
Biological Therapies in the Treatment of Cancer—Update and New Directions


Abstract
1. Introduction
2. A Strategy for Treating Cancer by Unblocking Effector Lymphocytes as a Type of Immunotherapy
3. Recombinant Antibodies in the Treatment of Cancer
4. Targeted Immunotherapy Based on Genetically Modified T Cells
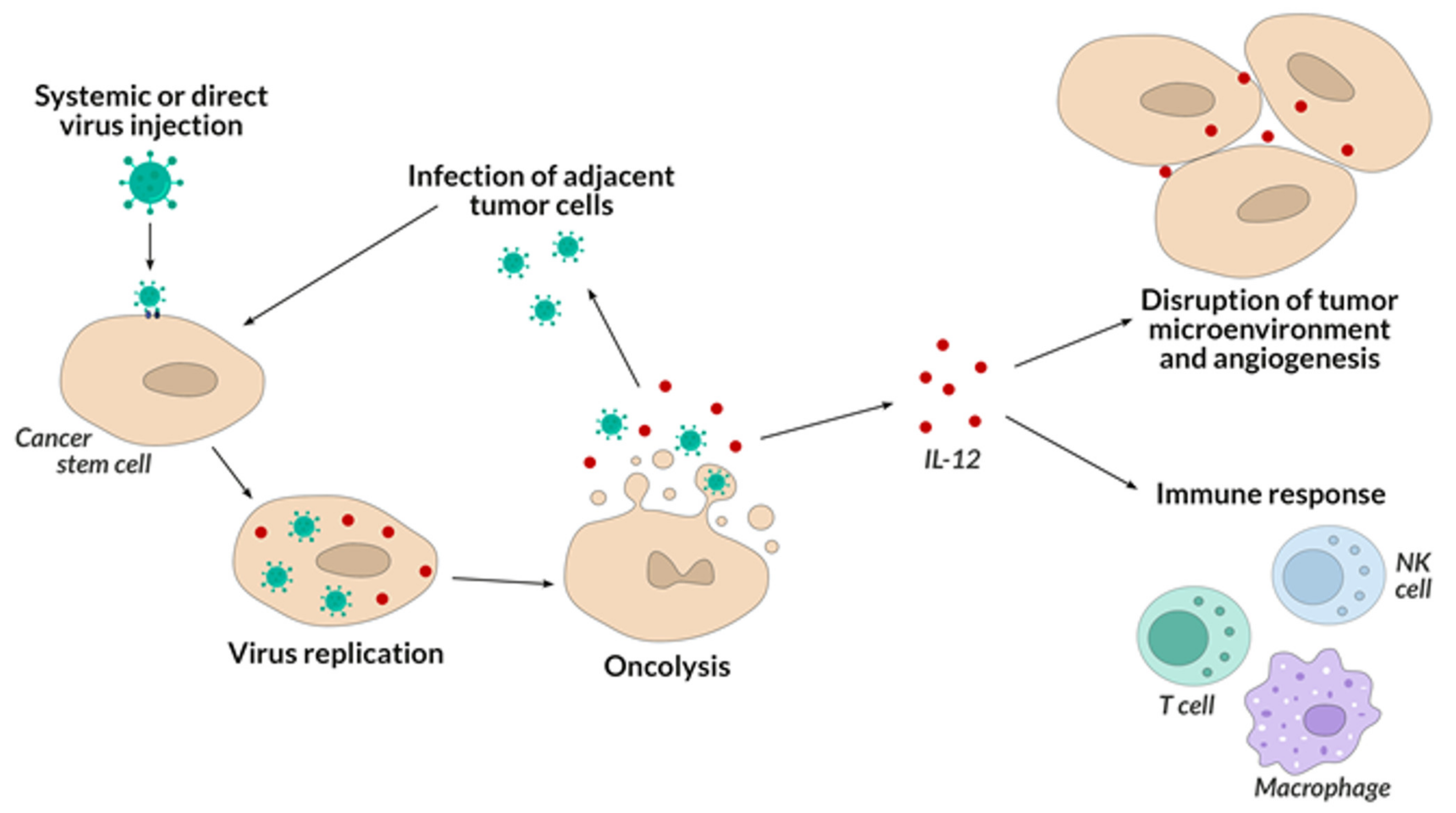
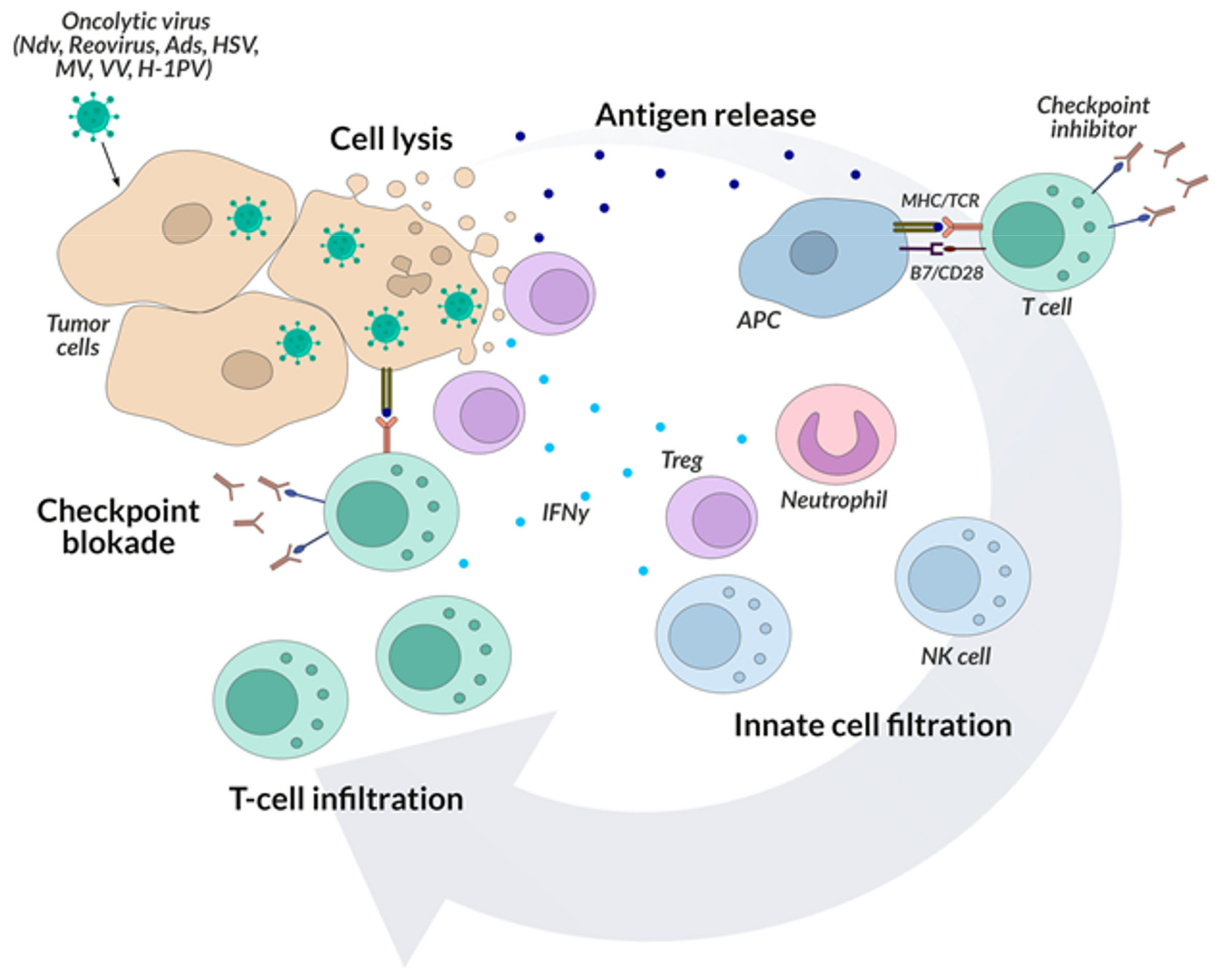
5. Oncolytic Viruses
6. Cancer Vaccines
7. Research on Improving the Effectiveness of Biological Therapies
7.1. Searching for Neo-Antigens
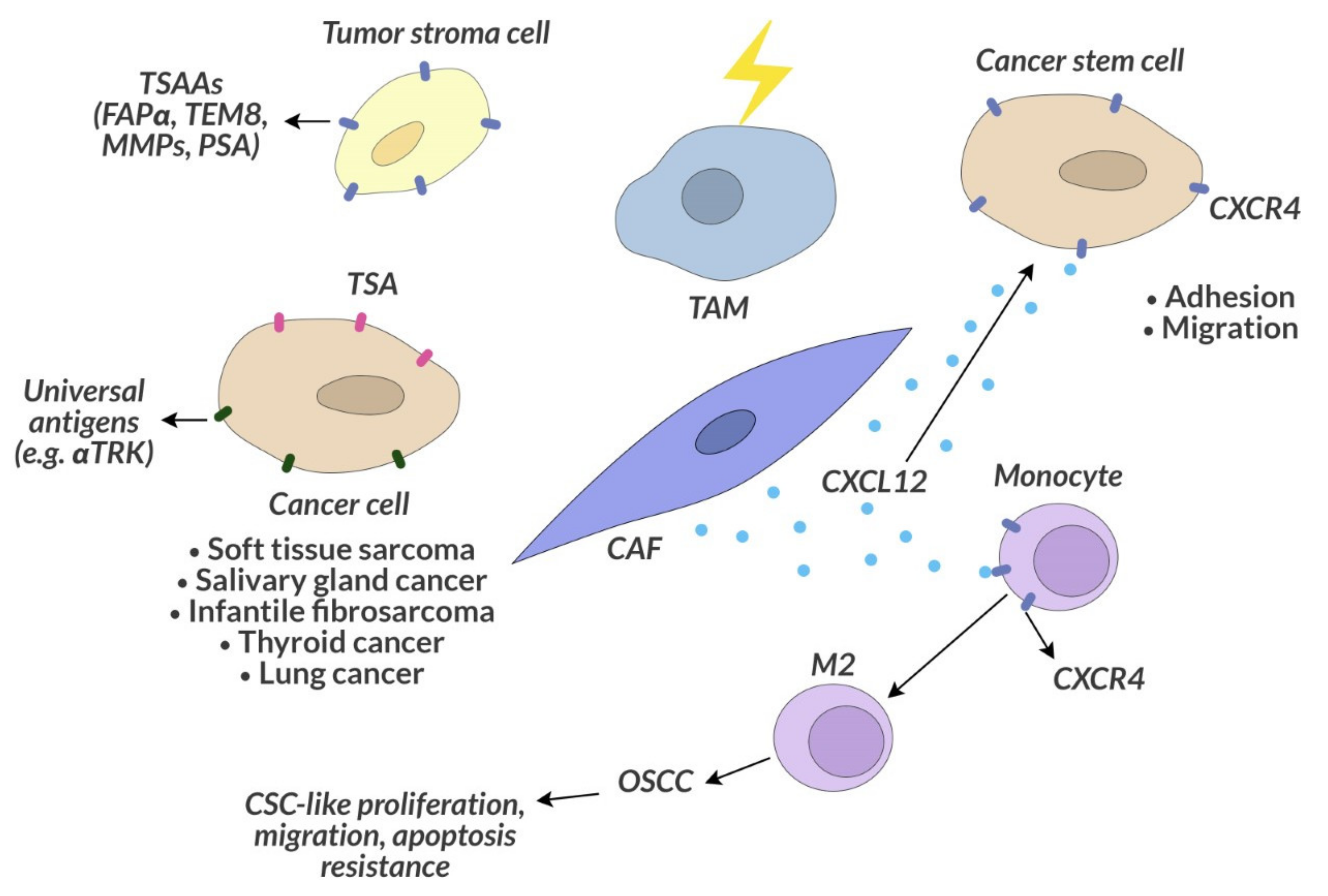
7.2. Tumor Stroma-Associated Antigens as a Target of Anticancer Therapy
7.3. Universal Therapeutic Goals
7.4. Cancer Stem Cells
7.5. Cancer Microenvironment
7.6. Release of LSCs from a Natural Niche as a Therapeutic Strategy
7.7. The Problem of Cancer Cell Heterogeneity
8. Directed Enzyme Prodrug Therapy in Treatment of Cancer
9. The Progress of Targeted Therapy
10. Applications and Potential of Biological Therapies
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- UICC. Global Cancer Control. Available online: https://www.uicc.org/news/globocan-2020-new-global-cancer-data (accessed on 24 September 2021).
- Walewski, J. Serendypity in a chase for a cure of cancer: Origin and perspectives of immunochemotherapy from nitrogen mustard to chimeric antygen receptors. Nowotwory 2015, 65, 96–102. [Google Scholar] [CrossRef]
- Han, X.J.; Ma, X.L.; Yang, L.; Wei, Y.Q.; Peng, Y.; Wei, X.W. Progress in neoantigen targeted cancer immunotherapies. Front. Cell Dev. Biol. 2020, 8, 728. [Google Scholar] [CrossRef] [PubMed]
- Kannaiyan, R.; Mahadevan, D. A comprehensive review of protein kinase inhibitors for cancer therapy. Expert Rev. Anticancer Ther. 2018, 218, 1249–1270. [Google Scholar] [CrossRef]
- Guilhot, F.; Schiffer, C.; Gambacorti-Passerini, C.; Niederwieser, D.; Resta, D.; Capdeville, R.; Zoellner, U.; Talpaz, M.; Druker, B.; Goldman, J.; et al. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N. Eng. J. Med. 2002, 346, 645–652. [Google Scholar]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Khozin, S.; Weinstock, C.; Blumenthal, G.M. Osimertinib for the treatment of metastatic EGFR T790M mutation-positive non-small cell lung cancer. Clin. Cancer Res. 2017, 23, 2131–2135. [Google Scholar] [CrossRef] [PubMed]
- Masui, H.; Kawamoto, T.; Sato, J.D.; Wolf, B.; Sato, G.; Mendelsohn, J. Growth inhibition of human tumor cells in athymic mice by anti-epidermal growth factor receptor monoclonal antibodies. Cancer Res. 1984, 44, 1002–1007. [Google Scholar] [CrossRef]
- Chames, P.; Van Regenmortel, M.; Weiss, E.; Baty, D. Therapeutic antibodies: Successes, limitations and hopes for the future. Br. J. Pharmacol. 2009, 157, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Tansey, E.M.; Catterall, P.P. Monoclonal antibodies: A witness seminar in contemporary medical history. Med. Hist. 1994, 38, 322–327. [Google Scholar] [CrossRef][Green Version]
- Neuberger, M.S.; Williams, G.T.; Mitchell, E.B.; Jouhal, S.S.; Flanagan, J.G.; Rabbitts, T.H. A hapten-specific chimaeric IgE antibody with human physiological effector function. Nature 1985, 314, 268–270. [Google Scholar] [CrossRef]
- Jones, P.T.; Dear, P.H.; Foote, J.; Neuberger, M.S.; Winter, G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 1986, 321, 522–525. [Google Scholar] [CrossRef]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef]
- Callahan, M.K.; Wolchok, J.D. At the bedside: CTLA-4- and PD-1-blocking antibodies in cancer immunotherapy. J. Leukoc. Biol. 2013, 94, 41–53. [Google Scholar] [CrossRef]
- Momtaz, P.; Postow, M.A. Immunologic checkpoints in cancer therapy: Focus on the programmed death-1 (PD-1) receptor pathway. Pharmgenomics Pers. Med. 2014, 7, 357–365. [Google Scholar] [PubMed]
- Asmar, R.; Yang, J.; Carvajal, R.D. Clinical utility of nivolumab in the treatment of advanced melanoma. Ther. Clin. Risk Manag. 2016, 12, 313–325. [Google Scholar] [PubMed]
- Lu, Y.C.; Robbins, P.F. Cancer immunotherapy targeting neoantigens. Sem. Immunol. 2016, 28, 22–27. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Instituteii. Available online: https://www.cancer.gov/about-cancer/treatment/types/immunotherapy/checkpoint-inhibitors (accessed on 24 September 2019).
- Xia, Y.; Medeiros, L.J.; Young, K.H. Immune checkpoint blockade: Releasing the brake towards hematological malignancies. Blood Rev. 2016, 30, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Grandjenette, C.; Dicato, M.; Diederich, M. Bispecific antibodies: An innovative arsenal to hunt, grab and destroy cancer cells. Curr. Pharm. Biotechnol. 2015, 16, 670–683. [Google Scholar] [PubMed]
- Lindhofer, H.; Mocikat, R.; Steipe, B.; Thierfelder, S. Preferential species-restricted heavy/light chain pairing in rat/mouse quadromas. Implications for a single-step purification of bispecific antibodies. J. Immunol. 1995, 155, 219–225. [Google Scholar] [PubMed]
- Smith, W.; Jarrett, A.L.; Beattie, R.E.; Corvalan, J.R.F. Immunoglobulins secreted by a hybrid-hybridoma: Analysis of chain assemblies. Hybridoma 1992, 11, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Chames, P.; Baty, D. Bispecific antibodies for cancer therapy. Curr. Opin. Drug Discov. Dev. 2009, 12, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Baeuerle, P.A.; Gires, O. EpCAM (CD326) finding its role in cancer. Br. J. Cancer 2007, 96, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Jager, M.; Schoberth, A.; Ruf, P.; Hess, J.; Lindhofer, H. The trifunctional antibody ertumaxomab destroys tumor cells that express low levels of human epidermal growth factor receptor 2. Cancer Res. 2009, 69, 4270–4276. [Google Scholar] [CrossRef] [PubMed]
- Zeidler, R.; Reisbach, G.; Wollenberg, B.; Lang, S.; Chaubal, S.; Schmitt, B.; Lindhofer, H. Simultaneous activation of T cells and accessory cells by a new class of intact bispecific antibody results in efficient tumor cell killing. J. Immunol. 1999, 163, 1246–1252. [Google Scholar]
- Heiss, M.M.; Murawa, P.; Koralewski, P.; Kutarska, E.; Kolesnik, O.O.; Ivanchenko, V.V.; Dudnichenko, A.S.; Aleknaviciene, B.; Razbadauskas, A.; Gore, M.; et al. The trifunctional antibody catumaxomab for the treatment of malignant ascites due to epithelial cancer: Results of a prospective randomized phase II/III trial. Int. J. Cancer 2010, 127, 2209–2221. [Google Scholar] [CrossRef]
- Heiss, M.M.; Strohlein, M.A.; Jager, M.; Kimmig, R.; Burges, A.; Schoberth, A.; Jauch, K.W.; Schildberg, F.W.; Lindhofer, H. Immunotherapy of malignant ascites with trifunctional antibodies. Int. J. Cancer 2005, 117, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Ströhlein, M.A.; Siegel, R.; Jäger, M.; Lindhofer, H.; Jauch, K.W.; Heiss, M.M. Induction of anti-tumor immunity by trifunctional antibodies in patients with peritoneal carcinomatosis. J. Exp. Clin. Cancer Res. 2009, 28, 18. [Google Scholar] [CrossRef] [PubMed]
- Kiewe, P.; Hasmüller, S.; Kahlert, S.; Heinrigs, M.; Rack, B.; Marmé, A.; Korfel, A.; Jäger, M.; Lindhofer, H.; Sommer, H.; et al. Phase I trial of the trifunctional anti-HER2 x anti-CD3 antibody ertumaxomab in metastatic breast cancer. Clin. Cancer Res. 2006, 12, 3085–3091. [Google Scholar] [CrossRef] [PubMed]
- Diermeier-Daucher, S.; Ortmann, O.; Buchholz, S.; Brockhoff, G. Trifunctional antibody ertumaxomab Non-immunological effects on Her2 receptor activity and downstream signaling. mAbs 2012, 4, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Bartlett, N.L.; Assouline, S.; Yoon, S.S.; Bosch, F.; Sehn, L.H.; Cheah, C.J.; Shadman, M.; Gregory, G.P.; Ku, M.; et al. Mosunetuzumab induces complete remissions in poor prognosis non-Hodgkin lymphoma patients, including those who are resistant to or relapsing after chimeric antigen receptor T-cell (CAR-T) therapies, and Is active in treatment through multiple lines. In Proceedings of the 2019 ASH Annual Meeting, Orlando, FL, USA, 8 December 2019. Abstract 6. [Google Scholar]
- Yuan, F.; Dellian, M.; Fukumura, D.; Leunig, M.; Berk, D.A.; Torchilin, V.P.; Jain, R.K. Vascular permeability in a human tumor xenograft: Molecular size dependence and cutoff size. Cancer Res. 1995, 55, 3752–3756. [Google Scholar]
- Fujimori, K.; Covell, D.G.; Fletcher, J.E.; Weinstein, J.N. A modeling analysis of monoclonal antibody percolation through tumors: A binding-site barrier. J. Nucl. Med. 1990, 31, 1191–1198. [Google Scholar]
- Adams, G.P.; Schier, R.; McCall, A.M.; Simmons, H.H.; Horak, E.M.; Alpaugh, R.K.; Marks, J.D.; Weiner, L.M. High affinity restricts the localization and tumor penetration of single-chain fv antibody molecules. Cancer Res. 2001, 61, 4750–4755. [Google Scholar]
- Muchekehu, R.; Liu, D.; Horn, M.; Campbell, L.; Del Rosario, J.; Bacica, M.; Moskowitz, H.; Osothprarop, T.; Dirksen, A.; Doppalapudi, V.; et al. The Effect of molecular weight, PK, and valency on tumor biodistribution and efficacy of antibody-based drugs. Transl. Oncol. 2013, 6, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Rubin, K.; Pietras, K.; Östman, A. High interstitial fluid pressure—An obstacle in cancer therapy. Nat. Rev. Cancer 2004, 4, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Cartron, G.; Dacheux, L.; Salles, G.; Solal-Celigny, P.; Bardos, P.; Colombat, P.; Watier, H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood 2002, 99, 754–758. [Google Scholar] [CrossRef]
- Shinkawa, T.; Nakamura, K.; Yamane, N.; Shoji-Hosaka, E.; Kanda, Y.; Sakurada, M.; Uchida, K.; Anazawa, H.; Satoh, M.; Yamasaki, M.; et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J. Biol. Chem. 2003, 278, 3466–3473. [Google Scholar] [CrossRef] [PubMed]
- Preithner, S.; Elm, S.; Lippold, S.; Locher, M.; Wolf, A.; da Silva, A.J.; Baeuerle, P.A.; Prang, N.S. High concentrations of therapeutic IgG1 antibodies are needed to compensate for inhibition of antibody-dependent cellular cytotoxicity by excess endogenous immunoglobulin G. Mol. Immunol. 2006, 43, 1183–1193. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Fc-receptors as regulators of immunity. Adv. Immunol. 2007, 96, 179–204. [Google Scholar] [PubMed]
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 2005, 23, 1126–1136. [Google Scholar] [CrossRef]
- Whitlow, M.; Filpula, D.; Rollence, M.L.; Feng, S.L.; Wood, J.F. Multivalent Fvs: Characterization of single-chain Fv oligomers and preparation of a bispecific Fv. Protein Eng. 1994, 7, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, F.; Kipriyanov, S.M.; Moldenhauer, G.; Little, M. Di, tri and tetrameric single chain Fv antibody fragments against human CD19: Effect of valency on cell binding. FEBS Lett. 1999, 453, 164–168. [Google Scholar] [CrossRef]
- Koren, T.; Volkel, T.; Kontermann, R.E. Bivalent and Bispecific Diabodies and Single-chain Diabodies. R. In Antibody Engineering; Kontermann, R.E., Dübel, S., Eds.; Springer: Berlin/Heidelberg, Germany, 2001; pp. 619–636. [Google Scholar]
- Kipriyanov, S.M.; Cochlovius, B.; Schafer, H.J.; Moldenhauer, G.; Bahre, A.; Le Gall, F.; Knackmuss, S.; Little, M. Synergistic antitumor effect of bispecific CD19 × CD3 and CD19 × CD16 diabodies in a preclinical model of non-Hodgkin’s lymphoma. J. Immunol. 2002, 169, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Brüsselbach, S.; Korn, T.; Völkel, T.; Müller, R.; Kontermann, R.E. Enzyme recruitment and tumor cell killing in vitro by a secreted bispecific single-chain diabody. Tumor Target. 1999, 4, 115–123. [Google Scholar]
- Baum, V.; Buhler, P.; Gierschner, D.; Herchenbach, D.; Fiala, G.J.; Schamel, W.W.; Wolf, P.; Elsasser-Beile, U. Antitumor activities of PSMAxCD3 diabodies by redirected T-cell lysis of prostatę cancer cells. Immunotherapy 2013, 5, 27–38. [Google Scholar] [CrossRef]
- Fortmuller, K.; Alt, K.; Gierschner, D.; Wolf, P.; Baum, V.; Freudenberg, N.; Wetterauer, U.; Elsasser-Beile, U.; Buhler, P. Effective targeting of prostate cancer by lymphocytes redirected by a PSMA x CD3 bispecific single-chain diabody. Prostate 2011, 71, 588–596. [Google Scholar] [CrossRef]
- Lieberman, J. The ABCs of granule-mediated cytotoxicity: New weapons in the arsenal. Nat. Rev. Immunol. 2003, 3, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Wischhusen, J.; Schneider, D.; Mittelbronn, M.; Meyermann, R.; Engelmann, H.; Jung, G.; Wiendl, H.; Weller, M. Death receptormediated apoptosis in human malignant glioma cells: Modulation by the CD40/CD40L system. J. Neuroimmunol. 2005, 162, 28–42. [Google Scholar] [CrossRef]
- Huse, M.; Catherine Milanoski, S.; Abeyweera, T.P. Building tolerance by dismantling synapses: Inhibitory receptor signaling in natural killer cells. Immunol. Rev. 2013, 251, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Topp, M.S.; Kufer, P.; Gokbuget, N.; Goebeler, M.; Klinger, M.; Neumann, S.; Horst, H.A.; Raff, T.; Viardot, A.; Schmid, M.; et al. Targeted therapy with the T-cellengaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J. Clin. Oncol. 2011, 29, 2493–2498. [Google Scholar] [CrossRef]
- Kantarjian, H.; Stein, A.; Gokbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.M.; Wei, A.; Dombret, H.; Foà, R.; Bassan, R.; et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef]
- Stein, A.S.; Larson, R.A.; Schuh, A.C.; Stevenson, W.; Lech-Maranda, E.; Tran, Q.; Zimmerman, Z.; Kormany, W.; Topp, M.S. Exposure-adjusted adverse events comparing blinatumomab with chemotherapy in advanced acute lymphoblastic leukemia. Blood Adv. 2018, 2, 1522–1531. [Google Scholar] [CrossRef] [PubMed]
- Klinger, M.; Brandl, C.; Zugmaier, G.; Hijazi, Y.; Bargou, R.C.; Topp, M.S.; Gokbuget, N.; Neumann, S.; Goebeler, M.; Viardot, A.; et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood 2012, 119, 6226–6633. [Google Scholar] [CrossRef]
- Loffler, A.; Kufer, P.; Lutterbuse, R.; Zettl, F.; Daniel, P.T.; Schwenkenbecher, J.M.; Riethmüller, G.; Dörken, B.; Bargou, R.C. A recombinant bispecific single-chain antibody, CD19 × CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood 2000, 95, 2098–2103. [Google Scholar] [CrossRef] [PubMed]
- Weiner, L.M.; Clark, J.I.; Davey, M.; Li, W.S.; Garcia de Palazzo, I.; Ring, D.B.; Alpaugh, R.K. Phase I trial of 2B1, a bispecific monoclonal antibody targeting c-erbB-2 and Fc gamma RIII. Cancer Res. 1995, 55, 4586–4593. [Google Scholar] [PubMed]
- Apel, A.; Ofran, Y.; Wolach, O.; Shimony, S.; Ram, R.; Levi, I.; Zektser, M.; Koren-Michowitz, M. Safety anf efficacy of blinatumomab: A real world data. Ann. Hematol. 2020, 99, 835–838. [Google Scholar] [CrossRef] [PubMed]
- Hummel, D.H.; Kufer, P.; Grüllich, C.; Deschler-Baier, B.; Chatterjee, M.; Goebeler, M.E.; Miller, K.; De Santis, M.; Loidl, W.C.; Buck, A.; et al. Phase I study of pasotuxizumab (AMG 212/BAY 2010112), a PSMA-targeting BiTE (Bispecific T-cell Engager) immune therapy for metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2020, 38, 124. [Google Scholar] [CrossRef]
- Clinical Trials. Available online: https://clinicaltrials.gov/ct2/show/NCT01723475 (accessed on 27 September 2019).
- Gleason, M.K.; Verneris, M.R.; Todhunter, D.A.; Zhang, B.; McCullar, V.; Zhou, S.X.; Panoskaltsis-Mortari, A.; Weiner, L.M.; Vallera, D.A.; Miller, J.S. Bispecific and trispecific killer cel engagers directly activate human NK cells through CD16 signaling and induce cytotoxicity and cytokine production. Mol. Cancer Ther. 2012, 11, 2674–2684. [Google Scholar] [CrossRef]
- Felices, M.; Lenvik, T.R.; Davis, Z.B.; Miller, J.S.; Daniel, A.; Vallera, D.A. Generation of BiKEs and TriKEs to improve NK cell-mediated targeting of tumor cells. Methods Mol. Biol. 2016, 1441, 333–346. [Google Scholar] [PubMed]
- Wolf, E.; Hofmeister, R.; Kufer, P.; Schlereth, B.; Baeuerle, P.A. BiTEs: Bispecific antibody constructs with unique anti-tumor activity. Drug Discov. Today 2005, 10, 1237–1244. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Reinhardt, C. Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res. 2009, 69, 4941–4944. [Google Scholar] [CrossRef]
- Lorenczewski, G.; Friedrich, M.; Kischel, R.; Dahlhoff, C.; Anlahr, J.; Balázs, M.; Rock, D.; Boyle, M.; Goldstein, R.; Coxon, A.; et al. Generation of a half-life extended anti-CD19 BiTE® antibody construct compatible with once-weekly dosing for treatment of CD19-positive malignancies. Blood 2017, 130, 2815. [Google Scholar]
- Moore, P.A.; Zhang, W.; Rainey, G.J.; Burke, S.; Li, H.; Huang, L.; Gorlatov, S.; Veri, M.C.; Aggarwal, S.; Yang, Y.; et al. Application of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B-cell lymphoma. Blood 2011, 117, 4542–4551. [Google Scholar] [CrossRef] [PubMed]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Pfeerle, A.; Nicholas, D.; Huntington, N.D. You have got a fast CAR: Chimeric antigen receptor NK cells in cancer therapy. Cancers 2020, 12, 706. [Google Scholar]
- Savoldo, B.; Ramos, C.A.; Liu, E.; Mims, M.P.; Keating, M.J.; Carrum, G.; Kamble, R.T.; Bollard, C.M.; Gee, A.P.; Mei, Z.; et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J. Clin. Investig. 2011, 121, 1822–1826. [Google Scholar] [CrossRef]
- Guedan, S.; Posey, A.D., Jr.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3, e96976. [Google Scholar] [CrossRef]
- Stoiber, S.; Cadilha, B.L.; Benmebarek, M.R.; Lesch, S.; Endres, S.; Kobold, S. Limitations in the design of chimeric antigen receptors for cancer therapy. Cells 2019, 8, 472. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Condomines, M.; van der Stegen, S.J.C.; Perna, F.; Kloss, C.C.; Gunset, G.; Plotkin, J.; Sadelain, M. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. 2015, 28, 415–428. [Google Scholar] [CrossRef]
- Van der Stegen, S.J.; Hamieh, M.; Sadelain, M. The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov. 2015, 14, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 16, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Santomasso, B.; Bachier, C.; Westin, J.; Rezvani, K.; Shpall, E.J. The Other Side of CAR T-Cell Therapy: Cytokine Release Syndrome, Neurologic Toxicity, and Financial Burden. In ASCO Educational Book, Proceedings of the 55th Annual Meeting of American Society of Clinical Oncology, Chicago, IL, USA, 31 May–4 June 2019; Pennell, N., Rugo, H.S., Eds.; American Society of Clinical Oncology: Alexandria, WV, USA, 2019; Volume 39, pp. 433–444. [Google Scholar]
- Koneru, M.; Purdon, T.J.; Spriggs, D.; Koneru, S.; Brentjens, R.J. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. OncoImmunology 2015, 4, e994446. [Google Scholar] [CrossRef]
- Zajca, C.U.; Dobersbergera, M.; Schaffnerc, I.; Mlynekd, G.; Pühringerd, D.; Salzera, B.; Djinovic-Carugod, K.; Steinbergerf, P.; De Sousa Linhares, A.; Yangg, N.J.; et al. A conformation-specific ON-switch for controlling CAR T cells with an orally available drug. Proc. Natl. Acad. Sci. USA 2020, 117, 4926–14935. [Google Scholar] [CrossRef]
- Jones, B.S.; Lamb, L.S.; Goldman, F.; Di Stasi, A. Improving the safety of cell therapy products by suicide gene transfer. Front. Pharmacol. 2014, 5, 254. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Hao, H.; Yang, G.; Zhang, Y.; Fu, Y. Immunotherapy with CAR-modified T cells: Toxicities and overcoming strategies. J. Immunol. Res. 2018, 17, 2386187. [Google Scholar] [CrossRef]
- Diaconu, I.; Ballard, B.; Zhang, M.; Chen, Y.; West, J.; Dotti, G.; Savoldo, B. Inducible caspase-9 selectively modulates the toxicities of CD19-specific chimeric antigen receptor-modified T cells. Mol. Ther. 2017, 25, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Stavrou, M.; Philip, B.; Traynor-White, C.; Shimobi Onuoha, C.G.D.; Cordoba, S.; Thomas, S.; Pule, M. A rapamycin-activated caspase 9-based suicide gene. Mol. Ther. 2018, 26, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, F.; et al. Chimeric antigen receptor–modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 18, 1509–1518. [Google Scholar] [CrossRef]
- Singh, N.; Frey, N.V.; Grupp, S.A.; Maude, S.L. CAR T cell therapy in acute lymphoblastic leukemia and potential for chronic lymphocytic leukemia. Curr. Treat. Options Oncol. 2016, 17, 28. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Cao, L.; Xie, J.; Shi, N.; Zhang, Z.; Luo, Z.; Yue, D.; Zhang, Z.; Wang, L.; Han, W.; et al. Efficiency of CD19 chimeric antigen receptor-modified T cells for treatment of B cell malignancies in phase I clinical trials: A meta-analysis. Oncotarget 2015, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wei, G.; Liu, D. CD19: A biomarker for B cell development, lymphoma diagnosis and therapy. Exp. Hematol. Oncol. 2012, 1, 36. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Kuramitsu, S.; Posey, A.D.; June, C.H. Expanding the therapeutic window for CAR T cel therapy in solid tumors: The knowns and unknowns of CAR T cell biology. Front. Immunol. 2018, 9, 2486. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Chinnasamy, N.; Abate-Daga, D.; Gros, A.; Robbins, P.F.; Zheng, Z.; Dudley, M.E.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 2013, 36, 133–151. [Google Scholar] [CrossRef] [PubMed]
- Wickström, S.L.; Lövgren, T.; Volkmar, M.; Reinhold, B.; Duke-Cohan, J.S.; Hartmann, L.; Rebmann, J.; Mueller, A.; Melief, J.; Maas, R.; et al. Cancer neoepitopes for immunotherapy: Discordance between tumor-Infiltrating T cell reactivity and tumor MHC peptidome display. Front. Immunol. 2019, 10, 2766. [Google Scholar] [CrossRef]
- Verdegaal, E.M.E.; de Miranda, N.F.C.C.; Visser, M.; Harryvan, T.; van Buuren, M.M.; Andersen, R.S.; Hadrup, S.R.; van der Minne, C.E.; Schotte, R.; Spits, H.; et al. Neoantigen landscape dynamics during human melanoma–T cell interactions. Nature 2016, 536, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Ninomiya, S.; Narala, N.; Huye, L.; Yagyu, S.; Savoldo, B.; Dotti, G.; Heslop, E.H.; Brenner, M.K.; Rooney, C.M.; Ramos, C.A. Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood 2015, 18, 3905–3916. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Hou, L.; Liu, J.; Liu, Y.; Ning, J.; Zhao, Q.; Zhang, Y. Indoleamine 2,3-dioxygenase 1 inhibitor-loaded nanosheets enhance CAR-T cell function in esophageal squamous cell carcinoma. Front. Immunol. 2021, 22, 661357. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Javeed, A.; Ashraf, M.; Yuzhu, H.; Mukhtar, M.M. Multilevel pharmacological manipulation of adenosine–prostaglandin E2/cAMP nexus in the tumor microenvironment: A ‘two hit’therapeutic opportunity. Pharm. Res. 2013, 73, 8–19. [Google Scholar] [CrossRef]
- Gorby, C.; Bellón, J.S.; Wilmes, S.; Warda, W.; Pohler, E.; Fyfe, P.K.; Cozzani, A.; Ferrand, C.; Walter, M.R.; Mitra, S.; et al. Engineered IL-10 variants elicit potent immunomodulatory effects at low ligand doses. Sci. Signal. 2020, 13, eabc0653. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, H.; Luo, H.; Sun, Y.; Shi, B.; Sun, R.; Li, Z. An IL-4/21 Inverted cytokine receptor improving CAR-T cell potency in immunosuppressive solid-tumor microenvironment. Front. Immunol. 2019, 10, 1691. [Google Scholar] [CrossRef]
- Chruściel, E.; Urban-Wójciuk, Z.; Arcimowicz, Ł.; Kurkowiak, M.; Kowalski, J.; Gliwiński, M.; Marjański, T.; Rzyman, W.; Biernat, W.; Dziadziuszko, R.; et al. Adoptive cell therapy—Harnessing antigen-specific T cells to target solid tumours. Cancers 2020, 12, 683. [Google Scholar] [CrossRef]
- Gaud, G.; Lesourne, R.; Love, P.E. Regulatory mechanisms in T cell receptor signalling. Nat. Rev. Immunol. 2018, 18, 485–497. [Google Scholar] [CrossRef]
- Van den Berg, J.H.; Heemskerk, B.; van Rooij, N.; Gomez-Eerland, R.; Michels, S.; van Zon, M.; de Boer, R.; Bakker, N.A.M.; Jorritsma-Smit, A.; van Buuren, M.M.; et al. Tumor infiltrating lymphocytes (TIL) therapy in metastatic melanoma: Boosting of neoantigen-specific T cell reactivity and long-term followup. J. Immunother. Cancer 2020, 8, e000848. [Google Scholar] [CrossRef] [PubMed]
- Westergaard, M.C.W.; Andersen, R.; Chong, C.; Kjeldsen, J.W.; Pedersen, M.; Friese, C.; Hasselager, T.; Lajer, H.; Coukos, G.; Bassani-Sternberg, M.; et al. Tumour-reactive T cell subsets in the microenvironment of ovarian cancer. Br. J. Cancer 2019, 120, 424–434. [Google Scholar] [CrossRef]
- Seliktar-Ofir, S.; Merhavi-Shoham, E.; Itzhaki, O.; Yunger, S.; Markel, G.; Schachter, J.; Besser, M.J. Selection of shared and neoantigen-reactive T cells for adoptive cell therapy based on CD137 separation. Front. Immunol. 2017, 8, 1211. [Google Scholar] [CrossRef]
- Dai, H.; Wu, Z.; Jia, H.; Tong, C.; Guo, Y.; Ti, D.; Han, X.; Liu, Y.; Zhang, W.; Wang, C.; et al. Bispecific CAR-T cells targeting both CD19 and CD22 for therapy of adults with relapsed or refractory B cell acute lymphoblastic leukemia. J. Hematol. Oncol. 2020, 13, 30. [Google Scholar] [CrossRef] [PubMed]
- Ingle, G.S.; Chan, P.; Elliott, J.M.; Chang, W.S.; Koeppen, H.; Stephan, J.P.; Scales, S.J. High CD21 expression inhibits internalization of anti-CD19 antibodies and cytotoxicity of an anti-CD19-drug conjugate. Br. J. Haematol. 2008, 140, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Ruella, M.; Maus, M.V. Catch me if you can: Leukemia escape after CD19-directed T cell immunotherapies. Comput. Struct. Biotechnol. J. 2016, 14, 357–362. [Google Scholar] [CrossRef]
- Zheng, L.; Ren, L.; Kouhi, A.; Khawli, L.A.; Hu, P.; Kaslow, H.R.; Epstein, A.L. A humanized Lym-1 CAR with novel DAP10/DAP12 signaling domains demonstrates reduced tonic signaling and increased anti-tumor activity in B Cell Lymphoma models. Clin. Cancer Res. 2020, 26, 3694–3706. [Google Scholar] [CrossRef]
- DeNardo, G.L.; DeNardo, S.J.; Lamborn, K.R.; Goldstein, D.S.; Levy, N.B.; Lewis, J.P.; O’Grady, L.F.; Raventos, A.; Kroger, L.A.; Macey, D.J.; et al. Low-dose, fractionated radioimmunotherapy for B-cell malignancies using 131I-Lym-1 antibody. Cancer Biother. Radiopharm. 1998, 13, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Leivas, A.; Rio, P.; Mateos, R.; Paciello, M.L.; Garcia-Ortiz, A.; Fernandez, L.; Perez-Martinez, A.; Lee, D.A.; Powell, D.J.; Valeri, A.; et al. NKG2D-CAR transduced primary natural killer cells efficiently target multiple myeloma cells. Blood 2018, 132, 590. [Google Scholar] [CrossRef]
- Muik, A.; Stubbert, L.J.; Jahedi, R.Z.; Geib, Y.; Kimpel, J.; Dold, C.; Tober, R.; Volk, A.; Klein, S.; Dietrich, U.; et al. Re-engineering vesicular stomatitis virus to abrogate neurotoxicity, circumvent humoral immunity, and enhance oncolytic potency. Cancer Res. 2014, 74, 3567–3578. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.P.; Kim, Y.; Clements, D.R.; Konda, P.; Schuster, H.; Kowalewski, D.J.; Paulo, J.A.; Cohen, A.M.; Stevanovic, S.; Gygi, S.P.; et al. Therapy-Induced MHC I Ligands Shape Neo-Antitumor CD8 T Cell Responses during Oncolytic Virus-Based Cancer Immunotherapy. J. Proteome Res. 2019, 18, 2666–2675. [Google Scholar] [CrossRef]
- Gong, J.; Sachdev, E.; Mita, A.C.; Mita, M.M. Clinical development of reovirus for cancer therapy: An oncolytic virus with immune-mediated antitumor activity. World J. Methodol. 2016, 6, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.L.; Robinson, M.; Han, Z.Q.; Branston, R.H.; English, C.; Reay, P.; McGrath, Y.; Thomas, S.K.; Thornton, M.; Bullock, P.; et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003, 10, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.S.; Lorence, R.M.; Groene, W.S.; Bamat, M.K. Naturally oncolytic viruses. Curr. Opin. Mol. Ther. 2006, 8, 314–321. [Google Scholar]
- Anderson, B.D.; Nakamura, T.; Russell, S.J.; Peng, K.W. High CD46 receptor density determines preferential killing of tumor cells by oncolytic measles virus. Cancer Res. 2004, 64, 4919–4926. [Google Scholar] [CrossRef]
- Hu, J.C.C.; Coffin, R.S.; Davis, C.J. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophagecolony-stimulating factor. Clin. Cancer Res. 2006, 12, 6737–6747. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Li, Q.; Lao, X.; Yang, H.; Li, S. Transarterial injection of recombinant human type-5 adenovirus H101 in combination with transarterial chemoembolization (TACE) improves overall and progressive-free survival in unresectable hepatocellular carcinoma (HCC). BMC Cancer 2015, 15, 707. [Google Scholar] [CrossRef]
- Chen, N.; Zhang, Q.; Yu, Y.A.; Stritzker, J.; Brader, P.; Schirbel, A.; Samnick, S.; Serganova, I.; Blasberg, R.; Fong, Y.; et al. A novel recombinant vaccinia virus expressing the human norepinephrine transporter retains oncolytic potential and facilitates deep-tissue imaging. Mol. Med. 2009, 15, 144–151. [Google Scholar] [CrossRef]
- Andtbacka, R.H.; Ross, M.; Puzanov, I.; Milhem, M.; Collichio, F.; Delman, K.A.; Amatruda, T.; Zager, J.S.; Cranmer, L.; Hsueh, E.; et al. Patterns of clinical response with Talimogene Laherparepvec (T-VEC) in patients with melanoma treated in the OPTiM phase III clinical trial. Ann. Surg. Oncol. 2016, 23, 4169–4177. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Available online: https://www.fda.gov/downloads/biologicsbloodvaccines/cellulargenetherapyproducts/approvedproducts/ucm469575 (accessed on 16 December 2020).
- He, B.; Chou, J.; Brandimarti, R.; Mohr, I.; Gluzman, Y.; Roizman, B. Suppression of the phenotype of g(1)34.5- herpes simplex virus 1: Failure of activated RNA-dependent protein kinase to shutoff protein synthesis is associated with a deletion in the domain of the a47 gene. J. Virol. 1997, 71, 6049–6054. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene laher parepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Fang, H. Clinical trials with oncolytic adenovirus in China. Curr. Cancer Drug Targets 2007, 7, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, N.; Ge, Y.; Ennist, D.L.; Zhu, M.; Mina, M.; Ganesh, S.; Reddy, P.S.; Yu, D.C. CG0070, a conditionally replicating granulocyte macrophage colony-stimulating factor–armed oncolytic adenowirus for the treatment of bladder cancer. Clin. Cancer Res. 2006, 12, 305–313. [Google Scholar] [CrossRef]
- Zwicker, J.; Muller, R. Cell cycle-regulated transcription in mammalian cells. Prog. Cell Cycle Res. 1995, 1, 91–99. [Google Scholar]
- Dawn, E.; Post, D.E.; Van Meir, E.G. A novel hypoxia-inducible factor (HIF)activated oncolytic adenovirus for cancer therapy. Oncogene 2003, 22, 2065–2072. [Google Scholar]
- Rodriguez, R.; Schuur, E.R.; Lim, H.Y.; Henderson, G.A.; Simons, J.W.; Henderson, D.R. Prostate attenuated replication competent adenowirus (ARCA) CN706: A selective cytotoxic for prostatę-sepecific antygen-positive prostatę cancer cells. Cancer Res. 1997, 57, 2559–2563. [Google Scholar]
- Zheng, M.; Huang, J.; Tong, A.; Yang, H. Oncolytic viruses for cancer therapy: Barriers and recent advances. Mol. Ther. Oncolytics 2019, 15, 234–247. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Rruby, C.E.; Hughes, T.; Slingluff, C.L. Current status of granulocyte-macrophage colony-stimulating factor in the immunotherapy of melanoma. J. Immunother. Cancer 2014, 2, 11. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.J.; Chan, M.K.; Yu, Z.; Kim, T.H.; Bhargava, A.; Stiles, B.M.; Horsburgh, B.C.; Shah, J.P.; Ghossein, R.A.; Singh, B.; et al. Effective intravenous therapy of murine pulmonary metastases with an oncolytic herpes virus expressing interleukin 12. Clin. Cancer Res. 2004, 10, 251–259. [Google Scholar] [CrossRef][Green Version]
- Lou, E. Oncolytic herpes viruses as a potential mechanism for cancer therapy. Acta Oncol. 2003, 42, 660–671. [Google Scholar] [CrossRef]
- Taguchi, S.; Fukuhara, H.; Todo, T. Oncolytic virus therapy in Japan: Progress in clinical trials and future perspectives. Jpn. J. Clin. Oncol. 2019, 49, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Cockle, J.V.; Brüning-Richardson, A.; Scott, K.J.; Thompson, J.; Kottke, T.; Morrison, E.; Ismail, A.; Carcaboso, A.M.; Rose, A.; Selby, P.; et al. Oncolytic Herpes Simplex Virus inhibits pediatric brain tumor migration and invasion. Mol. Ther. Oncolytics 2017, 2, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.M.; Johnston, J.M.; Reddy, A.T.; Fiveash, J.; Madan-Swain, A.; Kachurak, K.; Bag, A.K.; Gillespie, G.Y.; Markert, J.M.; Friedman, G.K. Rationale and design of a phase I clinical trial to evaluate HSV G207 alone or with a single radiation dose in children with progressive or recurrent malignant supratentorial brain tumors. Hum. Gene Ther. Clin. Dev. 2017, 28, 7–16. [Google Scholar] [CrossRef]
- Patel, D.M.; Foreman, P.M.; Nabors, L.B.; Riley, K.O.; Gillespie, G.Y.; Markert, J.M. Design of a phase I clinical trial to evaluate M032, a genetically engineered HSV-1 expressing IL-12, in patients with recurrent/progressive glioblastoma multiforme, anaplastic astrocytoma, or gliosarcoma. Hum. Gene Ther. Clin. Dev. 2016, 27, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Blass, E.; Ott, P.A. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat. Rev. Clin. Oncol. 2021, 18, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Franzoi, M.A.; Romano, E.; Piccart, M. Immunotherapy for early breast cancer: Too soon, too superficial, or just right? Ann. Oncol. 2021, 32, 323–336. [Google Scholar] [CrossRef]
- Chaurasiya, S.; Chen, N.G.; Fong, Y. Oncolytic viruses and immunity. Curr. Opin. Immunol. 2018, 51, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Haines, B.B.; Denslow, A.; Grzesik, P.; Lee, J.S.; Farkaly, T.; Hewett, J.; Wambua, D.; Kong, L.; Behera, P.; Jacques, J.; et al. ONCR-177, an Oncolytic HSV-1 Designed to Potently Activate Systemic Antitumor Immunity. Cancer Immunol. Res. 2021, 9, 291–308. [Google Scholar] [CrossRef] [PubMed]
- Silk, A.W.; Kaufman, H.; Gabrail, N.; Mehnert, J.; Bryan, J.; Norrell, J.; Medina, D.; Bommareddy, P.; Shafren, D.; Grose, M.; et al. Phase 1b study of intratumoral Coxsackievirus A21 (CVA21) and systemic pembrolizumab in advanced melanoma patients: Interim results of the CAPRA clinical trial. Cancer Res. 2017, 77, CT026. [Google Scholar]
- Puzanov, I.; Milhem, M.M.; Minor, D.; Hamid, O.; Li, A.; Chen, L.; Chastain, M.; Gorski, K.S.; Anderson, A.; Chou, J.; et al. Talimogene Laherparepvec in Combination With Ipilimumab in Previously Untreated, Unresectable Stage IIIB-IV Melanoma. J. Clin. Oncol. 2016, 34, 2619–2626. [Google Scholar] [CrossRef]
- Chiang, C.L.; Coukos, G.; Kandalaft, L.E. Whole tumor antigen vaccines: Where are we? Vaccines 2015, 3, 344–372. [Google Scholar] [CrossRef] [PubMed]
- Lutz, E.; Yeo, C.J.; Lillemoe, K.D.; Biedrzycki, B.; Kobrin, B.; Herman, J.; Sugar, E.; Piantadosi, S.; Cameron, J.L.; Solt, S.; et al. A lethally irradiated allogeneic granulocyte-macrophage colony stimulating factor secreting tumor vaccine for pancreatic adenocarcinoma. A phase II trial of safety, efficacy, and immune activation. Ann. Surg. 2011, 253, 328–335. [Google Scholar] [PubMed]
- Melief, C.J.M.; Welters, M.J.P.; Vergote, I.; Kroep, J.R.; Kenter, G.G.; Ottevanger, P.B.; Tjalma, W.A.A.; Denys, H.; van Poelgeest, M.I.E.; Nijman, H.W.; et al. Strong vaccine responses during chemotherapy are associated with prolonged cancer survival. Sci. Transl. Med. 2020, 12, eaaz8235. [Google Scholar] [CrossRef] [PubMed]
- Moderna. Available online: https://www.modernatx.com/pipeline/therapeuticareas/immuno-oncology (accessed on 21 September 2021).
- Chick, R.; Faries, M.; Hale, D.; Bohan, P.; Hickerson, A.; Vreeland, T.; Myers, J.W.; Cindass, J.L.; Brown, T.A.; Hyngstrom, J.R.; et al. Multiinstitutional, prospective, randomized, double-blind, placebo-controlled phase IIb trial of the tumor lysate, particle-loaded, dendritic cell (TLPLDC) vaccine to prevent recurrence in high-risk melanoma patients: A subgroup analysis. J. Clin. Oncol. 2020, 38, 63. [Google Scholar] [CrossRef]
- Hollingsworth, R.E.; Jansen, K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 2019, 4, 7. [Google Scholar] [CrossRef]
- Liu, J.; Miao, L.; Sui, J.; Hao, Y.; Huang, G. Nanoparticle cancer vaccines: Design considerations and recent advances. Asian J. Pharm. Sci. 2020, 15, 576–590. [Google Scholar] [CrossRef]
- Osipov, A.; Murphy, A.; Zheng, L. From immune checkpoints to vaccines: The past, present and future of cancer immunotherapy. Adv. Cancer Res. 2019, 143, 63–144. [Google Scholar]
- Carralot, J.P.; Weide, B.; Schoor, O.; Probst, J.; Scheel, B.; Teufel, R.; Hoerr, I.; Garbe, C.; Rammensee, H.G.; Pascolo, S. Production and characterization of amplified tumor-derived cRNA libraries to be used as vaccines against metastatic melanomas. Genet. Vaccines Ther. 2005, 3, 6. [Google Scholar] [CrossRef]
- Chang, S.S.; O’Keefe, D.S.; Bacich, D.J.; Reuter, V.E.; Heston, W.D.W.; Gaudin, P.B. Prostate-specific membrane antigen is produced in tumor-associated neovasculature. Clin. Cancer Res. 1999, 5, 2674–2681. [Google Scholar] [PubMed]
- Kandalaft, L.E.; Motz, G.T.; Busch, J.; Coukos, G. Angiogenesis and the tumor vasculature as antitumor immune modulators: The role of vascular endothelial growth factor and endothelin. Curr. Top. Microbiol. Immunol. 2011, 344, 129–148. [Google Scholar] [PubMed]
- Gulley, J.L.; Borre, M.; Vogelzang, N.J.; Ng, S.; Agarwal, N.; Parker, C.C.; Pook, D.W.; Rathenborg, P.; Flaig, T.W.; Carles, J.; et al. Phase III trial of PROSTVAC in asymptomatic or minimally symptomatic metastatic castration-resistant prostate cancer. J. Clin. Oncol. 2019, 37, 1051–1061. [Google Scholar] [CrossRef]
- Morse, M.A.; Gwin, W.R.; Mitchell, D.A. Vaccine Therapies for Cancer: Then and Now. Targeted Oncol. 2021, 16, 121–152. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Dannull, J.; Heiser, A.; Yancey, D.; Pruitt, S.; Madden, J.; Coleman, D.; Niedzwiecki, D.; Gilboa, E.; Vieweg, J. Immunological and clinical responses in metastatic renal cancer patients vaccinated with tumor RNA-transfected dendritic cells. Cancer Res. 2003, 63, 2127–2133. [Google Scholar] [PubMed]
- Lim, M.; Badruddoza, A.Z.M.; Firdous, J.; Azad, M.; Mannan, A.; Al-Hilal, T.A.; Cho, C.S.; Islam, M.A. Engineered nanodelivery systems to improve DNA vaccine technologies. Pharmaceutics 2020, 12, 30. [Google Scholar] [CrossRef]
- Mueller, M.; Reichardt, W.; Koerner, J.; Groettrup, M. Coencapsulation of tumor lysate and CpG-ODN in PLGA-microspheres enables successful immunotherapy of prostate carcinoma in TRAMP mice. J. Control. Release 2012, 162, 159–166. [Google Scholar] [CrossRef]
- Trimble, C.L.; Morrow, M.P.; Kraynyak, K.A.; Shen, X.; Dallas, M.; Yan, J.; Edwards, L.; Parker, R.L.; Denny, L.; Giffear, M.; et al. Safety, efficacy, and immunogenicity of VGX-3100, a therapeutic synthetic DNA vaccine targeting human papillomavirus 16 and 18 E6 and E7 proteins for cervical intraepithelial neoplasia 2/3: A randomised, double-blind, placebo-controlled phase 2b trial. Lancet 2015, 86, 2078–2088. [Google Scholar] [CrossRef]
- Warminski, M.; Sikorski, P.J.; Kowalska, J.; Jemielity, J. Applications of Phosphate Modification and Labeling to Study (m)RNA Caps. Top. Curr. Chem. 2017, 375, 16. [Google Scholar] [CrossRef]
- Kowalska, J.; Lewdorowicz, M.; Zuberek, J.; Grudzien-Nogalska, E.; Bojarska, E.; Stepinski, J.; Rhoads, R.E.; Darzynkiewicz, E.; Davis, R.E.; Jemielity, J.E. Synthesis and characterization of mRNA cap analogs containing phosphorothioate substitutions that bind tightly to eIF4E and are resistant to the decapping pyrophosphatase DcpS. RNA 2008, 14, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, A.N.; Diken, M.; Kreiter, S.; Selmi, A.; Kowalska, J.; Jemielity, J.; Darzynkiewicz, E.; Huber, C.; Türeci, Ö.; Sahin, U. Phosphorothioate cap analogs increase stability and translational efficiency of RNA vaccines in immature dendritic cells and induce superior immune responses in vivo. Gene Ther. 2010, 17, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Calvo Tardon, M.; Allard, M.; Dutoit, V.; Dietrich, P.Y.; Walker, P.R. Peptides as cancer vaccines. Curr. Opin. Pharmacol. 2019, 47, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Bowen, W.S.; Svrivastava, A.K.; Batra, L.; Barsoumian, H.; Shirwan, H. Current challenges for cancer vaccine adjuvant development. Expert Rev. Vaccines 2018, 17, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Fraser, C.K.; Diener, K.R.; Brown, M.P.; Hayball, J.D. Improving vaccines by incorporating immunological coadjuvants. Expert Rev. Vaccines 2007, 6, 559–578. [Google Scholar]
- Naumann, K.; Wehner, R.; Schwarze, A.; Petzold, C.; Schmitz, M.; Rohayem, J. Activation of dendritic cells by the novel Toll-like receptor 3 agonist RGC100. Clin. Dev. Immunol. 2013, 2013, 283649. [Google Scholar] [CrossRef]
- Available online: https://www.drugs.com/newdrugs/fda-approves-provenge-cellular-immunotherapy-men-advanced-prostate-cancer-2130.html (accessed on 21 September 2021).
- Higano, C.S.; Schellhammer, P.F.; Small, E.J.; Burch, P.A.; Nemunaitis, J.; Yuh, L.; Provost, N.; Frohlich, M.W. Integrated data from 2 randomized, double-blind, placebo-controlled, phase 3 trials of active cellular immunotherapy with sipuleucel-T in advanced prostate cancer. Cancer 2009, 115, 3670–3679. [Google Scholar] [CrossRef] [PubMed]
- Pol, J.G.; Acuna, S.A.; Yadollahi, B.; Tang, N.; Stephenson, K.B.; Atherton, M.J.; Hanwell, D.; El-Warrak, A.; Goldstein, A.; Moloo, B.; et al. Preclinical evaluation of a MAGE-A3 vaccination utilizing the oncolytic Maraba virus currently in first-in-human trials. Oncoimmunology 2018, 8, e1512329. [Google Scholar] [CrossRef]
- Bonehill, A.; Heirman, C.; Tuyaerts, S.; Michiels, A.; Breckpot, K.; Brasseur, F.; Zhang, Y.; Van Der Bruggen, P.; Thielemans, K. Messenger RNA-electroporated dendritic cells presenting MAGE-A3 simultaneously in HLA class I and class II molecules. J. Immunol. 2004, 172, 6649–6657. [Google Scholar] [CrossRef]
- Verbeke, R.; Lentacker, I.; Breckpot, K.; Janssens, J.; Van Calenbergh, S.; De Smedt, S.C.; Dewitte, H. Broadening the Message: A Nanovaccine Co-loaded with Messenger RNA and -GalCer Induces Antitumor Immunity through Conventional and Natural Killer T Cells. ACS Nano 2019, 13, 1655–1669. [Google Scholar] [CrossRef]
- Richman, L.P.; Vonderheide, R.H.; Rech, A.J. Neoantigen dissimilarity to the self-proteome predicts immunogenicity and response to immune checkpoint blockade. Cell Syst. 2019, 9, 375–382. [Google Scholar] [CrossRef]
- Olweus, J.; Lund-Johansen, F. Finding, Neo (antigens, that is). Blood 2019, 134, 108–109. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef]
- Hu-Lieskovan, S.; Ott, P.A.; Naing, A.; Besada, R.H.; Gates, S.J.; Kohler, V.R.; Curran, R.R.; Bushway, M.E.; Scherer, J.; Balogh, K.N.; et al. The personalized vaccine, NEO-PV-01 with anti-PD1, induces neoantigen-specific de novo immune responses in patients with advanced metastatic melanoma: Association with clinical outcomes. Cancer Res. 2019, 79, 942. [Google Scholar]
- Yin, Z.; Jiang, K.; Li, R.; Dong, C.; Wang, L. Multipotent mesenchymal stromal cells play critical roles in hepatocellular carcinoma initiation, progression and therapy. Mol. Cancer 2018, 17, 178. [Google Scholar] [CrossRef]
- Hofmeister, V.; Vetter, C.; Schrama, D.; Bröcker, E.B.; Becker, J.C. Tumor stroma-associated antigens for anti-cancer immunotherapy. Cancer Immunol. Immunother. 2006, 55, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, H.; Liu, I.; Yu, J.; Ren, X. Fibroblast activation protein: A potential therapeutic target in cancer. Cancer Biol. Ther. 2012, 13, 123–129. [Google Scholar] [CrossRef]
- Høye, A.M.; Tolstrup, S.D.; Horton, E.R.; Nicolau, M.; Frost, H.; Woo, J.H.; Mauldin, J.P.; Frankel, A.E.; Cox, T.R.; Erler, J.T. Tumor endothelial marker 8 promotes cancer progression and metastasis. Oncotarget 2018, 9, 30173–30188. [Google Scholar] [CrossRef]
- Nanda, A.; Karim, B.; Peng, Z.; Liu, G.; Qiu, W.; Gan, C.; Vogelstein, B.; Croix, B.S.; Kinzler, K.W.; Huso, D.L. Tumor endothelial marker 1 (Tem1) functions in the growth and progression of abdominal tumors. Proc. Natl. Acad. Sci. USA 2006, 103, 3351–3356. [Google Scholar] [CrossRef]
- Quintero-Fabián, S.; Arreola, R.; Becerril-Villanueva, E.; Torres-Romero, J.C.; Arana-Argáez, V.; Lara-Riegos, J.; Ramírez-Camacho, M.A.; Alvarez-Sánchez, M.E. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front. Oncol. 2019, 9, 1370. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Reuter, V.E.; Heston, W.D.; Bander, N.H.; Grauer, L.S.; Gaudin, P.B. Five different anti-prostate-specific membrane antigen (PSMA) antibodies confirm PSMA expression in tumor-associated neovasculature. Cancer Res. 1999, 59, 3192–3198. [Google Scholar]
- Amatu, A.; Sartore-Bianchi, A.; Siena, S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open 2016, 1, e000023. [Google Scholar] [CrossRef] [PubMed]
- Nakagawara, A. Trk receptor tyrosine kinases: A bridge between cancer and neural development. Cancer Lett. 2001, 169, 107–114. [Google Scholar] [CrossRef]
- Available online: www.ema.europa.eu/en/documents/product-information/vitrakvi-epar-product-information.en.pdf (accessed on 27 September 2021).
- Bi, J.; Ichu, T.A.; Zanca, C.; Yang, H.; Zhang, W.; Gu, Y.; Chowdhry, S.; Reed, A.; Ikegami, S.; Turner, K.M.; et al. Oncogene Amplification in Growth Factor Signaling Pathways Renders Cancers Dependent on Membrane Lipid Remodeling. Cell Metab. 2019, 30, 525–538.e8. [Google Scholar] [CrossRef]
- Carrà, G.; Cartellà, A.; Maffeo, B.; Morotti, A. Strategies For Targeting Chronic Myeloid Leukaemia Stem Cells. Blood Lymphat. Cancer 2019, 9, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Pellicano, F.; Park, L.; Hopcroft, L.E.M.; Shah, M.M.; Jackson, L.; Scott, M.T.; Clarke, C.J.; Sinclair, A.; Abraham, S.A.; Hair, A.; et al. Hsa-mir183/EGR1—Mediated regulation of E2F1 is required for CML stem/progenitor cell survival. Blood 2018, 131, 1532–1544. [Google Scholar] [CrossRef] [PubMed]
- Warfvinge, R.; Geironson, L.; Sommarin, M.N.E.; Lang, S.; Karlsson, C.; Roschupkina, T.; Stenke, L.; Stentoft, J.; Olsson-Strömberg, U.; Hjorth-Hansen, H.; et al. Single-cell molecular analysis defines therapy response and immunophenotype of stem cell subpopulations in CML. Blood 2017, 129, 2384–2394. [Google Scholar] [CrossRef]
- Serna, N.; Álamo, P.; Ramesh, P.; Vinokurova, D.; Sánchez-García, L.; Unzueta, U.; Gallardo, A.; Céspedes, M.V.; Vázquez, E.; Villaverde, A.; et al. Nanostructured toxins for the selective destruction of drug resistant human CXCR4+ colorectal cancer stem cells. J. Control. Release 2020, 320, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Gao, Z.; Duan, Y.; Yuan, W.; Ke, Y. Notch signaling plays a crucial role in cancer stem-like cells maintaining stemness and mediating chemotaxis in renal cell carcinoma. J. Exp. Clin. Cancer Res. 2017, 36, 41. [Google Scholar] [CrossRef]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Xing, F.; Saidou, J.; Watabe, K. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front. Biosci. 2010, 15, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Lia, X.; Bub, W.; Mengb, L.; Liub, X.; Wanga, S.; Jianga, L.; Rena, M.; Fana, Y.; Sun, H. CXCL12/CXCR4 pathway orchestrates CSC-like properties by CAF recruited tumor associated macrophage in OSCC. Exp. Cell Res. 2019, 378, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Colmone, A.; Amorim, M.; Pontier, A.L.; Wang, S.; Jablonski, E.; Sipkins, D.A. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science 2008, 322, 1861–1865. [Google Scholar] [CrossRef] [PubMed]
- Uy, G.L.; Rettig, M.P.; Motabi, I.H.; McFarland, K.; Trinkaus, K.M.; Hladnik, L.M.; Kulkarni, S.; Abboud, C.N.; Cashen, A.F.; Stockerl-Goldstein, K.E.; et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood 2012, 119, 3917–3924. [Google Scholar] [CrossRef]
- Villatoro, A.; Konieczny, J.; Cuminetti, V.; Arranz, L. Leukemia stem cell release from the stem cell niche to treat acute myeloid leukemia. Front. Cell Dev. Biol. 2020, 8, 607. [Google Scholar] [CrossRef]
- Ishikawa, F.; Yoshida, S.; Saito, Y.; Hijikata, A.; Kitamura, H.; Tanaka, S.; Nakamura, R.; Tanaka, T.; Tomiyama, H.; Saito, N.; et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat. Biotechnol. 2007, 25, 1315–1321. [Google Scholar] [CrossRef]
- Winkler, T.G.; Barbier, V.; Nowlan, B.; Jacobsen, R.N.; Forristal, C.E.; Patton, J.T.; Magnani, J.L.; Lévesque, J.P. Vascular niche E-selectin regulates hematopietic stem cell dormancy, self renewal and chemoresistance. Nat. Med. 2012, 18, 1651–1657. [Google Scholar] [CrossRef]
- Zhai, Y.; Zhang, J.; Wang, H.; Lu, W.; Liu, S.; Yu, Y.; Weng, W.; Ding, Z.; Zhu, Q.; Shi, J. Growth differentiation factor 15 contributes to cancer-associated fibroblasts-mediated chemo-protection of AML cells. J. Exp. Clin. Cancer Res. 2016, 35, 147. [Google Scholar] [CrossRef]
- Yu, Z.; Liu, L.; Shu, Q.; Li, D.; Wang, R. Leukemia stem cells promote chemoresistance by inducing downregulation of lumican in mesenchymal stem cells. Oncol. Lett. 2019, 18, 4317–4327. [Google Scholar] [CrossRef] [PubMed]
- Dragu, D.L.; Necula, L.G.; Bleotu, C.; Diaconu, C.C.; Chivu-Economescu, M. Therapies targeting cancer stem cells: Current trends and future challenges. World J. Stem Cells 2015, 7, 1185–1201. [Google Scholar]
- Ievesque, J.P.; Hendy, J.; Takamatsu, Y.; Simmons, P.J.; Bendall, L.J. Disruption of the CXCR4/CXCL12 chemotactic interaction during hematopietic stem cell mobilization induced by GCSF or cyclophosphamide. J. Clin. Investig. 2003, 111, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Cuadron, D.; Boluda, B.; Martinez, P.; Bergua, J.; Rodriguez-Veiga, R.; Esteve, J.; Vives, S.; Serrano, J.; Vidriales, B.; Salamero, O.; et al. A phase I-II study of plerixafor in combination with fludarabine, idarubicin and G-CSF (PLERIFLAG regimen) for the treatment of patients with the first early-relapsed or refractory acute myeloid leukemia. Ann. Hematol. 2018, 97, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Borthakur, G.; Ofran, Y.; Nagler, A.; Rowe, J.M.; Foran, J.M.; Uy, G.L.; DiPersio, J.F.; Altman, J.K.; Frankfurt, O.; Tallman, M.S.; et al. The peptidic CXCR4 antagonist, BL-8040, significantly reduces bone marrow immature leukemia progenitors by inducing differentiation, apoptosis and mobilization: Results of the dose esxcalation clinical trial in acute myeloid leukemia. Blood 2015, 126, 2546. [Google Scholar] [CrossRef]
- Borthakur, G.; Tallman, M.S.; Ofran, Y.; Foran, J.; Uy, G.; DiPersio, J.; Nagler, A.; Rowe, J.; Showel, M.M.; Altman, J.K.; et al. The CXCR4 inhibitor BL-8040 in combination with cytarabine results in a significantly extended overall survival of relapsed/refractory AML patients. EHA Libr. 2018, 215317, S993. [Google Scholar]
- Winkler, I.G.; Barbier, V.; Pattabiraman, D.R.; Gonda, T.J.; Magnani, J.L.; Levesque, J.P. Vascular niche E-selectin protects acute myeloid leukaemia stem cells from chemotherapy. Blood 2014, 124, 620. [Google Scholar] [CrossRef]
- DeAngelo, D.J.; Jonas, B.A.; Liesveld, J.L.; Bixby, D.L.; Advani, A.S.; Marlton, P. Uproleselan (GMI-1271), an E-selectin antagonist, improves the efficacy and safety of chemotherapy in relapsed/refractory (R/R) and newly diagnosed older patients with acute myeloid leukemia: Final, correlative, and subgroup analyses. Blood 2018, 132, 331. [Google Scholar] [CrossRef]
- Fujita, M.; Davari, P.; Takada, Y.K.; Takada, Y. Stromal cell-derived factor-1 (CXCL12) activates integrins by direct binding to an allosteric ligand-binding site (site 2) of integrins aithout CXCR4. Biochem. J. 2020, 475, 723–732. [Google Scholar] [CrossRef]
- Matsunaga, T.; Takemoto, N.; Sato, T.; Takimoto, R.; Tanaka, I.; Fujimi, A.; Akiyama, T.; Kuroda, H.; Kawano, Y.; Kobune, M.; et al. Ineraction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat. Med. 2003, 9, 1158–1165. [Google Scholar] [CrossRef]
- Arcangeli, M.L.; Bardin, F.; Frontera, V.; Bidaut, G.; Obrados, E.; Adams, R.H.; Chabannon, C.; Aurrand-Lions, M. Function of Jam-B/Jam-C interaction in homing and mobilization of human and mouse hematopoietic stem and progenitor cells. Stem Cells 2014, 32, 1043–1054. [Google Scholar] [CrossRef]
- Lim, Z.F.; Patrick, P.C. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J. Hematol. Oncol. 2019, 12, 134. [Google Scholar] [CrossRef] [PubMed]
- Janne, P.A.; Yang, J.C.; Kim, D.W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.J.; Kim, S.W.; Su, W.C.; Horn, L.; et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Bagshawe, K.D. Antibody directed enzymes revive anti-cancer prodrugs concept. Br. J. Cancer 1987, 56, 531–532. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wei, S.L.; Chen, B.M.; Chern, J.W.; Wu, M.F.; Liu, P.W.; Roffler, S.R. Bystander killing of tumour cells by antibody-targeted enzymatic activation of a glucuronide prodrug. Br. J. Cancer 1999, 79, 1378–1385. [Google Scholar] [CrossRef]
- Sharma, S.K.; Bagshawe, K.D. Antibody Directed Enzyme Prodrug Therapy (ADEPT): Trials and tribulations. Adv. Drug Deliv. Rev. 2017, 118, 2–7. [Google Scholar] [CrossRef]
- Sherwood, R.F.; Melton, R.G.; Alwan, S.M.; Hughes, P. Purification and properties of carboxypeptidase g2 from pseudomonas sp. strain rs-16. Use of a novel triazine dye affinity method. Eur. J. Biochem. 1985, 148, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Springer, C.J.; Bagshawe, K.D.; Sharma, S.K.; Searle, F.; Boden, J.A.; Antoniw, P.; Burke, P.J.; Rogers, G.T.; Sherwood, R.F.; Melton, R.G. Ablation of human choriocarcinoma xenografts in nude mice by antibody-directed enzyme prodrug therapy (adept) with three novel compounds. Eur. J. Cancer 1991, 27, 1361–1366. [Google Scholar] [CrossRef]
- Sharma, S.K.; Bagshawe, K.D.; Springer, C.J.; Burke, P.J.; Rogers, G.T.; Boden, J.A.; Antoniw, P.; Melton, R.G.; Sherwood, R.F. Antibody directed enzyme prodrug therapy (adept): A three phase system. Dis. Markers 1991, 9, 225–231. [Google Scholar] [PubMed]
- Sharma, S.K.; Bagshawe, K.D.; Burke, P.J.; Boden, R.W.; Rogers, G.T. Inactivation and clearance of an anti-CEA carboxypeptidase g2 conjugate in blood after localisation in a xenograft model. Br. J. Cancer 1990, 61, 659–662. [Google Scholar] [CrossRef]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef]
- Bjørnstad, R.; Aesoy, R.; Bruserud, Y.; Brenner, A.K.; Giraud, F.; Dowling, T.H.; Gausdal, G.; Moreau, P.; Døskeland, S.O.; Anizon, F.; et al. A Kinase Inhibitor with Anti-Pim Kinase Activity is a Potent and Selective Cytotoxic Agent Toward Acute Myeloid Leukemia. Mol. Cancer Ther. 2019, 18, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.H.; Li, J.J.; Xie, F.W.; Chen, J.F.; Yu, Y.H.; Ouyang, X.N.; Liang, H. Expression of Pim-1 in Tumors, Tumor Stroma and Tumor-Adjacent Mucosa Co-determines the Prognosis of Colon Cancer Patients. PLoS ONE 2013, 8, e76693. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, S.M.; Barrette, T.R.; Ghosh, D.; Shah, R.; Varambally, S.; Kurachi, K.; Pienta, K.J.; Rubin, M.A.; Chinnaiyan, A.M. Delineation of Prognostic Biomarkers in Prostate Cancer. Nature 2001, 412, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Sawaguchi, Y.; Yamazaki, R.; Nishiyama, Y.; Mae, M.; Abe, A.; Nishiyama, H.; Nishisaka, F.; Ibuki, T.; Sasai, T.; Matsuzaki, T. Novel Pan-Pim Kinase Inhibitors With Imidazopyridazine and Thiazolidinedione Structure Exert Potent Antitumor Activities. Front. Pharmacol. 2021, 12, 672536. [Google Scholar] [CrossRef]
- Zainab, R.; Kaleem, A.; Ponczek, M.B.; Abdullah, R.; Iqtedar, M.; Hoessli, D.C. Finding inhibitors for PCSK9 using computational methods. PLoS ONE 2021, 16, e0255523. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S. Bioinformatics in drug development and assessment. Drug Metab. Rev. 2005, 37, 279–310. [Google Scholar] [CrossRef]
- FDA Grants Accelerated Approval to Amivantamab-Vmjw for Metastatic Non-Small Cell Lung Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-amivantamab-vmjwmetastatic-non-small-cell-lung-cancer (accessed on 23 July 2021).
- Park, K.; John, T.; Kim, S.-W.; Lee, J.S.; Shu, C.A.; Kim, D.-W.; Viteri Ramirez, S.; Spira, A.I.; Sabari, J.K.; Han, J.-Y.; et al. Amivantamab (JNJ-61186372), an Anti-EGFR-MET Bispecific Antibody, in Patients with EGFR Exon 20 Insertion (Exon20ins)-Mutated Non-Small Cell Lung Cancer (NSCLC). J. Clin. Oncol. 2020, 38, 9512. [Google Scholar] [CrossRef]
- Alsina, M.; Varga, A.; Amatu, A.; Schellens, J.H.M.; Witteveen, P.O.; Boni, V.; Moreno, V.; Bol, K.; Lourbakos, A.; Ferrer, M.M.; et al. Phase I/II Study of Single Agent MCLA-128, a Full Length IgG1 Bispecific Antibody Targeting the HER3 Pathway: Overall Safety at the Recommended Phase II Dose (R2PD) and Preliminary Activity in HER2+ Metastatic Gastric/Gastroesophageal Junction Cancer (GC/GEJ). Ann. Oncol. 2018, 29, viii223–viii224. [Google Scholar]
- Hamilton, E.P.; Petit, T.; Pistilli, B.; Goncalves, A.; Ferreira, A.A.; Dalenc, F.; Cardoso, F.; Mita, M.M.; Dezentjé, V.O.; Manso, L.; et al. Clinical Activity of MCLA-128 (Zenocutuzumab), Trastuzumab, and Vinorelbine in HER2 Amplified Metastatic Breast Cancer (MBC) Patients (Pts) Who Had Progressed on Anti-HER2 ADCs. J. Clin. Oncol. 2020, 38, 3093. [Google Scholar] [CrossRef]
- Szöőr, A.; Szöllősi, J.; Vereb, G. From antibodies to living drugs: Quo vadis cancer immunotherapy? Biol. Futura 2021, 72, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Yu, J.X.; Hubbard-Lucey, V.M.; Neftelinov, S.T.; Hodge, J.P.; Lin, Y. Trial watch: The clinical trial landscape for PD1/PDL1 immune checkpoint inhibitors. Nat. Rev. Drug Discov. 2018, 17, 854–855. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477. [Google Scholar] [CrossRef]
- Sevenich, L. Turning “cold” into “hot” tumors—Opportunities and challenges for radio-immunotherapy against primary and metastatic brain cancers. Front. Oncol. 2019, 9, 163. [Google Scholar] [CrossRef]
- O’Leary, M.C.; Lu, X.; Huang, Y.; Lin, X.; Mahmood, I.; Przepiorka, D.; Gavin, D.; Lee, S.; Liu, K.; George, B.; et al. FDA approval summary: Tisagenlecleucel for treatment of patients with relapsed or refractory B-cell precursor acute lymphoblastic leukemia. Clin. Cancer Res. 2019, 25, 1142–1146. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Grupp, S.A. Tisagenlecleucel in children and young adults with b-cell lymphoblastic leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, C.A.; Hunter, B.D.; Redd, R.; Rodig, S.J.; Chen, P.H.; Wright, K.; Lipschitz, M.; Ritz, J.; Kamihara, Y.; Armand, P.; et al. Axicabtagene Ciloleucel in the Non-Trial Setting: Outcomes and Correlates of Response, Resistance, and Toxicity. J. Clin. Oncol. 2020, 38, 3095–3106. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Nastoupil, L.; Westin, J.; Lee, H.J.; Navsaria, L.; Steiner, R.E.; Ahmed, S.; Moghrabi, O.; Oriabure, O.; Chen, W.; et al. Outcomes and management of patients with mantle cell lymphoma after progression on brexucabtagene autoleucel therapy. Br. J. Haematol. 2021, 192, e38–e42. [Google Scholar] [CrossRef]
- Mian, A.; Hill, B.T. Brexucabtagene autoleucel for the treatment of relapsed/refractory mantle cell lymphoma. Expert Opin. Biol. Ther. 2021, 21, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-idecabtagene-vicleucel-multiple-myeloma (accessed on 27 March 2021).
- Santos Apolonio, J.; Lima de Souza Gonçalves, V.; Cordeiro Santos, M.L.; Silva Luz, M.; Silva Souza, J.V.; Rocha Pinheiro, S.L.; de Souza, W.R.; Sande Loureiro, M.; de Melo, F.F. Oncolytic virus therapy in cancer: A current review. World J. Virol. 2021, 10, 229–255. [Google Scholar] [CrossRef] [PubMed]
- Mondal, M.; Guo, J.; He, P.; Zhou, D. Recent advances of oncolytic virus in cancer therapy. Hum. Vaccines Immunother. 2020, 16, 2389–2402. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Immune Checkpoint | Stage of the Immune Response | Place of Action | Presence on Cells | Inhibitors of Immune Checkpoint |
|---|---|---|---|---|
| CTLA-4 | Early activation phase | Lymph nodes | Activated T and B cells | Ipilimumab |
| PD-1 | The effector phase | Periferal tissue, cancer imcroenvironment | T and B cells, natural killer cells, myeloid-derived suppressor cells | Nivolumab, Pembrolizumab, |
| PD-L1 | The effector phase | Periferal tissue, cancer imcroenvironment | Antigen presenting cells, T and B cells, natural killer cells, myeloid-derived suppressor cells, hematopoietic cells, cancer cells | Atezolizumab, Durvalumab, Avelumab |
| Type of Vaccine | Problems in Applicaion | Clinical Application | Origin |
|---|---|---|---|
| Dendritic cells | Huge cost | PROVENGE®, castration-resistant prostate cancer, approved by FDA | From patient’s PBMCs |
| Peptide/protein | Limited efficacy (intracellular processing) | No application | Synthetic peptides |
| DNA | Risk of insertional mutagenesis, limited efficacy | No application | From autologus tumor cells |
| mRNA | Instability (enzymatic degradation) | No application | From autologus tumor cells, tumor cRNA libraries |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papież, M.A.; Krzyściak, W. Biological Therapies in the Treatment of Cancer—Update and New Directions. Int. J. Mol. Sci. 2021, 22, 11694. https://doi.org/10.3390/ijms222111694
Papież MA, Krzyściak W. Biological Therapies in the Treatment of Cancer—Update and New Directions. International Journal of Molecular Sciences. 2021; 22(21):11694. https://doi.org/10.3390/ijms222111694
Chicago/Turabian StylePapież, Monika A., and Wirginia Krzyściak. 2021. "Biological Therapies in the Treatment of Cancer—Update and New Directions" International Journal of Molecular Sciences 22, no. 21: 11694. https://doi.org/10.3390/ijms222111694
APA StylePapież, M. A., & Krzyściak, W. (2021). Biological Therapies in the Treatment of Cancer—Update and New Directions. International Journal of Molecular Sciences, 22(21), 11694. https://doi.org/10.3390/ijms222111694