
The Mechanism of Mitochondrial Injury in Alpha-1 Antitrypsin Deficiency Mediated Liver Disease

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Hepatic Accumulation of Human ZAAT in Pi*Z Transgenic Mice Leads to Lipid Accumulation in the Liver of Transgenic Mice
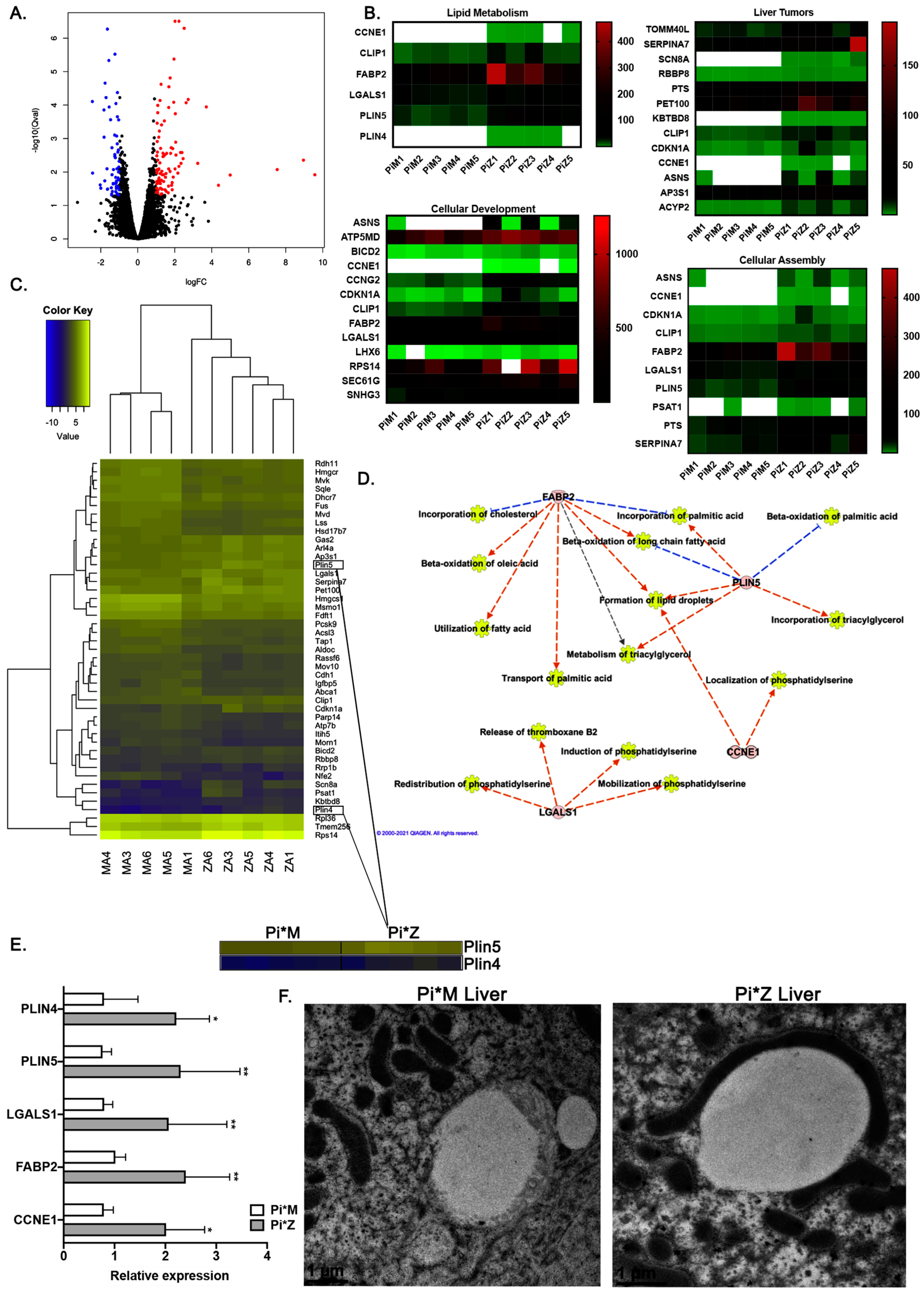
2.2. Transcriptome Analyses Suggest Pi*Z Transgenic Mice Acquire Mitochondrial Abnormalities and Changes in Lipid Metabolism Pathways
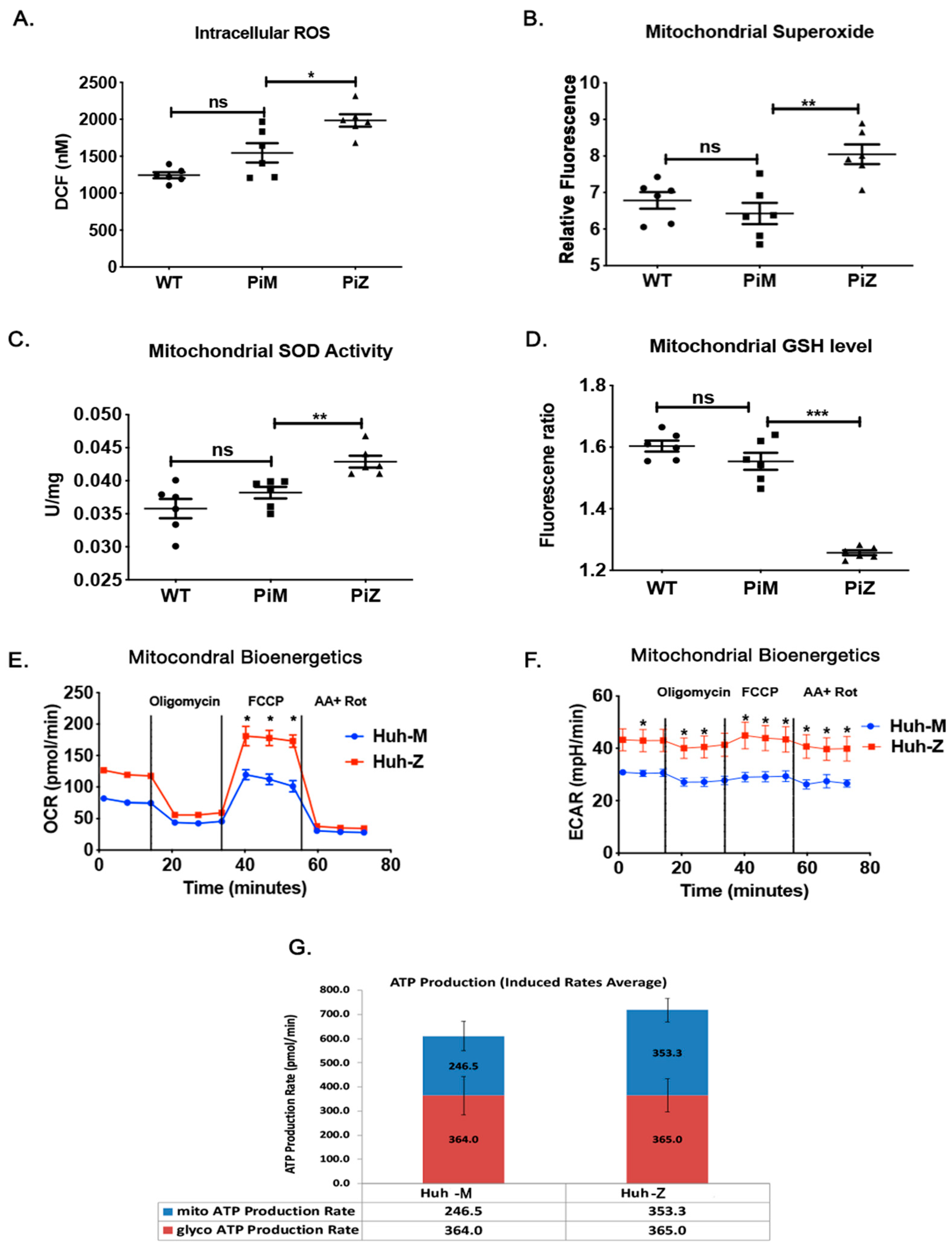
2.3. Hepatic Human ZAAT Accumulation Is Associated with Mitochondrial Dysfunction in the Liver of Transgenic Mice
2.4. Hepatic Human ZAAT Accumulation Impairs Mitochondrial Membrane Integrity within the Liver of Transgenic Mice
2.5. AAT Protein Associates with Mitochondria In Vitro and In Vivo
2.6. AAT Imports to the Mitochondria Using Hepatic Mitochondrial Import Machinery
2.7. ZAAT Knockdown Reduces the Lipid Content of Hepatocytes and Restores Mitochondrial Function in ZAAT-Expressing Hepatocytes
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Production, Maintenance, and Identification of Transgenic Mice
4.3. Human AAT Transgenic Mouse Liver Tissue RNA Sequencing
4.4. Mitochondrial Extraction
4.5. Mitochondrial DNA Isolation and Assessment
4.6. Superoxide Measurement
4.7. ROS Assay
4.8. Antibodies
4.9. Histology, Histochemistry, and Immunohistochemistry
4.10. Cholesterol Quantitation of Mouse Liver Tissues
4.11. Triglyceride Quantitation of Mouse Liver Tissues
4.12. Mitochondrial SOD-2 Activity Assay
4.13. Mitochondrial GSH Measurement
4.14. Measurement of Mitochondrial Respiration
4.15. Mitochondrial Membrane Integrity Measurement
4.16. Lipid Droplet Staining and Quantification
4.17. Immunoblotting and Co-Immunoprecipitation
4.18. Immunostaining and Immunofluorescence Microscopy
4.19. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kalsheker, N.A. α1-Antitrypsin deficiency: Best clinical practice. J. Clin. Pathol. 2009, 62, 865–869. [Google Scholar] [CrossRef]
- Perlmutter, D.H. Liver injury in alpha1-antitrypsin deficiency: An aggregated protein induces mitochondrial injury. J. Clin. Investig. 2002, 110, 1579–1583. [Google Scholar] [CrossRef]
- Fregonese, L.; Stolk, J. Hereditary alpha-1-antitrypsin deficiency and its clinical consequences. Orphanet J. Rare Dis. 2008, 3, 16. [Google Scholar] [CrossRef]
- Stockley, R.A.; Turner, A.M. α-1-Antitrypsin deficiency: Clinical variability, assessment, and treatment. Trends Mol. Med. 2014, 20, 105–115. [Google Scholar] [CrossRef]
- Greene, C.M.; McElvaney, N.G. Z alpha-1 antitrypsin deficiency and the endoplasmic reticulum stress response. World J. Gastrointest. Pharmacol. Ther. 2010, 1, 94–101. [Google Scholar] [CrossRef]
- Greene, C.M.; Marciniak, S.J.; Teckman, J.; Ferrarotti, I.; Brantly, M.L.; Lomas, D.A.; Stoller, J.K.; McElvaney, N.G. Alpha1-Antitrypsin deficiency. Nat. Rev. Dis. Primers 2016, 2, 16051. [Google Scholar] [CrossRef]
- Brode, S.K.; Ling, S.C.; Chapman, K.R. Alpha-1 antitrypsin deficiency: A commonly overlooked cause of lung disease. Can. Med. Assoc. J. 2012, 184, 1365–1371. [Google Scholar] [CrossRef]
- Carlson, J.A.; Rogers, B.B.; Sifers, R.N.; Finegold, M.J.; Clift, S.M.; DeMayo, F.J.; Bullock, D.W.; Woo, S.L. Accumulation of PiZ alpha 1-antitrypsin causes liver damage in transgenic mice. J. Clin. Investig. 1989, 83, 1183–1190. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Rockenstein, E.; Crews, L.; Masliah, E. Role of protein aggregation in mitochondrial dysfunction and neurodegeneration in Alzheimer’s and Parkinson’s diseases. Neuromol. Med. 2003, 4, 21–36. [Google Scholar] [CrossRef]
- Pieczenik, S.R.; Neustadt, J. Mitochondrial dysfunction and molecular pathways of disease. Exp. Mol. Pathol. 2007, 83, 84–92. [Google Scholar] [CrossRef]
- Teckman, J.H.; An, J.-K.; Blomenkamp, K.; Schmidt, B.; Perlmutter, D. Mitochondrial autophagy and injury in the liver in α1-antitrypsin deficiency. Am. J. Physiol. Liver Physiol. 2004, 286, G851–G862. [Google Scholar] [CrossRef]
- Degli Esposti, D.; Hamelin, J.; Bosselut, N.; Saffroy, R.; Sebagh, M.; Pommier, A.; Martel, C.; Lemoine, A. Mitochondrial Roles and Cytoprotection in Chronic Liver Injury. Biochem. Res. Int. 2012, 2012, 387626. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Dolgacheva, L.P. Interaction of misfolded proteins and mitochondria in neurodegenerative disorders. Biochem. Soc. Trans. 2017, 45, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Jarc, E.; Petan, T. Lipid Droplets and the Management of Cellular Stress. Yale J. Biol Med. 2019, 92, 435–452. [Google Scholar]
- Teckman, J.H.; An, J.K.; Loethen, S.; Perlmutter, D.H. Fasting in alpha1-antitrypsin deficient liver: Constitutive [correction of consultative] activation of autophagy. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G1156–G1165. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Moncan, M.; Mnich, K.; Blomme, A.; Almanza, A.; Samali, A.; Gorman, A.M. Regulation of lipid metabolism by the unfolded protein response. J. Cell. Mol. Med. 2021, 25, 1359–1370. [Google Scholar] [CrossRef]
- Khodayari, N.; Wang, R.L.; Marek, G.; Krotova, K.; Kirst, M.; Liu, C.; Rouhani, F.; Brantly, M. SVIP regulates Z variant alpha-1 antitrypsin retro-translocation by inhibiting ubiquitin ligase gp78. PLoS ONE 2017, 12, e0172983. [Google Scholar] [CrossRef]
- Soni, K.G.; Mardones, G.A.; Sougrat, R.; Smirnova, E.; Jackson, C.L.; Bonifacino, J.S. Coatomer-dependent protein delivery to lipid droplets. J. Cell Sci. 2009, 122, 1834–1841. [Google Scholar] [CrossRef]
- Rambold, A.S.; Cohen, S.; Lippincott-Schwartz, J. Fatty Acid Trafficking in Starved Cells: Regulation by Lipid Droplet Lipolysis, Autophagy, and Mitochondrial Fusion Dynamics. Dev. Cell 2015, 32, 678–692. [Google Scholar] [CrossRef]
- Wang, H.; Sreenivasan, U.; Hu, H.; Saladino, A.; Polster, B.M.; Lund, L.M.; Gong, D.-W.; Stanley, W.C.; Sztalryd, C. Perilipin 5, a lipid droplet-associated protein, provides physical and metabolic linkage to mitochondria. J. Lipid Res. 2011, 52, 2159–2168. [Google Scholar] [CrossRef]
- Ruan, L.; Zhou, C.; Jin, E.; Kucharavy, A.; Zhang, Y.; Wen, Z.; Florens, L.; Li, R. Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nat. Cell Biol. 2017, 543, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Ruther, U.; Tripodi, M.; Cortcsc, R.; Wagner, E.F. The human alpha-1-antitrypsin gene is efficiently expressed from two tissue-specific promotors in transgenic mice. Nucleic Acids Res. 1987, 15, 7519–7529. [Google Scholar] [CrossRef][Green Version]
- Clark, V.C.; Dhanasekaran, R.; Brantly, M.; Rouhani, F.; Schreck, P.; Nelson, D.R. Liver Test Results Do Not Identify Liver Disease in Adults With α1-Antitrypsin Deficiency. Clin. Gastroenterol. Hepatol. 2012, 10, 1278–1283. [Google Scholar] [CrossRef]
- Malintan, N.T.; Buckingham, S.D.; Lomas, D.A.; Sattelle, D.B. Calcium signalling in mammalian cell lines expressing wild type and mutant human α1-Antitrypsin. Sci. Rep. 2019, 9, 17293. [Google Scholar] [CrossRef]
- Tan, Y.; Jin, Y.; Wang, Q.; Huang, J.; Wu, X.; Ren, Z. Perilipin 5 Protects against Cellular Oxidative Stress by Enhancing Mitochondrial Function in HepG2 Cells. Cells 2019, 8, 1241. [Google Scholar] [CrossRef] [PubMed]
- Marcus, N.Y.; Blomenkamp, K.; Ahmad, M.; Teckman, J.H. Oxidative stress contributes to liver damage in a murine model of alpha-1-antitrypsin deficiency. Exp. Biol. Med. 2012, 237, 1163–1172. [Google Scholar] [CrossRef]
- Yan, H.P.; Li, M.; Lu, X.L.; Zhu, Y.M.; Ou-Yang, W.-X.; Xiao, Z.H.; Qiu, J.; Li, S.J. Use of plasma mitochondrial DNA levels for determining disease severity and prognosis in pediatric sepsis: A case control study. BMC Pediatr. 2018, 18, 267. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Ni, H.; Li, J.; Zhuang, X.; Zhang, J.; Qi, Z.; Chen, Q.; Wen, Z.; Shi, H.; Luo, X.; et al. The Impact of Circulating Mitochondrial DNA on Cardiomyocyte Apoptosis and Myocardial Injury After TLR4 Activation in Experimental Autoimmune Myocarditis. Cell. Physiol. Biochem. 2017, 42, 713–728. [Google Scholar] [CrossRef]
- Gohil, V.M.; Greenberg, M.L. Mitochondrial membrane biogenesis: Phospholipids and proteins go hand in hand. J. Cell Biol. 2009, 184, 469–472. [Google Scholar] [CrossRef]
- Cregan, S.P.; Dawson, V.; Slack, R.S. Role of AIF in caspase-dependent and caspase-independent cell death. Oncogene 2004, 23, 2785–2796. [Google Scholar] [CrossRef]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Anandatheerthavarada, H.K. Mitochondrial trafficking of APP and alpha synuclein: Relevance to mitochondrial dysfunction in Alzheimer’s and Parkinson’s diseases. Biochim. Biophys. Acta 2010, 1802, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Kueppers, F.; Andrake, M.D.; Xu, Q.; Jr, R.L.D.; Kim, J.; Sanders, C.L. Protein modeling to assess the pathogenicity of rare variants of SERPINA1 in patients suspected of having Alpha 1 Antitrypsin Deficiency. BMC Med. Genet. 2019, 20, 125. [Google Scholar] [CrossRef]
- Hill, K.; Model, K.; Ryan, M.T.; Dietmeier, K.; Martin, F.; Wagner, R.; Pfanner, N. Tom40 forms the hydrophilic channel of the mitochondrial import pore for preproteins [see comment]. Nature 1998, 395, 516–521. [Google Scholar] [CrossRef]
- Uren, R.T.; Dewson, G.; Bonzon, C.; Lithgow, T.; Newmeyer, D.D.; Kluck, R.M. Mitochondrial release of pro-apoptotic proteins: Electrostatic interactions can hold cytochrome c but not Smac/DIABLO to mitochondrial membranes. J. Biol. Chem. 2005, 280, 2266–2274. [Google Scholar] [CrossRef] [PubMed]
- Teckman, J.H.; Blomenkamp, K.S. Pathophysiology of Alpha-1 Antitrypsin Deficiency Liver Disease. Methods Mol. Biol. 2017, 1639, 1–8. [Google Scholar] [CrossRef]
- Benador, I.Y.; Veliova, M.; Mahdaviani, K.; Petcherski, A.; Wikstrom, J.D.; Assali, E.A.; Acin-Perez, R.; Shum, M.; Oliveira, M.F.; Cinti, S.; et al. Mitochondria Bound to Lipid Droplets Have Unique Bioenergetics, Composition, and Dynamics that Support Lipid Droplet Expansion. Cell Metab. 2018, 27, 869–885.e6. [Google Scholar] [CrossRef] [PubMed]
- Tyedmers, J.; Mogk, A.; Bukau, B. Cellular strategies for controlling protein aggregation. Nat. Rev. Mol. Cell Biol. 2010, 11, 777–788. [Google Scholar] [CrossRef]
- Vevea, J.D.; Garcia, E.J.; Chan, R.B.; Zhou, B.; Schultz, M.; Di Paolo, G.; McCaffery, J.M.; Pon, L.A. Role for Lipid Droplet Biogenesis and Microlipophagy in Adaptation to Lipid Imbalance in Yeast. Dev. Cell 2015, 35, 584–599. [Google Scholar] [CrossRef]
- Bosma, M.; Dapito, D.H.; Drosatos-Tampakaki, Z.; Huiping-Son, N.; Huang, L.-S.; Kersten, S.; Drosatos, K.; Goldberg, I.J. Sequestration of fatty acids in triglycerides prevents endoplasmic reticulum stress in an in vitro model of cardiomyocyte lipotoxicity. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2014, 1841, 1648–1655. [Google Scholar] [CrossRef]
- Prentki, M.; Madiraju, S.R. Glycerolipid/free fatty acid cycle and islet beta-cell function in health, obesity and diabetes. Mol. Cell. Endocrinol. 2012, 353, 88–100. [Google Scholar] [CrossRef]
- Hidvegi, T.; Mukherjee, A.; Ewing, M.; Kemp, C.; Perlmutter, D.H. The Role of Autophagy in Alpha-1-Antitrypsin Deficiency. Methods Enzymol. 2011, 499, 33–54. [Google Scholar] [CrossRef] [PubMed]
- Senoner, T.; Schindler, S.; Stättner, S.; Öfner, D.; Troppmair, J.; Primavesi, F. Associations of Oxidative Stress and Postoperative Outcome in Liver Surgery with an Outlook to Future Potential Therapeutic Options. Oxidative Med. Cell. Longev. 2019, 2019, 3950818. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Huang, Q.; Long, X.; Guo, X.; Sun, X.; Jin, X.; Li, Z.; Ren, T.; Yuan, P.; Huang, X.; et al. Mitochondrial elongation-mediated glucose metabolism reprogramming is essential for tumour cell survival during energy stress. Oncogene 2017, 36, 4901–4912. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, S.J.; Lomas, D.A. Alpha1-Antitrypsin Deficiency and Autophagy. N. Engl. J. Med. 2010, 363, 1863–1864. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Jiang, Q.; Ding, X.; Xu, W.; Wang, D.W.; Chen, M. The ER Stress-Mediated Mitochondrial Apoptotic Pathway and MAPKs Modulate Tachypacing-Induced Apoptosis in HL-1 Atrial Myocytes. PLoS ONE 2015, 10, e0117567. [Google Scholar] [CrossRef] [PubMed]
- Hidvegi, T.; Schmidt, B.Z.; Hale, P.; Perlmutter, D.H. Accumulation of mutant alpha1-antitrypsin Z in the endoplasmic reticulum activates caspases-4 and -12, NFkappaB, and BAP31 but not the unfolded protein response. J. Biol. Chem. 2005, 280, 39002–39015. [Google Scholar] [CrossRef] [PubMed]
- Bajt, M.L.; Cover, C.; Lemasters, J.J.; Jaeschke, H. Nuclear Translocation of Endonuclease G and Apoptosis-Inducing Factor during Acetaminophen-Induced Liver Cell Injury. Toxicol. Sci. 2006, 94, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, H.D.; Bergmann, A. The Role of Apoptosis-Induced Proliferation for Regeneration and Cancer. Cold Spring Harb. Perspect. Biol. 2012, 4, a008797. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial Import and Accumulation of alpha-Synuclein Impair Complex I in Human Dopaminergic Neuronal Cultures and Parkinson Disease Brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef]
- Sivasothy, P.; Dafforn, T.R.; Gettins, P.G.; Lomas, D.A. Pathogenic alpha 1-antitrypsin polymers are formed by reactive loop-beta-sheet A linkage. J. Biol. Chem. 2000, 275, 33663–33668. [Google Scholar] [CrossRef]
- Sifers, R.N. Intracellular Processing of 1-Antitrypsin. Proc. Am. Thorac. Soc. 2010, 7, 376–380. [Google Scholar] [CrossRef]
- Khodayari, N.; Marek, G.; Lu, Y.; Krotova, K.; Wang, R.L.; Brantly, M. Erdj3 Has an Essential Role for Z Variant Alpha-1-Antitrypsin Degradation. J. Cell. Biochem. 2017, 118, 3090–3101. [Google Scholar] [CrossRef]
- Fan, A.C.; Young, J.C. Function of cytosolic chaperones in Tom70-mediated mitochondrial import. Protein Pept. Lett. 2011, 18, 122–131. [Google Scholar] [CrossRef]
- Lavie, J.; De Belvalet, H.; Sonon, S.; Ion, A.M.; Dumon, E.; Melser, S.; Lacombe, D.; Dupuy, J.-W.; Lalou, C.; Bénard, G. Ubiquitin-Dependent Degradation of Mitochondrial Proteins Regulates Energy Metabolism. Cell Rep. 2018, 23, 2852–2863. [Google Scholar] [CrossRef]
- Clark, V.C.; Marek, G.; Liu, C.; Collinsworth, A.; Shuster, J.; Kurtz, T.; Nolte, J.; Brantly, M. Clinical and histologic features of adults with alpha-1 antitrypsin deficiency in a non-cirrhotic cohort. J. Hepatol. 2018, 69, 1357–1364. [Google Scholar] [CrossRef]
- Cho, A.; Haruyama, N.; Kulkarni, A.B. Generation of transgenic mice. Curr. Protoc. Cell Biol. 2009, 42, 19.11.1–19.11.22. [Google Scholar] [CrossRef]
- FastQC. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 26 April 2010).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- hg19. Available online: http://hgdownload.soe.ucsc.edu/downloads.html#human (accessed on 1 February 2009).
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khodayari, N.; Wang, R.L.; Oshins, R.; Lu, Y.; Millett, M.; Aranyos, A.M.; Mostofizadeh, S.; Scindia, Y.; Flagg, T.O.; Brantly, M. The Mechanism of Mitochondrial Injury in Alpha-1 Antitrypsin Deficiency Mediated Liver Disease. Int. J. Mol. Sci. 2021, 22, 13255. https://doi.org/10.3390/ijms222413255
Khodayari N, Wang RL, Oshins R, Lu Y, Millett M, Aranyos AM, Mostofizadeh S, Scindia Y, Flagg TO, Brantly M. The Mechanism of Mitochondrial Injury in Alpha-1 Antitrypsin Deficiency Mediated Liver Disease. International Journal of Molecular Sciences. 2021; 22(24):13255. https://doi.org/10.3390/ijms222413255
Chicago/Turabian StyleKhodayari, Nazli, Rejean L. Wang, Regina Oshins, Yuanqing Lu, Michael Millett, Alek M. Aranyos, Sayedamin Mostofizadeh, Yogesh Scindia, Tammy O. Flagg, and Mark Brantly. 2021. "The Mechanism of Mitochondrial Injury in Alpha-1 Antitrypsin Deficiency Mediated Liver Disease" International Journal of Molecular Sciences 22, no. 24: 13255. https://doi.org/10.3390/ijms222413255
APA StyleKhodayari, N., Wang, R. L., Oshins, R., Lu, Y., Millett, M., Aranyos, A. M., Mostofizadeh, S., Scindia, Y., Flagg, T. O., & Brantly, M. (2021). The Mechanism of Mitochondrial Injury in Alpha-1 Antitrypsin Deficiency Mediated Liver Disease. International Journal of Molecular Sciences, 22(24), 13255. https://doi.org/10.3390/ijms222413255