
Necroptosis Inhibition by Hydrogen Sulfide Alleviated Hypoxia-Induced Cardiac Fibroblasts Proliferation via Sirtuin 3


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
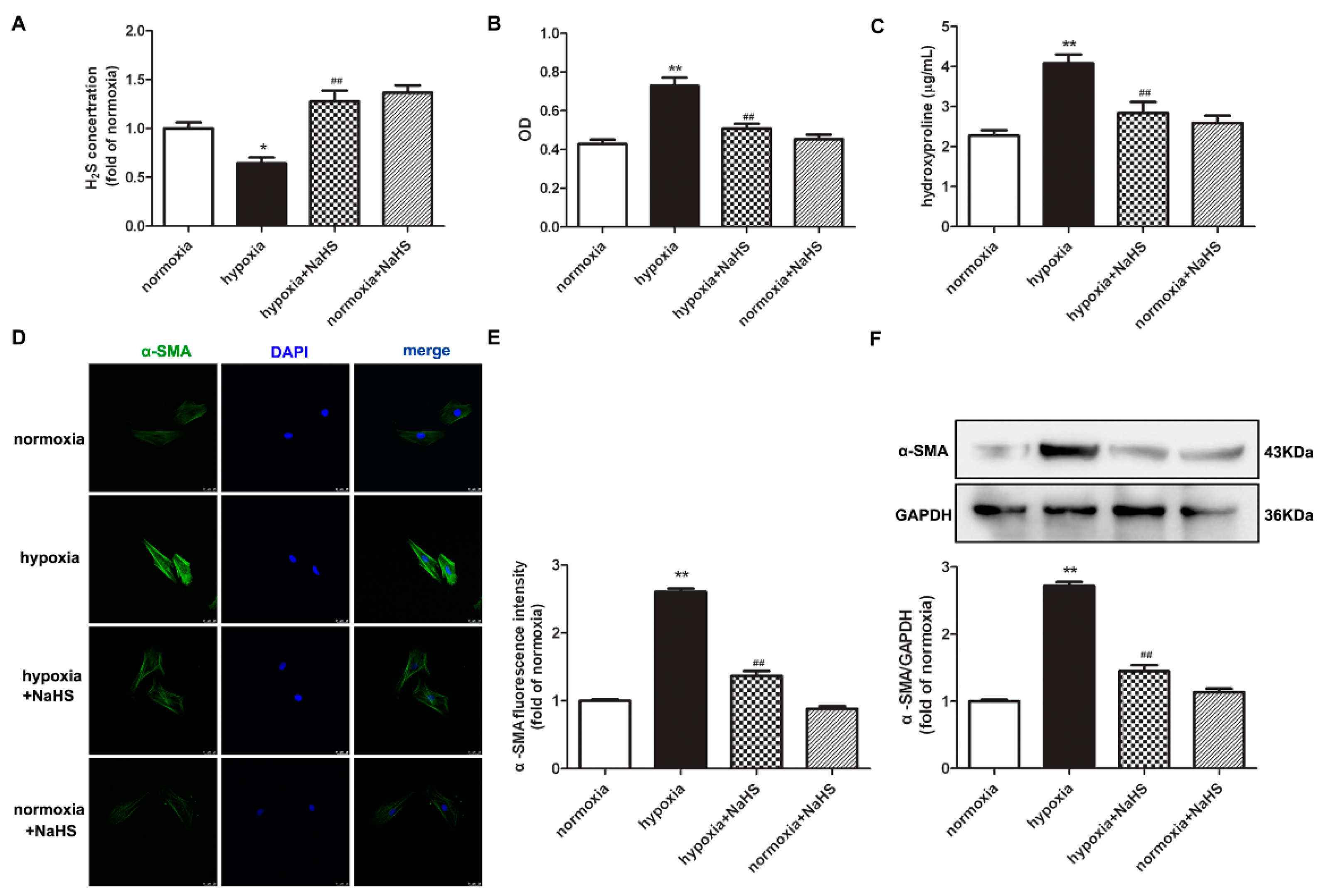
2.1. NaHS Decreases Cell Number and Hydroxyproline Content in Cardiac Fibroblasts with Hypoxia
2.2. NaHS Suppresses α-SMA Expression in Cardiac Fibroblasts with Hypoxia
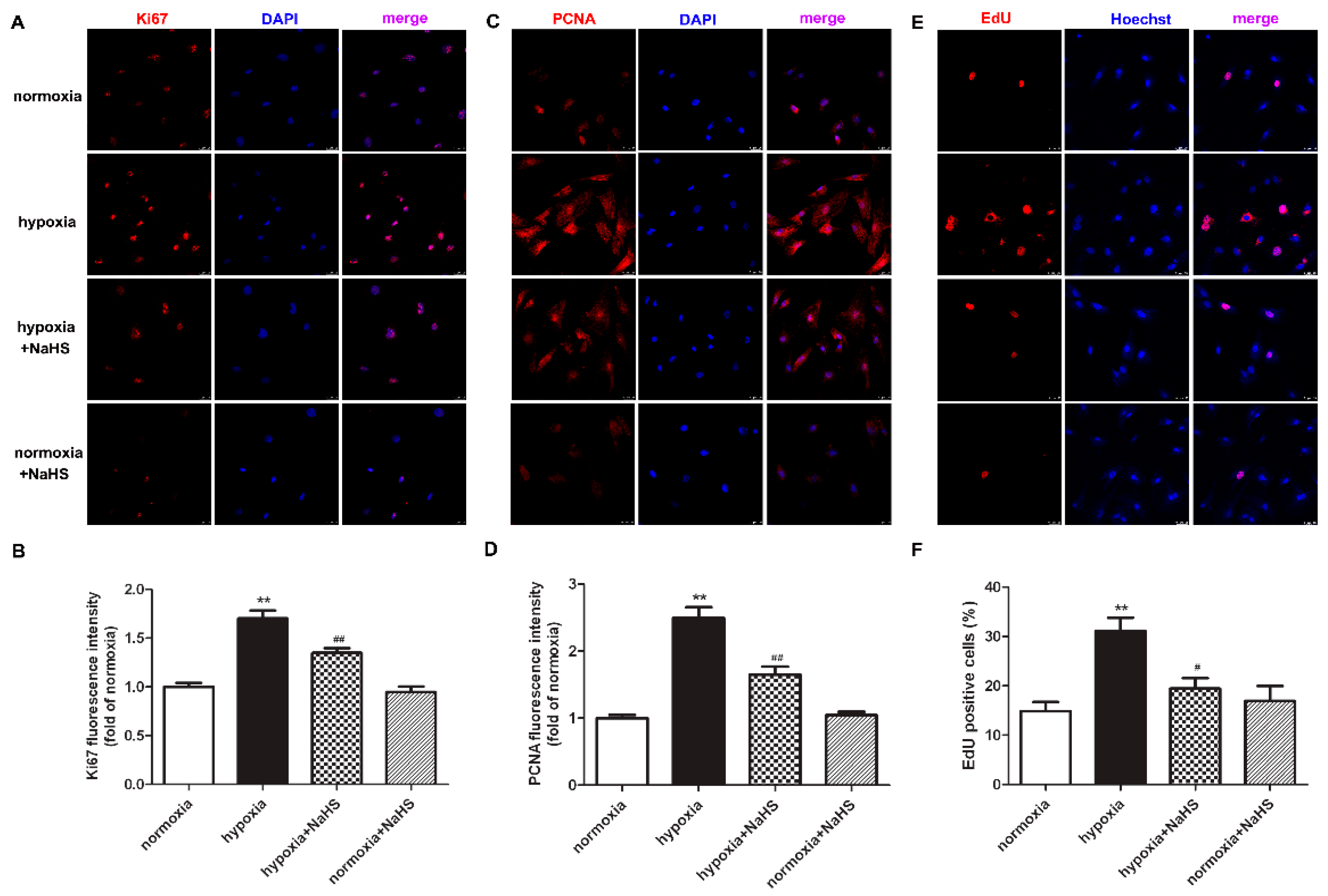
2.3. NaHS Inhibits Ki67 and PCNA Expression in Cardiac Fibroblasts with Hypoxia
2.4. NaHS Reduces EdU Positive Cardiac Fibroblasts with Hypoxia
2.5. NaHS Represses Collagen Synthesis in Cardiac Fibroblasts with Hypoxia
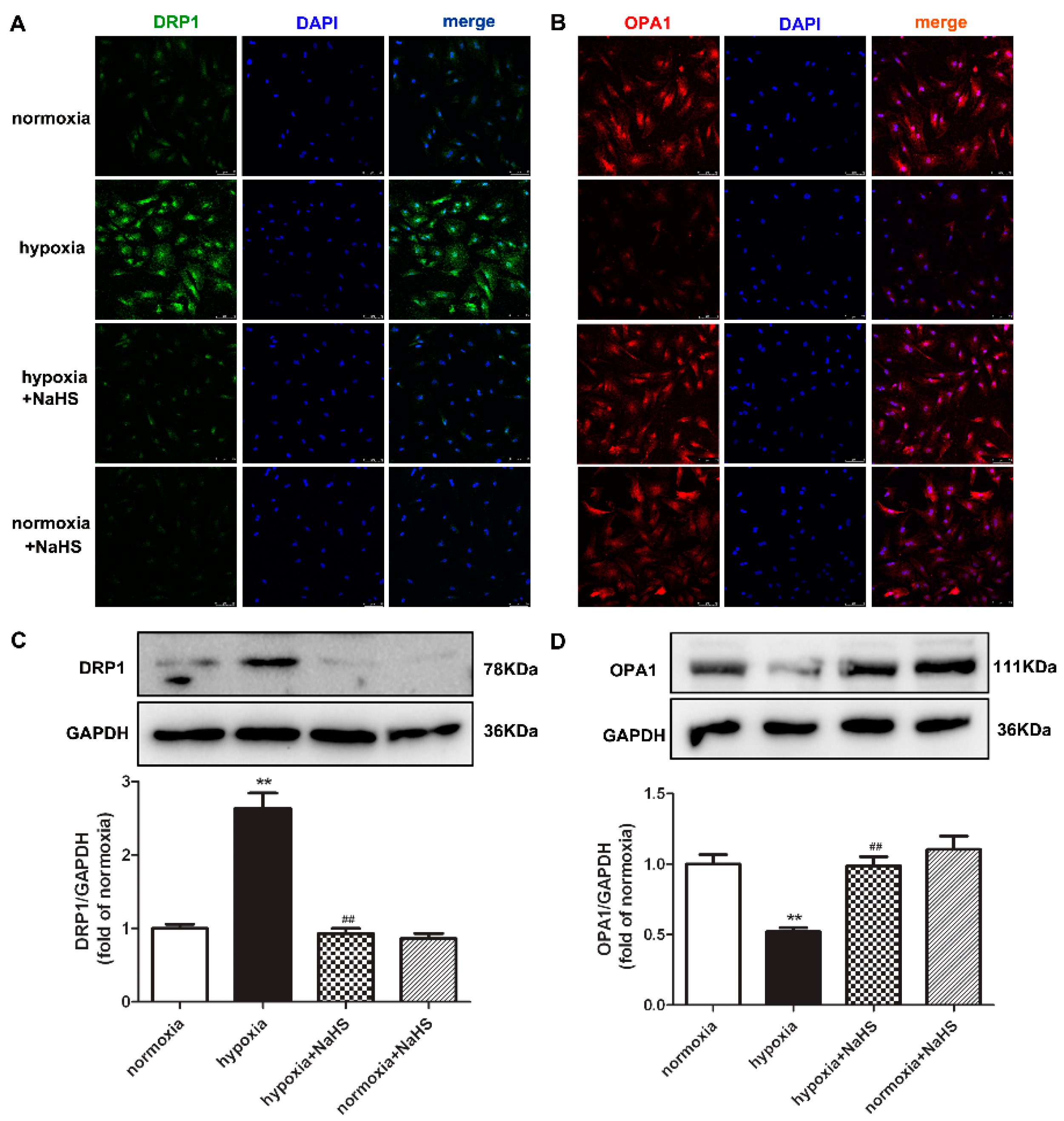
2.6. NaHS Restores DRP1 and OPA1 Protein Expression in Cardiac Fibroblasts with Hypoxia
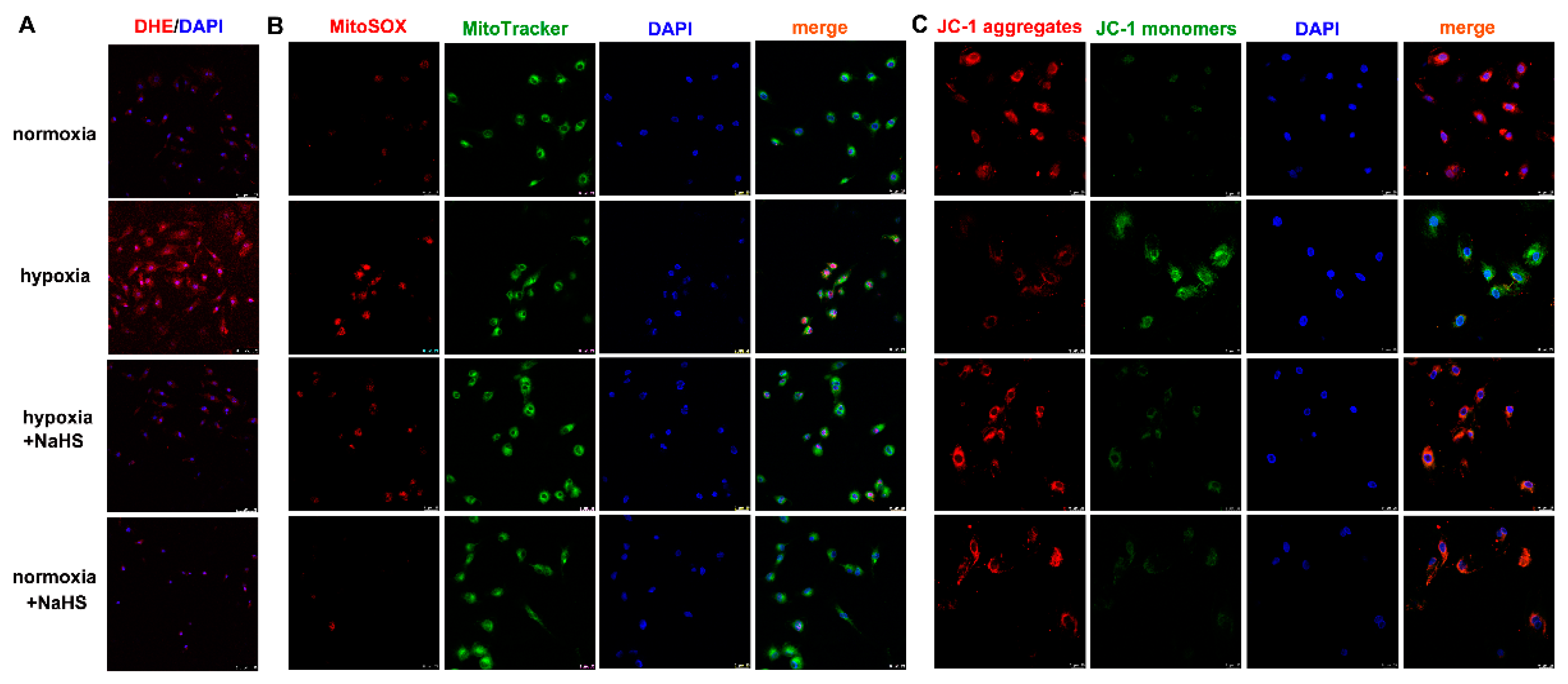
2.7. NaHS Alleviates Oxidative Stress in Cardiac Fibroblasts with Hypoxia
2.8. NaHS Improves Mitochondrial Membrane Potential in Cardiac Fibroblasts with Hypoxia
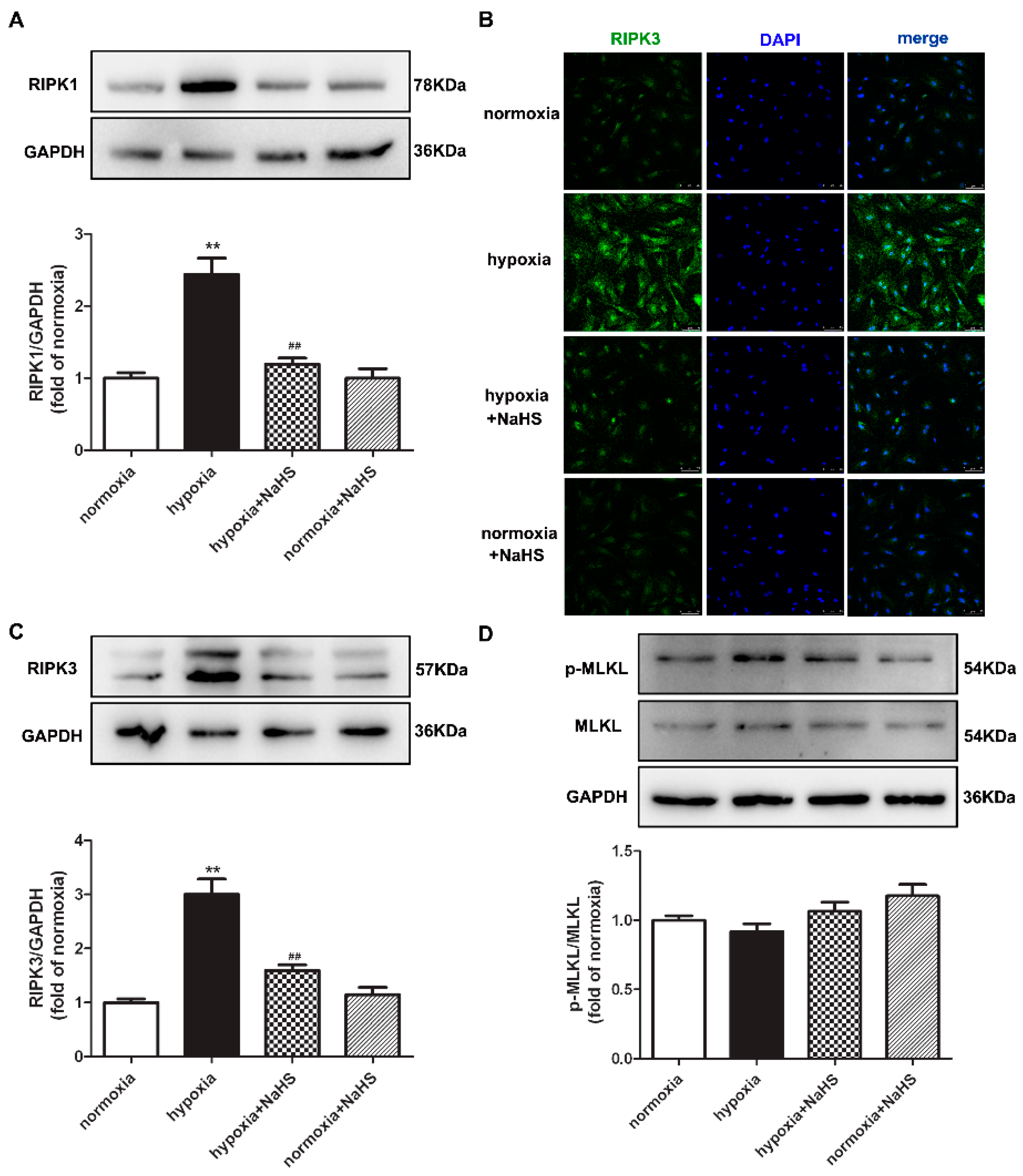
2.9. NaHS Attenuates Necroptosis in Cardiac Fibroblasts with Hypoxia
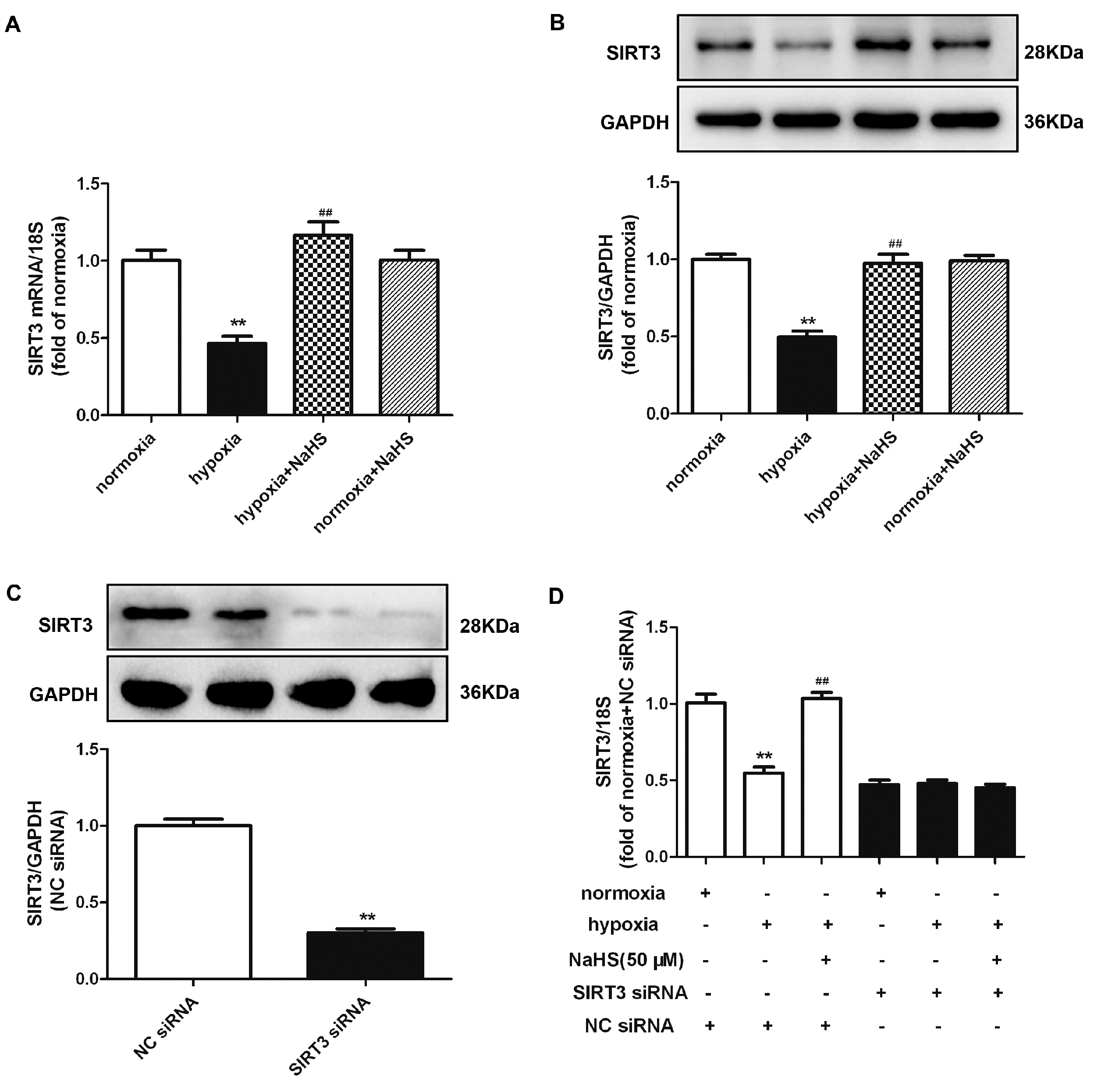
2.10. NaHS Enhances SIRT3 Expression in Cardiac Fibroblasts with Hypoxia
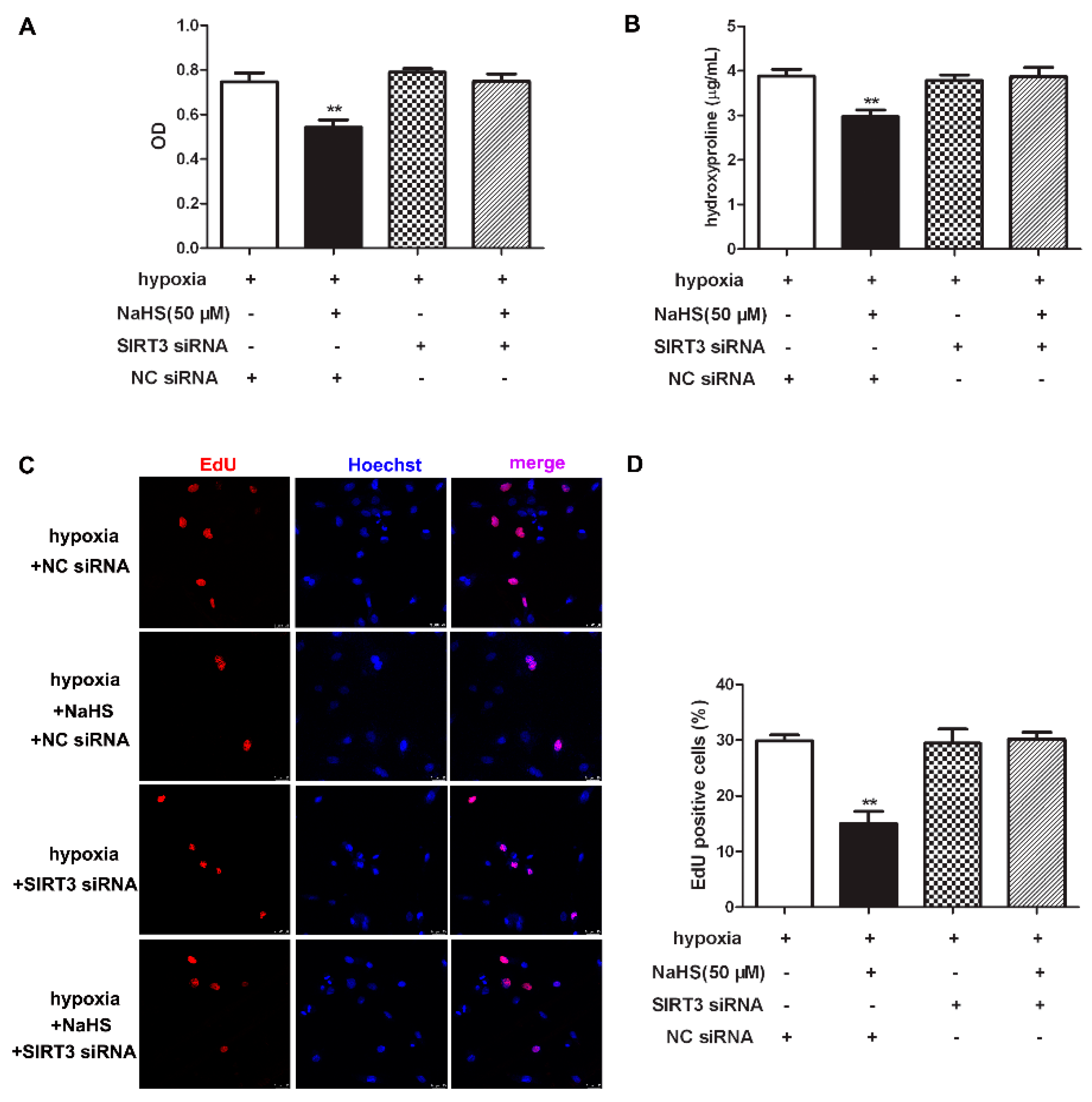
2.11. NaHS Fails to Inhibit Cell Proliferation after SIRT3 Was Knockdown in Cardiac Fibroblasts with Hypoxia
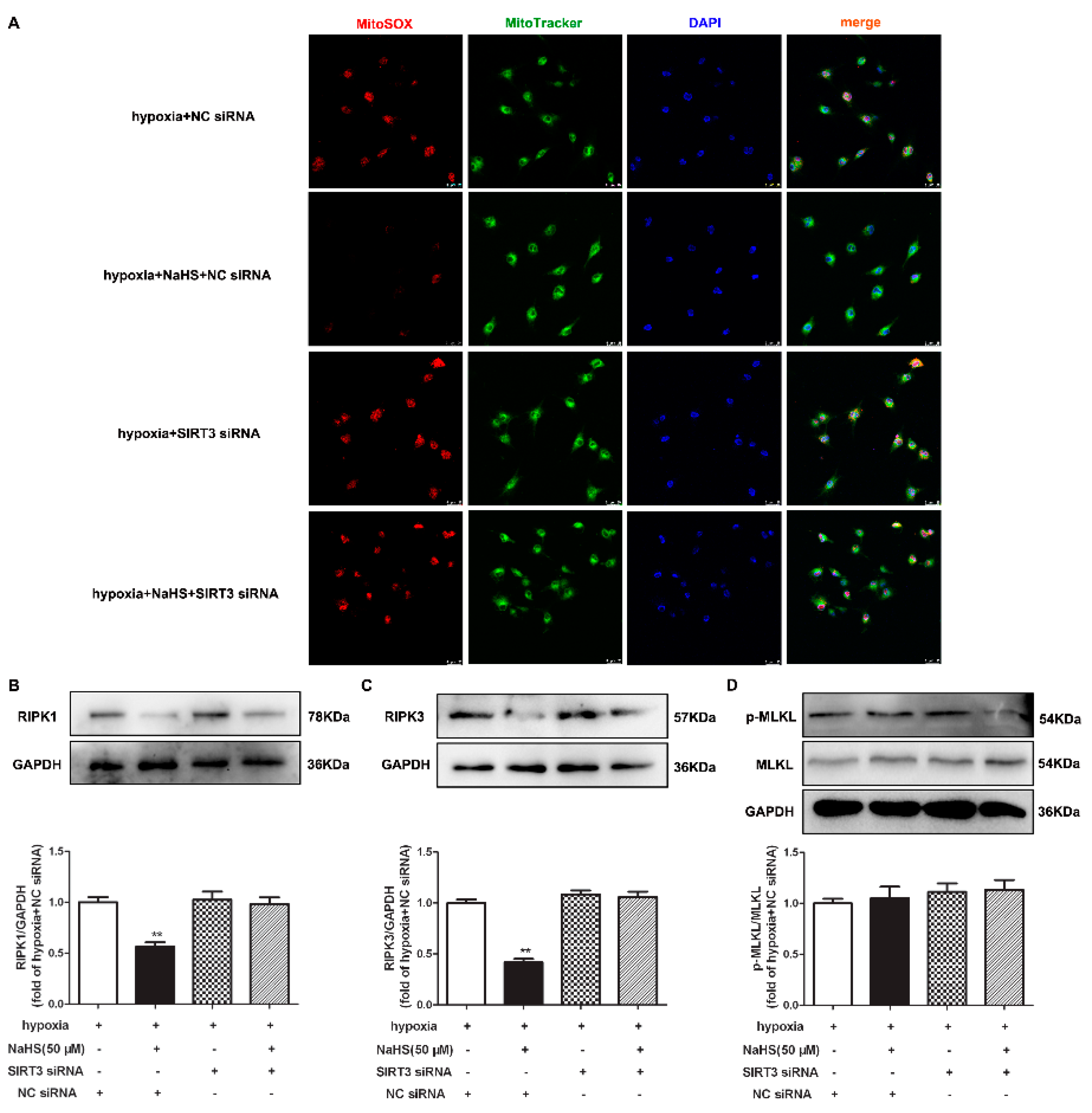
2.12. NaHS Fails to Alleviate Mitochondrial Oxidative Stress and Attenuate Necroptosis after SIRT3 Was Knocked Down in Cardiac Fibroblasts with Hypoxia
3. Discussion
4. Materials and Methods
4.1. Primary Cardiac Fibroblasts Culture and Treatment
4.2. Measurement of H2S in the Culture Medium
4.3. siRNA Transfection
4.4. Cell Number Assay
4.5. Hydroxyproline Content Determination
4.6. EdU (5-Ethynyl-2′-deoxyuridine) Staining
4.7. JC-1 Staining
4.8. DHE Staining
4.9. MitoSOX Staining
4.10. Real-Time PCR
4.11. Western Blot
4.12. Immunofluorescence
4.13. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Smit, M.; Coetzee, A.R.; Lochner, A. The Pathophysiology of Myocardial Ischemia and Perioperative Myocardial Infarction. J. Cardiothorac. Vasc. Anesth. 2020, 34, 2501–2512. [Google Scholar] [CrossRef]
- Zhu, H.; Toan, S.; Mui, D.; Zhou, H. Mitochondrial quality surveillance as a therapeutic target in myocardial infarction. Acta Physiol. 2021, 231, e13590. [Google Scholar] [CrossRef] [PubMed]
- Talman, V.; Ruskoaho, H. Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res. 2016, 365, 563–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, G.; Liu, J.; Liu, S.; Song, Q.; Liu, L.; Xie, L.; Han, Y.; Ji, Y. Hydrogen sulfide pretreatment improves mitochondrial function in myocardial hypertrophy via a SIRT3-dependent manner. Br. J. Pharmacol. 2018, 175, 1126–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Yu, J.; Chen, Y.; Liu, L.; Xu, M.; Sun, L.; Luo, H.; Wang, Y.; Meng, G. Exogenous Hydrogen Sulfide Supplement Attenuates Isoproterenol-Induced Myocardial Hypertrophy in a Sirtuin 3-Dependent Manner. Oxid. Med. Cell. Longev. 2018, 2018, 9396089. [Google Scholar] [CrossRef]
- Xie, L.; Feng, H.; Li, S.; Meng, G.; Liu, S.; Tang, X.; Ma, Y.; Han, Y.; Xiao, Y.; Gu, Y.; et al. SIRT3 Mediates the Antioxidant Effect of Hydrogen Sulfide in Endothelial Cells. Antioxid. Redox Signal. 2016, 24, 329–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, G.; Xiao, Y.; Ma, Y.; Tang, X.; Xie, L.; Liu, J.; Gu, Y.; Yu, Y.; Park, C.M.; Xian, M.; et al. Hydrogen Sulfide Regulates Kruppel-Like Factor 5 Transcription Activity via Specificity Protein 1 S-Sulfhydration at Cys664 to Prevent Myocardial Hypertrophy. J. Am. Heart Assoc. 2016, 5, e004160. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Gu, Y.; Wen, M.; Zhao, S.; Wang, W.; Ma, Y.; Meng, G.; Han, Y.; Wang, Y.; Liu, G.; et al. Hydrogen Sulfide Induces Keap1 S-sulfhydration and Suppresses Diabetes-Accelerated Atherosclerosis via Nrf2 Activation. Diabetes 2016, 65, 3171–3184. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Han, Y.; Li, L.; Lu, H.; Meng, G.; Li, X.; Shirhan, M.; Peh, M.T.; Xie, L.; Zhou, S.; et al. The hydrogen sulfide donor, GYY4137, exhibits anti-atherosclerotic activity in high fat fed apolipoprotein E(-/-) mice. Br. J. Pharmacol. 2013, 169, 1795–1809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, G.; Wang, J.; Xiao, Y.; Bai, W.; Xie, L.; Shan, L.; Moore, P.K.; Ji, Y. GYY4137 protects against myocardial ischemia and reperfusion injury by attenuating oxidative stress and apoptosis in rats. J. Biomed. Res. 2015, 29, 203–213. [Google Scholar]
- Sun, H.J.; Wu, Z.Y.; Nie, X.W.; Wang, X.Y.; Bian, J.S. Implications of hydrogen sulfide in liver pathophysiology: Mechanistic insights and therapeutic potential. J. Adv. Res. 2021, 27, 127–135. [Google Scholar] [CrossRef]
- Ngowi, E.E.; Sarfraz, M.; Afzal, A.; Khan, N.H.; Khattak, S.; Zhang, X.; Li, T.; Duan, S.F.; Ji, X.Y.; Wu, D.D. Roles of Hydrogen Sulfide Donors in Common Kidney Diseases. Front. Pharmacol. 2020, 11, 564281. [Google Scholar] [CrossRef]
- Khattak, S.; Zhang, Q.Q.; Sarfraz, M.; Muhammad, P.; Ngowi, E.E.; Khan, N.H.; Rauf, S.; Wang, Y.Z.; Qi, H.W.; Wang, D.; et al. The Role of Hydrogen Sulfide in Respiratory Diseases. Biomolecules 2021, 11, 682. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xiang, H.; Liu, J.; Chen, Y.; He, R.R.; Liu, B. Mitochondrial Sirtuin 3: New emerging biological function and therapeutic target. Theranostics 2020, 10, 8315–8342. [Google Scholar] [CrossRef]
- Chen, Y.; Hua, Y.; Li, X.; Arslan, I.M.; Zhang, W.; Meng, G. Distinct Types of Cell Death and the Implication in Diabetic Cardiomyopathy. Front. Pharmacol. 2020, 11, 42. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.E.; Price, D.R.; Ryter, S.W.; Choi, A.M.K. Necroptosis: A crucial pathogenic mediator of human disease. JCI Insight 2019, 4, e128834. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xu, F.; Zhang, L.; Wang, X.; Wang, Y.; Woo, A.Y.; Zhu, W. GRK2/beta-arrestin mediates arginine vasopressin-induced cardiac fibroblast proliferation. Clin. Exp. Pharmacol. Physiol. 2017, 44, 285–293. [Google Scholar] [CrossRef]
- Gu, J.; Chen, X.; Jin, Y.; Liu, M.; Xu, Q.; Liu, X.; Luo, Z.; Ling, S.; Liu, N.; Liu, S. A Neonatal Mouse Model for Pressure Overload: Myocardial Response Corresponds to Severity. Front. Cardiovasc. Med. 2021, 8, 660246. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Long, L.; Cheng, Y.; Chu, J.; Shen, Z.; Liu, L.; Li, J.; Xie, Q.; Liu, H.; Wu, M.; et al. Qingda granule attenuates cardiac fibrosis via suppression of the TGF-beta1/Smad2/3 signaling pathway in vitro and in vivo. Biomed. Pharmacother. 2021, 137, 111318. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Gong, W.; Zhang, S.; Shen, J.; Liu, X.; Wang, Y.; Chen, Y.; Meng, G. Protective role of sirtuin3 against oxidative stress and NLRP3 inflammasome in cholesterol accumulation and foam cell formation of macrophages with ox-LDL-stimulation. Biochem. Pharmacol. 2021, 192, 114665. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, X.; Hua, Y.; Ding, Y.; Meng, G.; Zhang, W. RIPK3-Mediated Necroptosis in Diabetic Cardiomyopathy Requires CaMKII Activation. Oxid. Med. Cell. Longev. 2021, 2021, 6617816. [Google Scholar]
- Nakada, Y.; Canseco, D.C.; Thet, S.; Abdisalaam, S.; Asaithamby, A.; Santos, C.X.; Shah, A.M.; Zhang, H.; Faber, J.E.; Kinter, M.T.; et al. Hypoxia induces heart regeneration in adult mice. Nature 2017, 541, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Liu, X.; Zhang, J.; Dong, G.; Xiao, W.; Xu, X. Hydrogen Sulfide Alleviates Skeletal Muscle Fibrosis via Attenuating Inflammation and Oxidative Stress. Front. Physiol. 2020, 11, 533690. [Google Scholar] [CrossRef]
- Li, Y.; Chandra, T.P.; Song, X.; Nie, L.; Liu, M.; Yi, J.; Zheng, X.; Chu, C.; Yang, J. H2S improves doxorubicin-induced myocardial fibrosis by inhibiting oxidative stress and apoptosis via Keap1-Nrf2. Technol. Health Care 2021, 29, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xing, Q.Q.; Tu, J.K.; Tang, W.B.; Yuan, X.N.; Xie, Y.Y.; Wang, W.; Peng, Z.Z.; Huang, L.; Xu, H.; et al. Involvement of hydrogen sulfide in the progression of renal fibrosis. Chin. Med. J. 2019, 132, 2872–2880. [Google Scholar] [CrossRef]
- Cadenas, S. ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free Radic. Biol. Med. 2018, 117, 76–89. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, A.; Wu, Y.; Guan, W.; Xiong, B.; Peng, X.; Wei, X.; Chen, C.; Liu, Z. Stachydrine ameliorates carbon tetrachloride-induced hepatic fibrosis by inhibiting inflammation, oxidative stress and regulating MMPs/TIMPs system in rats. Biomed. Pharmacother. 2018, 97, 1586–1594. [Google Scholar] [CrossRef]
- Zhao, L.; Wu, D.; Sang, M.; Xu, Y.; Liu, Z.; Wu, Q. Stachydrine ameliorates isoproterenol-induced cardiac hypertrophy and fibrosis by suppressing inflammation and oxidative stress through inhibiting NF-kappaB and JAK/STAT signaling pathways in rats. Int. Immunopharmacol. 2017, 48, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Gu, J.; Wang, J.; Wu, Y.; Yang, A.; Chen, T.; Zhou, T.; Liu, Z. Physcion 8-O-beta-glucopyranoside ameliorates liver fibrosis through inflammation inhibition by regulating SIRT3-mediated NF-kappaB P65 nuclear expression. Int. Immunopharmacol. 2021, 90, 107206. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Ding, Y.; Dai, G.L.; Zhang, Y.; Xu, M.T.; Shen, J.R.; Chen, T.T.; Chen, Y.; Meng, G.L. Sirtuin 3 deficiency exacerbates diabetic cardiomyopathy via necroptosis enhancement and NLRP3 activation. Acta Pharmacol. Sin. 2021, 42, 230–241. [Google Scholar] [CrossRef]
- Xu, X.; Chen, C.; Lu, W.J.; Su, Y.L.; Shi, J.Y.; Liu, Y.C.; Wang, L.; Xiao, C.X.; Wu, X.; Lu, Q. Pyrroloquinoline quinone can prevent chronic heart failure by regulating mitochondrial function. Cardiovasc. Diagn. Ther. 2020, 10, 453–469. [Google Scholar] [CrossRef]
- Su, H.; Zeng, H.; Liu, B.; Chen, J.X. Sirtuin 3 is essential for hypertension-induced cardiac fibrosis via mediating pericyte transition. J. Cell. Mol. Med. 2020, 24, 8057–8068. [Google Scholar] [CrossRef]
- Palomer, X.; Roman-Azcona, M.S.; Pizarro-Delgado, J.; Planavila, A.; Villarroya, F.; Valenzuela-Alcaraz, B.; Crispi, F.; Sepulveda-Martinez, A.; Miguel-Escalada, I.; Ferrer, J.; et al. SIRT3-mediated inhibition of FOS through histone H3 deacetylation prevents cardiac fibrosis and inflammation. Signal. Transduct. Target Ther. 2020, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Yan, F.; Li, J.; Zhang, C.; Bu, P. SIRT3 attenuates AngII-induced cardiac fibrosis by inhibiting myofibroblasts transdifferentiation via STAT3-NFATc2 pathway. Am. J. Transl. Res. 2017, 9, 3258–3269. [Google Scholar]
- Fu, W.; Li, H.; Fu, H.; Zhao, S.; Shi, W.; Sun, M.; Li, Y. The SIRT3 and SIRT6 Promote Prostate Cancer Progression by Inhibiting Necroptosis-Mediated Innate Immune Response. J. Immunol. Res. 2020, 2020, 8820355. [Google Scholar] [CrossRef]
- Sun, L.; Chen, Y.; Luo, H.; Xu, M.; Meng, G.; Zhang, W. Ca2+/calmodulin-dependent protein kinase II regulation by inhibitor 1 of protein phosphatase 1 alleviates necroptosis in high glucose-induced cardiomyocytes injury. Biochem. Pharmacol. 2019, 163, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ren, S.; Lu, Y.; Chen, X.; Qu, J.; Ma, X.; Deng, Q.; Hu, Z.; Jin, Y.; Zhou, Z.; et al. Inhibition of Syk promotes chemical reprogramming of fibroblasts via metabolic rewiring and H S production. EMBO J. 2021, 40, e106771. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Gong, W.; Xu, M.; Zhang, S.; Shen, J.; Zhu, M.; Wang, Y.; Chen, Y.; Shi, J.; Meng, G. Necroptosis Inhibition by Hydrogen Sulfide Alleviated Hypoxia-Induced Cardiac Fibroblasts Proliferation via Sirtuin 3. Int. J. Mol. Sci. 2021, 22, 11893. https://doi.org/10.3390/ijms222111893
Zhang Y, Gong W, Xu M, Zhang S, Shen J, Zhu M, Wang Y, Chen Y, Shi J, Meng G. Necroptosis Inhibition by Hydrogen Sulfide Alleviated Hypoxia-Induced Cardiac Fibroblasts Proliferation via Sirtuin 3. International Journal of Molecular Sciences. 2021; 22(21):11893. https://doi.org/10.3390/ijms222111893
Chicago/Turabian StyleZhang, Yue, Weiwei Gong, Mengting Xu, Shuping Zhang, Jieru Shen, Mingxian Zhu, Yuqin Wang, Yun Chen, Jiahai Shi, and Guoliang Meng. 2021. "Necroptosis Inhibition by Hydrogen Sulfide Alleviated Hypoxia-Induced Cardiac Fibroblasts Proliferation via Sirtuin 3" International Journal of Molecular Sciences 22, no. 21: 11893. https://doi.org/10.3390/ijms222111893
APA StyleZhang, Y., Gong, W., Xu, M., Zhang, S., Shen, J., Zhu, M., Wang, Y., Chen, Y., Shi, J., & Meng, G. (2021). Necroptosis Inhibition by Hydrogen Sulfide Alleviated Hypoxia-Induced Cardiac Fibroblasts Proliferation via Sirtuin 3. International Journal of Molecular Sciences, 22(21), 11893. https://doi.org/10.3390/ijms222111893