
Prediction of Peptide Detectability Based on CapsNet and Convolutional Block Attention Module


Abstract
:1. Introduction
2. Results
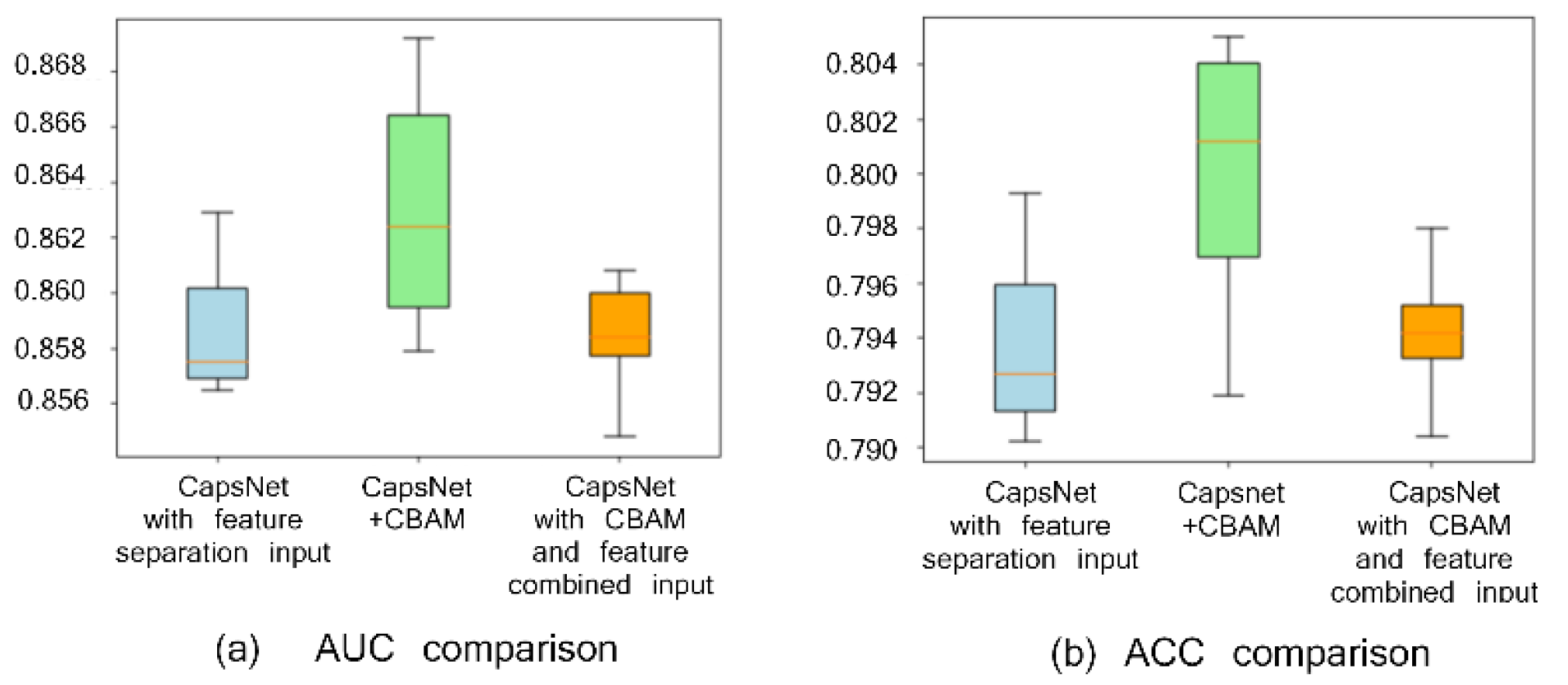
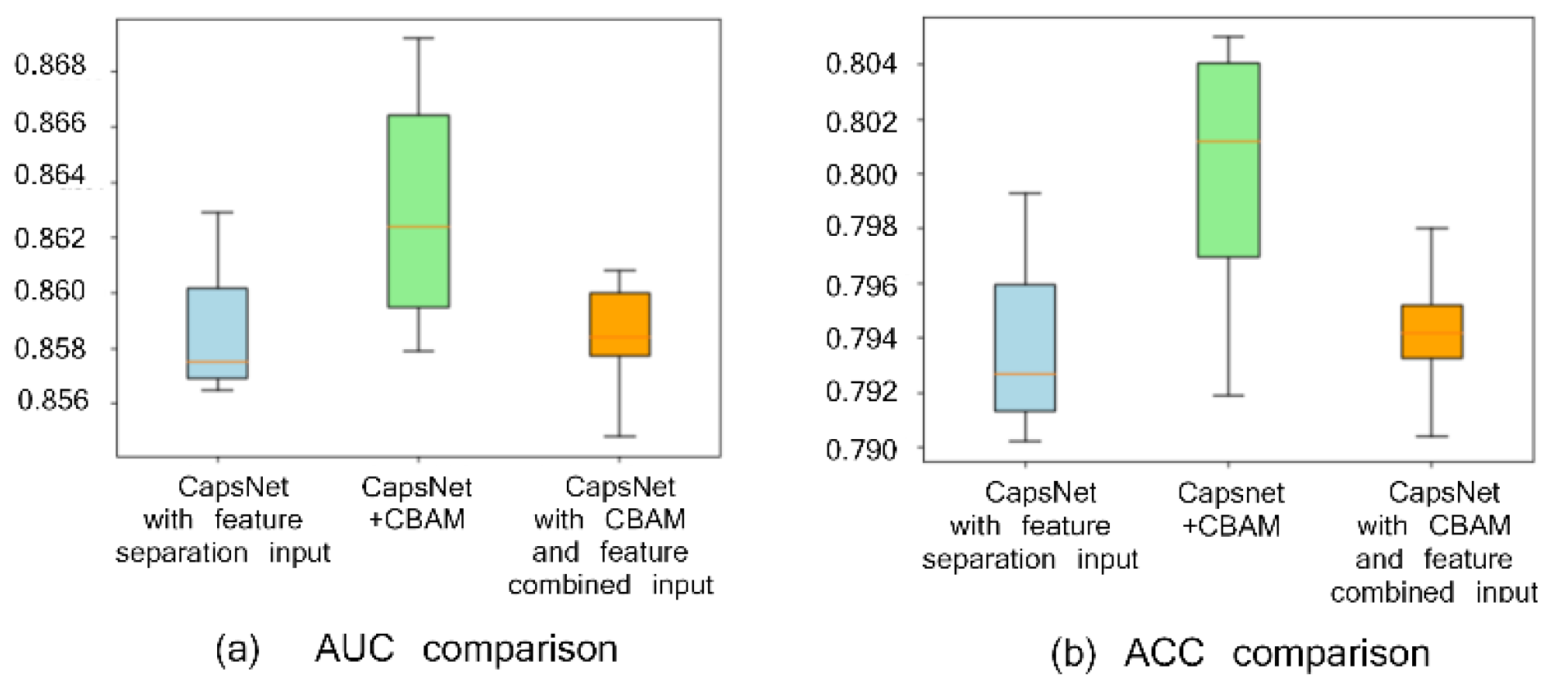
2.1. Comparison with or without CBAM Module and with or without Feature Separation Input
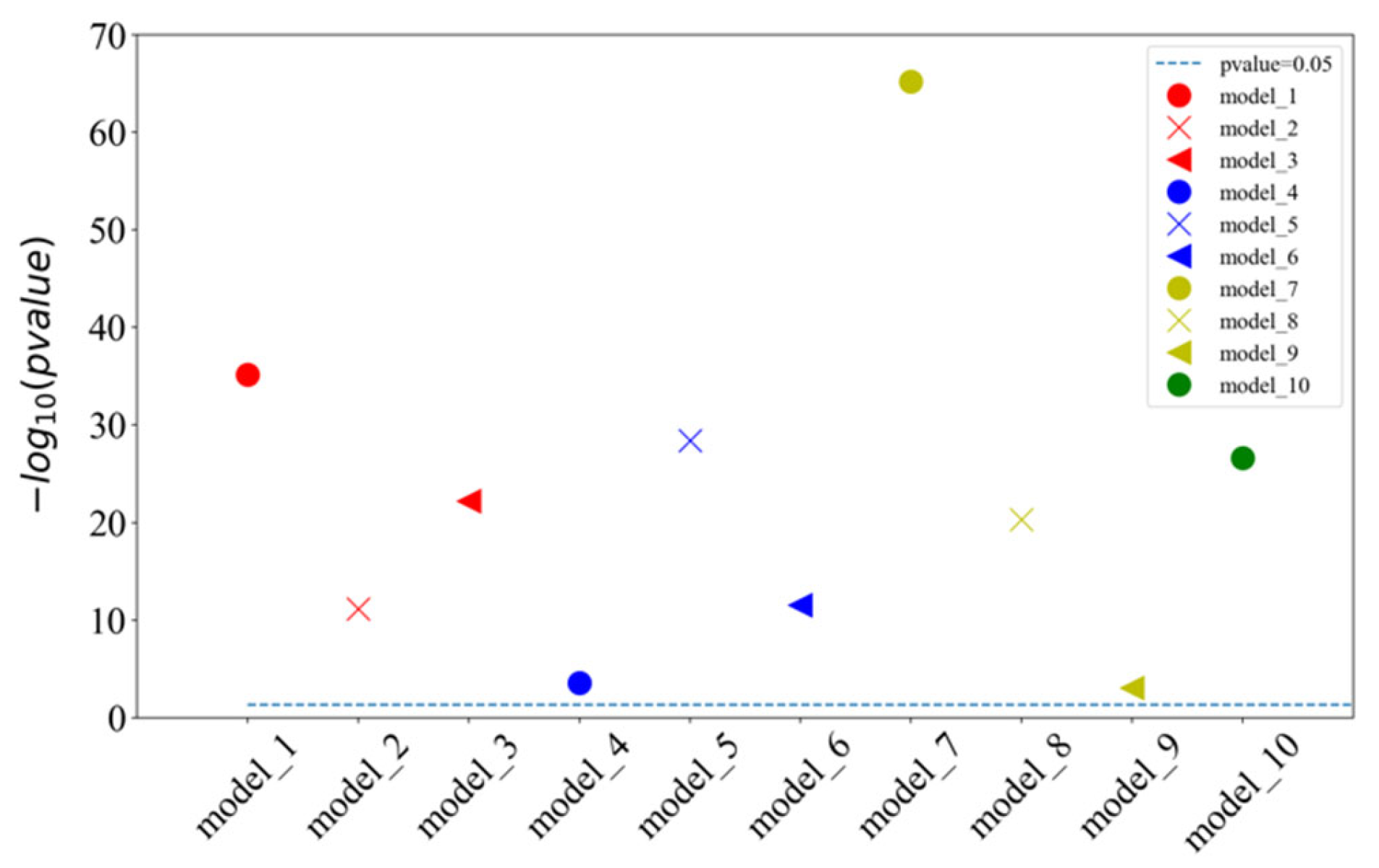
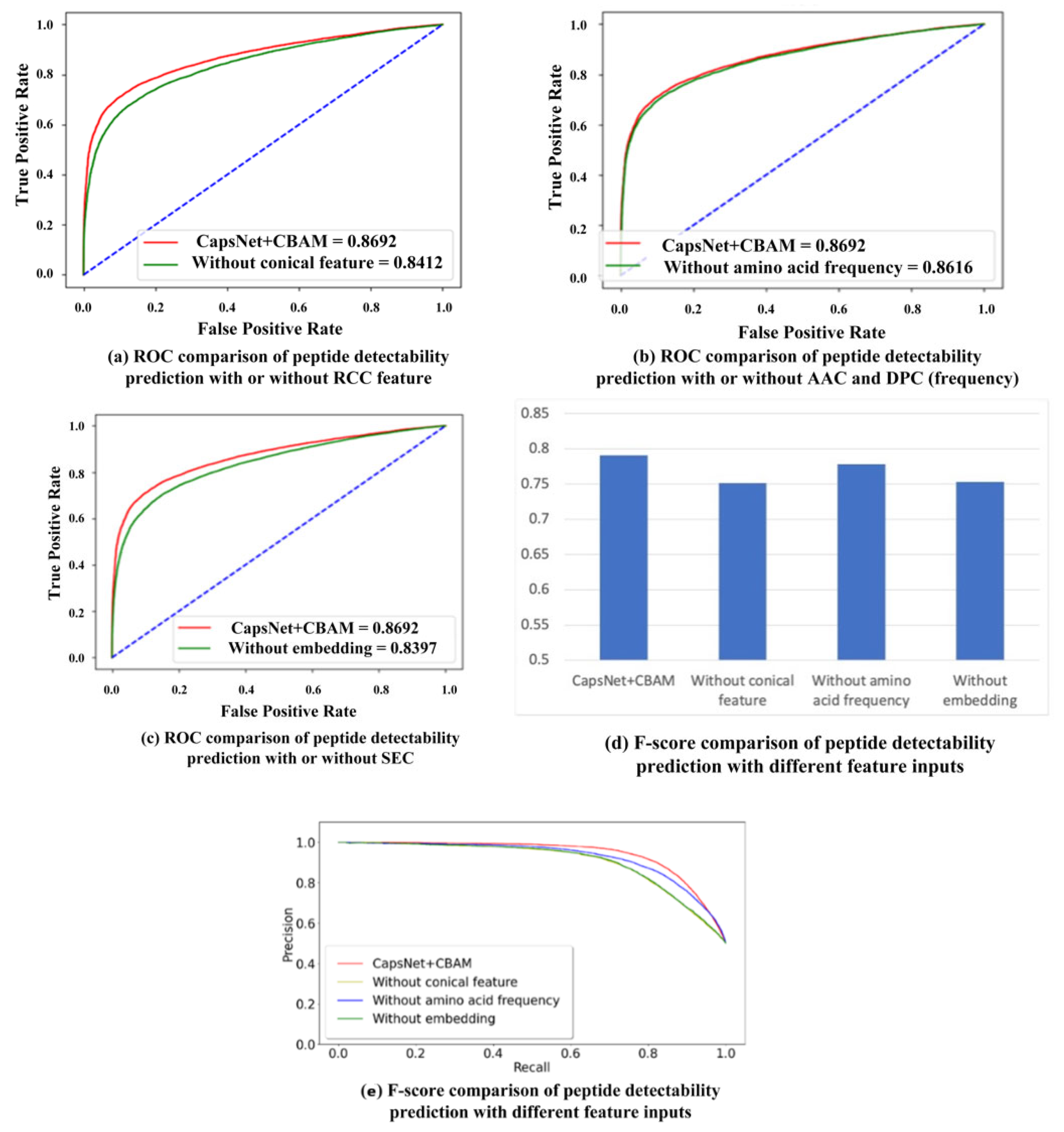
2.2. Comparison with Different Input Features
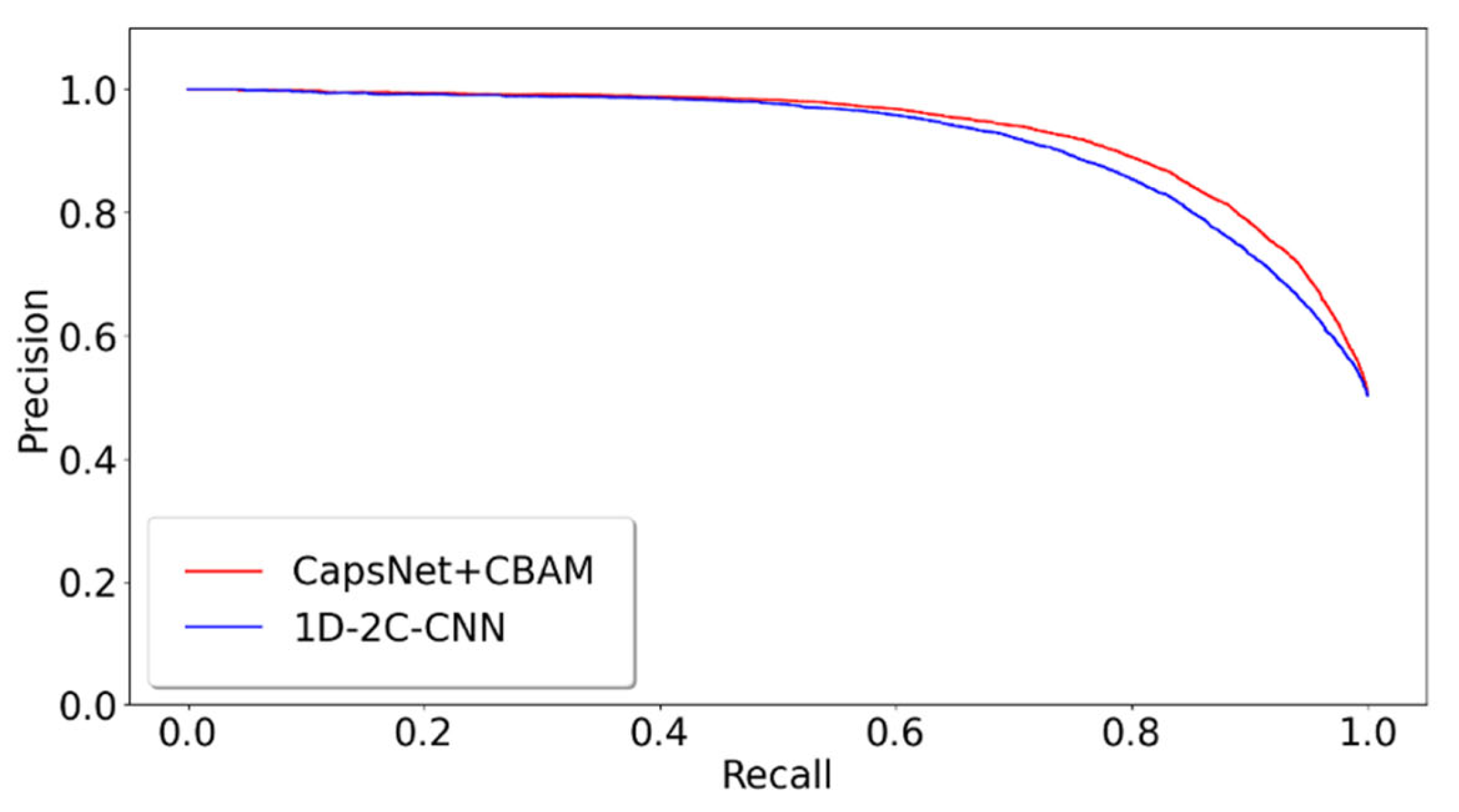
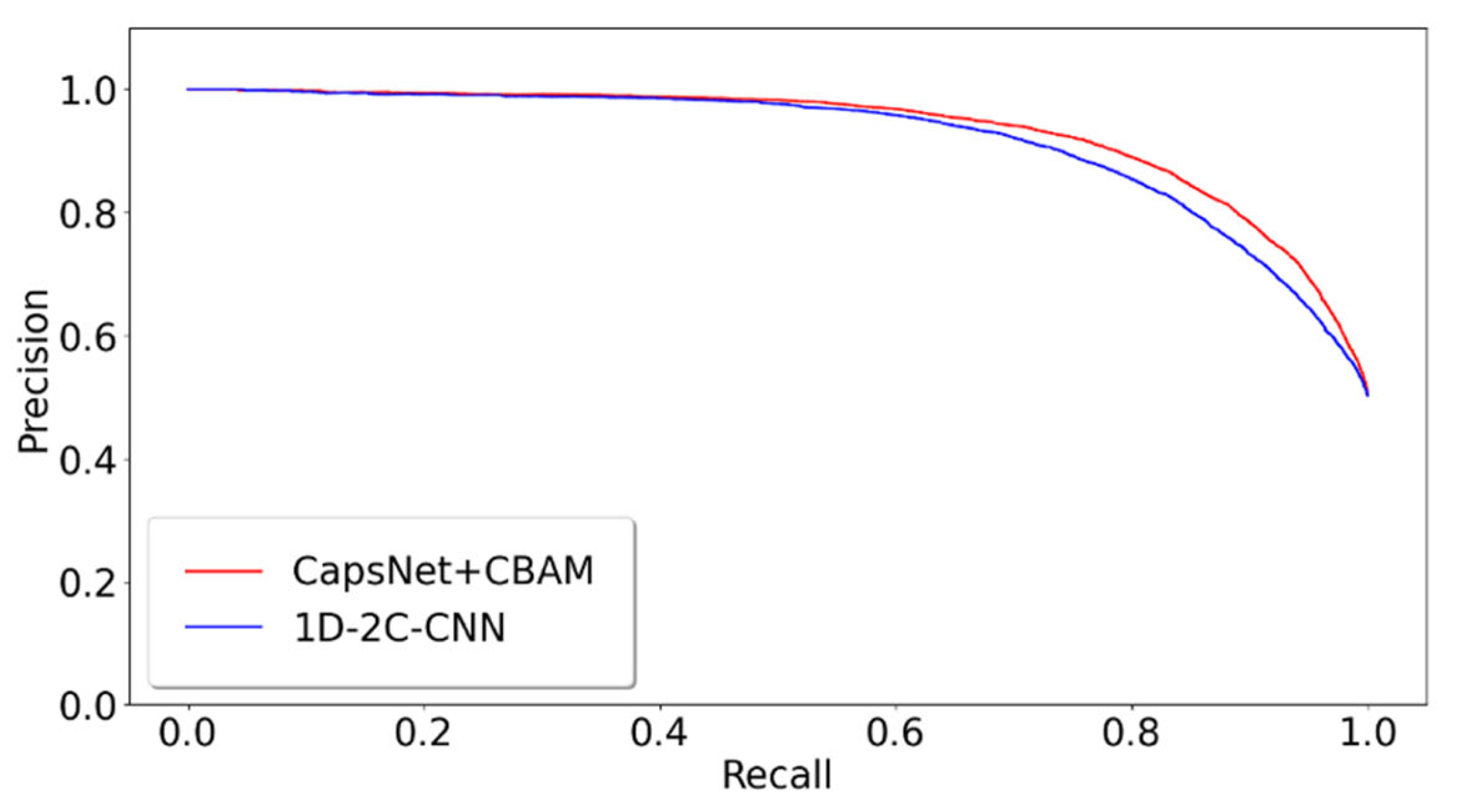
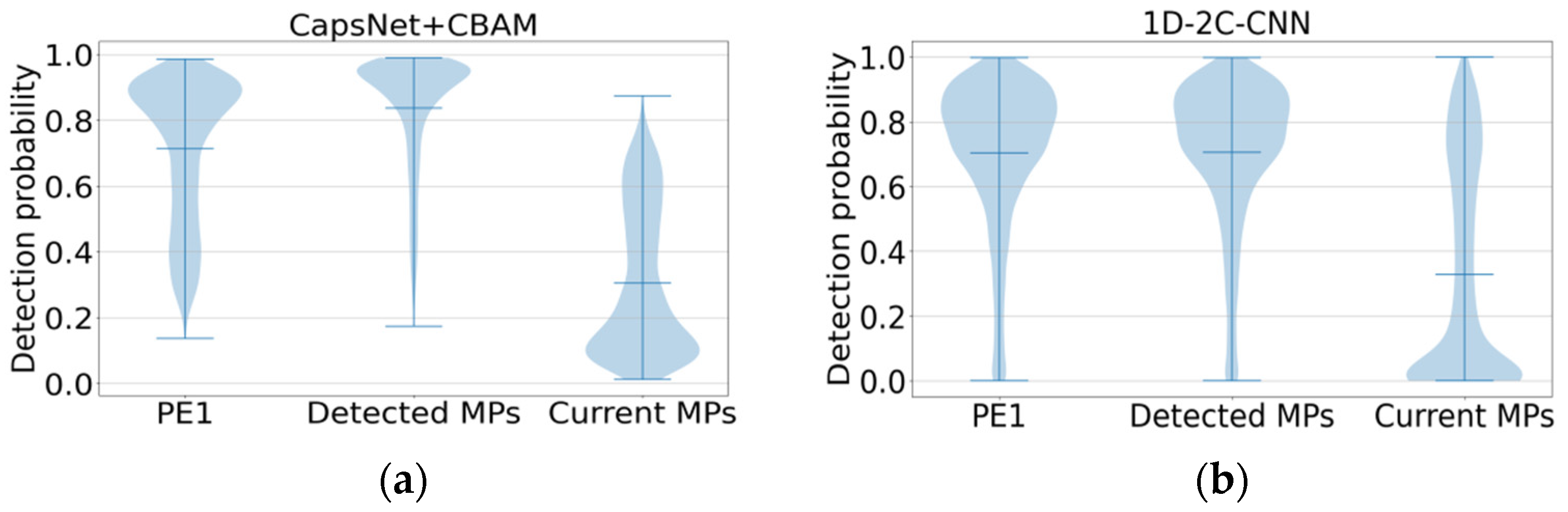
2.3. Different Prediction Method Comparison for the GPMDB Dataset Test
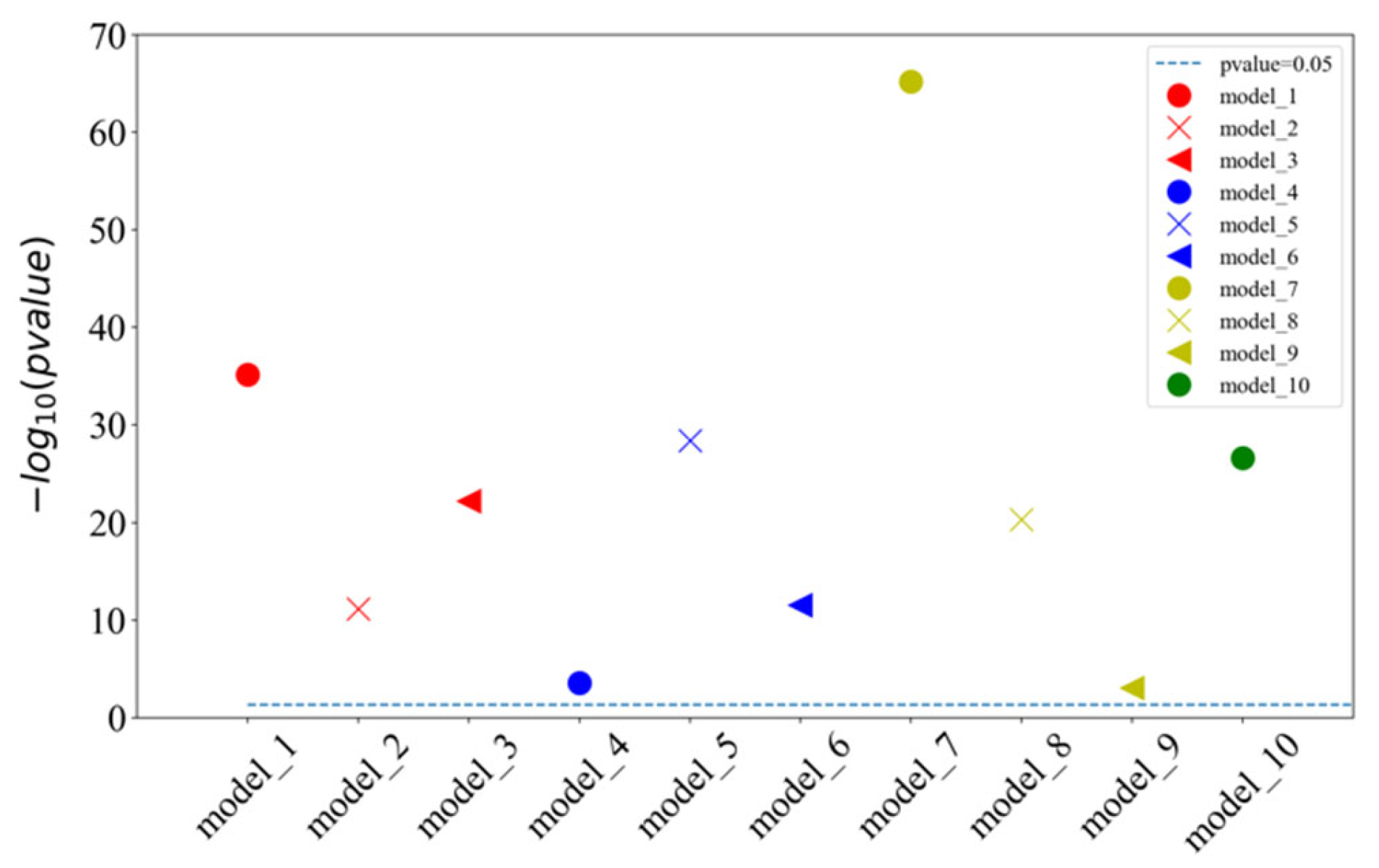
2.4. Additional Benchmarks for Testing
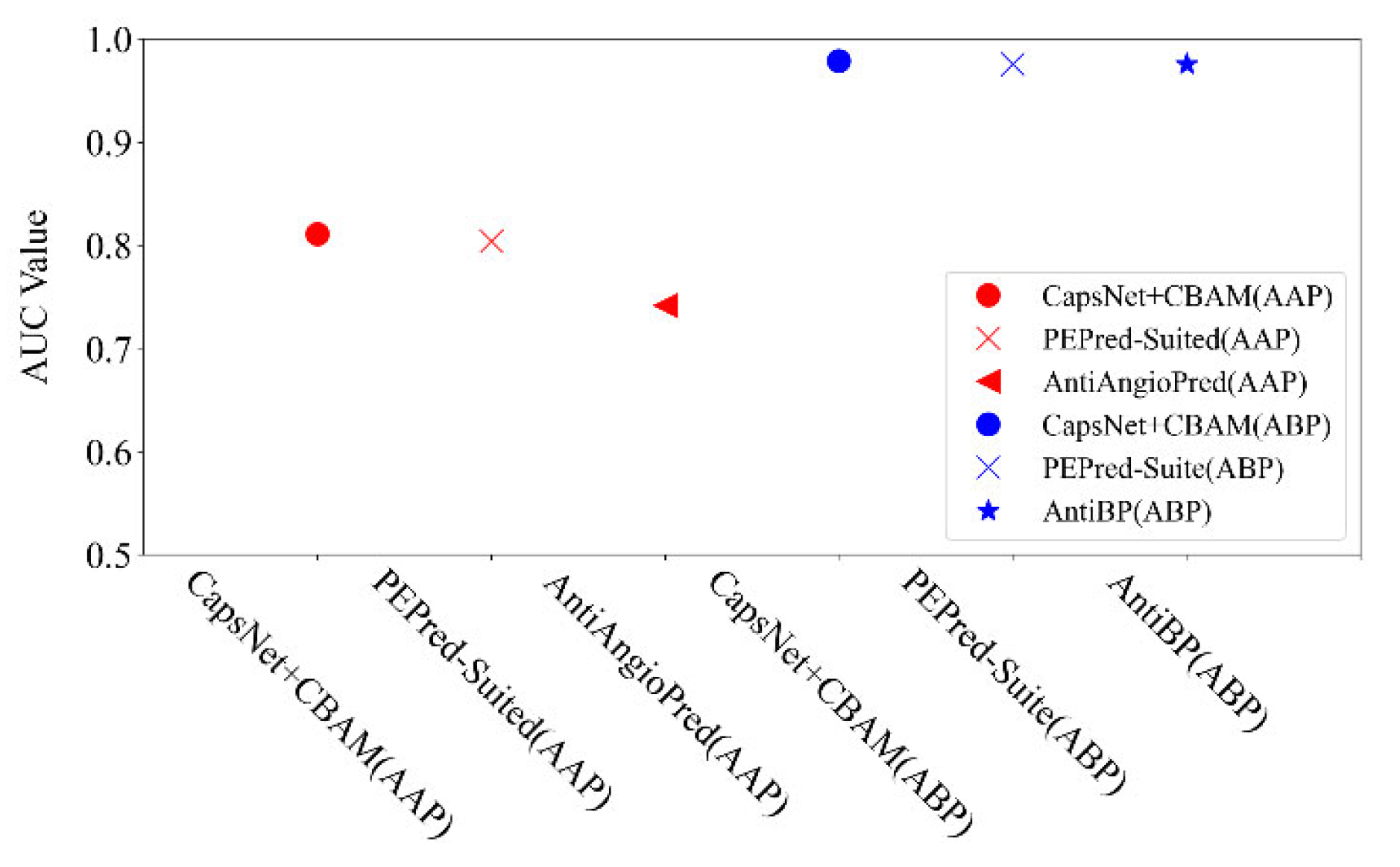
2.5. Additional Datasets for Testing
3. Discussion
4. Materials and Methods
4.1. Dataset
4.2. Feature Selection
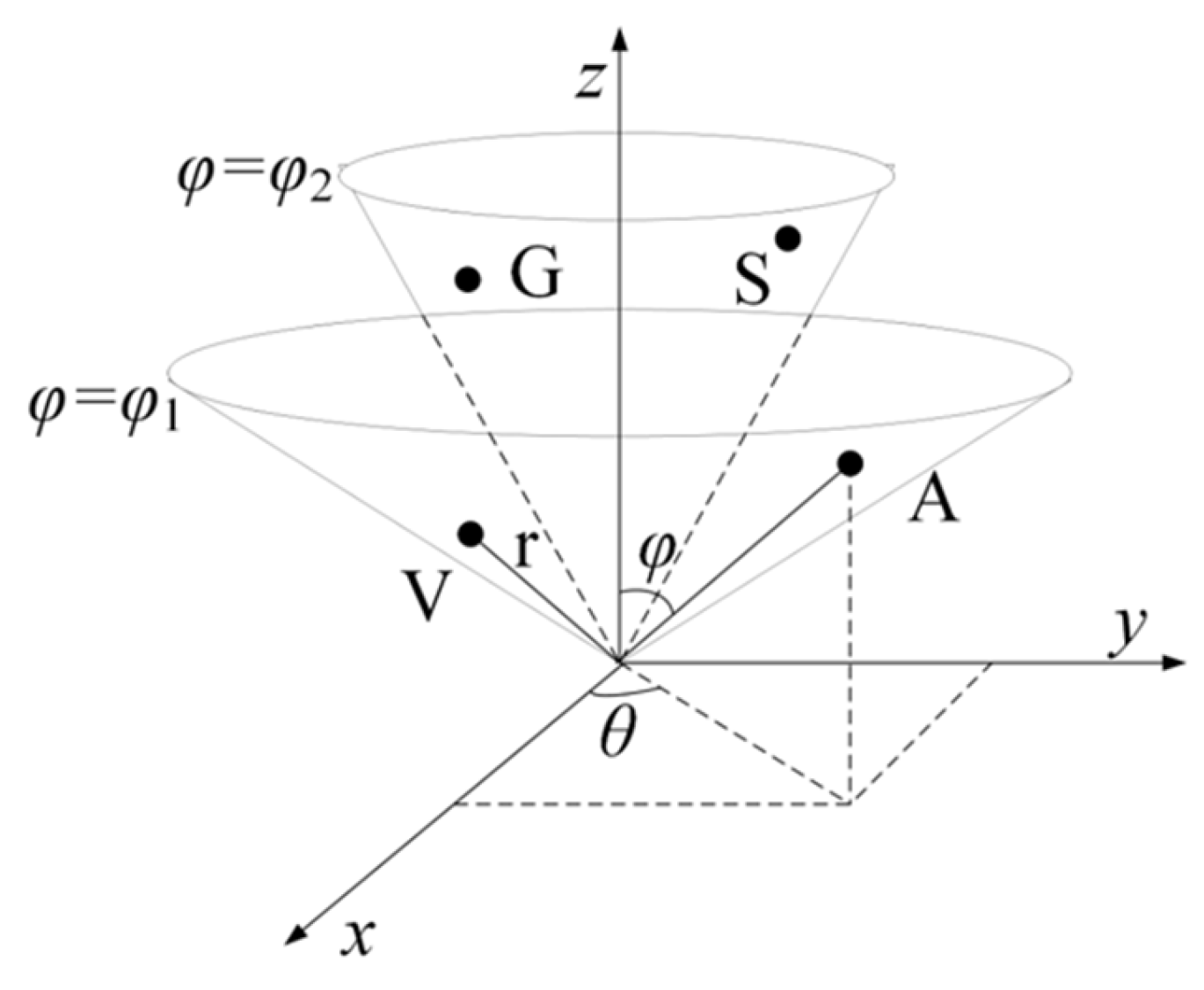
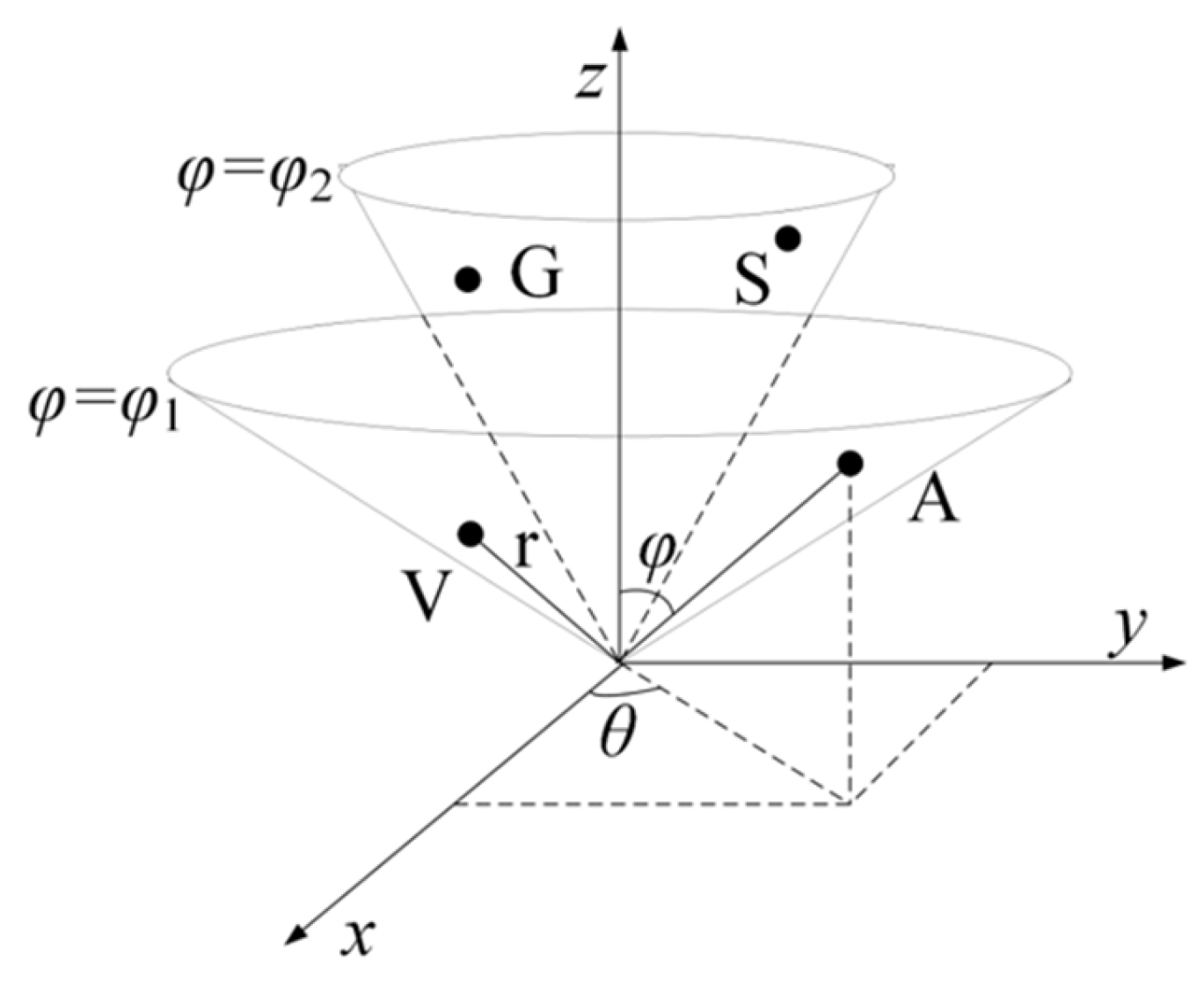
4.2.1. RCC Feature Based on Physicochemical Properties of Amino Acids
4.2.2. Amino Acid Composition and Dipeptide Composition
4.2.3. Neural Network Embedding
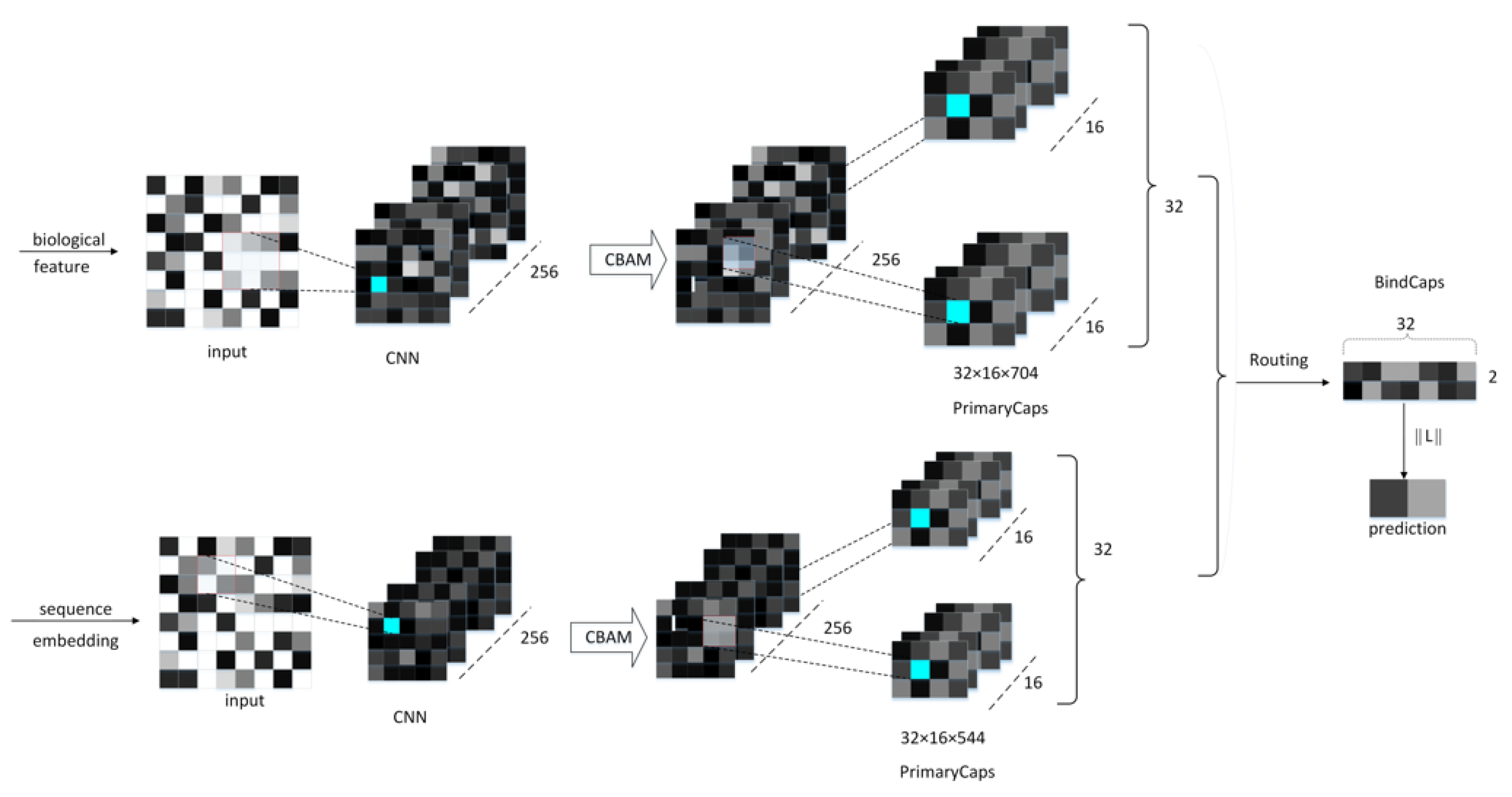
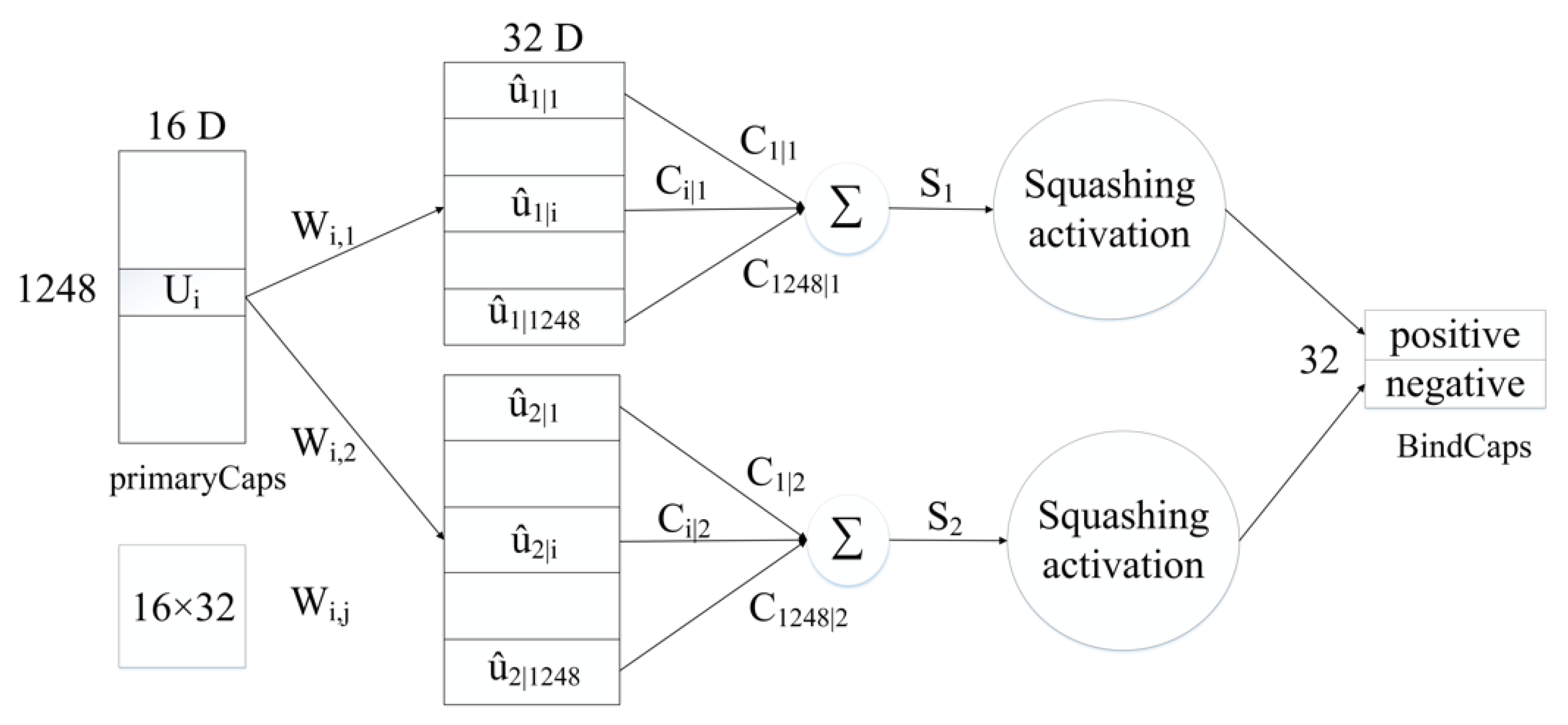
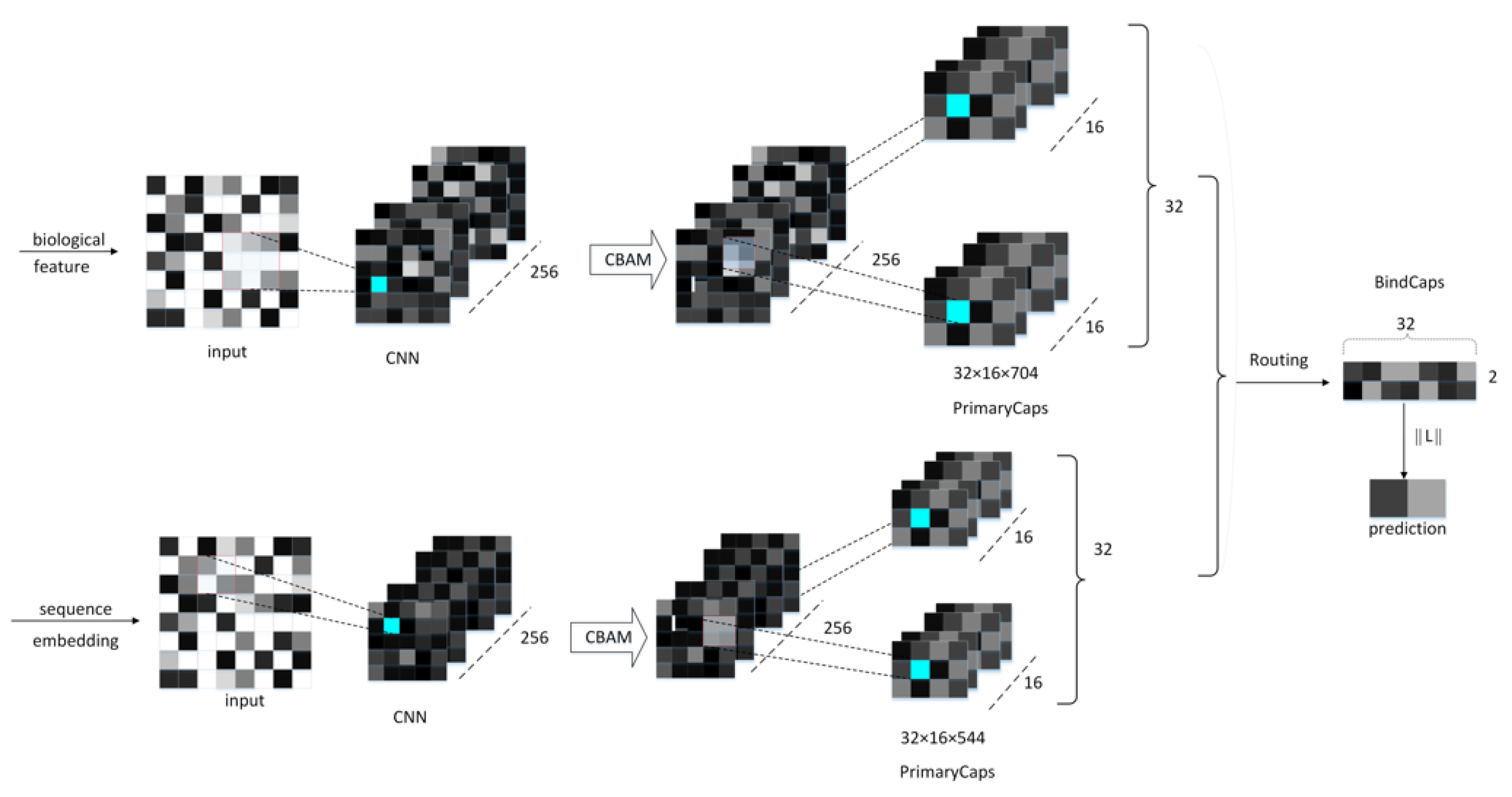
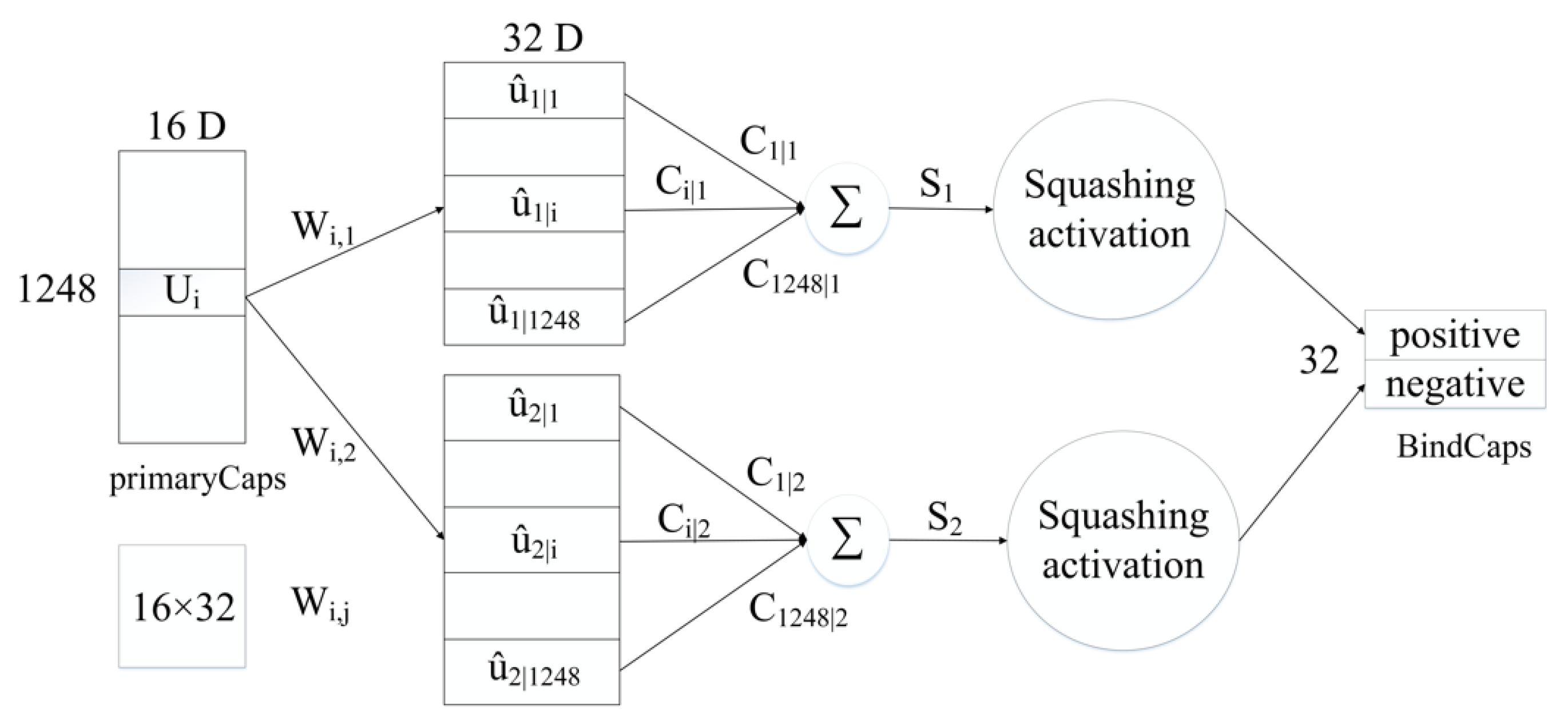
4.3. Neural Network Architecture
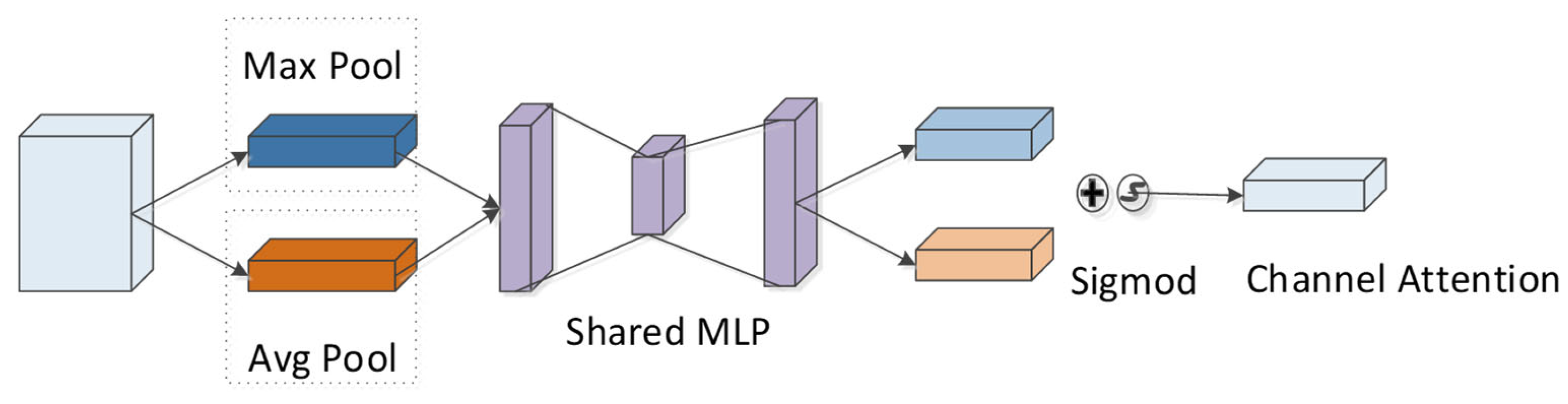
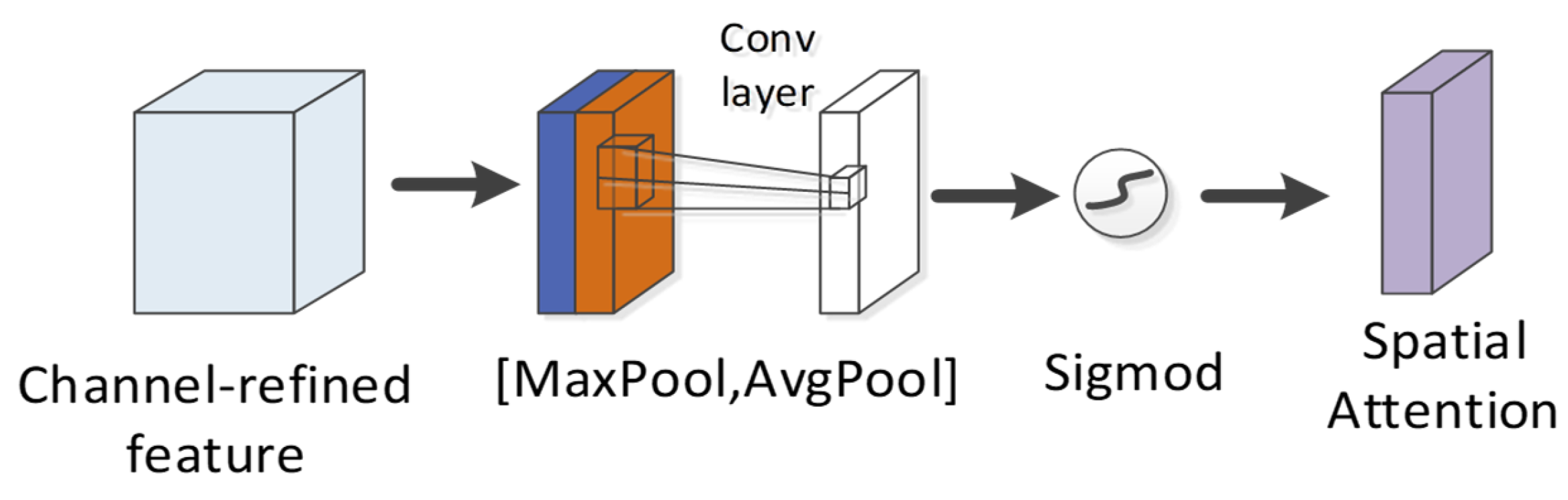
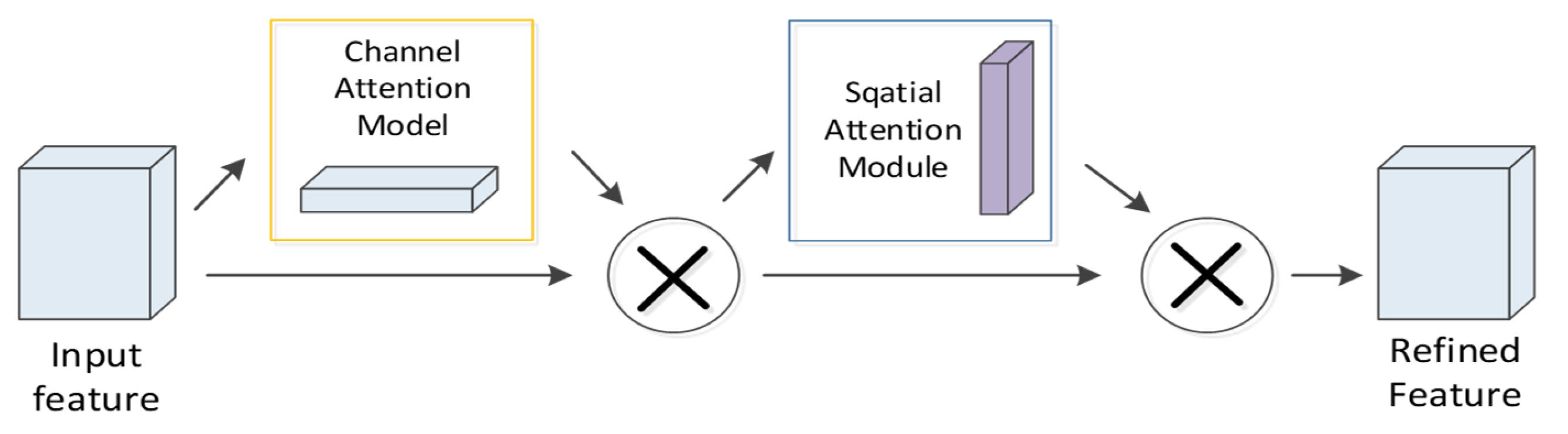
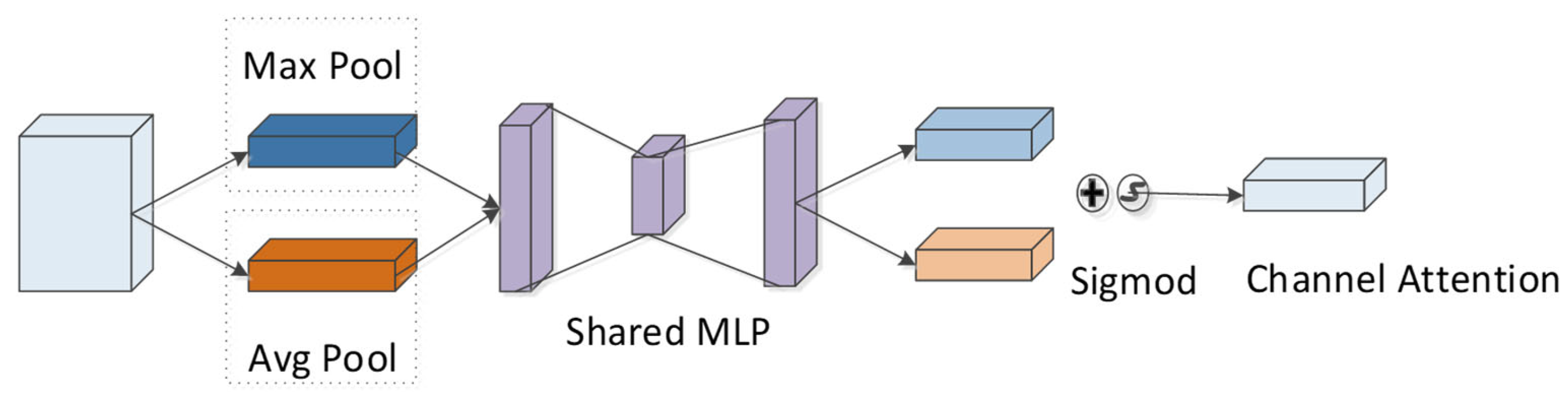
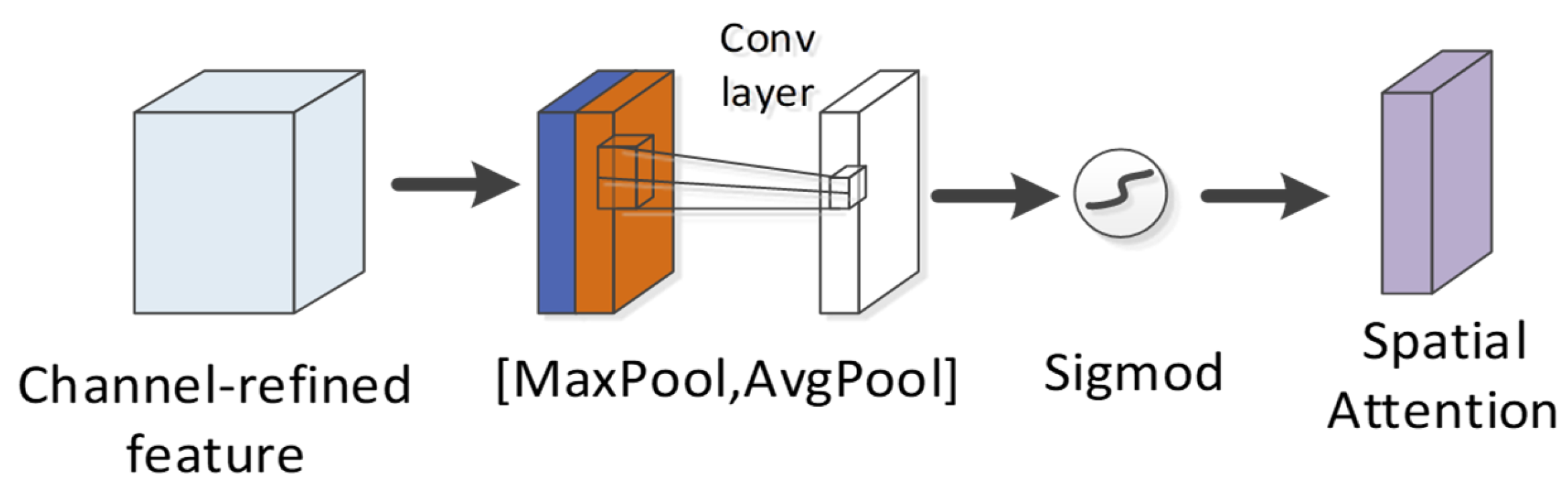
4.4. CBAM Module
4.5. Network Parameter Setting and Feature Transfer
4.6. Evaluation Index
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nilsson, T.; Mann, M.; Aebersold, R.; Yates, J.R.; Bairoch, A.; Bergeron, J. Mass spectrometry in high-throughput proteomics: Ready for the big time. Nat. Methods 2010, 7, 681–685. [Google Scholar] [CrossRef]
- Alves, P.; Arnold, R.J.; Novotny, M.V.; Radivojac, P.; Reilly, J.P.; Tang, H. Advancement in protein inference from shotgun proteomics using peptide detectability. Biocomputing 2007, 12, 409–420. [Google Scholar]
- Craig, R.; Cortens, J.P.; Beavis, R.C. Open source system for analyzing, validating, and storing protein identification data. J. Proteome Res. 2004, 3, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Jarnuczak, A.F.; Lee, D.; Lawless, C.; Holman, S.W.; Eyers, C.E.; Hubbard, S.J. Analysis of intrinsic peptide detectability via integrated label-free and srm-based absolute quantitative proteomics. J. Proteome Res. 2016, 15, 2945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.F.; Arnold, R.J.; Tang, H.; Radivojac, P. The importance of peptide detectability for protein identification, quantification, and experiment design in ms/ms proteomics. J. Proteome Res. 2010, 9, 6288–6297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.; Rao, B.; Liu, L.; Cui, L.; Xiao, G.; Su, R.; Wei, L. PepFormer: End-to-End Transformer-Based Siamese Network to Predict and Enhance Peptide Detectability Based on Sequence Only. Anal. Chem. 2021, 93, 6481–6490. [Google Scholar] [CrossRef]
- Le, N.Q.K.; Huynh, T.T. Identifying SNAREs by incorporating deep learning architecture and amino acid embedding representation. Front. Physiol. 2019, 10, 1501. [Google Scholar] [CrossRef] [PubMed]
- Le, N.Q.K.; Ho, Q.T.; Nguyen, T.T.D.; Ou, Y.Y. A transformer architecture based on BERT and 2D convolutional neural network to identify DNA enhancers from sequence information. Briefings Bioinf. 2021, 5, 5. [Google Scholar]
- Guruceaga, E.; Garin-Muga, A.; Prieto, G.; Bejarano, B.; Marcilla, M.; Marín-Vicente, C.; Segura, V. Enhanced missing proteins detection in nci60 cell lines using an integrative search engine approach. J. Proteome Res. 2017, 16, 4374–4390. [Google Scholar] [CrossRef]
- Shuichi, K.; Piotr, P.; Maria, P.; Andrzej, K.; Toshiaki, K.; Minoru, K. Aaindex: Amino acid index database, progress report 2008. Nucleic Acids Res. 2008, 36, D202–D205. [Google Scholar]
- Zimmer, D.; Schneider, K.; Sommer, F.; Schroda, M.; Mühlhaus, T. Artificial intelligence understands peptide observability and assists with absolute protein quantification. Front. Plant Sci. 2018, 9, 1559. [Google Scholar] [CrossRef]
- Wei, L.; Zhou, C.; Su, R.; Zou, Q. PEPred-Suite: Improved and robust prediction of therapeutic peptides using adaptive feature representation learning. Bioinformatics 2019, 35, 4272–4280. [Google Scholar] [CrossRef]
- Zhang, Y.P.; Zou, Q. PPTPP: A novel therapeutic peptide prediction method using physicochemical property encoding and adaptive feature representation learning. Bioinformatics 2020, 36, 3982–3987. [Google Scholar] [CrossRef] [PubMed]
- Hinton, G.E.; Salakhutdinov, R.R. Reducing the dimensionality of data with neural networks. Science 2006, 313, 504–507. [Google Scholar] [CrossRef] [Green Version]
- Serrano, G.; Guruceaga, E.; Segura, V. DeepMSPeptide: Peptide detectability prediction using deep learning. Bioinformatics 2020, 36, 1279–1280. [Google Scholar] [CrossRef]
- Zhang, D.; Xu, Z.C.; Su, W.; Yang, Y.H.; Lv, H.; Yang, H.; Lin, H. iCarPS: A computational tool for identifying protein carbonylation sites by novel encoded features. Bioinformatics 2021, 37, 171–177. [Google Scholar] [CrossRef]
- Sabour, S.; Frosst, N.; Hinton, G.E. Dynamic routing between capsules. arXiv 2017, arXiv:1710.09829. [Google Scholar]
- Woo, S.; Park, J.; Lee, J.Y.; Kweon, I.S. Cbam: Convolutional block attention module. In Proceedings of the European Conference on Computer Vision (ECCV), Munich, Germany, 8–14 September 2018; pp. 3–19. [Google Scholar]
- Gaudet, P.; Michel, P.A.; Zahn-Zabal, M.; Britan, A.; Cusin, I.; Domagalski, M.; Bairoch, A. The neXtProt knowledgebase on human proteins: 2017 update. Nucleic Acids Res. 2017, 45, D177–D182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramaprasad, A.S.; Singh, S.; Gajendra, P.S.R.; Venkatesan, S. AntiAngioPred: A server for prediction of anti-angiogenic peptides. PLoS ONE 2015, 10, e0136990. [Google Scholar]
- Lata, S.; Sharma, B.K.; Raghava, G.P. Analysis and prediction of antibacterial peptides. BMC Bioinf. 2007, 8, 263. [Google Scholar] [CrossRef] [Green Version]
- Manavalan, B.; Subramaniyam, S.; Shin, T.H.; Kim, M.O.; Lee, G. Machine-learning-based prediction of cell-penetrating peptides and their uptake efficiency with improved accuracy. J. Proteome Res. 2018, 17, 2715–2726. [Google Scholar] [CrossRef]
- Gautam, A.; Chaudhary, K.; Kumar, R.; Sharma, A.; Kapoor, P.; Tyagi, A.; Raghava, G.P. In silico approaches for designing highly effective cell penetrating peptides. J. Transl. Med. 2013, 11, 74. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, P.; Bautista, M.; Gonzalez, J.; Escalera, S. Beyond one-hot encoding: Lower dimensional target embedding. Image Vision Comput. 2018, 75, 21–31. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Shen, L.; Sun, G. Squeeze-and-excitation networks. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Salt Lake City, UT, USA, 18–22 June 2018; pp. 7132–7141. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Le, N.Q.K.; Do, D.T.; Hung, T.N.K.; Lam, L.H.T.; Huynh, T.T.; Nguyen, N.T.K. A computational framework based on ensemble deep neural networks for essential genes identification. Int. J. Mol. Sci. 2020, 21, 9070. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | AUC | Accuracy | Specificity | Sensitivity | F-Score |
|---|---|---|---|---|---|
| CapsNet + CBAM | |||||
| Without conical feature | |||||
| Without amino acid frequency | |||||
| Without embedding |
| Model | AUC | Accuracy | Specificity | Sensitivity | F-Score |
|---|---|---|---|---|---|
| CapsNet + CBAM | |||||
| 1D-2C-CNN [15] | 0.8570 | 0.7953 | 0.8880 | 0.7027 | 0.7744 |
| RF [9] | 0.7549 | 0.6924 | 0.7746 | 0.6103 | 0.6649 |
| SvmR [9] | 0.7384 | 0.6813 | 0.7830 | 0.5797 | 0.6453 |
| DNN [11] | 0.7360 | 0.6692 | 0.6813 | 0.6572 | 0.6659 |
| C5 [9] | 0.7312 | 0.6644 | 0.6513 | 0.6775 | 0.6687 |
| Pls [9] | 0.6350 | 0.6043 | 0.6396 | 0.5690 | 0.5898 |
| Glm [9] | 0.6349 | 0.6036 | 0.6426 | 0.5646 | 0.5875 |
| Gaussian [16] | 0.6342 | 0.5983 | 0.6121 | 0.5845 | 0.5927 |
| Groups | Description | Amino Acids |
|---|---|---|
| Class Ⅰ | Non-polar residues | A, V, L, I, P, F, W, M |
| Class Ⅱ | Polar residues | G, S, T, C, Y, N, Q |
| Class Ⅲ | Basic residues | K, R, H |
| Class Ⅳ | Acidic residues | D, E |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, M.; Duan, Y.; Li, Z.; Zhang, Y. Prediction of Peptide Detectability Based on CapsNet and Convolutional Block Attention Module. Int. J. Mol. Sci. 2021, 22, 12080. https://doi.org/10.3390/ijms222112080
Yu M, Duan Y, Li Z, Zhang Y. Prediction of Peptide Detectability Based on CapsNet and Convolutional Block Attention Module. International Journal of Molecular Sciences. 2021; 22(21):12080. https://doi.org/10.3390/ijms222112080
Chicago/Turabian StyleYu, Minzhe, Yushuai Duan, Zhong Li, and Yang Zhang. 2021. "Prediction of Peptide Detectability Based on CapsNet and Convolutional Block Attention Module" International Journal of Molecular Sciences 22, no. 21: 12080. https://doi.org/10.3390/ijms222112080
APA StyleYu, M., Duan, Y., Li, Z., & Zhang, Y. (2021). Prediction of Peptide Detectability Based on CapsNet and Convolutional Block Attention Module. International Journal of Molecular Sciences, 22(21), 12080. https://doi.org/10.3390/ijms222112080