
Experimental Characterization of the Interaction between the N-Terminal SH3 Domain of Crkl and C3G




Abstract
:1. Introduction
2. Results and Discussion
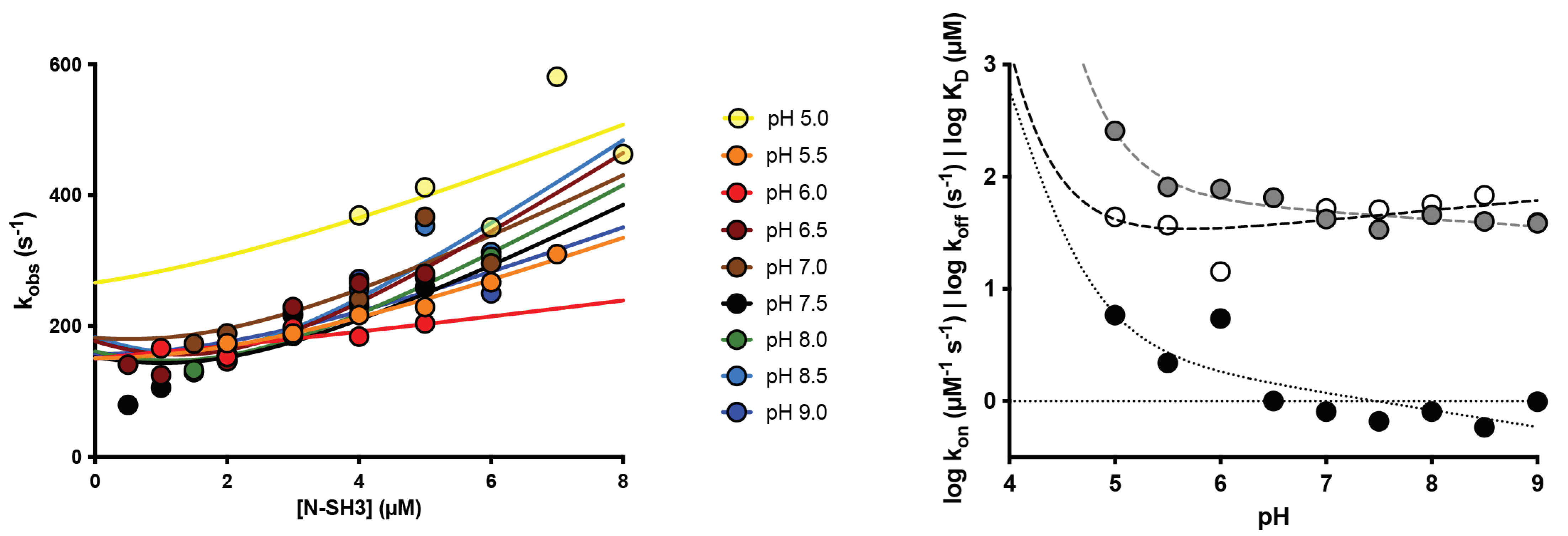
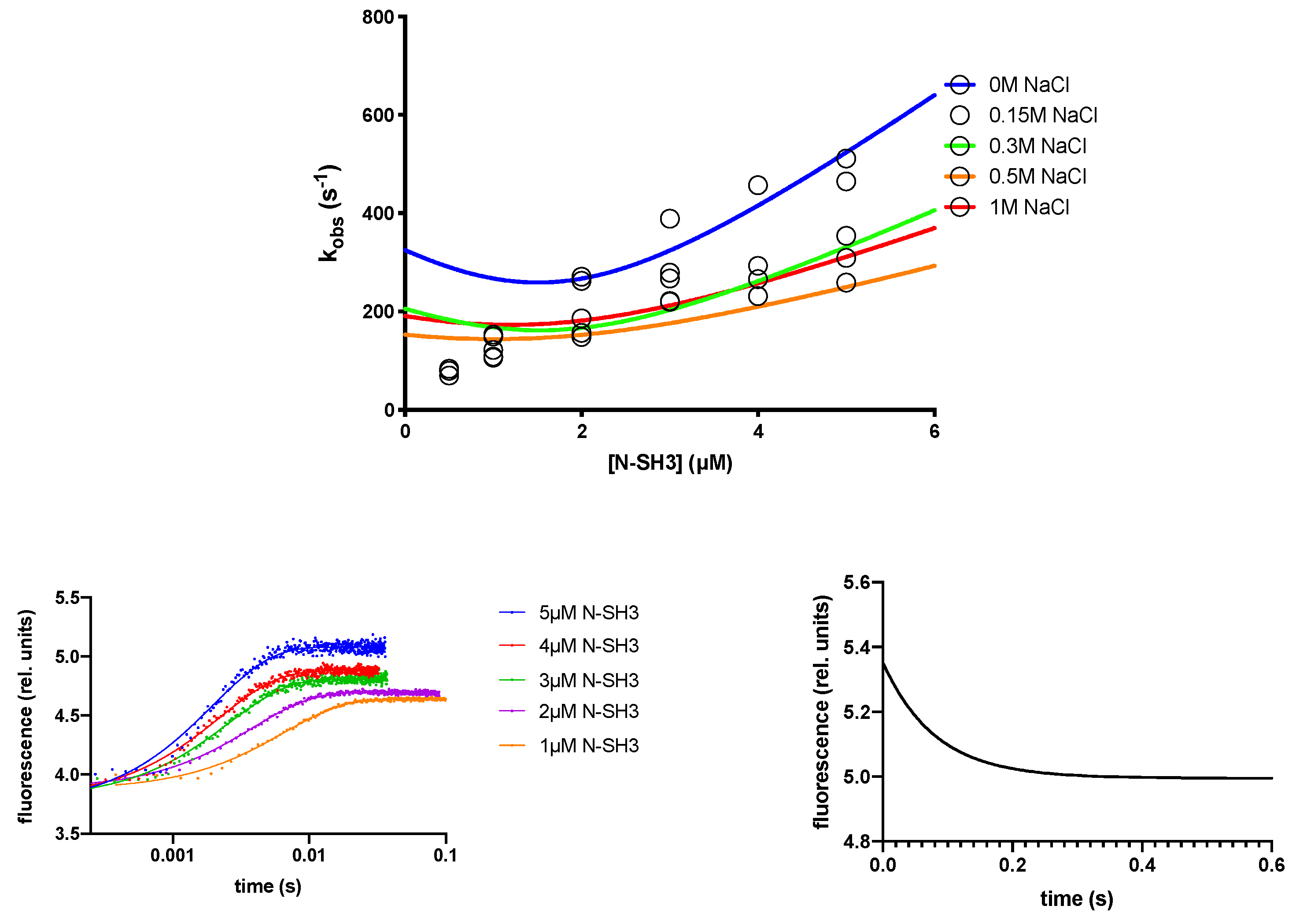
2.1. The Recognition Event of C3G by the N-SH3 Domain Is Electrostatically Driven
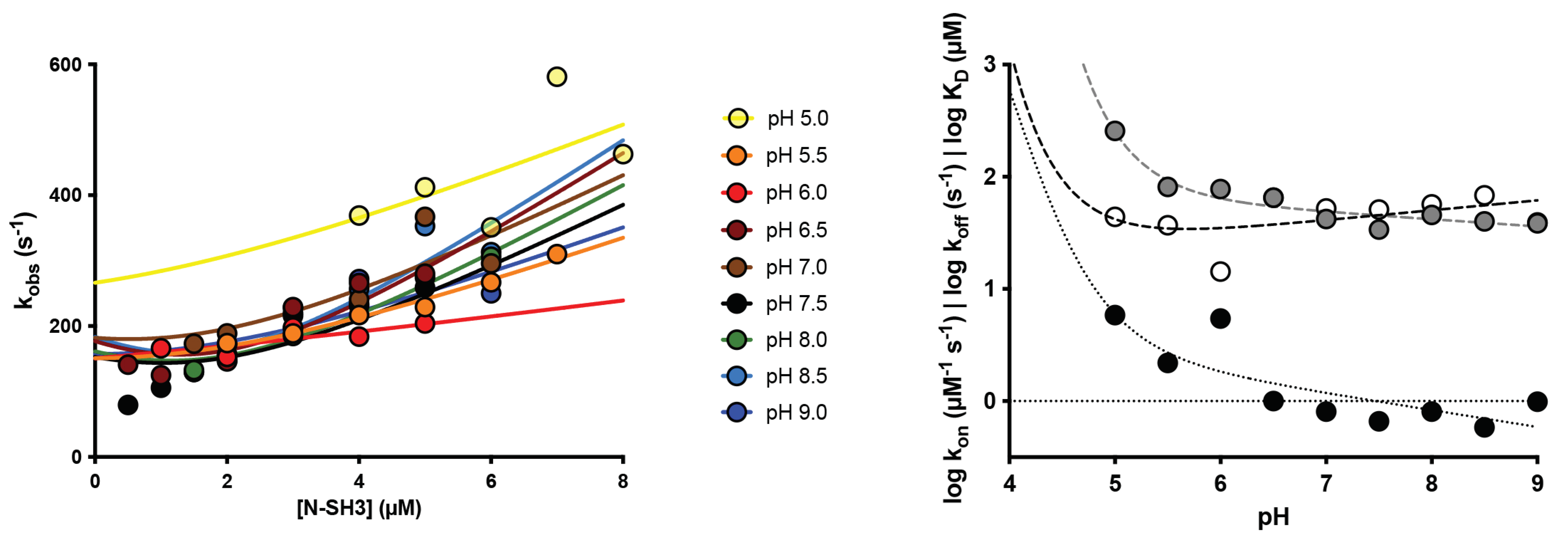
2.2. Protonation of Negatively Charged Residues Abolishes Binding
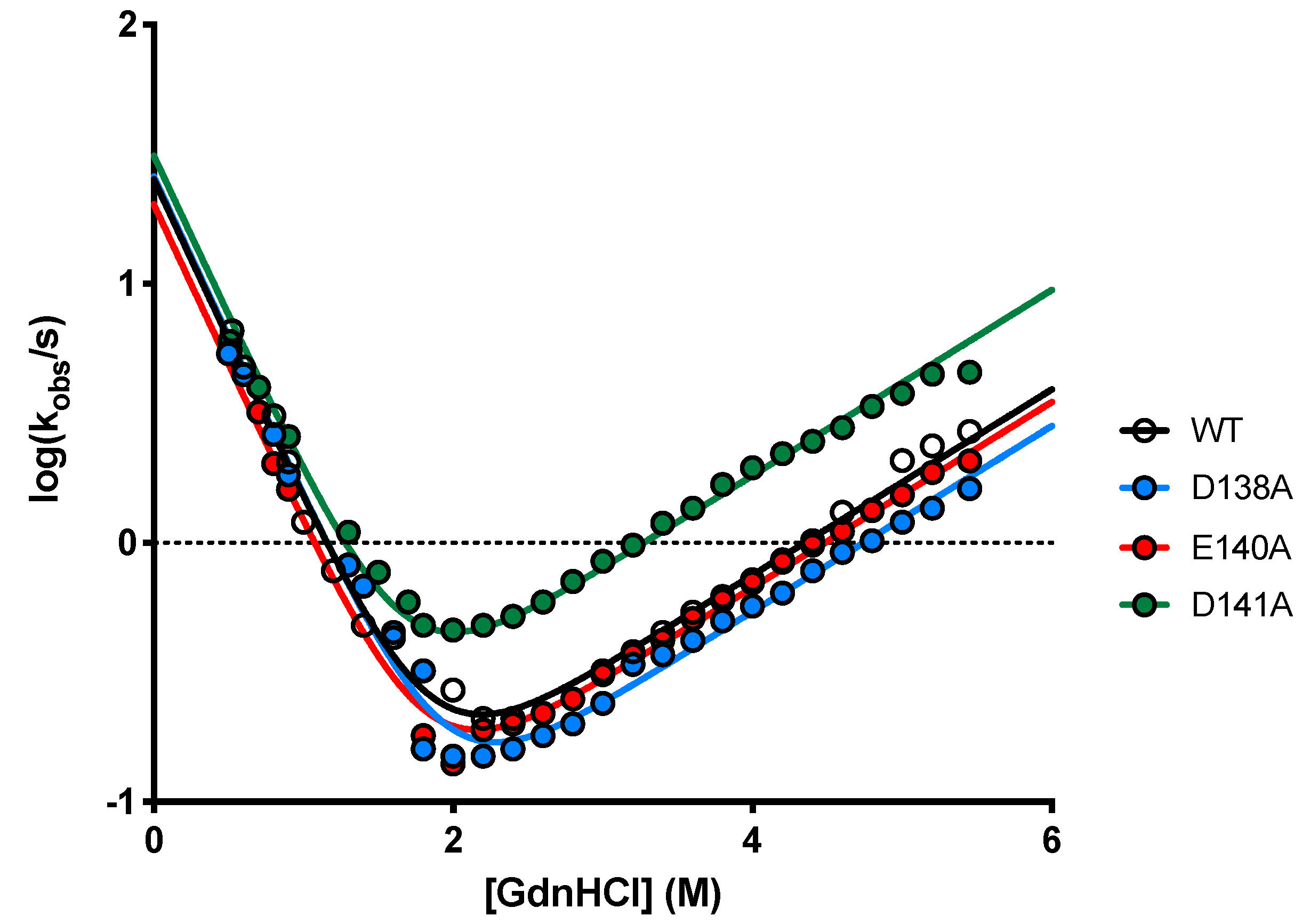
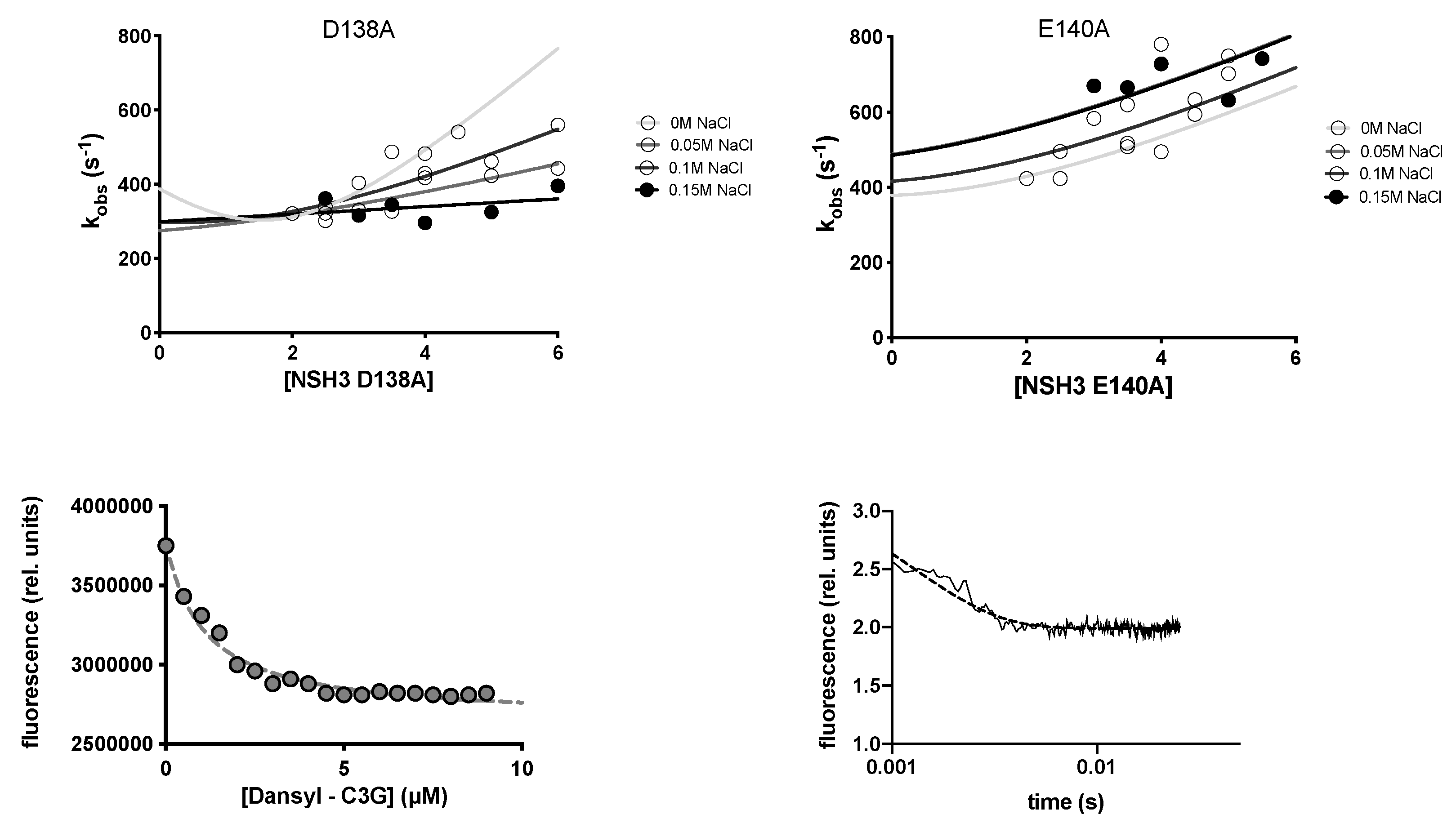
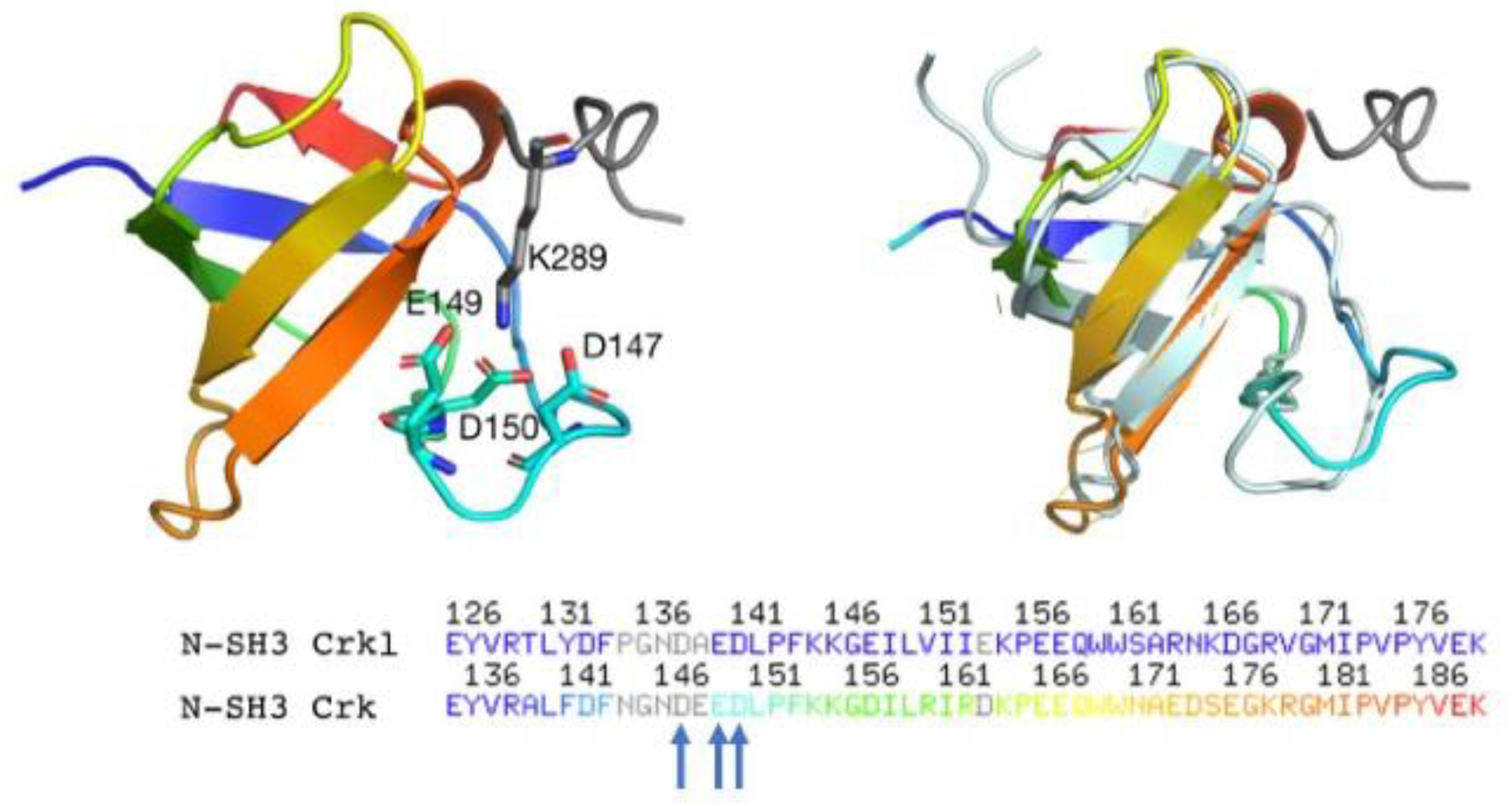
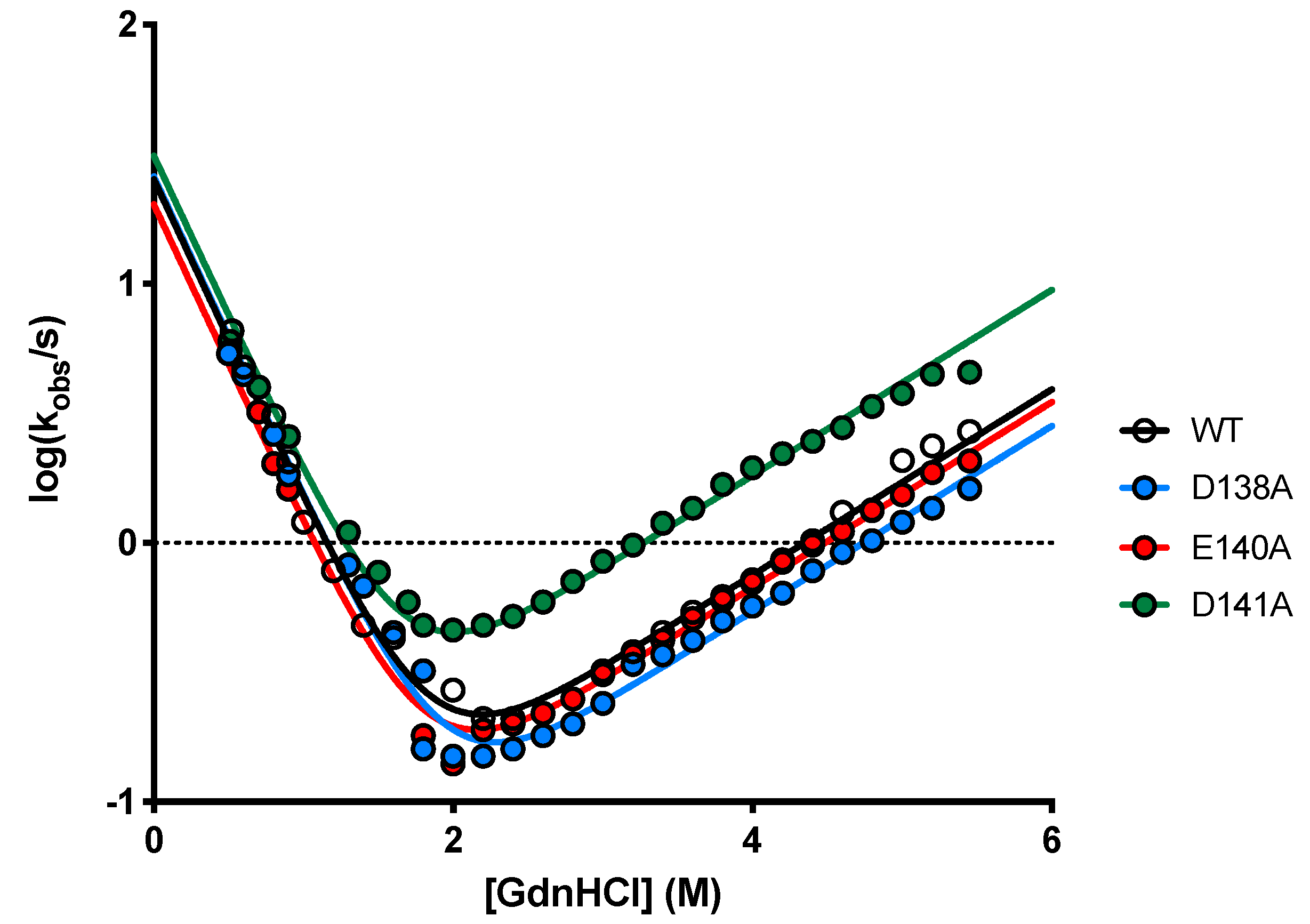
2.3. Determining the Role of D138, E140 and D141 by Site-Directed Mutagenesis
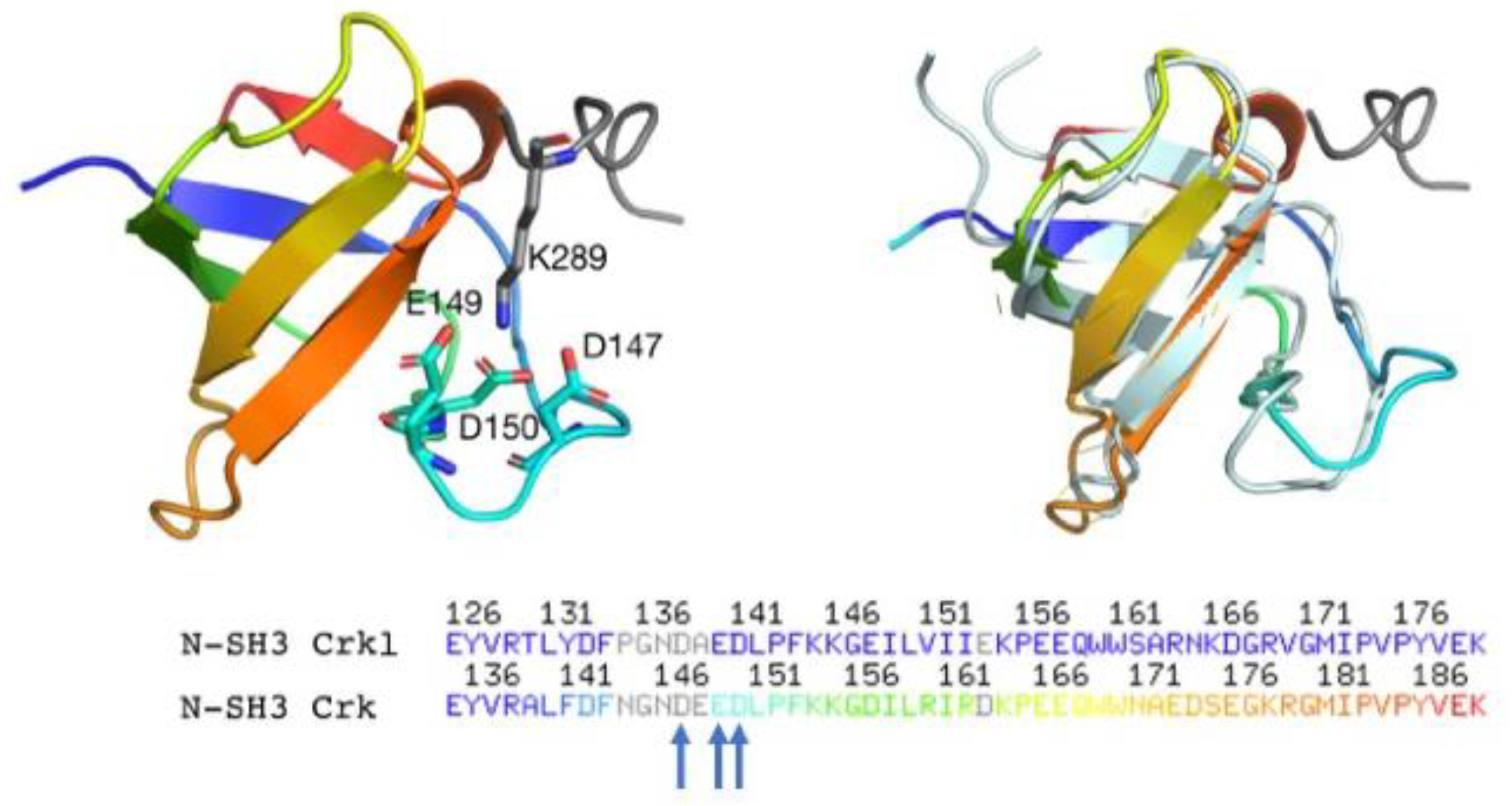
2.4. Determinants of N-SH3 Domain Binding Selectivity
3. Conclusions
4. Materials and Methods
4.1. Site-Directed Mutagenesis
4.2. Protein Expression and Purification
4.3. Stopped-Flow (un)folding Experiments
4.4. Stopped-Flow Kinetic Binding and Displacement Experiments
4.5. Equilibrium Binding Experiment of D141A
Author Contributions
Funding
Conflicts of Interest
References
- Park, T. Crk and CrkL as Therapeutic Targets for Cancer Treatment. Cells 2021, 10, 739. [Google Scholar] [CrossRef]
- Nichols, G.L.; Raines, M.A.; Vera, J.C.; Lacomis, L.; Tempst, P.; Golde, D.W. Identification of CRKL as the Constitutively Phosphorylated 39-KD Tyrosine Phosphoprotein in Chronic Myelogenous Leukemia Cells. Blood 1994, 84, 2912–2918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, R.; Mayer, B.J.; Cicchetti, P.; Baltimore, D. Identification of a Ten-Amino Acid Proline-Rich SH3 Binding Site. Science 1993, 259, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Chen, J.K.; Feng, S.; Dalgarno, D.C.; Brauer, A.W.; Schreiber, S.L. Structural Basis for the Binding of Proline-Rich Peptides to SH3 Domains. Cell 1994, 76, 933–945. [Google Scholar] [CrossRef]
- Barnett, P.; Bottger, G.; Klein, A.T.; Tabak, H.F.; Distel, B. The Peroxisomal Membrane Protein Pex13p Shows a Novel Mode of SH3 Interaction. EMBO J. 2000, 19, 6382–6391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, M.; Nagata, K.; Hachimori, Y.; Horiuchi, M.; Ogura, K.; Mandiyan, V.; Schlessinger, J.; Inagaki, F. Novel Recognition Mode between Vav and Grb2 SH3 Domains. EMBO J. 2001, 20, 2995–3007. [Google Scholar] [CrossRef] [Green Version]
- Lim, W.A.; Richards, F.M.; Fox, R.O. Structural Determinants of Peptide-Binding Orientation and of Sequence Specificity in SH3 Domains. Nature 1994, 372, 375–379. [Google Scholar] [CrossRef]
- Uemura, N.; Griffin, J.D. The Adapter Protein Crkl Links Cbl to C3G after Integrin Ligation and Enhances Cell Migration. J. Biol. Chem. 1999, 274, 37525–37532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparks, A.B.; Rider, J.E.; Hoffman, N.G.; Fowlkes, D.M.; Quillam, L.A.; Kay, B.K. Distinct Ligand Preferences of Src Homology 3 Domains from Src, Yes, Abl, Cortactin, P53bp2, PLCgamma, Crk, and Grb2. Proc. Natl. Acad. Sci. USA 1996, 93, 1540–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, M.; Ota, S.; Tanimura, R.; Nakamura, H.; Matuoka, K.; Takenawa, T.; Nagashima, K.; Kurata, T. Interaction between the Amino-Terminal SH3 Domain of CRK and Its Natural Target Proteins. J. Biol. Chem. 1996, 271, 14468–14472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knudsen, B.S.; Feller, S.M.; Hanafusa, H. Four Proline-Rich Sequences of the Guanine-Nucleotide Exchange Factor C3G Bind with Unique Specificity to the First Src Homology 3 Domain of Crk. J. Biol. Chem. 1994, 269, 32781–32787. [Google Scholar] [CrossRef]
- Gotoh, T.; Hattori, S.; Nakamura, S.; Kitayama, H.; Noda, M.; Takai, Y.; Kaibuchi, K.; Matsui, H.; Hatase, O.; Takahashi, H. Identification of Rap1 as a Target for the Crk SH3 Domain-Binding Guanine Nucleotide-Releasing Factor C3G. Mol. Cell Biol. 1995, 15, 6746–6753. [Google Scholar] [CrossRef] [Green Version]
- Bos, J.L.; de Rooij, J.; Reedquist, K.A. Rap1 Signalling: Adhering to New Models. Nat. Rev. Mol. Cell. Biol. 2001, 2, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Raaijmakers, J.H.; Bos, J.L. Specificity in Ras and Rap Signaling. J. Biol. Chem. 2009, 284, 10995–10999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichiba, T.; Hashimoto, Y.; Nakaya, M.; Kuraishi, Y.; Tanaka, S.; Kurata, T.; Mochizuki, N.; Matsuda, M. Activation of C3G Guanine Nucleotide Exchange Factor for Rap1 by Phosphorylation of Tyrosine 504. J. Biol. Chem. 1999, 274, 14376–14381. [Google Scholar] [CrossRef] [Green Version]
- Sakkab, D.; Lewitzky, M.; Posern, G.; Schaeper, U.; Sachs, M.; Birchmeier, W.; Feller, S.M. Signaling of Hepatocyte Growth Factor/Scatter Factor (HGF) to the Small GTPase Rap1 via the Large Docking Protein Gab1 and the Adapter Protein CRKL. J. Biol. Chem. 2000, 275, 10772–10778. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, B.S.; Zheng, J.; Feller, S.M.; Mayer, J.P.; Burrell, S.K.; Cowburn, D.; Hanafusa, H. Affinity and Specificity Requirements for the First Src Homology 3 Domain of the Crk Proteins. EMBO J. 1995, 14, 2191–2198. [Google Scholar] [CrossRef]
- Wu, X.; Knudsen, B.; Feller, S.M.; Zheng, J.; Sali, A.; Cowburn, D.; Hanafusa, H.; Kuriyan, J. Structural Basis for the Specific Interaction of Lysine-Containing Proline-Rich Peptides with the N-Terminal SH3 Domain of c-Crk. Structure 1995, 3, 215–226. [Google Scholar] [CrossRef] [Green Version]
- Malatesta, F. The Study of Bimolecular Reactions under Non-Pseudo-First Order Conditions. Biophys. Chem. 2005, 116, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Gianni, S.; Engström, A.; Larsson, M.; Calosci, N.; Malatesta, F.; Eklund, L.; Ngang, C.C.; Travaglini-Allocatelli, C.; Jemth, P. The Kinetics of PDZ Domain-Ligand Interactions and Implications for the Binding Mechanism. J. Biol. Chem. 2005, 280, 34805–34812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonini, E.; Brunori, M. Hemoglobin and Myoglobin in Their Reactions with Ligands; North-Holland: Amsterdam, The Netherlands, 1971. [Google Scholar]
- Schreiber, G.; Haran, G.; Zhou, H.-X. Fundamental Aspects of Protein−Protein Association Kinetics. Chem. Rev. 2009, 109, 839–860. [Google Scholar] [CrossRef] [Green Version]
- Pal, D.; Chakrabarti, P. Cis Peptide Bonds in Proteins: Residues Involved, Their Conformations, Interactions and Locations. J. Mol. Biol. 1999, 294, 271–288. [Google Scholar] [CrossRef] [Green Version]
- Brandl, M.; Weiss, M.S.; Jabs, A.; Sühnel, J.; Hilgenfeld, R. C-H...Pi-Interactions in Proteins. J. Mol. Biol. 2001, 307, 357–377. [Google Scholar] [CrossRef]
- Myers, J.K. Denaturant m Values and Heat Capacity Changes: Relation to Changes in Accessible Surface Areas of Protein Unfolding. Protein Sci. 1995, 4, 2138–2148. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.E.; Fersht, A.R. Folding of Chymotrypsin Inhibitor 2. 1. Evidence for a Two-State Transition. Biochemistry 1991, 30, 10428–10435. [Google Scholar] [CrossRef]
- Fersht, A.R.; Matouschek, A.; Serrano, L. The Folding of an Enzyme. I. Theory of Protein Engineering Analysis of Stability and Pathway of Protein Folding. J. Mol. Biol. 1992, 224, 771–782. [Google Scholar] [CrossRef]
- Cesareni, G.; Panni, S.; Nardelli, G.; Castagnoli, L. Can We Infer Peptide Recognition Specificity Mediated by SH3 Domains? FEBS Lett. 2002, 513, 38–44. [Google Scholar] [CrossRef] [Green Version]
- Kay, B.K. SH3 Domains Come of Age. FEBS Lett. 2012, 586, 2606–2608. [Google Scholar] [CrossRef] [Green Version]
- Saksela, K.; Permi, P. SH3 Domain Ligand Binding: What’s the Consensus and Where’s the Specificity? FEBS Lett. 2012, 586, 2609–2614. [Google Scholar] [CrossRef] [Green Version]
- Malagrinò, F.; Troilo, F.; Bonetti, D.; Toto, A.; Gianni, S. Mapping the Allosteric Network within a SH3 Domain. Sci. Rep. 2019, 9, 8279. [Google Scholar] [CrossRef]
- Toto, A.; Bonetti, D.; De Simone, A.; Gianni, S. Understanding the Mechanism of Binding between Gab2 and the C Terminal SH3 Domain from Grb2. Oncotarget 2017, 8, 82344–82351. [Google Scholar] [CrossRef] [Green Version]
- Gianni, S.; Haq, S.R.; Montemiglio, L.C.; Jürgens, M.C.; Engström, Å.; Chi, C.N.; Brunori, M.; Jemth, P. Sequence-Specific Long Range Networks in PSD-95/Discs Large/ZO-1 (PDZ) Domains Tune Their Binding Selectivity. J. Biol. Chem. 2011, 286, 27167–27175. [Google Scholar] [CrossRef] [Green Version]
- Chi, C.N.; Elfström, L.; Shi, Y.; Snäll, T.; Engström, A.; Jemth, P. Reassessing a Sparse Energetic Network within a Single Protein Domain. Proc. Natl. Acad. Sci. USA 2008, 105, 4679–4684. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N-SH3 WT versus C3G277–296 | |||
| [NaCl] | kon (µM−1 s−1) | koff (s−1) | KD (µM) |
| 0 M | 130 ± 7 | 12 ± 1 | 0.09 ± 0.05 |
| 0.15 M | 96 ± 8 | 23 ± 1 | 0.24 ± 0.06 |
| 0.3 M | 83 ± 7 | 32 ± 2 | 0.39 ± 0.14 |
| 0.5 M | 51 ± 4 | 34 ± 1 | 0.66 ± 0.12 |
| 1.0 M | 68 ± 5 | 51 ± 2 | 0.75 ± 0.20 |
| pH | kon (µM−1 s−1) | koff (s−1) | KD (µM) |
| 5.0 | 44 ± 15 | 257 ± 10 | 5.9 ± 0.8 |
| 5.5 | 37 ± 3 | 81 ± 3 | 2.2 ± 1.0 |
| 6.0 | 15 ± 4 | 78 ± 3 | 5.4 ± 1.5 |
| 6.5 | 65 ± 4 | 65 ± 2 | 1.0 ± 0.6 |
| 7.0 | 52 ± 5 | 42 ± 2 | 0.8 ± 0.5 |
| 7.5 | 51 ± 5 | 34 ± 2 | 0.7 ± 0.3 |
| 8.0 | 56 ± 5 | 46 ± 3 | 0.8 ± 0.4 |
| 8.5 | 68 ± 7 | 40 ± 3 | 0.6 ± 0.4 |
| 9.0 | 39 ± 4 | 39 ± 2 | 1.0 ± 0.4 |
| N-SH3 D138A versus C3G277–296 | |||
| [NaCl] | kon (µM−1 s−1) | koff (s−1) | KD (µM) |
| 0 M | 158 ± 12 | 139 ± 1 | 0.9 ± 0.3 |
| 0.05 M | 81 ± 8 | 212 ± 2 | 2.6 ± 0.6 |
| 0.10 M | 48 ± 8 | 183 ± 2 | 3.8 ± 0.3 |
| 0.15 M | 12 ± 5 | 254 ± 2 | 22.0 ± 2.0 |
| N-SH3 E140A versus C3G277–296 | |||
| [NaCl] | kon (µM−1 s−1) | koff (s−1) | KD (µM) |
| 0 M | 78 ± 6 | 254 ± 3 | 3.3 ± 0.7 |
| 0.05 M | 89 ± 8 | 330 ± 3 | 3.7 ± 0.6 |
| 0.10 M | 77 ± 8 | 302 ± 2 | 3.9 ± 0.8 |
| 0.15 M | 81 ± 8 | 339 ± 2 | 4.2 ± 1.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pagano, L.; Malagrinò, F.; Nardella, C.; Gianni, S.; Toto, A. Experimental Characterization of the Interaction between the N-Terminal SH3 Domain of Crkl and C3G. Int. J. Mol. Sci. 2021, 22, 13174. https://doi.org/10.3390/ijms222413174
Pagano L, Malagrinò F, Nardella C, Gianni S, Toto A. Experimental Characterization of the Interaction between the N-Terminal SH3 Domain of Crkl and C3G. International Journal of Molecular Sciences. 2021; 22(24):13174. https://doi.org/10.3390/ijms222413174
Chicago/Turabian StylePagano, Livia, Francesca Malagrinò, Caterina Nardella, Stefano Gianni, and Angelo Toto. 2021. "Experimental Characterization of the Interaction between the N-Terminal SH3 Domain of Crkl and C3G" International Journal of Molecular Sciences 22, no. 24: 13174. https://doi.org/10.3390/ijms222413174
APA StylePagano, L., Malagrinò, F., Nardella, C., Gianni, S., & Toto, A. (2021). Experimental Characterization of the Interaction between the N-Terminal SH3 Domain of Crkl and C3G. International Journal of Molecular Sciences, 22(24), 13174. https://doi.org/10.3390/ijms222413174