bahamense var. compressum (Dinophyceae) in vitro. Eur. J. Phycol. 2014, 49, 265–275. [Google Scholar] [CrossRef]
- Azanza, M.P.V.; Azanza, R.V.; Vargas, V.M.D.; Hedreyda, C.T. Bacterial endosymbionts of Pyrodinium bahamense var. compressum. Microb. Ecol. 2006, 52, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Silva, E.S. Intracellular bacteria: The origin of dinoflagellate toxicity. J. Environ. Pathol. Toxicol. Oncol. 1990, 10, 124–128. [Google Scholar]
- Onda, D.F.L.; Azanza, R.V.; Lluisma, A.O. Potential DMSP-degrading Roseobacter clade dominates endosymbiotic microflora of Pyrodinium bahamense var. compressum (Dinophyceae) in vitro. Arch. Microbiol. 2015, 197, 965–971. [Google Scholar] [CrossRef]
- Law, S.V.; Rodrigues, K.F.; Jani, J.; Anton, A.; Chin, G.J.W.L. A metagenomic study of bacterial communities associated with the saxitoxin producing dinoflagellate, Pyrodinium bahamense var. compressum. Malays. J. Microbiol. 2020, 16, 176–183. [Google Scholar] [CrossRef]
- Lin, S. Genomic understanding of dinoflagellates. Res. Microbiol. 2011, 162, 551–569. [Google Scholar] [CrossRef] [PubMed]
- Nand, A.; Zhan, Y.; Salazar, O.R.; Aranda, M.; Voolstra, C.R.; Dekker, J. Genetic and spatial organization of the unusual chromosomes of the dinoflagellate Symbiodinium microadriaticum. Nat. Genet. 2021, 53, 618–629. [Google Scholar] [CrossRef]
- Wang, D.Z.; Dong, H.P.; Li, C.; Xie, Z.X.; Lin, L.; Hong, H.S. Identification and characterization of cell wall proteins of a toxic dinoflagellate Alexandrium catenella using 2-D DIGE and MALDI TOF-TOF mass spectrometry. Evid. Based Complement. Altern. Med. 2011, 2011, 984080. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, S.F.; Lin, L.; Wang, D.Z. Comparative transcriptome analysis of a toxin-producing dinoflagellate Alexandrium catenella and its non-toxic mutant. Mar. Drugs 2014, 12, 5698–5718. [Google Scholar] [CrossRef]
- Morse, D.; Tse, S.P.K.; Lo, S.C.L. Exploring dinoflagellate biology with high-throughput proteomics. Harmful Algae 2018, 75, 16–26. [Google Scholar] [CrossRef]
- Vingiani, G.M.; Stalberga, D.; De Luca, P.; Ianora, A.; De Luca, D.; Lauritano, C. De novo transcriptome of the non-saxitoxin producing Alexandrium tamutum reveals new insights on harmful dinoflagellates. Mar. Drugs 2020, 18, 386. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Z.; Lin, L.; Wang, M.H.; Li, C.; Hong, H.S. Proteomic analysis of a toxic dinoflagellate Alexandrium catenella under different growth phases and conditions. Chin. Sci. Bull. 2012, 57, 3328–3341. [Google Scholar] [CrossRef]
- Chan, L.L.; Sit, W.H.; Lam, P.K.S.; Hsieh, D.P.H.; Hodgkiss, I.J.; Wan, J.M.F.; Ho, A.Y.T.; Choi, N.M.C.; Wang, D.Z.; Dudgeon, D. Identification and characterization of a “biomarker of toxicity” from the proteome of the paralytic shellfish toxin-producing dinoflagellate Alexandrium tamarense (Dinophyceae). Proteomics 2006, 6, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Subong, B.J.J.; Benico, G.A.; Sulit, A.K.L.; Mendoza, C.O.; Cruz, L.J.; Azanza, R.V.; Jimenez, E.C. Toxicity and protein expression of Alexandrium species collected in the Philippine waters. Philipp. J. Sci. 2017, 146, 425–436. [Google Scholar]
- Subong, B.J.J.; Lluisma, A.O.; Azanza, R.V.; Salvador-Reyes, L.A. Differentiating two closely related Alexandrium species using comparative quantitative proteomics. Toxins 2020, 13, 7. [Google Scholar] [CrossRef]
- Nagappan, S.; Devendran, S.; Tsai, P.C.; Jayaraman, H.; Alagarsamy, V.; Pugazhendhi, A.; Ponnusamy, V.K. Metabolomics integrated with transcriptomics and proteomics: Evaluation of systems reaction to nitrogen deficiency stress in microalgae. Process Biochem. 2020, 91, 1–14. [Google Scholar] [CrossRef]
- Sudasinghe, N.; Reddy, H.; Csakan, N.; Deng, S.; Lammers, P.; Schaub, T. Temperature-dependent lipid conversion and nonlipid composition of microalgal hydrothermal liquefaction oils monitored by Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Bioenergy Res. 2015, 8, 1962–1972. [Google Scholar] [CrossRef]
- Schulze, C.; Strehle, A.; Merdivan, S.; Mundt, S. Carbohydrates in microalgae: Comparative determination by TLC, LC-MS without derivatization, and the photometric thymol-sulfuric acid method. Algal Res. 2017, 25, 372–380. [Google Scholar] [CrossRef]
- Zhang, Y.; Fonslow, B.R.; Shan, B.; Baek, M.C.; Yates, J.R. Protein analysis by shotgun/bottom-up proteomics. Chem. Rev. 2013, 113, 2343–2394. [Google Scholar] [CrossRef]
- Elliott, M.H.; Smith, D.S.; Parker, C.E.; Borchers, C. Current trends in quantitative proteomics. J. Mass Spectrom. 2009, 44, 1637–1660. [Google Scholar] [CrossRef]
- Chen, X.; Wei, S.; Ji, Y.; Guo, X.; Yang, F. Quantitative proteomics using SILAC: Principles, applications, and developments. Proteomics 2015, 15, 1637–1660. [Google Scholar] [CrossRef]
- Lin, S.; Cheng, S.; Song, B.; Zhong, X.; Lin, X.; Li, W.; Li, L.; Zhang, Y.; Zhang, H.; Ji, Z.; et al. The Symbiodinium kawagutii genome illuminates dinoflagellate gene expression and coral symbiosis. Science 2015, 350, 691–694. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.F.; Zhang, Y.; Lin, L.; Wang, D.Z. iTRAQ-Based quantitative proteomic analysis of a toxigenic dinoflagellate Alexandrium catenella at different stages of toxin biosynthesis during the cell cycle. Mar. Drugs 2018, 16, 491. [Google Scholar] [CrossRef]
- Zhu, W.; Smith, J.W.; Huang, C.M. Mass spectrometry-based label-free quantitative proteomics. J. Biomed. Biotechnol. 2010, 2010, 840518. [Google Scholar] [CrossRef]
- Neilson, K.A.; Ali, N.A.; Muralidharan, S.; Mirzaei, M.; Mariani, M.; Assadourian, G.; Lee, A.; Van Sluyter, S.C.; Haynes, P.A. Less label, more free: Approaches in label-free quantitative mass spectrometry. Proteomics 2011, 11, 535–553. [Google Scholar] [CrossRef]
- Yang, I.; John, U.; Beszteri, S.; Glöckner, G.; Krock, B.; Goesmann, A.; Cembella, A.D. Comparative gene expression in toxic versus non-toxic strains of the marine dinoflagellate Alexandrium minutum. BMC Genom. 2010, 11, 248. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Stephens, T.G.; González-Pech, R.A.; Beltran, V.H.; Lapeyre, B.; Bongaerts, P.; Cooke, I.; Aranda, M.; Bourne, D.G.; Forêt, S.; et al. Symbiodinium genomes reveal adaptive evolution of functions related to coral-dinoflagellate symbiosis. Commun. Biol. 2018, 1, 95. [Google Scholar] [CrossRef] [PubMed]
- Burgess, T.L.; Kelly, R.B. Constitutive and regulated secretion of proteins. Annu. Rev. Cell Biol. 1987, 3, 243–293. [Google Scholar] [CrossRef]
- Taroncher-Oldenburg, C.; Anderson, D.M. Identification and characterization of three differentially expressed genes, encoding S-adenosylhomocysteine hydrolase, methionine aminopeptidase, and a histone-like protein, in the toxic dinoflagellate Alexandrium fundyense. Appl. Environ. Microbiol. 2000, 66, 2105–2112. [Google Scholar] [CrossRef] [PubMed]
- du Plessis, L.; Škunca, N.; Dessimoz, C. The what, where, how and why of gene ontology-A primer for bioinformaticians. Brief. Bioinform. 2011, 12, 723–735. [Google Scholar] [CrossRef]
- Santos, M.A.G.; Azanza, R.V. Responses of Pyrodinium bahamense var. compressum and associated cultivable bacteria to antibiotic treatment. J. Appl. Phycol. 2012, 24, 825–835. [Google Scholar] [CrossRef]
- Wang, D.Z.; Gao, Y.; Lin, L.; Hong, H.S. Comparative proteomic analysis reveals proteins putatively involved in toxin biosynthesis in the marine dinoflagellate Alexandrium catenella. Mar. Drugs 2013, 11, 213–232. [Google Scholar] [CrossRef]
- Fu, J.; Momčilović, I.; Prasad, P.V.V. Roles of protein synthesis Elongation Factor EF-Tu in heat tolerance in plants. J. Bot. 2012, 2012, 835836. [Google Scholar] [CrossRef]
- Roderer, D.; Raunser, S. Tc toxin complexes: Assembly, membrane permeation, and protein translocation. Annu. Rev. Microbiol. 2019, 73, 247–265. [Google Scholar] [CrossRef] [PubMed]
- Reisch, C.R.; Moran, M.A.; Whitman, W.B. Bacterial catabolism of dimethylsulfoniopropionate (DMSP). Front. Microbiol. 2011, 2, 172. [Google Scholar] [CrossRef]
- Bullock, H.A.; Luo, H.; Whitman, W.B. Evolution of dimethylsulfoniopropionate metabolism in marine phytoplankton and bacteria. Front. Microbiol. 2017, 8, 637. [Google Scholar] [CrossRef] [PubMed]
- Bentley, R.; Chasteen, T.G. Environmental VOSCs—Formation and degradation of dimethyl sulfide, methanethiol and related materials. Chemosphere 2004, 55, 291–317. [Google Scholar] [CrossRef]
- Wang, P.; Wang, Q.; Yang, Y.; Coward, J.K.; Nzila, A.; Sims, P.F.G.; Hyde, J.E. Characterisation of the bifunctional dihydrofolate synthase-folylpolyglutamate synthase from Plasmodium falciparum; a potential novel target for antimalarial antifolate inhibition. Mol. Biochem. Parasitol. 2010, 172, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.F.; Zhang, Y.; Xie, Z.X.; Zhang, H.; Lin, L.; Wang, D.Z. iTRAQ-based quantitative proteomic analysis of a toxigenic dinoflagellate Alexandrium catenella and its non-toxic mutant. Proteomics 2015, 15, 4041–4050. [Google Scholar] [CrossRef]
- Jiang, X.W.; Wang, J.; Gao, Y.; Chan, L.L.; Lam, P.K.S.; Gu, J.D. Relationship of proteomic variation and toxin synthesis in the dinoflagellate Alexandrium tamarense CI01 under phosphorus and inorganic nitrogen limitation. Ecotoxicology 2015, 24, 1744–1753. [Google Scholar] [CrossRef] [PubMed]
- Kellmann, R.; Mihali, T.K.; Young, J.J.; Pickford, R.; Pomati, F.; Neilan, B.A. Biosynthetic intermediate analysis and functional homology reveal a saxitoxin gene cluster in cyanobacteria. Appl. Environ. Microbiol. 2008, 74, 4044–4053. [Google Scholar] [CrossRef] [PubMed]
- Akbar, M.A.; Yusof, N.Y.M.; Tahir, N.I.; Ahmad, A.; Usup, G.; Sahrani, F.K.; Bunawan, H. Biosynthesis of saxitoxin in marine dinoflagellates: An omics perspective. Mar. Drugs 2020, 18, 103. [Google Scholar] [CrossRef] [PubMed]
- Bromke, M.A. Amino acid biosynthesis pathways in diatoms. Metabolites 2013, 3, 294–311. [Google Scholar] [CrossRef]
- Jiang, X.W.; Wang, J.; Chan, L.L.; Lam, P.K.S.; Gu, J.D. Comparison of three protein extraction procedures from toxic and non-toxic dinoflagellates for proteomics analysis. Ecotoxicology 2015, 24, 1395–1406. [Google Scholar] [CrossRef]
- De Mello Serrano, G.C.; Kiyota, E.; Zanata, N.; Arruda, P. Lysine degradation through the saccharopine pathway in bacteria: LKR and SDH in bacteria and its relationship to the plant and animal enzymes. FEBS Lett. 2012, 586, 905–1011. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Lin, S.; Zhang, H.; Koerting, C.; Yu, Z. Utilization of urea and expression profiles of related genes in the dinoflagellate Prorocentrum donghaiense. PLoS ONE 2017, 12, e0187837. [Google Scholar] [CrossRef] [PubMed]
- Morse, D.; Salois, P.; Markovic, P.; Hastings, J.W. A nuclear-encoded form II RuBisCO in dinoflagellates. Science 1995, 268, 1622–1624. [Google Scholar] [CrossRef]
- Howe, C.J.; Nisbet, R.E.R.; Barbrook, A.C. The remarkable chloroplast genome of dinoflagellates. J. Exp. Bot. 2008, 59, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Hackett, J.D.; Anderson, D.M.; Erdner, D.L.; Bhattacharya, D. Dinoflagellates: A remarkable evolutionary experiment. Am. J. Bot. 2004, 91, 1523–1534. [Google Scholar] [CrossRef]
- Steidinger, K.A.; Tester, L.S.; Taylor, F.J.R. A redescription of Pyrodinium bahamense var. compressa (Böhm) stat. nov. from Pacific red tides. Phycologia 1980, 19, 329–334. [Google Scholar] [CrossRef]
- Phlips, E.J.; Badylak, S.; Youn, S.; Kelley, K. The occurrence of potentially toxic dinoflagellates and diatoms in a subtropical lagoon, the Indian River Lagoon, Florida, USA. Harmful Algae 2004, 3, 39–49. [Google Scholar] [CrossRef]
- Soler-Figueroa, B.M.; Otero, E. Seasonal changes in bioluminescence and dinoflagellate composition in a tropical bioluminescent bay, Bahía Fosforescente, La Parguera, Puerto Rico. J. Exp. Mar. Bio. Ecol. 2016, 483, 120–129. [Google Scholar] [CrossRef]
- Seliger, H.H.; Biggley, W.H.; Swift, E. Absolute values of photon emission from the marine dinoflagellates Pyrodinium bahamense, Gonyaulax polyedra and Pyrocystis lunuu. Photochem. Photobiol. 1969, 10, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Biggley, W.H.; Swift, E.; Buchanan, R.J.; Seliger, H.H. Stimulable and spontaneous bioluminescence in the marine dinoflagellates, Pyrodinium bahamense, Gonyaulax polyedra, and Pyrocystis lunula. J. Gen. Physiol. 1969, 54, 96–122. [Google Scholar] [CrossRef] [PubMed]
- Uribe, P.; Fuentes, D.; Valdés, J.; Shmaryahu, A.; Zúñiga, A.; Holmes, D.; Valenzuela, P.D.T. Preparation and analysis of an expressed sequence tag library from the toxic dinoflagellate Alexandrium catenella. Mar. Biotechnol. 2008, 10, 692–700. [Google Scholar] [CrossRef]
- Latz, M.I.; Rohr, J. Luminescent response of the red tide dinoflagellate Lingulodinium polyedrum to laminar and turbulent flow. Limnol. Oceanogr. 1999, 44, 1423–1435. [Google Scholar] [CrossRef]
- Hastings, J. Circadian rhythms in dinoflagellates: What is the purpose of synthesis and destruction of proteins? Microorganisms 2013, 1, 26–32. [Google Scholar] [CrossRef]
- Abrahams, M.V.; Townsend, L.D. Bioluminescence in dinoflagellates: A test of the burglar alarm hypothesis. Ecology 1993, 74, 258–260. [Google Scholar] [CrossRef]
- Akimoto, H.; Wu, C.; Kinumi, T.; Ohmiya, Y. Biological rhythmicity in expressed proteins of the marine dinoflagellate Lingulodinium polyedrum demonstrated by chronological proteomics. Biochem. Biophys. Res. Commun. 2004, 315, 306–312. [Google Scholar] [CrossRef]
- Akimoto, H.; Kinumi, T.; Ohmiya, Y. Circadian rhythm of a TCA cycle enzyme is apparently regulated at the translational level in the dinoflagellate Lingulodinium polyedrum. J. Biol. Rhythm. 2005, 20, 479–489. [Google Scholar] [CrossRef]
- Usup, G.; Hamid, S.Z.; Chiet, P.K.; Wah, C.K.; Ahmad, A. Marked differences in fatty acid profiles of some planktonic and benthic marine dinoflagellates from Malaysian waters. Phycologia 2008, 47, 105–111. [Google Scholar] [CrossRef]
- Leblond, J.D.; Anderson, B.; Kofink, D.; Logares, R.; Rengefors, K.; Kremp, A. Fatty acid and sterol composition of two evolutionarily closely related dinoflagellate morphospecies from cold Scandinavian brackish and freshwaters. Eur. J. Phycol. 2006, 41, 303–311. [Google Scholar] [CrossRef]
- Hixson, S.M.; Arts, M.T. Climate warming is predicted to reduce omega-3, long-chain, polyunsaturated fatty acid production in phytoplankton. Glob. Chang. Biol. 2016, 22, 2744–2755. [Google Scholar] [CrossRef]
- Parrish, C.C.; Abrajano, T.A.; Budge, S.M.; Helleur, R.J.; Hudson, E.D.; Pulchan, K.; Ramos, C. Lipid and Phenolic Biomarkers in Marine Ecosystems: Analysis and Applications. In Marine Chemistry; Wangersky, P.J., Ed.; Springer: Berlin/Heidelberg, Germany, 2005; pp. 194–223. [Google Scholar]
- Wen, Z.Y.; Chen, F. Heterotrophic production of eicosapentaenoic acid by microalgae. Biotechnol. Adv. 2003, 21, 27–294. [Google Scholar] [CrossRef]
- Ikawa, M. Algal polyunsaturated fatty acids and effects on plankton ecology and other organisms. UNH Cent. Freshw. Biol. Res. 2004, 6, 17–44. [Google Scholar]
- Mardones, J.I.; Dorantes-Aranda, J.J.; Nichols, P.D.; Hallegraeff, G.M. Fish gill damage by the dinoflagellate Alexandrium catenella from Chilean fjords: Synergistic action of ROS and PUFA. Harmful Algae 2015, 49, 40–49. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, L.; Qiu, S.; Ge, S. Determination of microalgal lipid content and fatty acid for biofuel production. Biomed Res. Int. 2018, 2018, 1503126. [Google Scholar] [CrossRef]
- Nelson, D.; Cox, M. Lehninger Principles of Biochemistry, 4th ed.; Nelso, D., Cox, M., Eds.; W.H. Freeman: New York, NY, USA, 2004. [Google Scholar]
- Yu, L.; Zhang, Y.; Li, M.; Wang, C.; Lin, X.; Li, L.; Shi, X.; Guo, C.; Lin, S. Comparative metatranscriptomic profiling and microRNA sequencing to reveal active metabolic pathways associated with a dinoflagellate bloom. Sci. Total Environ. 2020, 699, 134323. [Google Scholar] [CrossRef] [PubMed]
- Fernie, A.R.; Carrari, F.; Sweetlove, L.J. Respiratory metabolism: Glycolysis, the TCA cycle and mitochondrial electron transport. Curr. Opin. Plant Biol. 2004, 7, 254–261. [Google Scholar] [CrossRef]
- Burriesci, M.S.; Raab, T.K.; Pringle, J.R. Evidence that glucose is the major transferred metabolite in dinoflagellate-cnidarian symbiosis. J. Exp. Biol. 2012, 215, 3467–3477. [Google Scholar] [CrossRef] [PubMed]
- Metón, I.; Fernández, F.; Baanante, I.V. Short- and long-term effects of refeeding on key enzyme activities in glycolysis-gluconeogenesis in the liver of gilthead seabream (Sparus aurata). Sci. Total. Environ. 2003, 225, 99–107. [Google Scholar] [CrossRef]
- Chen, C.Y.; Zhao, X.Q.; Yen, H.W.; Ho, S.H.; Cheng, C.L.; Lee, D.J.; Bai, F.W.; Chang, J.S. Microalgae-based carbohydrates for biofuel production. Biochem. Eng. J. 2013, 78, 1–10. [Google Scholar] [CrossRef]
- Okuda, K. Structure and phylogeny of cell coverings. J. Plant Res. 2002, 115, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Domozych, D.S.; Sørensen, I.; Willats, W.G.T. The distribution of cell wall polymers during antheridium development and spermatogenesis in the Charophycean green alga, Chara corallina. Ann. Bot. 2009, 104, 1045–1056. [Google Scholar] [CrossRef]
- Estevez, J.M.; Fernández, P.V.; Kasulin, L.; Dupree, P.; Estevez, J.M. Chemical and in situ characterization of macromolecular components of the cell walls from the green seaweed Codium fragile. Glycobiology 2009, 19, 1045–1056. [Google Scholar] [CrossRef]
- Templeton, D.W.; Quinn, M.; Van Wychen, S.; Hyman, D.; Laurens, L.M.L. Separation and quantification of microalgal carbohydrates. J. Chromatogr. A 2012, 1270, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Nevo, Z.; Sharon, N. The cell wall of Peridinium westii, a non cellulosic glucan. BBA Biomembr. 1969, 173, 161–175. [Google Scholar] [CrossRef]
- Delattre, C.; Fenoradosoa, T.A.; Michaud, P. Galactans: An overview of their most important sourcing and applications as natural polysaccharides. Braz. Arch. Biol. Technol. 2011, 54, 1075–1092. [Google Scholar] [CrossRef]
- Azanza Corrales, R.; Hall, S. Isolation and culture of Pyrodinium bahamense var. compressum from the Philippines. Dev. Mar. Biol. 1993, 1993, 724–730. [Google Scholar]
- AOAC Official Method. 930-15. In Official Methods of Analysis of the Association of Official Analytical Chemists; Latimer, G.W., Ed.; AOAC International: Gaithersburg, MD, USA, 2016. [Google Scholar]
- AOAC Official Method. 942-05. In Official Methods of Analysis of the Association of Official Analytica Chemistsl; Latimer, G.W., Ed.; AOAC International: Gaithersburg, MD, USA, 2016. [Google Scholar]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- AOAC Official Method. 969-33. In Official Methods of Analysis of the Association of Official Analytical Chemists; Horwitz, W., Ed.; AOAC International: Gaithersburg, MD, USA, 2000. [Google Scholar]
- AOAC Official Method. 963-22. In Official Methods of Analysis of the Association of Official Analytical Chemists; Horwitz, W., Ed.; AOAC International: Gaithersburg, MD, USA, 2000. [Google Scholar]
- Masuko, T.; Minami, A.; Iwasaki, N.; Majima, T.; Nishimura, S.I.; Lee, Y.C. Carbohydrate analysis by a phenol-sulfuric acid method in microplate format. Anal. Biochem. 2005, 339, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, L.; Bleasby, A. EMBOSS: The European molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Krey, J.F.; Wilmarth, P.A.; Shin, J.B.; Klimek, J.; Sherman, N.E.; Jeffery, E.D.; Choi, D.; David, L.L.; Barr-Gillespie, P.G. Accurate label-free protein quantitation with high- and low-resolution mass spectrometers. J. Proteome Res. 2014, 13, 1034–1044. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Cox, J. Perseus: A bioinformatics platform for integrative analysis of proteomics data in cancer research. In Cancer Systems Biology; Humana Press: Totowa, NJ, USA, 2018; Volume 1711, pp. 133–148. [Google Scholar]
- Bogdanow, B.; Zauber, H.; Selbach, M. Systematic errors in peptide and protein identification and quantification by modified peptides. Mol. Cell. Proteom. 2016, 15, 2791–2801. [Google Scholar] [CrossRef] [PubMed]
- Ctortecka, C.; Palve, V.; Kuenzi, B.M.; Fang, B.; Sumi, N.J.; Izumi, V.; Novakova, S.; Kinose, F.; Remsing Rix, L.L.; Haura, E.B.; et al. Functional proteomics and deep network interrogation reveal a complex mechanism of action of midostaurin in lung cancer cells. Mol. Cell. Proteom. 2018, 17, 2434–2447. [Google Scholar] [CrossRef] [PubMed]
- Rohrbough, J.G.; Breci, L.; Merchant, N.; Miller, S.; Haynes, P.A. Verification of single-peptide protein identifications by the application of complementary database search algorithms. J. Biomol. Tech. 2006, 17, 327. [Google Scholar]
- Suriyanarayanan, T.; Qingsong, L.; Kwang, L.T.; Mun, L.Y.; Seneviratne, C.J. Quantitative proteomics of strong and weak biofilm formers of Enterococcus faecalis reveals novel regulators of biofilm formation. Mol. Cell. Proteom. 2018, 17, 643–654. [Google Scholar] [CrossRef]
- Kammers, K.; Cole, R.N.; Tiengwe, C.; Ruczinski, I. Detecting significant changes in protein abundance. EuPA Open Proteom. 2015, 7, 11–19. [Google Scholar] [CrossRef]
- Savitski, M.M.; Wilhelm, M.; Hahne, H.; Kuster, B.; Bantscheff, M. A scalable approach for protein false discovery rate estimation in large proteomic data sets. Mol. Cell. Proteom. 2015, 14, 2394–2404. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, A.; Storey, J.D.; Llinás, M.; Singh, M. Beyond the e-value: Stratified statistics for protein domain prediction. PLoS Comput. Biol. 2015, 11, e1004509. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subong, B.J.J.; Malto, Z.B.L.; Lluisma, A.O.; Azanza, R.V.; Salvador-Reyes, L.A. Biochemical Mapping of Pyrodinium bahamense Unveils Molecular Underpinnings behind Organismal Processes. Int. J. Mol. Sci. 2021, 22, 13332. https://doi.org/10.3390/ijms222413332
Subong BJJ, Malto ZBL, Lluisma AO, Azanza RV, Salvador-Reyes LA. Biochemical Mapping of Pyrodinium bahamense Unveils Molecular Underpinnings behind Organismal Processes. International Journal of Molecular Sciences. 2021; 22(24):13332. https://doi.org/10.3390/ijms222413332
Chicago/Turabian StyleSubong, Bryan John J., Zabrina Bernice L. Malto, Arturo O. Lluisma, Rhodora V. Azanza, and Lilibeth A. Salvador-Reyes. 2021. "Biochemical Mapping of Pyrodinium bahamense Unveils Molecular Underpinnings behind Organismal Processes" International Journal of Molecular Sciences 22, no. 24: 13332. https://doi.org/10.3390/ijms222413332
APA StyleSubong, B. J. J., Malto, Z. B. L., Lluisma, A. O., Azanza, R. V., & Salvador-Reyes, L. A. (2021). Biochemical Mapping of Pyrodinium bahamense Unveils Molecular Underpinnings behind Organismal Processes. International Journal of Molecular Sciences, 22(24), 13332. https://doi.org/10.3390/ijms222413332