
Conditioned Media of Adipose-Derived Stem Cells Suppresses Sidestream Cigarette Smoke Extract Induced Cell Death and Epithelial-Mesenchymal Transition in Lung Epithelial Cells

 ,
, 
Abstract
1. Introduction
2. Results
2.1. CSE Induces Lung Epithelial Cell Death in Concentration- and Serum-Dependent Manners
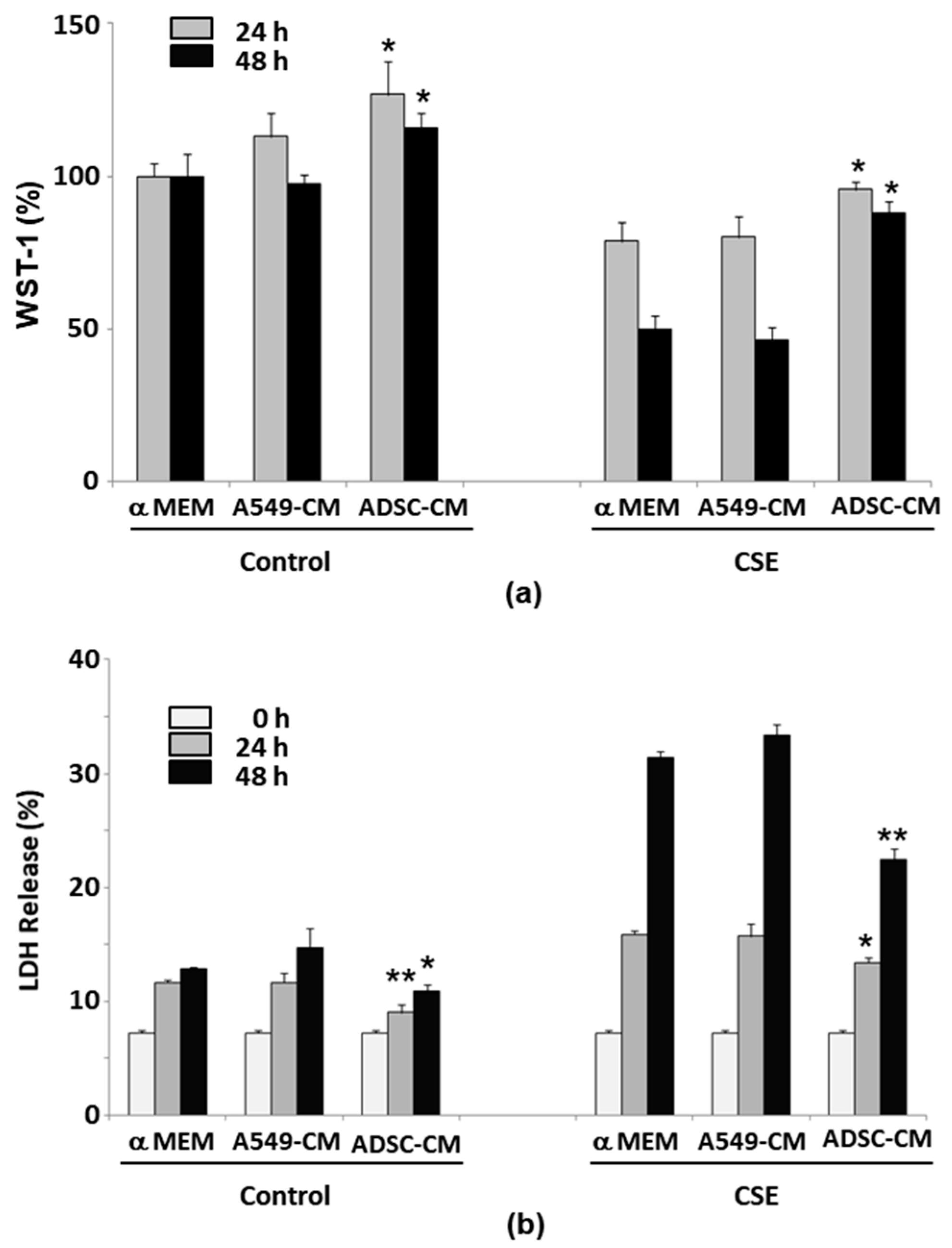
2.2. ADSC-Conditioned Medium Protected Cells from CSE-Induced Epithelial Cell Death
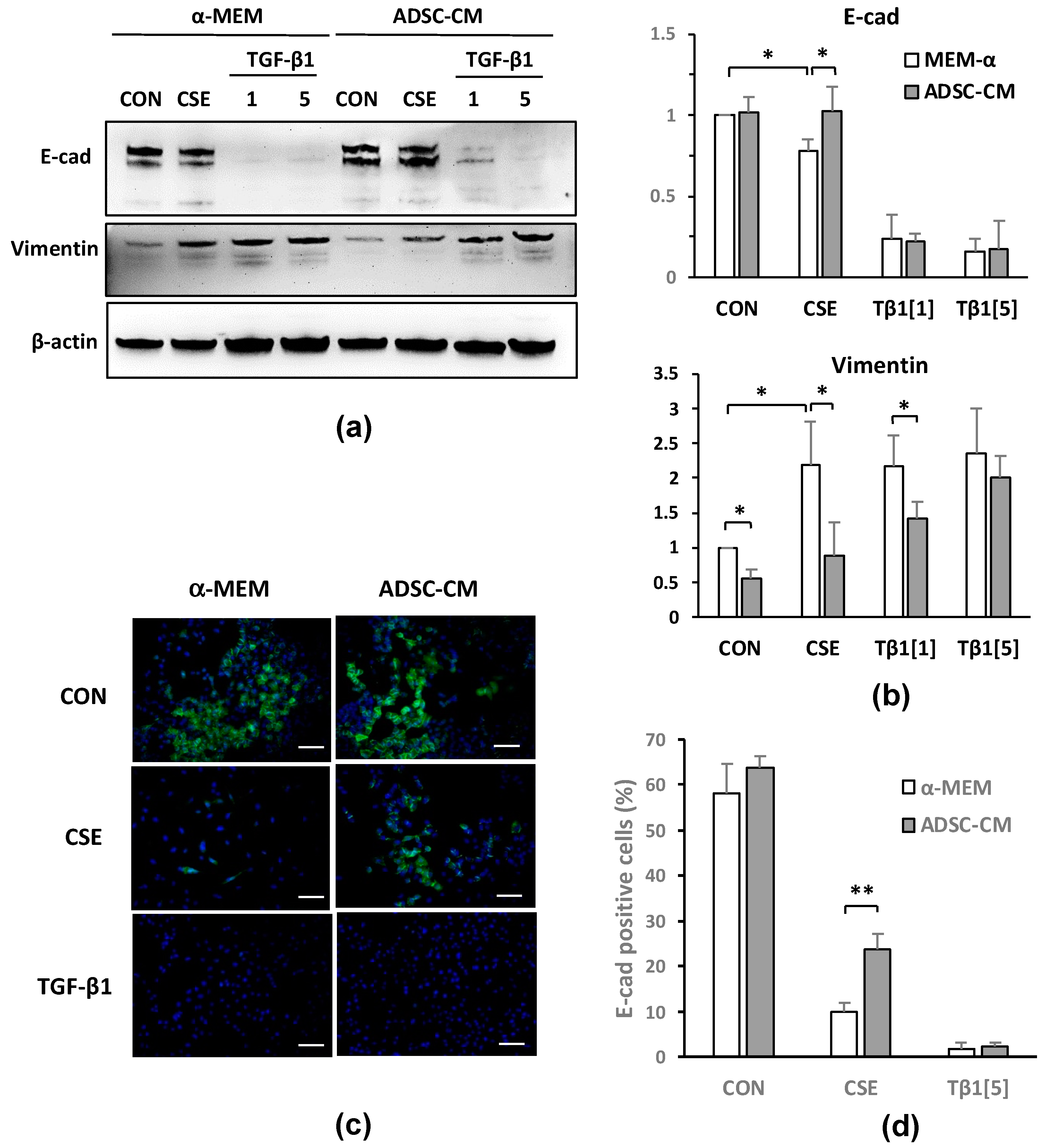
2.3. ADSC-CM Reduces CSE-Induced EMT in A549 Cells
2.4. Gene Expression Profiles of A549 Cells Responding to CSE or TGF-β1
2.5. ADSC-CM Inhibits Both CSE and TGF-β1 Induced Epithelial Cell Migration
3. Discussion
4. Materials and Methods
4.1. Chemicals and Culture Medium
4.2. Preparation of ADSC-Conditioned Medium
4.3. Cell Injury Induced by Cigarette Smoke Extract Exposure
4.4. Gene Expression Analysis
4.5. Immunoblotting and Fluorescence Detection
4.6. Cell Migration Assay
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TGF | Transforming Growth Factor |
| TNF | Tumor Necrosis Factor |
| ADSC-CM | Adipose Derived Stem Cell-Conditioned Medium |
| CSE | Cigarette Smoke Extract |
| COPD | Chronic Obstructive Pulmonary Disease |
| URA | Upstream Regulator Analysis |
| EMT | Epithelial-to-Mesenchymal Transition |
| LDH | Lactate Dehydrogenase |
| MSC | Mesenchymal Stem Cell |
References
- Mannino, D.M.; Buist, A.S. Global burden of COPD: Risk factors, prevalence, and future trends. Lancet 2007, 370, 765–773. [Google Scholar] [CrossRef]
- Adcock, I.M.; Caramori, G.; Barnes, P.J. Chronic obstructive pulmonary disease and lung cancer: New molecular insights. Respir. Int. Rev. Thorac. Dis. 2011, 81, 265–284. [Google Scholar] [CrossRef]
- Hartman, T.E.; Tazelaar, H.D.; Swensen, S.J.; Muller, N.L. Cigarette smoking: CT and pathologic findings of associated pulmonary diseases. Radiogr. Rev. Publ. Radiol. Soc. North Am. Inc 1997, 17, 377–390. [Google Scholar] [CrossRef]
- Burns, D.M. Cigarettes and cigarette smoking. Clin. Chest Med. 1991, 12, 631–642. [Google Scholar] [CrossRef]
- Church, D.F.; Pryor, W.A. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ. Health Perspect. 1985, 64, 111–126. [Google Scholar] [CrossRef]
- Chow, C.K. Cigarette smoking and oxidative damage in the lung. Ann. N. Y. Acad. Sci. 1993, 686, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Deliconstantinos, G.; Villiotou, V.; Stavrides, J.C. Scavenging effects of hemoglobin and related heme containing compounds on nitric oxide, reactive oxidants and carcinogenic volatile nitrosocompounds of cigarette smoke. A new method for protection against the dangerous cigarette constituents. Anticancer Res. 1994, 14, 2717–2726. [Google Scholar] [PubMed]
- Gardi, C.; Valacchi, G. Cigarette smoke and ozone effect on murine inflammatory responses. Ann. N. Y. Acad. Sci. 2012, 1259, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Rom, O.; Avezov, K.; Aizenbud, D.; Reznick, A.Z. Cigarette smoking and inflammation revisited. Respir. Physiol. Neurobiol. 2013, 187, 5–10. [Google Scholar] [CrossRef]
- Hoshino, Y.; Mio, T.; Nagai, S.; Miki, H.; Ito, I.; Izumi, T. Cytotoxic effects of cigarette smoke extract on an alveolar type II cell-derived cell line. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 281, L509–L516. [Google Scholar] [CrossRef] [PubMed]
- Walser, T.; Cui, X.; Yanagawa, J.; Lee, J.M.; Heinrich, E.; Lee, G.; Sharma, S.; Dubinett, S.M. Smoking and lung cancer: The role of inflammation. Proc. Am. Thorac. Soc. 2008, 5, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Eapen, M.S.; Sharma, P.; Gaikwad, A.V.; Lu, W.; Myers, S.; Hansbro, P.M.; Sohal, S.S. Epithelial-mesenchymal transition is driven by transcriptional and post transcriptional modulations in COPD: Implications for disease progression and new therapeutics. Int. J. Chronic Obstr. Pulm. Dis. 2019, 14, 1603–1610. [Google Scholar] [CrossRef] [PubMed]
- Zahedi, A.; Phandthong, R.; Chaili, A.; Remark, G.; Talbot, P. Epithelial-to-mesenchymal transition of A549 lung cancer cells exposed to electronic cigarettes. Lung Cancer 2018, 122, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Agraval, H.; Yadav, U.C.S. MMP-2 and MMP-9 mediate cigarette smoke extract-induced epithelial-mesenchymal transition in airway epithelial cells via EGFR/Akt/GSK3β/β-catenin pathway: Amelioration by fisetin. Chem. Biol. Interact. 2019, 314, 108846. [Google Scholar] [CrossRef]
- New, M.L.; White, C.M.; McGonigle, P.; McArthur, D.G.; Dwyer-Nield, L.D.; Merrick, D.T.; Keith, R.L.; Tennis, M.A. Prostacyclin and EMT Pathway Markers for Monitoring Response to Lung Cancer Chemoprevention. Cancer Prev. Res. 2018, 11, 643–654. [Google Scholar] [CrossRef]
- Milara, J.; Peiró, T.; Serrano, A.; Cortijo, J. Epithelial to mesenchymal transition is increased in patients with COPD and induced by cigarette smoke. Thorax 2013, 68, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, S.; Sajjan, U.S. Repair and Remodeling of airway epithelium after injury in Chronic Obstructive Pulmonary Disease. Curr. Respir. Care Rep. 2013, 2, 145–154. [Google Scholar] [CrossRef]
- Liu, M.; Zhou, C.; Zheng, J. Cigarette smoking impairs the response of EGFR-TKIs therapy in lung adenocarcinoma patients by promoting EGFR signaling and epithelial-mesenchymal transition. Am. J. Transl. Res. 2015, 7, 2026–2035. [Google Scholar]
- Zou, Y.; Li, S.; Zou, W.; Hu, G.; Zhou, Y.; Peng, G.; He, F.; Li, B.; Ran, P. Upregulation of gelatinases and epithelial-mesenchymal transition in small airway remodeling associated with chronic exposure to wood smoke. PLoS ONE 2014, 9, e96708. [Google Scholar] [CrossRef]
- Milara, J.; Peiró, T.; Serrano, A.; Guijarro, R.; Zaragozá, C.; Tenor, H.; Cortijo, J. Roflumilast N-oxide inhibits bronchial epithelial to mesenchymal transition induced by cigarette smoke in smokers with COPD. Pulm. Pharmacol. Ther. 2014, 28, 138–148. [Google Scholar] [CrossRef]
- Zhao, Y.; Xu, Y.; Li, Y.; Xu, W.; Luo, F.; Wang, B.; Pang, Y.; Xiang, Q.; Zhou, J.; Wang, X.; et al. NF-κB-mediated inflammation leading to EMT via miR-200c is involved in cell transformation induced by cigarette smoke extract. Toxicol. Sci. Off. J. Soc. Toxicol. 2013, 135, 265–276. [Google Scholar] [CrossRef]
- Zhang, L.; Gallup, M.; Zlock, L.; Basbaum, C.; Finkbeiner, W.E.; McNamara, N.A. Cigarette smoke disrupts the integrity of airway adherens junctions through the aberrant interaction of p120-catenin with the cytoplasmic tail of MUC1. J. Pathol. 2013, 229, 74–86. [Google Scholar] [CrossRef]
- Shen, H.J.; Sun, Y.H.; Zhang, S.J.; Jiang, J.X.; Dong, X.W.; Jia, Y.L.; Shen, J.; Guan, Y.; Zhang, L.H.; Li, F.F.; et al. Cigarette smoke-induced alveolar epithelial-mesenchymal transition is mediated by Rac1 activation. Biochim. Biophys. Acta 2014, 1840, 1838–1849. [Google Scholar] [CrossRef] [PubMed]
- Eurlings, I.M.; Reynaert, N.L.; van den Beucken, T.; Gosker, H.R.; de Theije, C.C.; Verhamme, F.M.; Bracke, K.R.; Wouters, E.F.; Dentener, M.A. Cigarette smoke extract induces a phenotypic shift in epithelial cells; involvement of HIF1α in mesenchymal transition. PLoS ONE 2014, 9, e107757. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, H.; Borok, Z.; Davies, K.J.; Ursini, F.; Forman, H.J. Cigarette smoke extract stimulates epithelial-mesenchymal transition through Src activation. Free Radic. Biol. Med. 2012, 52, 1437–1442. [Google Scholar] [CrossRef]
- Araya, J.; Cambier, S.; Markovics, J.A.; Wolters, P.; Jablons, D.; Hill, A.; Finkbeiner, W.; Jones, K.; Broaddus, V.C.; Sheppard, D.; et al. Squamous metaplasia amplifies pathologic epithelial-mesenchymal interactions in COPD patients. J. Clin. Investig. 2007, 117, 3551–3562. [Google Scholar] [CrossRef] [PubMed]
- Sari, E.; Oztay, F.; Tasci, A.E. Vitamin D modulates E-cadherin turnover by regulating TGF-β and Wnt signalings during EMT-mediated myofibroblast differentiation in A459 cells. J. Steroid Biochem. Mol. Biol. 2020, 202, 105723. [Google Scholar] [CrossRef]
- Salvi, S.S.; Barnes, P.J. Chronic obstructive pulmonary disease in non-smokers. Lancet 2009, 374, 733–743. [Google Scholar] [CrossRef]
- Center for Disease Control and Prevention: Health Effects of Secondhand Smoke. Available online: https://www.cdc.gov/tobacco/data_statistics/fact_sheets/secondhand_smoke/health_effects/ (accessed on 27 September 2020).
- Schick, S.; Glantz, S. Philip Morris toxicological experiments with fresh sidestream smoke: More toxic than mainstream smoke. Tob. Control 2005, 14, 396–404. [Google Scholar] [CrossRef]
- Kim, M.S.; Huang, Y.; Lee, J.; Zhong, X.; Jiang, W.W.; Ratovitski, E.A.; Sidransky, D. Cellular transformation by cigarette smoke extract involves alteration of glycolysis and mitochondrial function in esophageal epithelial cells. Int. J. Cancer 2010, 127, 269–281. [Google Scholar] [CrossRef]
- Schweitzer, K.S.; Johnstone, B.H.; Garrison, J.; Rush, N.I.; Cooper, S.; Traktuev, D.O.; Feng, D.; Adamowicz, J.J.; Van Demark, M.; Fisher, A.J.; et al. Adipose stem cell treatment in mice attenuates lung and systemic injury induced by cigarette smoking. Am. J. Respir. Crit. Care Med. 2011, 183, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Michaeloudes, C.; Zhang, Y.; Wiegman, C.H.; Adcock, I.M.; Lian, Q.; Mak, J.C.W.; Bhavsar, P.K.; Chung, K.F. Mesenchymal stem cells alleviate oxidative stress-induced mitochondrial dysfunction in the airways. J. Allergy Clin. Immunol. 2018, 141, 1634–1645.e1635. [Google Scholar] [CrossRef] [PubMed]
- Huh, J.W.; Kim, S.Y.; Lee, J.H.; Lee, J.S.; Van Ta, Q.; Kim, M.; Oh, Y.M.; Lee, Y.S.; Lee, S.D. Bone marrow cells repair cigarette smoke-induced emphysema in rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, L255–L266. [Google Scholar] [CrossRef] [PubMed]
- Weiss, D.J.; Casaburi, R.; Flannery, R.; LeRoux-Williams, M.; Tashkin, D.P. A placebo-controlled, randomized trial of mesenchymal stem cells in COPD. Chest 2013, 143, 1590–1598. [Google Scholar] [CrossRef]
- Cortes-Dericks, L.; Galetta, D. The therapeutic potential of mesenchymal stem cells in lung cancer: Benefits, risks and challenges. Cell Oncol. 2019, 42, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.S.; Park, B.S.; Sung, J.H. The wound-healing and antioxidant effects of adipose-derived stem cells. Expert Opin. Biol. Ther. 2009, 9, 879–887. [Google Scholar] [CrossRef]
- Kim, W.S.; Park, B.S.; Sung, J.H.; Yang, J.M.; Park, S.B.; Kwak, S.J.; Park, J.S. Wound healing effect of adipose-derived stem cells: A critical role of secretory factors on human dermal fibroblasts. J. Dermatol. Sci. 2007, 48, 15–24. [Google Scholar] [CrossRef]
- Kim, W.S.; Park, B.S.; Kim, H.K.; Park, J.S.; Kim, K.J.; Choi, J.S.; Chung, S.J.; Kim, D.D.; Sung, J.H. Evidence supporting antioxidant action of adipose-derived stem cells: Protection of human dermal fibroblasts from oxidative stress. J. Dermatol. Sci. 2008, 49, 133–142. [Google Scholar] [CrossRef]
- Kim, S.Y.; Lee, J.H.; Kim, H.J.; Park, M.K.; Huh, J.W.; Ro, J.Y.; Oh, Y.M.; Lee, S.D.; Lee, Y.S. Mesenchymal stem cell-conditioned media recovers lung fibroblasts from cigarette smoke-induced damage. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 302, L891–L908. [Google Scholar] [CrossRef]
- Bajetto, A.; Thellung, S.; Dellacasagrande, I.; Pagano, A.; Barbieri, F.; Florio, T. Cross talk between mesenchymal and glioblastoma stem cells: Communication beyond controversies. Stem Cells Transl. Med. 2020, 9, 1310–1330. [Google Scholar] [CrossRef]
- Pommier, R.M.; Gout, J.; Vincent, D.F.; Cano, C.E.; Kaniewski, B.; Martel, S.; Rodriguez, J.; Fourel, G.; Valcourt, U.; Marie, J.C.; et al. The human NUPR1/P8 gene is transcriptionally activated by transforming growth factor beta via the SMAD signalling pathway. Biochem J 2012, 445, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.W.; Mitra, A.; Fillmore, R.A.; Jiang, W.G.; Samant, R.S.; Fodstad, O.; Shevde, L.A. NUPR1 interacts with p53, transcriptionally regulates p21 and rescues breast epithelial cells from doxorubicin-induced genotoxic stress. Curr. Cancer Drug Targets 2008, 8, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Greathouse, K.L.; White, J.R.; Vargas, A.J.; Bliskovsky, V.V.; Beck, J.A.; von Muhlinen, N.; Polley, E.C.; Bowman, E.D.; Khan, M.A.; Robles, A.I.; et al. Interaction between the microbiome and TP53 in human lung cancer. Genome Biol. 2018, 19, 123. [Google Scholar] [CrossRef]
- Kong, F.F.; Zhu, Y.L.; Yuan, H.H.; Wang, J.Y.; Zhao, M.; Gong, X.D.; Liu, F.; Zhang, W.Y.; Wang, C.R.; Jiang, B. FOXM1 regulated by ERK pathway mediates TGF-beta1-induced EMT in NSCLC. Oncol. Res 2014, 22, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Gachechiladze, M.; Skarda, J. The role of BRCA1 in non-small cell lung cancer. Biomed. Pap. Med. Fac. Univ. Palacky. Olomouc Czech Repub. 2012, 156, 200–203. [Google Scholar] [CrossRef]
- Song, X.; Lu, F.; Liu, R.Y.; Lei, Z.; Zhao, J.; Zhou, Q.; Zhang, H.T. Association between the ATF3 gene and non-small cell lung cancer. Thorac. Cancer 2012, 3, 217–223. [Google Scholar] [CrossRef]
- Miki, D.; Kubo, M.; Takahashi, A.; Yoon, K.A.; Kim, J.; Lee, G.K.; Zo, J.I.; Lee, J.S.; Hosono, N.; Morizono, T.; et al. Variation in TP63 is associated with lung adenocarcinoma susceptibility in Japanese and Korean populations. Nat. Genet. 2010, 42, 893–896. [Google Scholar] [CrossRef]
- Wang, Y.; Broderick, P.; Matakidou, A.; Vijayakrishnan, J.; Eisen, T.; Houlston, R.S. Variation in TP63 is associated with lung adenocarcinoma in the UK population. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1453–1462. [Google Scholar] [CrossRef]
- Gao, Z.; Liu, R.; Ye, N.; Liu, C.; Li, X.; Guo, X.; Zhang, Z.; Li, X.; Yao, Y.; Jiang, X. FOXO1 Inhibits Tumor Cell Migration via Regulating Cell Surface Morphology in Non-Small Cell Lung Cancer Cells. Cell Physiol. Biochem. Int. J. Exp. Cell Physiol. Biochem. Pharmacol. 2018, 48, 138–148. [Google Scholar] [CrossRef]
- Zhang, L.; Bu, L.; Hu, J.; Xu, Z.; Ruan, L.; Fang, Y.; Wang, P. HDAC1 knockdown inhibits invasion and induces apoptosis in non-small cell lung cancer cells. Biol. Chem. 2018, 399, 603–610. [Google Scholar] [CrossRef]
- Lee, J.W.; Soung, Y.H.; Kim, S.Y.; Nam, S.W.; Park, W.S.; Wang, Y.P.; Jo, K.H.; Moon, S.W.; Song, S.Y.; Lee, J.Y.; et al. ERBB2 kinase domain mutation in the lung squamous cell carcinoma. Cancer Lett. 2006, 237, 89–94. [Google Scholar] [CrossRef]
- Georgakilas, A.G.; Martin, O.A.; Bonner, W.M. p21: A Two-Faced Genome Guardian. Trends Mol. Med. 2017, 23, 310–319. [Google Scholar] [CrossRef]
- Wu, A.; Wu, B.; Guo, J.; Luo, W.; Wu, D.; Yang, H.; Zhen, Y.; Yu, X.; Wang, H.; Zhou, Y.; et al. Elevated expression of CDK4 in lung cancer. J. Transl Med. 2011, 9, 38. [Google Scholar] [CrossRef]
- Kuo, K.T.; Huang, W.C.; Bamodu, O.A.; Lee, W.H.; Wang, C.H.; Hsiao, M.; Wang, L.S.; Yeh, C.T. Histone demethylase JARID1B/KDM5B promotes aggressiveness of non-small cell lung cancer and serves as a good prognostic predictor. Clin. Epigenetics 2018, 10, 107. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Liang, Y.; Thakur, A.; Zhang, S.; Yang, T.; Chen, T.; Gao, L.; Chen, M.; Ren, H. Diagnostic significance of S100A2 and S100A6 levels in sera of patients with non-small cell lung cancer. Tumour. Biol. 2016, 37, 2299–2304. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, X.; Zhang, B.; Mao, W.; Liu, T.; Sun, M.; Wu, Y. TREM1: A positive regulator for inflammatory response via NF-kappaB pathway in A549 cells infected with Mycoplasma pneumoniae. Biomed. Pharm. 2018, 107, 1466–1472. [Google Scholar] [CrossRef]
- Zhu, M.L.; Kyprianou, N. Role of androgens and the androgen receptor in epithelial-mesenchymal transition and invasion of prostate cancer cells. FASEB J. 2010, 24, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Chai, D.M.; Qin, Y.Z.; Wu, S.W.; Ma, L.; Tan, Y.Y.; Yong, X.; Wang, X.L.; Wang, Z.P.; Tao, Y.S. WISP2 exhibits its potential antitumor activity via targeting ERK and E-cadherin pathways in esophageal cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 102. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Chang, J.S.; Syu, S.H.; Wong, T.S.; Chan, J.Y.; Tang, Y.C.; Yang, Z.P.; Yang, W.C.; Chen, C.T.; Lu, S.C.; et al. IL-1β promotes malignant transformation and tumor aggressiveness in oral cancer. J. Cell. Physiol. 2015, 230, 875–884. [Google Scholar] [CrossRef]
- Dai, L.; Li, J.; Tsay, J.J.; Yie, T.A.; Munger, J.S.; Pass, H.; Rom, W.N.; Tan, E.M.; Zhang, J.Y. Identification of autoantibodies to ECH1 and HNRNPA2B1 as potential biomarkers in the early detection of lung cancer. Oncoimmunology 2017, 6, e1310359. [Google Scholar] [CrossRef]
- Deskin, B.; Yin, Q.; Zhuang, Y.; Saito, S.; Shan, B.; Lasky, J.A. Inhibition of HDAC6 Attenuates Tumor Growth of Non-Small Cell Lung Cancer. Transl. Oncol. 2020, 13, 135–145. [Google Scholar] [CrossRef]
- Muramoto, K.; Tange, R.; Ishii, T.; Miyauchi, K.; Sato, T. Downregulation of Transcription Factor Sp1 Suppresses Malignant Properties of A549 Human Lung Cancer Cell Line with Decreased beta4-Galactosylation of Highly Branched N-Glycans. Biol Pharm. Bull. 2017, 40, 1282–1288. [Google Scholar] [CrossRef]
- Wang, X.M.; Li, J.; Yan, M.X.; Liu, L.; Jia, D.S.; Geng, Q.; Lin, H.C.; He, X.H.; Li, J.J.; Yao, M. Integrative analyses identify osteopontin, LAMB3 and ITGB1 as critical pro-metastatic genes for lung cancer. PLoS ONE 2013, 8, e55714. [Google Scholar] [CrossRef]
- Hu, B.; Li, X.; Chen, L.; Liu, Z. High Expression of CARM1 Inhibits Lung Cancer Progression by Targeting TP53 by Regulating CTNNB1. Lung 2020, 198, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Herbst, A.; Rahmig, K.; Stieber, P.; Philipp, A.; Jung, A.; Ofner, A.; Crispin, A.; Neumann, J.; Lamerz, R.; Kolligs, F.T. Methylation of NEUROG1 in serum is a sensitive marker for the detection of early colorectal cancer. Am. J. Gastroenterol. 2011, 106, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Puustinen, P.; Jaattela, M. KIAA1524/CIP2A promotes cancer growth by coordinating the activities of MTORC1 and MYC. Autophagy 2014, 10, 1352–1354. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Yu, D.; Zhou, J.; Zhuang, S.; Jiang, T. TGM2 interference regulates the angiogenesis and apoptosis of colorectal cancer via Wnt/beta-catenin pathway. Cell Cycle 2019, 18, 1122–1134. [Google Scholar] [CrossRef]
- Liu, B.; Liu, Q.; Song, Y.; Li, X.; Wang, Y.; Wan, S.; Zhang, Z.; Su, H. Polymorphisms of HIF1A gene are associated with prognosis of early stage non-small-cell lung cancer patients after surgery. Med. Oncol. 2014, 31, 877. [Google Scholar] [CrossRef][Green Version]
- Vu, T.; Jin, L.; Datta, P.K. Effect of Cigarette Smoking on Epithelial to Mesenchymal Transition (EMT) in Lung Cancer. J. Clin. Med. 2016, 5. [Google Scholar] [CrossRef]
- Xu, F.; Liu, X.C.; Li, L.; Ma, C.N.; Zhang, Y.J. Effects of TRPC1 on epithelial mesenchymal transition in human airway in chronic obstructive pulmonary disease. Medicine 2017, 96, e8166. [Google Scholar] [CrossRef]
- Jiang, G.; Liu, C.T.; Zhang, W.D. IL-17A and GDF15 are able to induce epithelial-mesenchymal transition of lung epithelial cells in response to cigarette smoke. Exp. Ther. Med. 2018, 16, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Guan, Y.; Shen, H.J.; Zhang, L.H.; Jiang, J.X.; Dong, X.W.; Shen, H.H.; Xie, Q.M. Akt/PKB signaling regulates cigarette smoke-induced pulmonary epithelial-mesenchymal transition. Lung Cancer 2018, 122, 44–53. [Google Scholar] [CrossRef]
- Mahmood, M.Q.; Walters, E.H.; Shukla, S.D.; Weston, S.; Muller, H.K.; Ward, C.; Sohal, S.S. β-catenin, Twist and Snail: Transcriptional regulation of EMT in smokers and COPD, and relation to airflow obstruction. Sci. Rep. 2017, 7, 10832. [Google Scholar] [CrossRef]
- Onzi, G.R.; Faccioni, J.L.; Pereira, L.C.; Thomé, M.P.; Bertoni, A.P.S.; Buss, J.H.; Fazolo, T.; Filippi-Chiela, E.; Wink, M.R.; Lenz, G. Adipose-derived stromal cell secretome disrupts autophagy in glioblastoma. J. Mol. Med. 2019, 97, 1491–1506. [Google Scholar] [CrossRef]
- Pietrobono, D.; Giacomelli, C.; Marchetti, L.; Martini, C.; Trincavelli, M.L. High Adenosine Extracellular Levels Induce Glioblastoma Aggressive Traits Modulating the Mesenchymal Stromal Cell Secretome. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Park, Y.M.; Yoo, S.H.; Kim, S.H. Adipose-derived stem cells induced EMT-like changes in H358 lung cancer cells. Anticancer Res. 2013, 33, 4421–4430. [Google Scholar]
- Du, X.; Qi, F.; Lu, S.; Li, Y.; Han, W. Nicotine upregulates FGFR3 and RB1 expression and promotes non-small cell lung cancer cell proliferation and epithelial-to-mesenchymal transition via downregulation of miR-99b and miR-192. Biomed. Pharmacother. Biomed. Pharmacother. 2018, 101, 656–662. [Google Scholar] [CrossRef]
- Li, L.; Pan, Y.; Mo, X.; Wei, T.; Song, J.; Luo, M.; Huang, G.; Teng, C.; Liang, K.; Mao, N.; et al. A novel metastatic promoter CEMIP and its downstream molecular targets and signaling pathway of cellular migration and invasion in SCLC cells based on proteome analysis. J. Cancer Res. Clin. Oncol. 2020, 146, 2519–2534. [Google Scholar] [CrossRef] [PubMed]
- Habiel, D.M.; Camelo, A.; Espindola, M.; Burwell, T.; Hanna, R.; Miranda, E.; Carruthers, A.; Bell, M.; Coelho, A.L.; Liu, H.; et al. Divergent roles for Clusterin in Lung Injury and Repair. Sci. Rep. 2017, 7, 15444. [Google Scholar] [CrossRef]
- Huang, S.C.; Wu, T.C.; Yu, H.C.; Chen, M.R.; Liu, C.M.; Chiang, W.S.; Lin, K.M. Mechanical strain modulates age-related changes in the proliferation and differentiation of mouse adipose-derived stromal cells. BMC Cell Biol. 2010, 11, 18. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-H.; Pan, K.-L.; Tang, Y.-C.; Tsai, M.-H.; Cheng, A.-J.; Shen, M.-Y.; Cheng, Y.-M.; Huang, T.-T.; Lin, P. LDOC1 silenced by cigarette exposure and involved in oral neoplastic transformation. Oncotarget 2015, 6, 25188–25201. [Google Scholar] [CrossRef]
- Wang, C.-K.; Lee, H.-L.; Chang, H.; Tsai, M.-H.; Kuo, Y.-C.; Lin, P. Enhancement between environmental tobacco smoke and arsenic on emphysema-like lesions in mice. J. Hazard. Mater. 2012, 221–222, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.-C.; Lin, C.-J.; Liu, H.-J.; Li, L.-A. Health risk of metal exposure via inhalation of cigarette sidestream smoke particulate matter. Environ. Sci. Pollut. Res. 2019, 26, 10835–10845. [Google Scholar] [CrossRef] [PubMed]
- Kasai, H.; Allen, J.T.; Mason, R.M.; Kamimura, T.; Zhang, Z. TGF-β1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir. Res. 2005, 6, 56. [Google Scholar] [CrossRef] [PubMed]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2013, 30, 523–530. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Upstream Regulator | Molecule Type | Predicted State | z-Score | p-Value of Overlap |
|---|---|---|---|---|
| NUPR1 | transcription regulator | Activated | 9.495 | 5.38 × 10−50 |
| TP53 | transcription regulator | Activated | 6.814 | 3.51 × 10−46 |
| E2F4 | transcription regulator | 1.65 × 10−38 | ||
| ERBB2 | kinase | Activated | 2.186 | 3.28 × 10−35 |
| TGFB1 | growth factor | Activated | 3.746 | 3.18 × 10−32 |
| FOXO1 | transcription regulator | −1.594 | 4.04 × 10−23 | |
| FOXM1 | transcription regulator | Inhibited | −4.121 | 5.29 × 10−21 |
| E2f | group | Inhibited | −3.317 | 2.44 × 10−20 |
| CCND1 | transcription regulator | −0.468 | 7.58 × 10−20 | |
| E2F1 | transcription regulator | 2.30 × 10−19 | ||
| CDKN1A | kinase | Activated | 4.263 | 5.50 × 10−17 |
| CDK4 | kinase | 1.78 × 10−16 | ||
| ESR1 | ligand-dependent nuclear receptor | −0.519 | 4.03 × 10−15 | |
| TP63 | transcription regulator | −0.619 | 7.11 × 10−15 | |
| KDM5B | transcription regulator | Activated | 4.874 | 1.41 × 10−14 |
| S100A6 | transporter | Inhibited | −3.742 | 1.60 × 10−14 |
| TREM1 | transmembrane receptor | 0.769 | 2.85 × 10−14 | |
| TNF | cytokine | Activated | 3.127 | 1.80 × 10−12 |
| E2F6 | transcription regulator | Activated | 3.162 | 7.50 × 10−12 |
| P38 MAPK | group | 1.399 | 9.32 × 10−12 | |
| AR | ligand-dependent nuclear receptor | −1.381 | 1.51 × 10−11 | |
| BRCA1 | transcription regulator | 1.564 | 5.50 × 10−11 | |
| HDAC1 | transcription regulator | 1.35 × 10−10 | ||
| ATF3 | transcription regulator | 1.134 | 1.52 × 10−10 | |
| estrogen receptor | group | Inhibited | −3.329 | 8.48 × 10−10 |
| Upstream Regulator | Molecule Type | Predicted State | z-Score | p-Value of Overlap |
|---|---|---|---|---|
| TGFB1 | growth factor | Activated | 6.637 | 1.43 × 10−36 |
| TNF | cytokine | 1.508 | 6.20 × 10−33 | |
| estrogen receptor | group | Inhibited | −3.797 | 3.75 × 10−29 |
| ERBB2 | kinase | Activated | 4.162 | 1.27 × 10−25 |
| JUN | transcription regulator | 0.595 | 7.24 × 10−25 | |
| WISP2 | growth factor | Inhibited | −2.947 | 7.22 × 10−18 |
| TREM1 | transmembrane receptor | −0.563 | 1.10 × 10−17 | |
| IL1A | cytokine | Activated | 2.028 | 4.32 × 10−16 |
| HNRNPA2B1 | other | 6.99 × 10−16 | ||
| ERK | group | 1.713 | 2.47 × 10−15 | |
| HDAC6 | transcription regulator | Activated | 3.375 | 3.41 × 10−15 |
| Cg | complex | Activated | 2.661 | 2.01 × 10−14 |
| SPDEF | transcription regulator | Inhibited | −3.174 | 5.92 × 10−14 |
| CTNNB1 | transcription regulator | 1.071 | 6.00 × 10−14 | |
| P38 MAPK | group | 1.489 | 1.06 × 10−13 | |
| COL18A1 | other | Inhibited | −3.022 | 1.08 × 10−13 |
| SP1 | transcription regulator | 0.862 | 1.57 × 10−13 | |
| ITGB1 | transmembrane receptor | 0.639 | 4.14 × 10−13 | |
| NEUROG1 | transcription regulator | −1.342 | 1.42 × 10−12 | |
| AR | ligand-dependent nuclear receptor | −0.101 | 2.34 × 10−12 | |
| TP63 | transcription regulator | 0.650 | 7.08 × 10−12 | |
| IL1B | cytokine | 0.154 | 9.64 × 10−12 | |
| KIAA1524 | other | Inhibited | −2.233 | 1.04 × 10−11 |
| TGM2 | enzyme | −0.168 | 1.47 × 10−11 | |
| HIF1A | transcription regulator | 1.337 | 2.12 × 10−11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, T.-Y.; Liu, C.-H.; Chen, T.-H.; Chen, M.-R.; Liu, S.-W.; Lin, P.; Lin, K.M.-C. Conditioned Media of Adipose-Derived Stem Cells Suppresses Sidestream Cigarette Smoke Extract Induced Cell Death and Epithelial-Mesenchymal Transition in Lung Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 12069. https://doi.org/10.3390/ijms222112069
Chen T-Y, Liu C-H, Chen T-H, Chen M-R, Liu S-W, Lin P, Lin KM-C. Conditioned Media of Adipose-Derived Stem Cells Suppresses Sidestream Cigarette Smoke Extract Induced Cell Death and Epithelial-Mesenchymal Transition in Lung Epithelial Cells. International Journal of Molecular Sciences. 2021; 22(21):12069. https://doi.org/10.3390/ijms222112069
Chicago/Turabian StyleChen, Tzu-Yin, Chia-Hao Liu, Tsung-Hsien Chen, Mei-Ru Chen, Shan-Wen Liu, Pinpin Lin, and Kurt Ming-Chao Lin. 2021. "Conditioned Media of Adipose-Derived Stem Cells Suppresses Sidestream Cigarette Smoke Extract Induced Cell Death and Epithelial-Mesenchymal Transition in Lung Epithelial Cells" International Journal of Molecular Sciences 22, no. 21: 12069. https://doi.org/10.3390/ijms222112069
APA StyleChen, T.-Y., Liu, C.-H., Chen, T.-H., Chen, M.-R., Liu, S.-W., Lin, P., & Lin, K. M.-C. (2021). Conditioned Media of Adipose-Derived Stem Cells Suppresses Sidestream Cigarette Smoke Extract Induced Cell Death and Epithelial-Mesenchymal Transition in Lung Epithelial Cells. International Journal of Molecular Sciences, 22(21), 12069. https://doi.org/10.3390/ijms222112069