
Genomic Regions Associated with Fusarium Wilt Resistance in Flax

 and
and
Abstract
:1. Introduction
2. Results
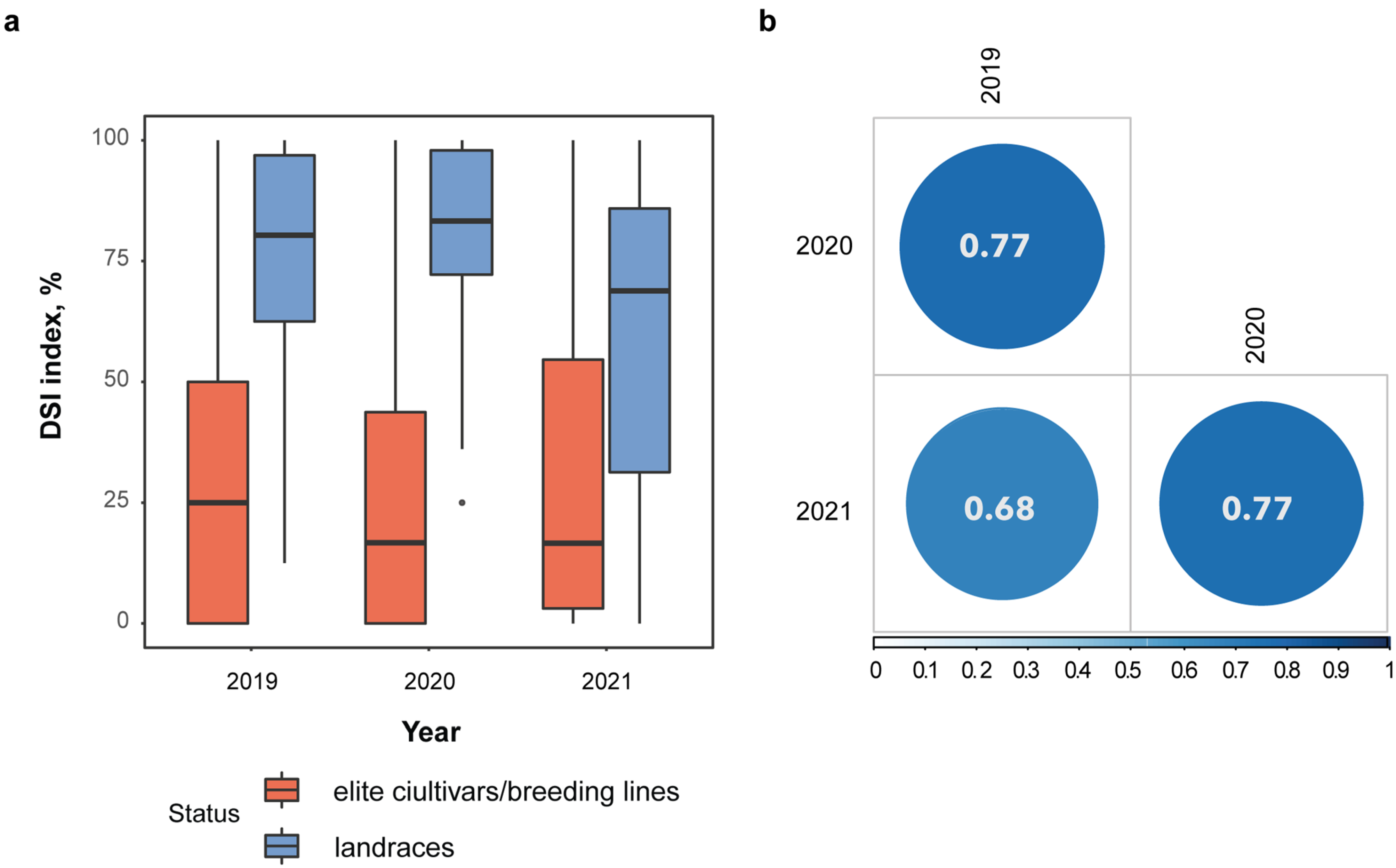
2.1. Evaluation Resistance to Fusarium Wilt in Flax
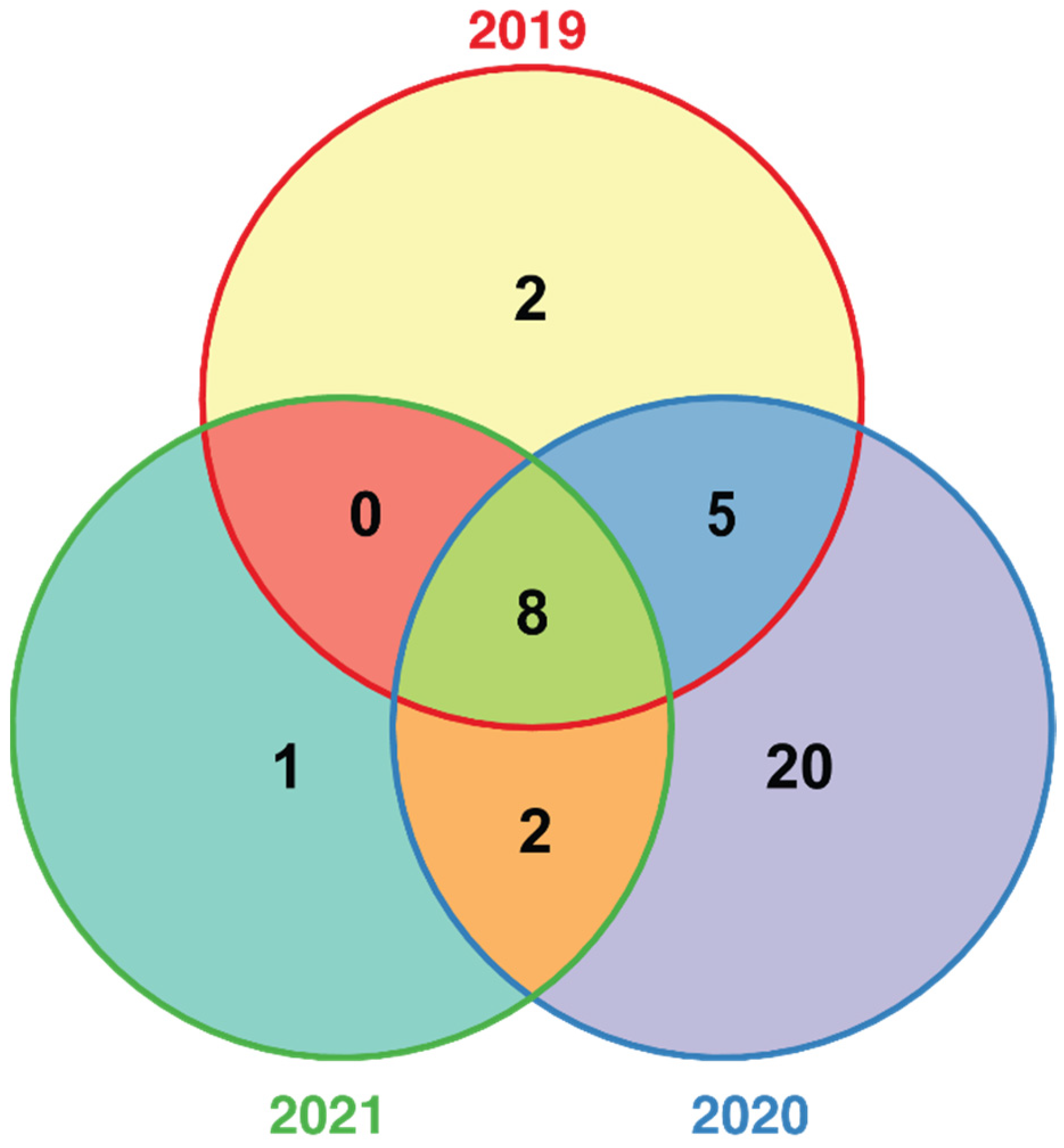
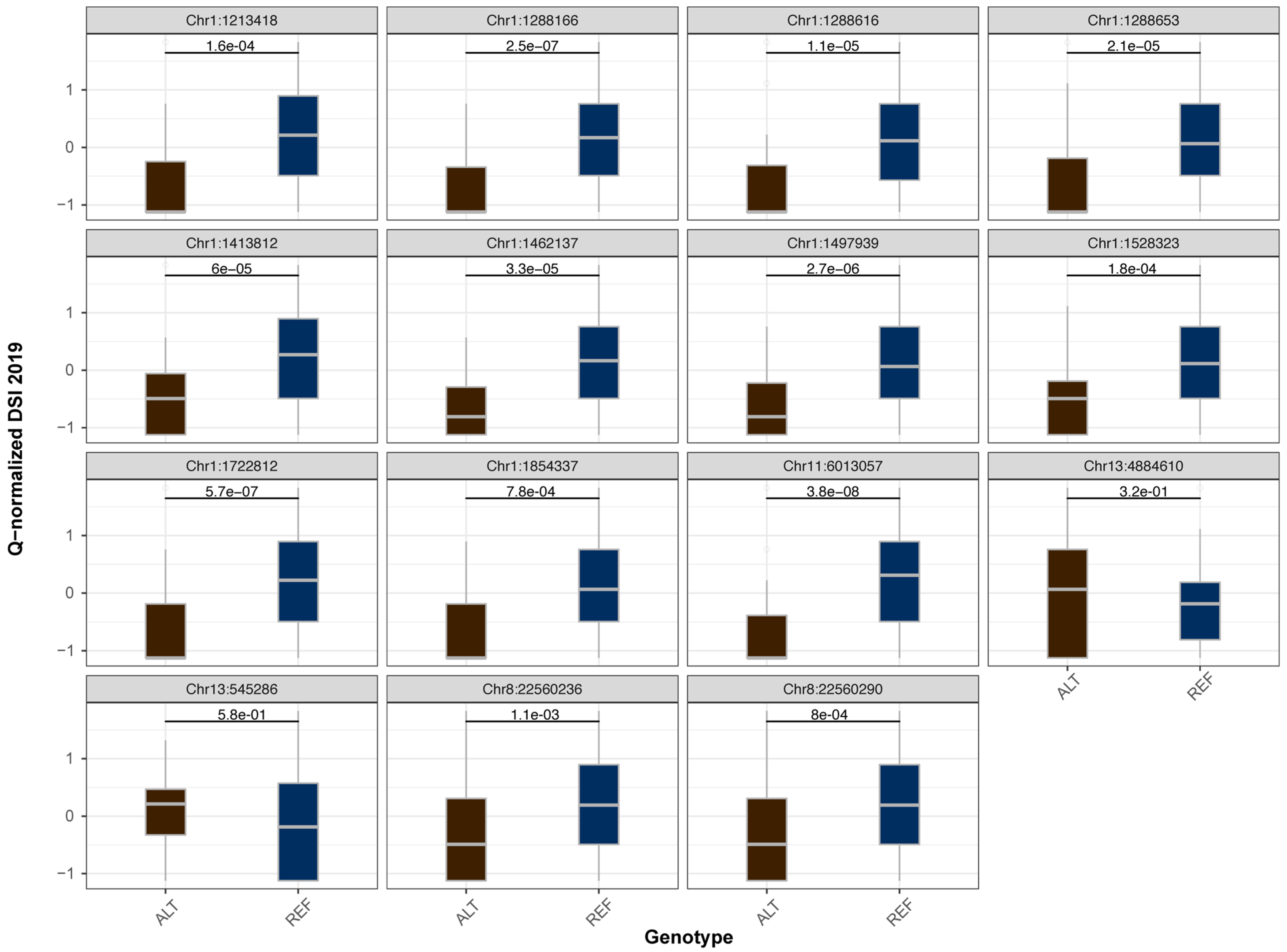
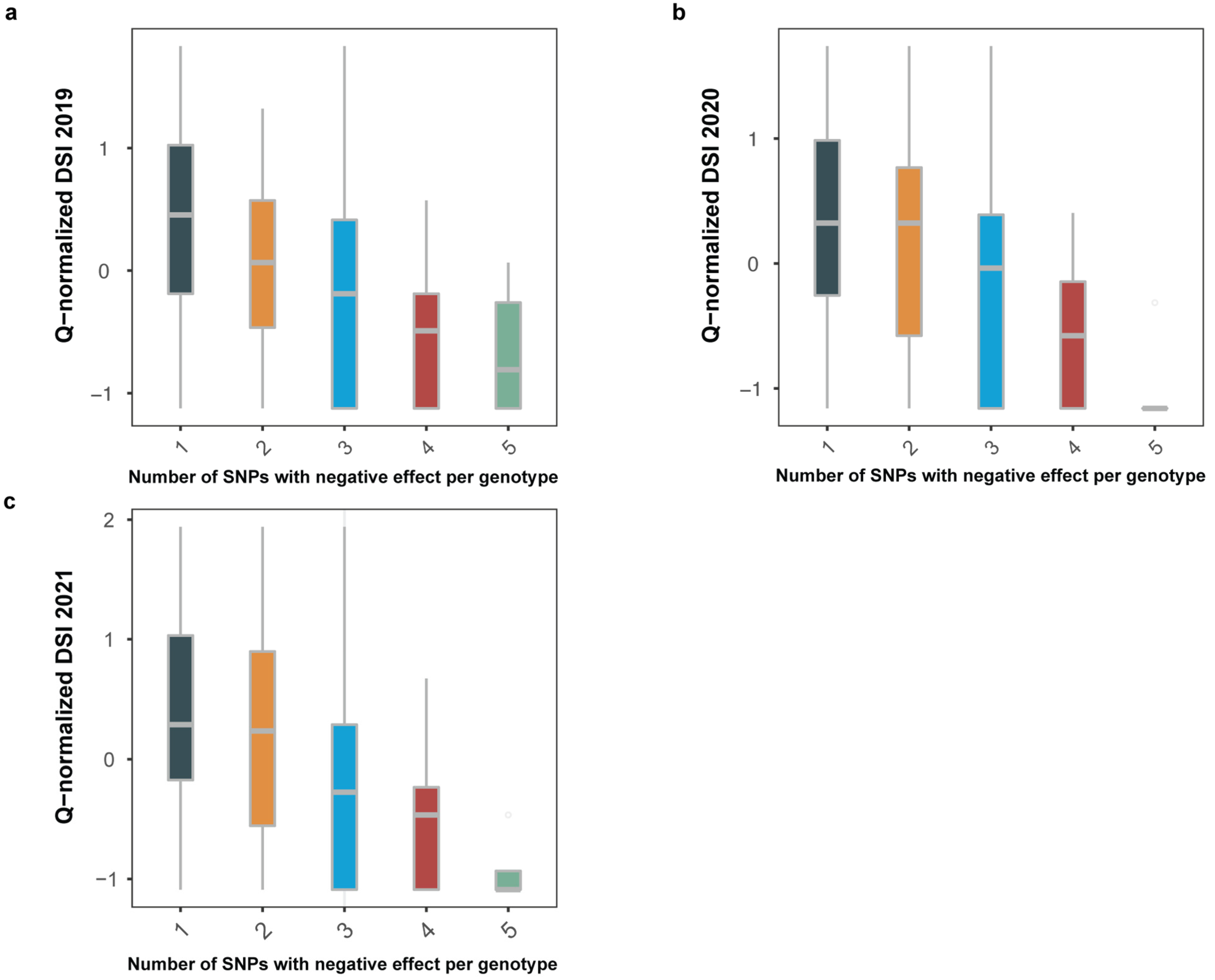
2.2. Association Mapping of Fusarium Wilt Resistance
2.3. Fusarium Wilt Resistance Candidate Genes
3. Discussion
4. Materials and Methods
4.1. Plant Material Collection and Phenotyping
4.2. DNA Sequencing and Variant Calling
4.3. Genetic Data Analyses
4.4. GWAS
4.5. Candidate Genes
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boyle, L.W. Histological Characters of Flax Roots in Relation to Resistance to Wilt and Root Rot; Technical Bulletin, No. 458; United States Department of Agriculture: Washington, DC, USA, 1934; pp. 1–18. [Google Scholar]
- Houston, B.R.; Knowles, P.F. Fifty-years survival of Flax Fusarium wilt in the absence of flax culture. Plant Dis. Rep. 1949, 33, 38–39. [Google Scholar]
- Saharan, G.; Mehta, N.; Sangwan, M. Fungal Diseases of Linseed. In Diseases of Oilseed Crops; Indus Publishing Company: New Delhi, India, 2005; pp. 176–201. [Google Scholar]
- Rashid, K.Y.; Kenaschuk, E.O. Effect of trifluralin on Fusarium wilt in flax. Can. J. Plant Sci. 1993, 73, 893–901. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.Y.; Chen, S.L.; Sun, Q.A.; He, D.T.; Wu, Y.N. Evaluation of Fusarium wilt resistance of flax varieties. Sci. Agric. Sin. 1993, 26, 44–49. [Google Scholar]
- Ondrej, M. Evaluation of flax genepool according to resistance to Fusarium wilt of flax and to mildew. Plant Genet. Resour. 1993, 92, 54–58. [Google Scholar]
- Rozhmina, T.A.; Loshakova, N.I. Samples of fiber and oil flax (Linum usitatissimum L.)—Sources of effective genes for resistance to Fusarium wilt and its dependence on temperature. Agric. Biol. 2016, 51, 310–317. [Google Scholar]
- Rozhmina, T.A. Identification of effective genes for resistance to Fusarium wilt in fiber flax varieties. Biol. Agric. 2017, 4, 10–12. [Google Scholar]
- Sánchez-Martín, J.; Keller, B. Contribution of recent technological advances to future resistance breeding. Theor. Appl. Genet. 2019, 132, 713–732. [Google Scholar] [CrossRef]
- Sánchez-Bayo, F. Impacts of Agricultural Pesticides on Terrestrial Ecosystems. In Ecological Impacts of Toxic Chemicals; Sánchez-Bayo, F., van den Brink, P.J., Mann, R.M., Eds.; Bentham Science Publishers Ltd.: Sharjah, United Arab Emirate, 2011; pp. 63–87. [Google Scholar]
- Kourelis, J.; van der Hoorn, R.A.L. Defended to the nines: 25 Years of resistance gene cloning identifies nine mechanisms for R protein function. Plant Cell 2018, 30, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.D.G.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [Green Version]
- Tsuda, K.; Katagiri, F. Comparing signaling mechanisms engaged in pattern-triggered and effector-triggered immunity. Curr. Opin. Plant. Biol. 2010, 13, 459–465. [Google Scholar] [CrossRef]
- Künstler, A.; Bacsó, R.; Gullner, G.; Hafez, Y.M.; Király, L. Staying alive—Is cell death dispensable for plant disease resistance during the hypersensitive response? Physiol. Mol. Plant Pathol. 2016, 93, 75–84. [Google Scholar] [CrossRef]
- Zhong, Y.; Cheng, Z.-M. A unique RPW8-encoding class of genes that originated in early land plants and evolved through domain fission, fusion, and duplication. Sci. Rep. 2016, 6, 32923. [Google Scholar] [CrossRef] [Green Version]
- Balint-Kurti, P. The plant hypersensitive response: Concepts, control and consequences. Mol. Plant Pathol. 2016, 20, 1163–1178. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Chhapekar, S.S.; Lu, L.; Oh, S.; Singh, S.; Kim, C.S.; Kim, S.; Choi, G.J.; Lim, Y.P.; Choi, S.R. Genome-wide identification and characterization of NBS-encoding genes in Raphanus sativus L. and their roles related to Fusarium oxysporum resistance. BMC Plant Biol. 2021, 21, 47. [Google Scholar] [CrossRef]
- Block, A.; Alfano, J.R. Plant targets for Pseudomonas syringae type III effectors: Virulence targets or guarded decoys? Curr. Opin. Microbiol. 2011, 14, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Dmitriev, A.A.; Krasnov, G.S.; Rozhmina, T.A.; Novakovskiy, R.O.; Snezhkina, A.V.; Fedorova, M.S.; Yurkevich, O.Y.; Muravenko, O.V.; Bolsheva, N.L.; Kudryavtseva, A.V.; et al. Differential gene expression in response to Fusarium oxysporum infection in resistant and susceptible genotypes of flax (Linum usitatissimum L.). BMC Plant Biol. 2017, 17 (Suppl. 2), 253. [Google Scholar] [CrossRef]
- Galindo-González, L.; Deyholos, M.K. RNA-seq Transcriptome Response of Flax (Linum usitatissimum L.) to the Pathogenic Fungus Fusarium oxysporum f.sp. lini. Front. Plant Sci. 2016, 7, 1766. [Google Scholar] [CrossRef] [Green Version]
- Boba, A.; Kostyn, K.; Kozak, B.; Zalewski, I.; Szopa, J.; Kulma, A. Transcriptomic profiling of susceptible and resistant flax seedlings after Fusarium oxysporum lini infection. PLoS ONE 2021, 16, e0246052. [Google Scholar] [CrossRef]
- Chandrawati, S.N.; Kumar, R.; Kumar, S.; Singh, P.K.; Yadav, V.K.; Ranade, S.A.; Yadav, H.K. Genetic diversity, population structure and association analysis in linseed (Linum usitatissimum L.). Physiol. Mol. Biol. Plants Int. J. Funct. Plant Biol. 2017, 23, 207–219. [Google Scholar] [CrossRef] [Green Version]
- Soto-Cerda, B.J.; Cloutier, S.; Quian, R.; Gajardo, H.A.; Olivos, M.; You, F.M. Genome-wide association analysis of mucilage and hull content in flax (Linum usitatissimum L.) seeds. Int. J. Mol. Sci. 2018, 19, 2870. [Google Scholar] [CrossRef] [Green Version]
- You, F.M.; Xiao, J.; Li, P.; Yao, Z.; Jia, G.; He, L.; Kumar, S.; Soto-Cerda, B.; Duguid, S.D.; Booker, H.M.; et al. Genome-wide association study and selection signatures detect genomic regions associated with seed yield and oil quality in flax. Int. J. Mol. Sci. 2018, 19, 2303. [Google Scholar] [CrossRef] [Green Version]
- Xie, D.; Dai, Z.; Yang, Z.; Tang, Q.; Deng, C.; Xu, Y.; Wang, J.; Chen, J.; Zhao, D.; Zhang, S.; et al. Combined genome-wide association analysis and transcriptome sequencing to identify candidate genes for flax seed fatty acid metabolism. Plant Sci. 2019, 286, 98–107. [Google Scholar] [CrossRef]
- Guo, D.; Jiang, H.; Yan, W.; Yang, L.; Ye, J.; Wang, Y.; Yan, Q.; Chen, J.; Gao, Y.; Duan, L.; et al. Resequencing 200 flax cultivated accessions identifies candidate genes related to seed size and weight and reveals signatures of artificial selection. Front. Plant Sci. 2020, 10, 1682. [Google Scholar] [CrossRef] [Green Version]
- Soto-Cerda, B.-J.; Aravena, G.; Cloutier, S. Genetic dissection of flowering time in flax (Linum usitatissimum L.) through single- and multi-locus genome-wide association studies. Mol. Genet. Genom. 2021, 296, 877–891. [Google Scholar] [CrossRef] [PubMed]
- Sertse, D.; You, F.M.; Ravichandran, S.; Soto-Cerda, B.J.; Duguid, S.; Cloutier, S. Loci harboring genes with important role in drought and related abiotic stress responses in flax revealed by multiple GWAS models. Appl. Genet. 2021, 134, 191–212. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Dai, Z.; Yang, Z.; Sun, J.; Zhao, D.; Yang, X.; Zhang, L.; Tang, Q.; Su, J. Genome-wide association study identifying candidate genes Influencing Important agronomic traits of flax (Linum usitatissimum L.) using SLAF-seq. Front. Plant Sci. 2018, 8, 2232. [Google Scholar] [CrossRef] [Green Version]
- Knowles, P.; Houston, B. Inheritance of Resistance to Fusarium Wilt of Flax in Dakota Selection 48–94. Agron. J. 1955, 47, 131–135. [Google Scholar] [CrossRef] [Green Version]
- Spielmeyer, W.; Green, A.; Bittisnich, D.; Mendham, N.; Lagudah, E. Identification of quantitative trait loci contributing to Fusarium wilt resistance on an AFLP linkage map of flax (Linum usitatissimum). Appl. Genet. 1998, 97, 633–641. [Google Scholar] [CrossRef]
- Spielmeyer, W.; Lagudah, E.; Mendham, N.; Green, A. Inheritance of resistance to flax wilt (Fusarium oxysporum f.sp. lini Schlecht) in a doubled haploid population of Linum usitatissimum L. Euphytica 1998, 101, 287–291. [Google Scholar]
- Caballo, C.; Castro, P.; Gil, J.; Millan, T.; Rubio, J.; Die, J.V. Candidate genes expression profiling during wilting in chickpea caused by Fusarium oxysporum f.sp. ciceris race 5. PLoS ONE 2019, 14, e0224212. [Google Scholar] [CrossRef]
- Gupta, P.K.; Kulwal, P.L.; Jaiswal, V. Association mapping in plants in the post-GWAS genomics era. Adv. Genet. 2019, 104, 75–154. [Google Scholar]
- Duk, M.; Kanapin, A.; Surkova, S.; Bankin, M.; Rozhmina, T.; Samsonova, A.; Samsonova, M. The genetic landscape of fiber flax. Front. Plant Sci. 2021, in press. [Google Scholar]
- Wang, J.; Zhang, Z. GAPIT Version 3: Boosting Power and Accuracy for Genomic Association and Prediction. Genom. Proteom. Bioinform. 2021, in press. [Google Scholar] [CrossRef]
- Flor, H. Tests for allelism of rust-resistance genes in flax. Crop Sci. 1965, 5, 415–418. [Google Scholar] [CrossRef]
- Wang, S.B.; Feng, J.Y.; Ren, W.L.; Huang, B.; Zhou, L.; Wen, Y.J.; Zhang, J.; Dunwell, J.M.; Xu, S.; Zhang, Y.M. Improving power and accuracy of genome-wide association studies via a multi-locus mixed linear model methodology. Sci. Rep. 2016, 6, 19444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berardini, T.Z.; Reiser, L.; Li, D.; Mezheritsky, Y.; Muller, R.; Strait, E.; Huala, E. The Arabidopsis information resource: Making and mining the “gold standard” annotated reference plant genome. Genesis 2015, 53, 474–485. [Google Scholar] [CrossRef] [Green Version]
- Porter, K.; Day, B. From filaments to function: The role of the plant actin cytoskeleton in pathogen perception, signaling and immunity. J Integr. Plant Biol. 2016, 58, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Skirpan, A.L.; McCubbin, A.G.; Ishimizu, T.; Wang, X.; Hu, Y.; Dowd, P.E.; Ma, H.; Kao, T. Isolation and characterization of kinase interacting protein 1, a pollen protein that interacts with the kinase domain of PRK1, a receptor-like kinase of petunia. Plant Physiol. 2001, 126, 1480–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romeis, T. Protein kinases in the plant defence response. Curr. Opin. Plant Biol. 2001, 4, 407–414. [Google Scholar] [CrossRef]
- Mizuno, Y.; Imano, S.; Camagna, M.; Suzuki, T.; Tanaka, A.; Sato, I.; Chiba, S.; Kawakita, K.; Takemoto, D. Nicotiana benthamiana exportin 1 is required for elicitor-induced phytoalexin production, cell death induction, and resistance against potato late blight pathogen Phytophthora infestans. J. Gen. Plant Pathol. 2019, 85, 347–355. [Google Scholar] [CrossRef]
- Sun, X.; Han, G.; Meng, Z.; Lin, L.; Sui, N. Roles of malic enzymes in plant development and stress responses. Plant Signal. Behav. 2019, 14, e1644596. [Google Scholar] [CrossRef] [PubMed]
- Jun, X.U.; Wang, X.Y.; Guo, W.Z. The cytochrome P450 superfamily: Key players in plant development and defense. J. Integr. Agric. 2015, 14, 1673–1686. [Google Scholar]
- Zhou, L.; Cheung, M.Y.; Li, M.W.; Fu, Y.; Sun, Z.; Sun, S.M.; Lam, H.M. Rice hypersensitive induced reaction protein 1 (OsHIR1) associates with plasma membrane and triggers hypersensitive cell death. BMC Plant Biol. 2010, 10, 29. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.W.; Kim, Y.J.; Hwang, B.K. The hypersensitive induced reaction and leucine-rich repeat proteins regulate plant cell death associated with disease and plant immunity. Mol. Plant-Microbe Interact. 2011, 24, 68–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusano, T.; Tateda, C.; Berberich, T.; Takahashi, Y. Voltage-dependent anion channels: Their roles in plant defense and cell death. Plant Cell Rep. 2009, 28, 1301–1308. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Inoue, H.; Tang, X.; Tan, Y.; Xu, X.; Wang, C.; Jiang, C.-J. Rice OsAAA-ATPase1 is Induced during Blast Infection in a Salicylic Acid-Dependent Manner, and Promotes Blast Fungus Resistance. Int. J. Mol. Sci. 2020, 21, 1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, Y.; Chane, A.; Jung, M.; Lee, Y. Recent Advances in Understanding the Roles of Pectin as an Active Participant in Plant Signaling Networks. Plants 2021, 10, 1712. [Google Scholar] [CrossRef]
- Li, Y.; Liu, P.; Takano, T.; Liu, S. A chloroplast-localized rubredoxin family protein gene from Puccinellia tenuiflora (PutRUB) increases NaCl and NaHCO3 tolerance by decreasing H2O2 accumulation. Int. J. Mol. Sci. 2016, 17, 804. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liu, P.P.; Ni, X. Molecular evolution and functional analysis of rubredoxin-like proteins in plants. BioMed Res. Int. 2019, 2019, 2932585. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, J.A.; Daudi, A.; Butt, V.S.; Paul Bolwell, G. Reactive oxygen species and their role in plant defence and cell wall metabolism. Planta 2012, 236, 765–779. [Google Scholar] [CrossRef]
- Pan, L.; Lv, S.; Yang, N.; Lv, Y.; Liu, Z.; Wu, J.; Wang, G. The multifunction of CLAVATA2 in plant development and immunity. Front. Plant Sci. 2016, 7, 1573. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, H.; He, S.; Meng, F.; Zhang, C.; Fan, S.; Wu, J.; Zhang, S.; Xu, P. GmSnRK1.1, a sucrose non-fermenting-1(SNF1)-related protein kinase, promotes soybean resistance to Phytophthora Sojae. Front. Plant Sci. 2019, 10, 996. [Google Scholar] [CrossRef] [Green Version]
- Loshakova, N.I.; Krylova, T.V.; Kudryavtseva, L.P. Guidelines for the Phytopathological Assessment of the Resistance of Fiber Flax to Diseases; Publishing house of the Russian Academy of Agricultural Sciences: Moscow, Russia, 2000. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Tello, D.; Gil, J.; Loaiza, C.D.; Riascos, J.J.; Cardozo, N.; Duitama, J. NGSEP3: Accurate variant calling across species and sequencing protocols. Bioinformatics 2019, 35, 4716–4723. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. 1000 Genomes Project Analysis Group. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Blighe, K.; Lun, A. PCAtools: Everything Principal Components Analysis. Available online: https://github.com/kevinblighe/PCAtools (accessed on 14 September 2021).
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X. rMVP: A Memory-efficient, Visualization-enhanced, and Parallel-accelerated tool for Genome-Wide Association Study. Genom. Proteom. Bioinform. 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Dong, S.-S.; Xu, J.-H.; He, W.-M.; Yang, T.-L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef] [PubMed]
- Hill, W.G.; Weir, B.S. Variances and covariances of squared linkage disequilibria in finite populations. Theor. Popul. Biol. 1988, 33, 54–78. [Google Scholar] [CrossRef]
- Mann, H.B.; Whitney, D.R. On a test of whether one of two random variables is stochastically larger than the other. Ann. Math. Stat. 1947, 18, 50–60. [Google Scholar] [CrossRef]
- You, F.; Cloutier, S. Mapping quantitative trait loci onto chromosome-scale pseudomolecules in flax. Methods Protoc. 2020, 3, 28. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosome | QTN Position | REF/ALT | Average PVE | Average Effect | Year | MAF | QTN Annotation | |
|---|---|---|---|---|---|---|---|---|
| ID | Number | |||||||
| CP027619.1 | 1 | 1213418 | C/T | 0.106 | −0.53 | 2019 2020 2021 | 0.138 | upstream_transcript_variant |
| CP027619.1 | 1 | 1288166 | A/G | 0.09 | −0.74 | 2019 2020 2021 | 0.093 | synonymous_variant |
| CP027619.1 | 1 | 1288616 | C/A | 0.10 | −0.72 | 2019 2020 2021 | 0.112 | synonymous_variant |
| CP027619.1 | 1 | 1288653 | T/C | 0.103 | −0.76 | 2019 2020 2021 | 0.103 | missense_variant |
| CP027619.1 | 1 | 1413812 | T/A | 0.102 | −0.69 | 2019 2020 2021 | 0.131 | intergenic_variant |
| CP027619.1 | 1 | 1462137 | A/G | 0.082 | −0.66 | 2019 2020 | 0.099 | upstream_transcript_variant |
| CP027619.1 | 1 | 1497939 | G/C | 0.075 | −0.62 | 2019 2020 | 0.103 | intergenic_variant |
| CP027619.1 | 1 | 1528323 | C/G | 0.078 | −0.64 | 2019 2020 | 0.101 | missense_variant |
| CP027619.1 | 1 | 1722812 | C/G | 0.105 | −0.68 | 2019 2020 2021 | 0.131 | intron_variant |
| CP027619.1 | 1 | 1854337 | T/C | 0.082 | 0.56 | 2019 2020 | 0.101 | upstream_transcript_variant |
| CP027632.1 | 8 | 22560236 | G/A | 0.064 | −0.53 | 2019 2020 | 0.185 | upstream_transcript_variant |
| CP027632.1 | 8 | 22560290 | C/A | 0.077 | 0.50 | 2019 2020 2021 | 0.19 | synonymous_variant |
| CP027621.1 | 11 | 6013057 | A/G | 0.097 | −0.64 | 2019 2020 2021 | 0.144 | intron_variant |
| CP027623 | 13 | 545286 | A/G | 0.07 | 0.52 | 2020 2021 | 0.127 | intergenic_variant |
| CP027623 | 13 | 4884610 | T/G | 0.067 | 1.35 | 2020 2021 | 0.457 | intergenic_variant |
| QTN | Candidate Gene | Position | A. thaliana Orthologue | Protein |
|---|---|---|---|---|
| Chr11:288653 | Lus10025717 | Chr1:1285554-1289474 | AT2G22560 | KIP1-like protein |
| Chr1:1462137 | Lus10025756 | Chr1:1463118-1465114 | AT2G46950, CYP709B2 | Cytochrome P450, family 709, subfamily B, polypeptide 2 |
| Chr1:1528323 | Lus10025773 | Chr1:1527916-1531752 | AT1G53050 | Protein kinase superfamily protein |
| Chr1:1722812 | Lus10025823 | Chr1:1722251-1726433 | AT1G79750, NADP-ME4 | NADP-malic enzyme 4 |
| Chr1:1854337 | Lus10025852 | Chr1:1851791-1853596 | AT5G17890, CHS3, DAR4 | DA1-related protein 4, nucleotide-binding, leucine-rich repeat protein |
| Chr1:1854337 | Lus10025853 | Chr1:1854958-1870647 | AT5G17020, XPO1A | Exportin 1A |
| Chr8:22560236 | Lus10015356 | Chr8:22561110-22562338 | AT5G62740, HIR1 | SPFH/Band 7/PHB domain-containing, membrane-associated protein family protein |
| Chr8:22560236 | Lus10015357 | Chr8:22562785-22565427 | AT5G62740, VDAC1 | Voltage-dependent anion channel 1 |
| Chr8:22560236 | Lus10015344 | Chr8:22554684-22556269 | AT5G40010, AATP1 | AAA-ATPase 1 |
| Chr8:22560236 | Lus10015339 | Chr8:22569641-22570539 | AT3G01270 | Pectate lyase family protein |
| Chr11:6013057 | Lus10035917 | Chr11:6011724-6014460 | AT1G80230 | Rubredoxin-like superfamily protein |
| Chr13:4884610 | Lus10009330 | Chr13:4886276-4888681 | AT1G71400, RLP12 | Receptor-like protein 12, RLK |
| Chr13:4884610 | Lus10009332 | Chr13:4890865-4893541 | AT5G66880, SNRK2.3 | Sucrose nonfermenting 1(SNF1)-related protein kinase 2.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanapin, A.; Bankin, M.; Rozhmina, T.; Samsonova, A.; Samsonova, M. Genomic Regions Associated with Fusarium Wilt Resistance in Flax. Int. J. Mol. Sci. 2021, 22, 12383. https://doi.org/10.3390/ijms222212383
Kanapin A, Bankin M, Rozhmina T, Samsonova A, Samsonova M. Genomic Regions Associated with Fusarium Wilt Resistance in Flax. International Journal of Molecular Sciences. 2021; 22(22):12383. https://doi.org/10.3390/ijms222212383
Chicago/Turabian StyleKanapin, Alexander, Mikhail Bankin, Tatyana Rozhmina, Anastasia Samsonova, and Maria Samsonova. 2021. "Genomic Regions Associated with Fusarium Wilt Resistance in Flax" International Journal of Molecular Sciences 22, no. 22: 12383. https://doi.org/10.3390/ijms222212383
APA StyleKanapin, A., Bankin, M., Rozhmina, T., Samsonova, A., & Samsonova, M. (2021). Genomic Regions Associated with Fusarium Wilt Resistance in Flax. International Journal of Molecular Sciences, 22(22), 12383. https://doi.org/10.3390/ijms222212383