
Quantitative FRET Microscopy Reveals a Crucial Role of Cytoskeleton in Promoting PI(4,5)P2 Confinement

 ,
,  ,
,  , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
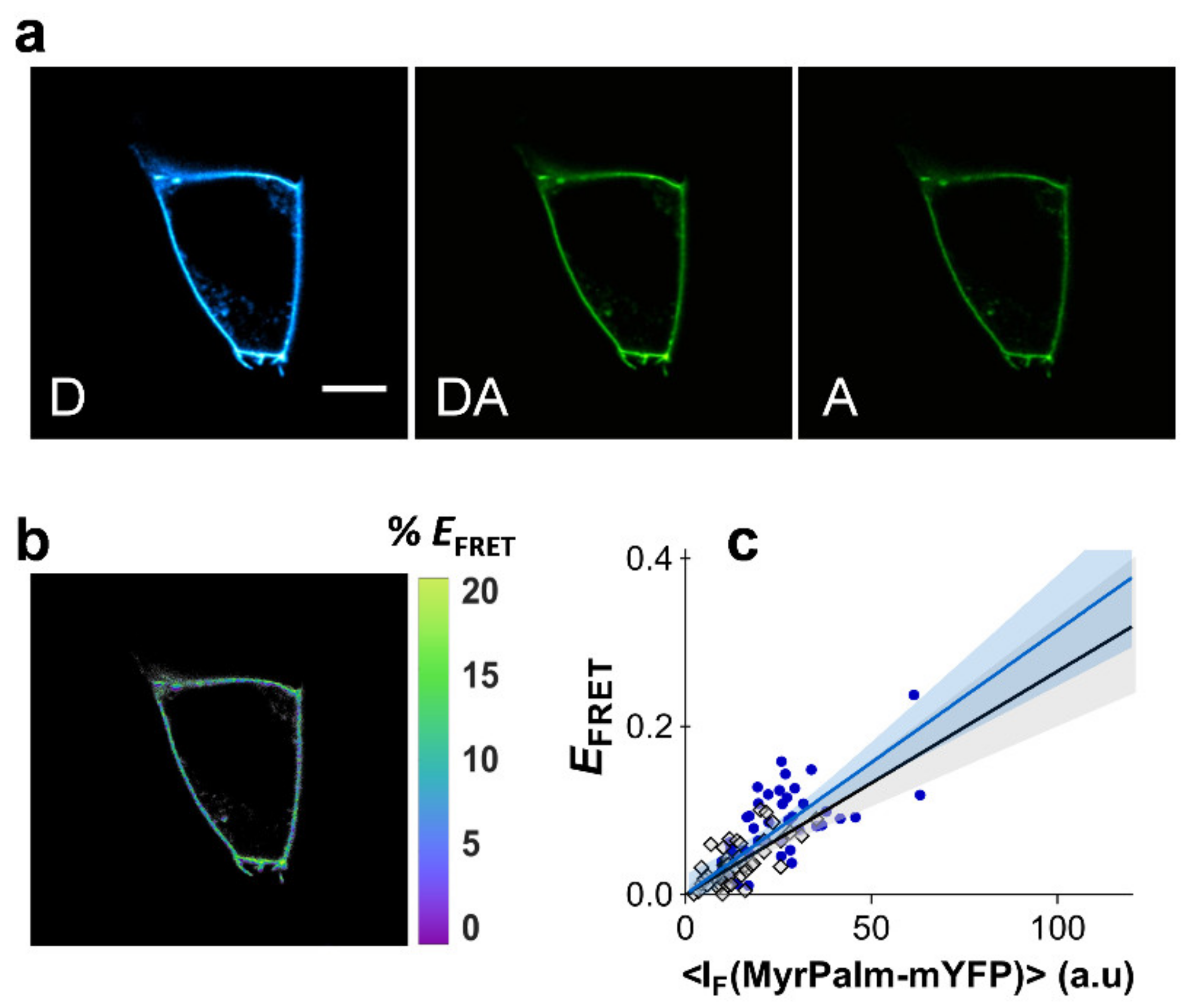
2.1. 2D FRET between Non-Compartmentalized Proteins Shows a Linear Dependence with Acceptor Concentration in the Low FRET Range
2.2. Bystander FRET in the Absence of Compartmentalization
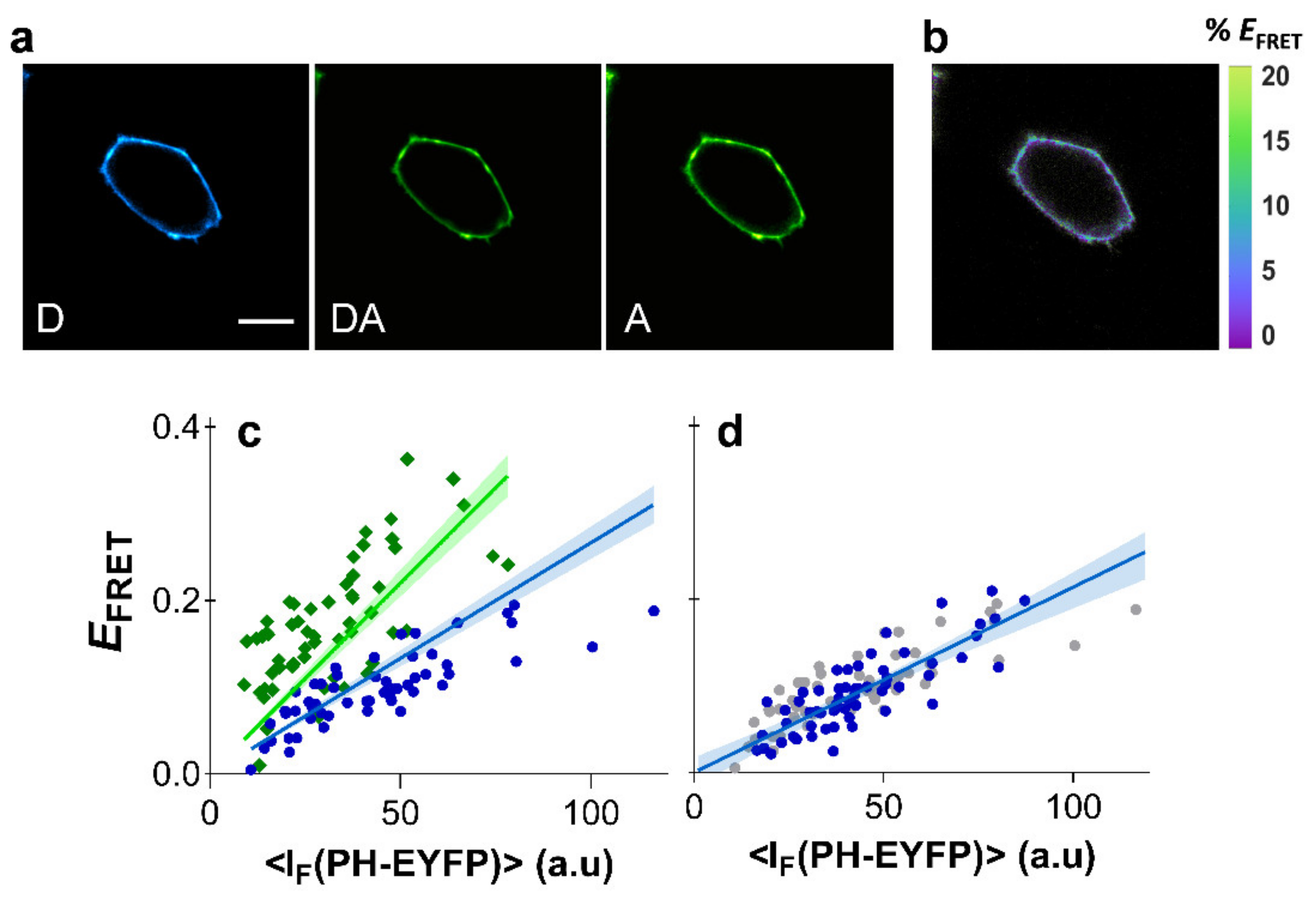
2.3. Clustering of PI(4,5)P2 in HEK293T Cells
2.4. Clustering of PI(4,5)P2 in HeLa Cells
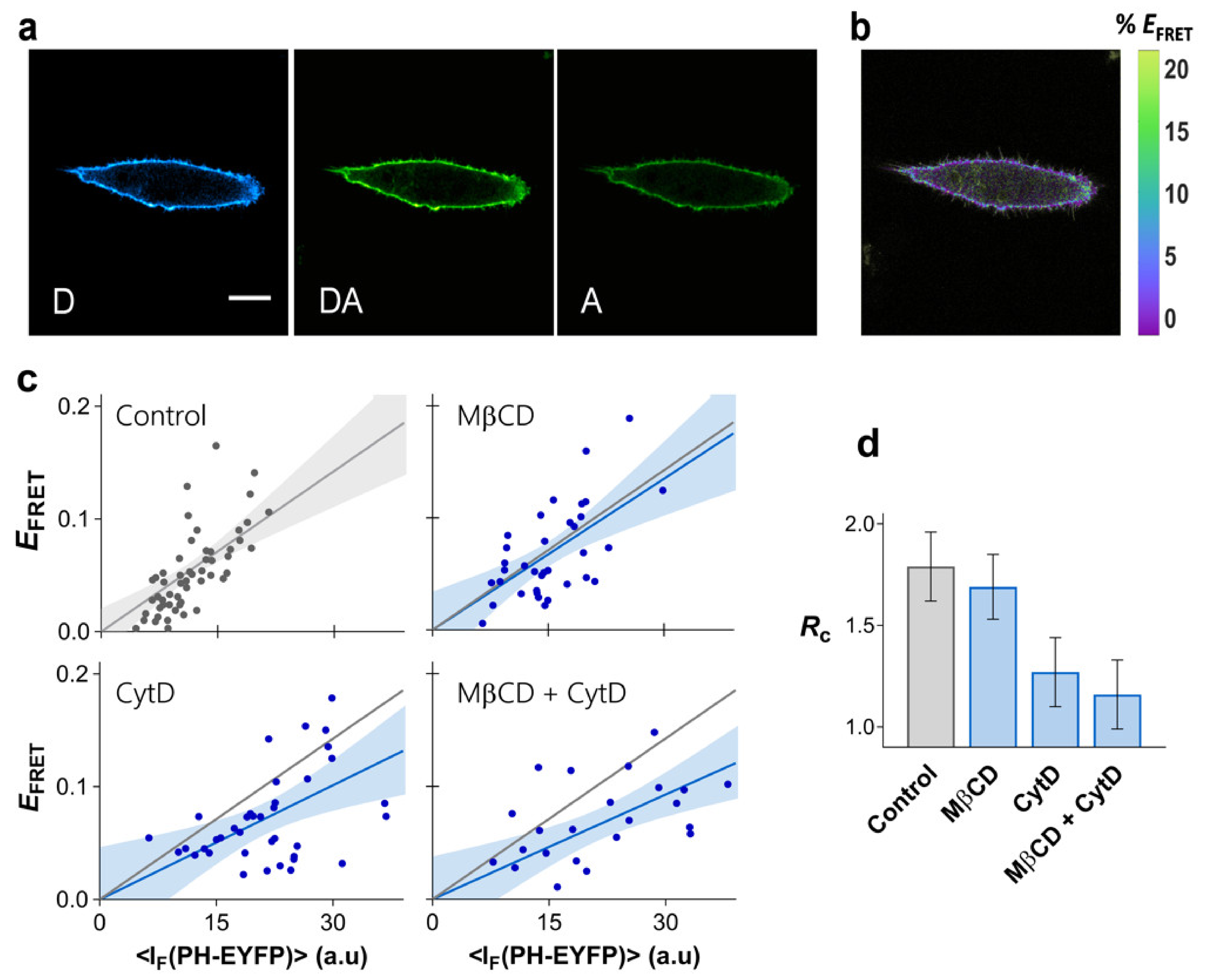
2.5. Determinants of PI-(4,5)P2 Clustering in HeLa Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Transfection
4.2. pDNA Constructs
4.3. Fluorescence Linear Dichroism Imaging
4.4. Three-Filter Cube FRET Microscopy
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferrell, J.E.; Huestis, W.H. Phosphoinositide metabolism and the morphology of human erythrocytes. J. Cell Biol. 1984, 98, 1992–1998. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Thapa, N.; Hedman, A.C.; Anderson, R.A. Phosphatidylinositol 4,5-bisphosphate: Targeted production and signaling. Bioessays 2013, 35, 513–522. [Google Scholar] [CrossRef]
- Moens, P.D.J.; Bagatolli, L.A. Profilin binding to sub-micellar concentrations of phosphatidylinositol (4,5) bisphosphate and phosphatidylinositol (3,4,5) trisphosphate. Biochim. Biophys. Acta 2007, 1768, 439–449. [Google Scholar] [CrossRef][Green Version]
- Gambhir, A.; Hangyás-Mihályné, G.; Zaitseva, I.; Cafiso, D.S.; Wang, J.; Murray, D.; Pentyala, S.N.; Smith, S.O.; McLaughlin, S. Electrostatic sequestration of PIP2 on phospholipid membranes by basic/aromatic regions of proteins. Biophys. J. 2004, 86, 2188–2207. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Collins, A.; Guo, L.; Smith-Dupont, K.B.; Gai, F.; Svitkina, T.; Janmey, P.A. Divalent cation-induced cluster formation by polyphosphoinositides in model membranes. J. Am. Chem. Soc. 2012, 134, 3387–3395. [Google Scholar] [CrossRef] [PubMed]
- Sarmento, M.J.; Coutinho, A.; Fedorov, A.; Prieto, M.; Fernandes, F. Membrane order is a key regulator of divalent cation-induced clustering of PI(3,5)P2 and PI(4,5)P2. Langmuir 2017, 33, 12463–12477. [Google Scholar] [CrossRef]
- Sarmento, M.J.; Coutinho, A.; Fedorov, A.; Prieto, M.; Fernandes, F. Ca2+ induces PI(4,5)P2 clusters on lipid bilayers at physiological PI(4,5)P2 and Ca2+ concentrations. Biochim. Biophys. Acta Biomembr. 2014, 1838, 822–830. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Vogt, V.M.; Feigenson, G.W. Multivalent Cation-Bridged PI(4,5)P2 Clusters Form at Very Low Concentrations. Biophys. J. 2018, 114, 2630–2639. [Google Scholar] [CrossRef]
- Borges-Araújo, L.; Fernandes, F. Structure and lateral organization of phosphatidylinositol 4,5-bisphosphate. Molecules 2020, 25, 3885. [Google Scholar] [CrossRef] [PubMed]
- Milosevic, I.; Sørensen, J.B.; Lang, T.; Krauss, M.; Nagy, G.; Haucke, V.; Jahn, R.; Neher, E. Plasmalemmal phosphatidylinositol-4,5-bisphosphate level regulates the releasable vesicle pool size in chromaffin cells. J. Neurosci. 2005, 25, 2557–2565. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, K.; Sugaya, T.; Umeda, M.; Yamamoto, S.; Terakawa, S.; Takahashi, M. The activation of exocytotic sites by the formation of phosphatidylinositol 4,5-bisphosphate microdomains at syntaxin clusters. J. Biol. Chem. 2005, 280, 17346–17352. [Google Scholar] [CrossRef]
- Huang, S.; Lifshitz, L.; Patki-Kamath, V.; Tuft, R.; Fogarty, K.; Czech, M.P. Phosphatidylinositol-4,5-bisphosphate-rich plasma membrane patches organize active zones of endocytosis and ruffling in cultured adipocytes. Mol. Cell. Biol. 2004, 24, 9102–9123. [Google Scholar] [CrossRef]
- Laux, T.; Fukami, K.; Thelen, M.; Golub, T.; Frey, D.; Caroni, P. GAP43, MARCKS, and CAP23 modulate PI(4,5)P(2) at plasmalemmal rafts, and regulate cell cortex actin dynamics through a common mechanism. J. Cell Biol. 2000, 149, 1455–1472. [Google Scholar] [CrossRef] [PubMed]
- Chierico, L.; Joseph, A.S.; Lewis, A.L.; Battaglia, G. Live cell imaging of membrane/cytoskeleton interactions and membrane topology. Sci. Rep. 2015, 4, 6056. [Google Scholar] [CrossRef] [PubMed]
- Van den Bogaart, G.; Meyenberg, K.; Risselada, H.J.; Amin, H.; Willig, K.I.; Hubrich, B.E.; Dier, M.; Hell, S.W.; Grubmüller, H.; Diederichsen, U.; et al. Membrane protein sequestering by ionic protein-lipid interactions. Nature 2011, 479, 552–555. [Google Scholar] [CrossRef]
- Wang, J.; Richards, D.A. Segregation of PIP2 and PIP3 into distinct nanoscale regions within the plasma membrane. Biol. Open 2012, 1, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Honigmann, A.; van den Bogaart, G.; Iraheta, E.; Risselada, H.J.; Milovanovic, D.; Mueller, V.; Müllar, S.; Diederichsen, U.; Fasshauer, D.; Grubmüller, H.; et al. Phosphatidylinositol 4,5-bisphosphate clusters act as molecular beacons for vesicle recruitment. Nat. Struct. Mol. Biol. 2013, 20, 679–686. [Google Scholar] [CrossRef]
- Favard, C.; Chojnacki, J.; Merida, P.; Yandrapalli, N.; Mak, J.; Eggeling, C.; Muriaux, D. HIV-1 Gag specifically restricts PI(4,5)P2 and cholesterol mobility in living cells creating a nanodomain platform for virus assembly. Sci. Adv. 2019, 5, eaaw8651. [Google Scholar] [CrossRef]
- Abe, M.; Makino, A.; Hullin-Matsuda, F.; Kamijo, K.; Ohno-Iwashita, Y.; Hanada, K.; Mizuno, H.; Miyawaki, A.; Kobayashi, T. A role for sphingomyelin-rich lipid domains in the accumulation of phosphatidylinositol-4,5-bisphosphate to the cleavage furrow during cytokinesis. Mol. Cell. Biol. 2012, 32, 1396–1407. [Google Scholar] [CrossRef]
- Myeong, J.; Park, C.G.; Suh, B.C.; Hille, B. Compartmentalization of phosphatidylinositol 4,5-bisphosphate metabolism into plasma membrane liquid-ordered/raft domains. Proc. Natl. Acad. Sci. USA 2021, 118, e2025343118. [Google Scholar] [CrossRef]
- Cheng, J.; Takenawa, T.; Tauchi-Sato, K.; Fujimoto, T.; Fujita, A. A distinct pool of phosphatidylinositol 4,5-bisphosphate in caveolae revealed by a nanoscale labeling technique. Proc. Natl. Acad. Sci. USA 2009, 106, 9256–9261. [Google Scholar]
- Ji, C.; Zhang, Y.; Xu, P.; Xu, T.; Lou, X. Nanoscale landscape of phosphoinositides revealed by specific pleckstrin homology (PH) domains using single-molecule superresolution imaging in the plasma membrane. J. Biol. Chem. 2015, 290, 26978–26993. [Google Scholar] [CrossRef] [PubMed]
- Van Rheenen, J.; Achame, E.M.; Janssen, H.; Calafat, J.; Jalink, K. PIP2 signaling in lipid domains: A critical re-evaluation. EMBO J. 2005, 24, 1664–1673. [Google Scholar] [CrossRef] [PubMed]
- Van Rheenen, J.; Jalink, K. Agonist-induced PIP2 hydrolysis inhibits cortical actin dynamics: Regulation at a global but not at a micrometer scale. Mol. Biol. Cell 2002, 13, 3257–3267. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Vogt, V.M.; Feigenson, G.W. PI(4,5)P2 clustering and its impact on biological functions. Annu. Rev. Biochem. 2021, 90, 681–707. [Google Scholar] [CrossRef]
- Loura, L.M.S.; Fernandes, F.; Prieto, M. Membrane microheterogeneity: Förster resonance energy transfer characterization of lateral membrane domains. Eur. Biophys. J. 2010, 39, 589–607. [Google Scholar] [CrossRef]
- Falkenburger, B.H.; Jensen, J.B.; Hille, B. Kinetics of M1 muscarinic receptor and G protein signaling to phospholipase C in living cells. J. Gen. Physiol. 2010, 135, 81–97. [Google Scholar] [CrossRef]
- Golebiewska, U.; Nyako, M.; Woturski, W.; Zaitseva, I.; McLaughlin, S. Diffusion coefficient of fluorescent phosphatidylinositol 4,5-bisphosphate in the plasma membrane of cells. Mol. Biol. Cell 2008, 19, 1663–1669. [Google Scholar] [CrossRef]
- Hammond, G.R.V.; Sim, Y.; Lagnado, L.; Irvine, R.F. Reversible binding and rapid diffusion of proteins in complex with inositol lipids serves to coordinate free movement with spatial information. J. Cell Biol. 2009, 184, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Van der Wal, J.; Habets, R.; Várnai, P.; Balla, T.; Jalink, K. Monitoring agonist-induced phospholipase C activation in live cells by fluorescence resonance energy transfer. J. Biol. Chem. 2001, 276, 15337–15344. [Google Scholar] [CrossRef] [PubMed]
- Varnai, P.; Thyagarajan, B.; Rohacs, T.; Balla, T. Rapidly inducible changes in phosphatidylinositol 4,5-bisphosphate levels influence multiple regulatory functions of the lipid in intact living cells. J. Cell Biol. 2006, 175, 377–382. [Google Scholar] [CrossRef]
- Várnai, P.; Lin, X.; Lee, S.B.; Tuymetova, G.; Bondeva, T.; Spät, A.; Rhee, S.G.; Hajnóczky, G.; Balla, T. Inositol lipid binding and membrane localization of isolated Pleckstrin Homology (PH) domains. J. Biol. Chem. 2002, 277, 27412–27422. [Google Scholar] [CrossRef] [PubMed]
- Kenworthy, A.K.; Edidin, M. Distribution of a glycosylphosphatidylinositol-anchored protein at the apical surface of MDCK cells examined at a resolution of <100 A using imaging fluorescence resonance energy transfer. J. Cell Biol. 1998, 142, 69–84. [Google Scholar] [PubMed]
- Kenworthy, A.K.; Petranova, N.; Edidin, M. High-Resolution FRET Microscopy of Cholera Toxin B-Subunit and GPI-anchored Proteins in Cell Plasma Membranes. Mol. Biol. Cell 2000, 11, 1645–1655. [Google Scholar] [CrossRef]
- King, C.; Raicu, V.; Hristova, K. Understanding the FRET signatures of interacting membrane proteins. J. Biol. Chem. 2017, 292, 5291–5310. [Google Scholar] [CrossRef]
- King, C.; Sarabipour, S.; Byrne, P.; Leahy, D.J.; Hristova, K. The FRET signatures of noninteracting proteins in membranes: Simulations and experiments. Biophys. J. 2014, 106, 1309–1317. [Google Scholar] [CrossRef]
- Fung, B.K.K.; Stryer, L. Surface density determination in membranes by fluorescence energy transfer. Biochemistry 1978, 17, 5241–5248. [Google Scholar] [CrossRef]
- Wolber, P.K.; Hudson, B.S. An analytic solution to the Förster energy transfer problem in two dimensions. Biophys. J. 1979, 28, 197–210. [Google Scholar] [CrossRef]
- Xu, C.; Watras, J.; Loew, L.M. Kinetic analysis of receptor-activated phosphoinositide turnover. J. Cell Biol. 2003, 161, 779–791. [Google Scholar] [CrossRef] [PubMed]
- Benninger, R.K.P.; Önfelt, B.; Neil, M.A.A.; Davis, D.M.; French, P.M.W. Fluorescence imaging of two-photon linear dichroism: Cholesterol depletion disrupts molecular orientation in cell membranes. Biophys. J. 2005, 88, 609–622. [Google Scholar] [CrossRef]
- Benninger, R.K.P. Fluorescence linear dichroism imaging for quantifying membrane order. In Methods in Membrane Lipids; Methods in Molecular BiologyTM; Dopico, A.M., Ed.; Springer: New York, NY, USA, 2015; Volume 400, pp. 161–179. ISBN 9781493917525. [Google Scholar]
- Zacharias, D.A.; Violin, J.D.; Newton, A.C.; Tsien, R.Y. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 2002, 296, 913–916. [Google Scholar] [CrossRef]
- Chen, H.; Puhl, H.L.; Koushik, S.V.; Vogel, S.S.; Ikeda, S.R. Measurement of FRET efficiency and ratio of donor to acceptor concentration in living cells. Biophys. J. 2006, 91, 39–41. [Google Scholar] [CrossRef] [PubMed]
- Raucher, D.; Stauffer, T.; Chen, W.; Shen, K.; Guo, S.; York, J.D.; Sheetz, M.P.; Meyer, T. Phosphatidylinositol 4,5-bisphosphate functions as a second messenger that regulates cytoskeleton–plasma membrane adhesion. Cell 2000, 100, 221–228. [Google Scholar] [CrossRef]
- Várnai, P.; Bondeva, T.; Tamás, P.; Tóth, B.; Buday, L.; Hunyady, L.; Balla, T. Selective cellular effects of overexpressed pleckstrin-homology domains that recognize PtdIns(3,4,5)P3 suggest their interaction with protein binding partners. J. Cell Sci. 2005, 118, 4879–4888. [Google Scholar] [CrossRef] [PubMed]
- Holz, R.W.; Hlubek, M.D.; Sorensen, S.D.; Fisher, S.K.; Balla, T.; Ozaki, S.; Prestwich, G.D.; Stuenkel, E.L.; Bittner, M.A. A pleckstrin homology domain specific for phosphatidylinositol 4, 5-bisphosphate (PtdIns-4,5-P2) and fused to green fluorescent protein identifies plasma membrane PtdIns-4,5-P2 as being important in exocytosis. J. Biol. Chem. 2000, 275, 17878–17885. [Google Scholar] [CrossRef] [PubMed]
- Szentpetery, Z.; Balla, A.; Kim, Y.; Lemmon, M.A.; Balla, T. Live cell imaging with protein domains capable of recognizing phosphatidylinositol 4,5-bisphosphate; a comparative study. BMC Cell Biol. 2009, 10, 67. [Google Scholar] [CrossRef]
- Falkenburger, B.H.; Jensen, J.B.; Hille, B. Kinetics of PIP2 metabolism and KCNQ2/3 channel regulation studied with a voltage-sensitive phosphatase in living cells. J. Gen. Physiol. 2010, 135, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Várnai, P.; Balla, T. Live cell imaging of phosphoinositide dynamics with fluorescent protein domains. Biochim. Biophys. Acta 2006, 1761, 957–967. [Google Scholar] [CrossRef]
- Dinic, J.; Ashrafzadeh, P.; Parmryd, I. Actin filaments attachment at the plasma membrane in live cells cause the formation of ordered lipid domains. Biochim. Biophys. Acta Biomembr. 2013, 1828, 1102–1111. [Google Scholar] [CrossRef]
- Golub, T.; Caroni, P. PI(4,5)P2-dependent microdomain assemblies capture microtubules to promote and control leading edge motility. J. Cell Biol. 2005, 169, 151–165. [Google Scholar] [CrossRef]
- Pike, L.J.; Miller, J.M. Cholesterol depletion delocalizes phosphatidylinositol bisphosphate and inhibits hormone-stimulated phosphatidylinositol turnover. J. Biol. Chem. 1998, 273, 22298–22304. [Google Scholar] [CrossRef]
- Cai, Z.; Li, F.; Gong, W.; Liu, W.; Duan, Q.; Chen, C.; Ni, L.; Xia, Y.; Cianflone, K.; Dong, N.; et al. Endoplasmic reticulum stress participates in aortic valve calcification in hypercholesterolemic animals. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2345–2354. [Google Scholar] [CrossRef]
- Scholze, M.J.; Barbieux, K.S.; De Simone, A.; Boumasmoud, M.; Süess, C.C.N.; Wang, R.; Gönczy, P. PI(4,5)P2 forms dynamic cortical structures and directs actin distribution as well as polarity in Caenorhabditis elegans embryos. Development 2018, 145, dev169144. [Google Scholar] [CrossRef]
- Logan, M.R.; Mandato, C.A. Regulation of the actin cytoskeleton by PIP2 in cytokinesis. Biol. Cell 2006, 98, 377–388. [Google Scholar] [CrossRef]
- Bezanilla, M.; Gladfelter, A.S.; Kovar, D.R.; Lee, W.-L. Cytoskeletal dynamics: A view from the membrane. J. Cell Biol. 2015, 209, 329–337. [Google Scholar] [CrossRef]
- Curthoys, N.M.; Parent, M.; Mlodzianoski, M.; Nelson, A.J.; Lilieholm, J.; Butler, M.B.; Valles, M.; Hess, S.T. Dances with membranes: Breakthroughs from super-resolution imaging. Curr. Top. Membr. 2015, 75, 59–123. [Google Scholar]
- Golebiewska, U.; Kay, J.G.; Masters, T.; Grinstein, S.; Im, W.; Pastor, R.W.; Scarlata, S.; McLaughlin, S. Evidence for a fence that impedes the diffusion of phosphatidylinositol 4,5-bisphosphate out of the forming phagosomes of macrophages. Mol. Biol. Cell 2011, 22, 3498–3507. [Google Scholar] [CrossRef]
- Chichili, G.R.; Rodgers, W. Cytoskeleton-membrane interactions in membrane raft structure. Cell. Mol. Life Sci. 2009, 66, 2319–2328. [Google Scholar] [CrossRef]
- Gowrishankar, K.; Ghosh, S.; Saha, S.; Rumamol, C.; Mayor, S.; Rao, M. Active remodeling of cortical actin regulates spatiotemporal organization of cell surface molecules. Cell 2012, 149, 1353–1367. [Google Scholar] [CrossRef]
- Gudheti, M.V.; Curthoys, N.M.; Gould, T.J.; Kim, D.; Gunewardene, M.S.; Gabor, K.A.; Gosse, J.A.; Kim, C.H.; Zimmerberg, J.; Hess, S.T. Actin mediates the nanoscale membrane organization of the clustered membrane protein influenza hemagglutinin. Biophys. J. 2013, 104, 2182–2192. [Google Scholar] [CrossRef]
- Jaumouillé, V.; Farkash, Y.; Jaqaman, K.; Das, R.; Lowell, C.A.; Grinstein, S. Actin cytoskeleton reorganization by syk regulates fcγ receptor responsiveness by increasing its lateral mobility and clustering. Dev. Cell 2014, 29, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Viola, A.; Gupta, N. Tether and trap: Regulation of membrane-raft dynamics by actin-binding proteins. Nat. Rev. Immunol. 2007, 7, 889–896. [Google Scholar] [CrossRef]
- Klarenbeek, J.B.; Goedhart, J.; Hink, M.A.; Gadella, T.W.J.; Jalink, K. A mTurquoise-based cAMP sensor for both FLIM and ratiometric read-out has improved dynamic range. PLoS ONE 2011, 6, e19170. [Google Scholar] [CrossRef]
- Megías, D.; Marrero, R.; Martínez Del Peso, B.; García, M.A.; Bravo-Cordero, J.-J.; García-Grande, A.; Santos, A.; Montoya, M.C. Novel lambda FRET spectral confocal microscopy imaging method. Microsc. Res. Tech. 2009, 72, 1–11. [Google Scholar] [CrossRef]
- Gordon, G.W.; Berry, G.; Liang, X.H.; Levine, B.; Herman, B. Quantitative fluorescence resonance energy transfer measurements using fluorescence microscopy. Biophys. J. 1998, 74, 2702–2713. [Google Scholar] [CrossRef]
- Kress, A.; Wang, X.; Ranchon, H.; Savatier, J.; Rigneault, H.; Ferrand, P.; Brasselet, S. Mapping the local organization of cell membranes using excitation-polarization-resolved confocal fluorescence microscopy. Biophys. J. 2013, 105, 127–136. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarmento, M.J.; Borges-Araújo, L.; Pinto, S.N.; Bernardes, N.; Ricardo, J.C.; Coutinho, A.; Prieto, M.; Fernandes, F. Quantitative FRET Microscopy Reveals a Crucial Role of Cytoskeleton in Promoting PI(4,5)P2 Confinement. Int. J. Mol. Sci. 2021, 22, 11727. https://doi.org/10.3390/ijms222111727
Sarmento MJ, Borges-Araújo L, Pinto SN, Bernardes N, Ricardo JC, Coutinho A, Prieto M, Fernandes F. Quantitative FRET Microscopy Reveals a Crucial Role of Cytoskeleton in Promoting PI(4,5)P2 Confinement. International Journal of Molecular Sciences. 2021; 22(21):11727. https://doi.org/10.3390/ijms222111727
Chicago/Turabian StyleSarmento, Maria J., Luís Borges-Araújo, Sandra N. Pinto, Nuno Bernardes, Joana C. Ricardo, Ana Coutinho, Manuel Prieto, and Fábio Fernandes. 2021. "Quantitative FRET Microscopy Reveals a Crucial Role of Cytoskeleton in Promoting PI(4,5)P2 Confinement" International Journal of Molecular Sciences 22, no. 21: 11727. https://doi.org/10.3390/ijms222111727
APA StyleSarmento, M. J., Borges-Araújo, L., Pinto, S. N., Bernardes, N., Ricardo, J. C., Coutinho, A., Prieto, M., & Fernandes, F. (2021). Quantitative FRET Microscopy Reveals a Crucial Role of Cytoskeleton in Promoting PI(4,5)P2 Confinement. International Journal of Molecular Sciences, 22(21), 11727. https://doi.org/10.3390/ijms222111727