
Targeting Oxidative Stress: Novel Coumarin-Based Inverse Agonists of GPR55

 and
and
Abstract
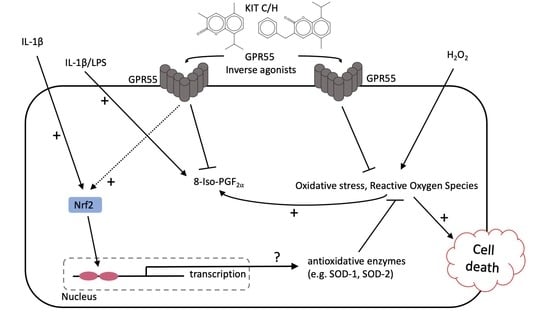
:
1. Introduction
2. Results
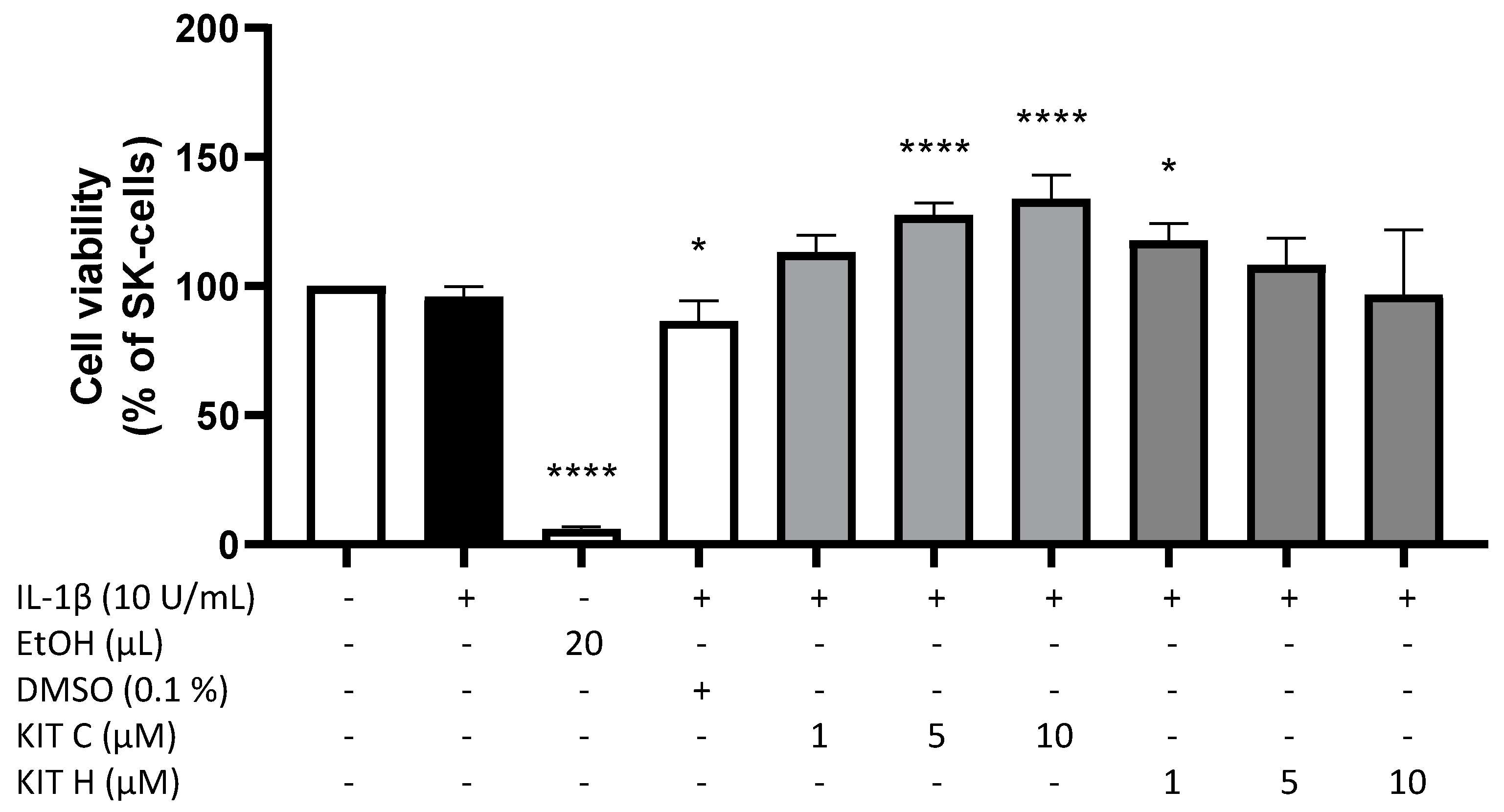
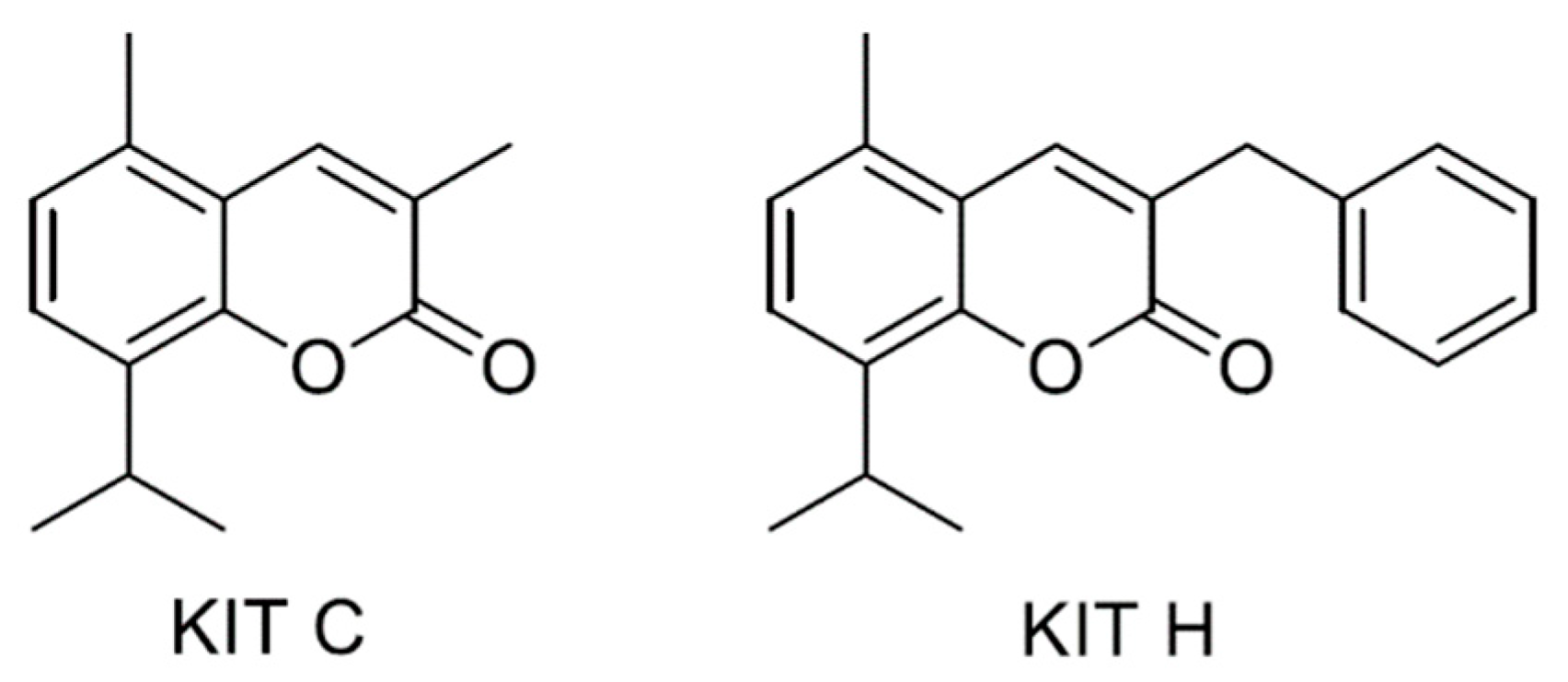
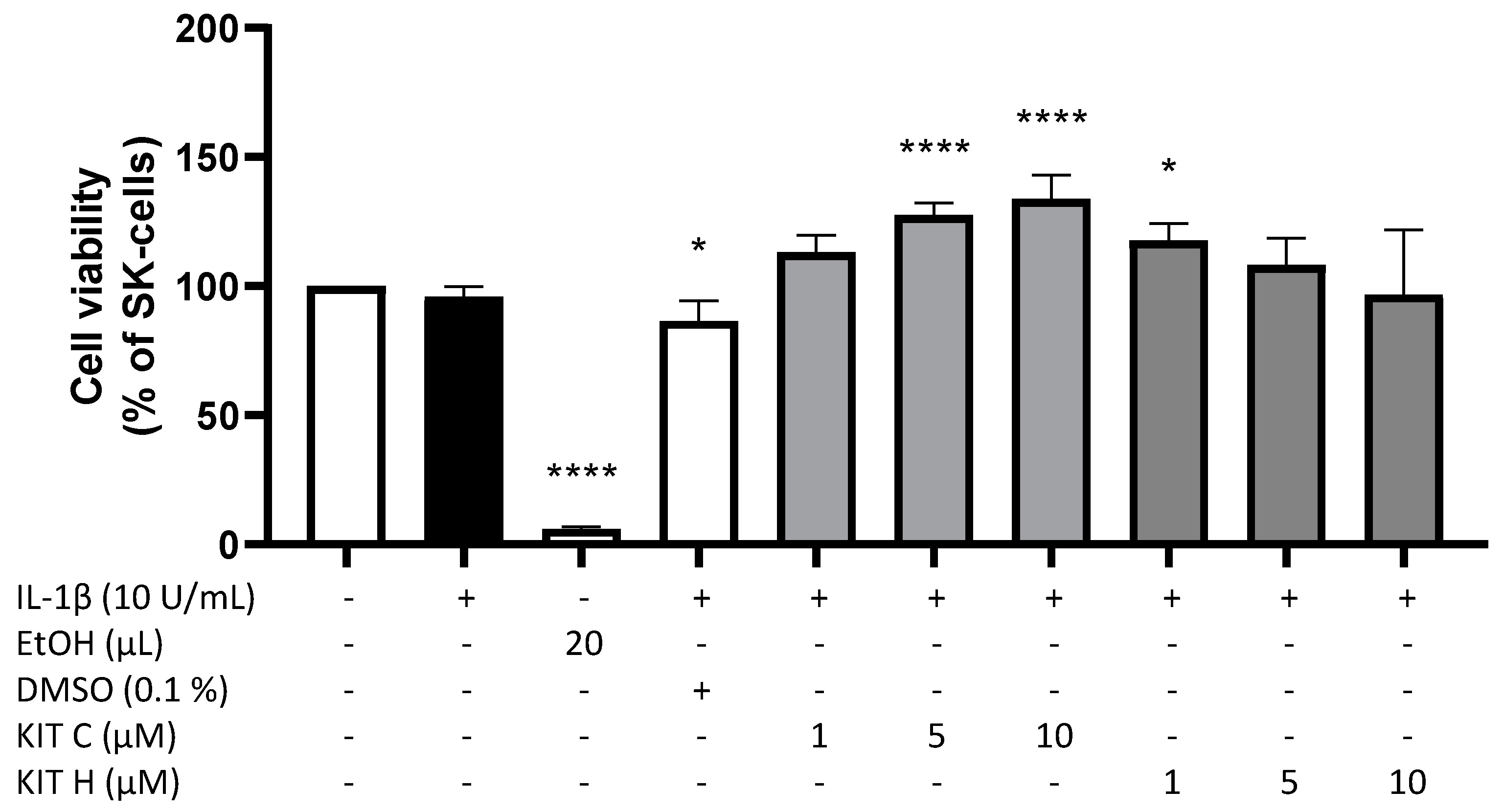
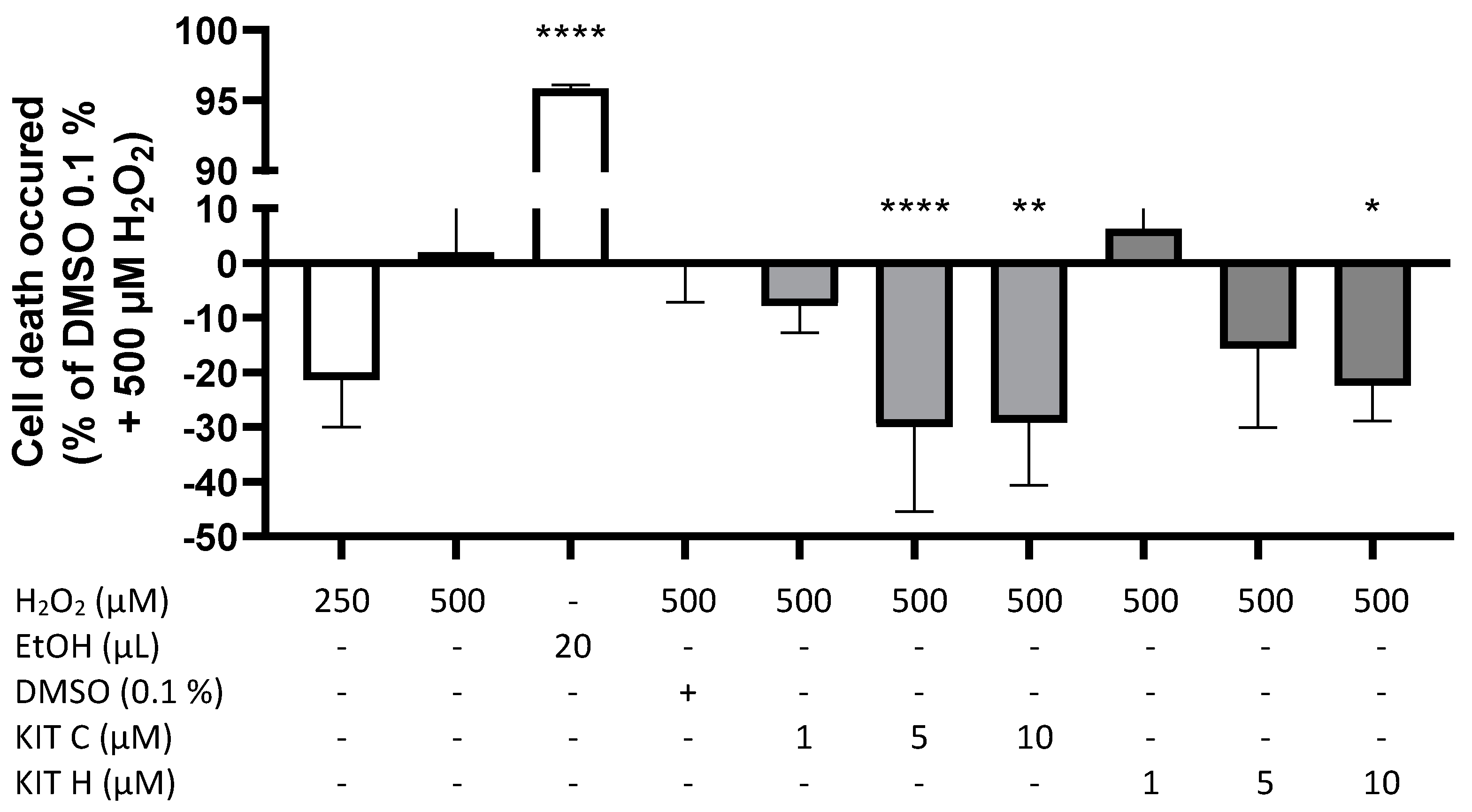
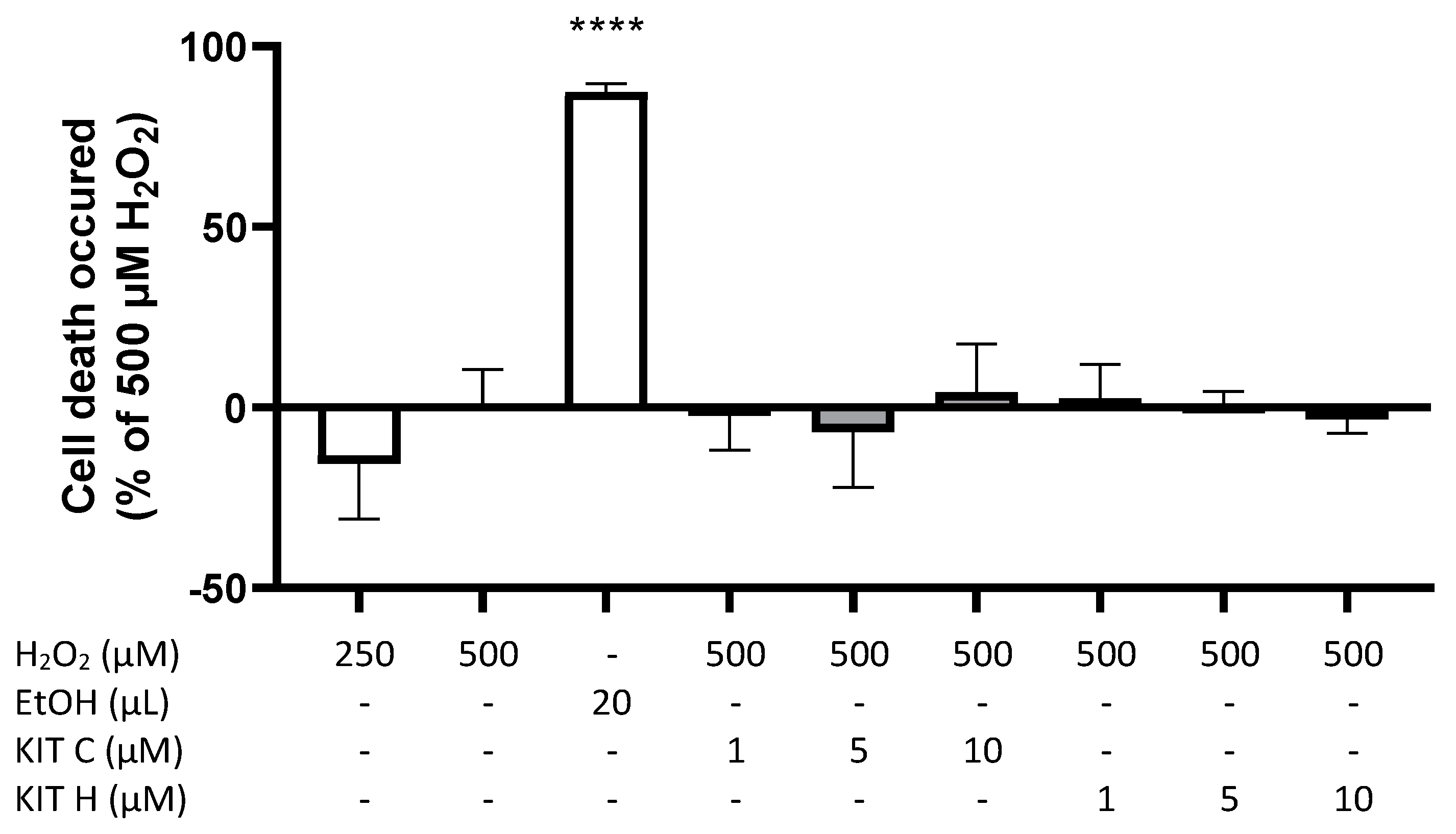
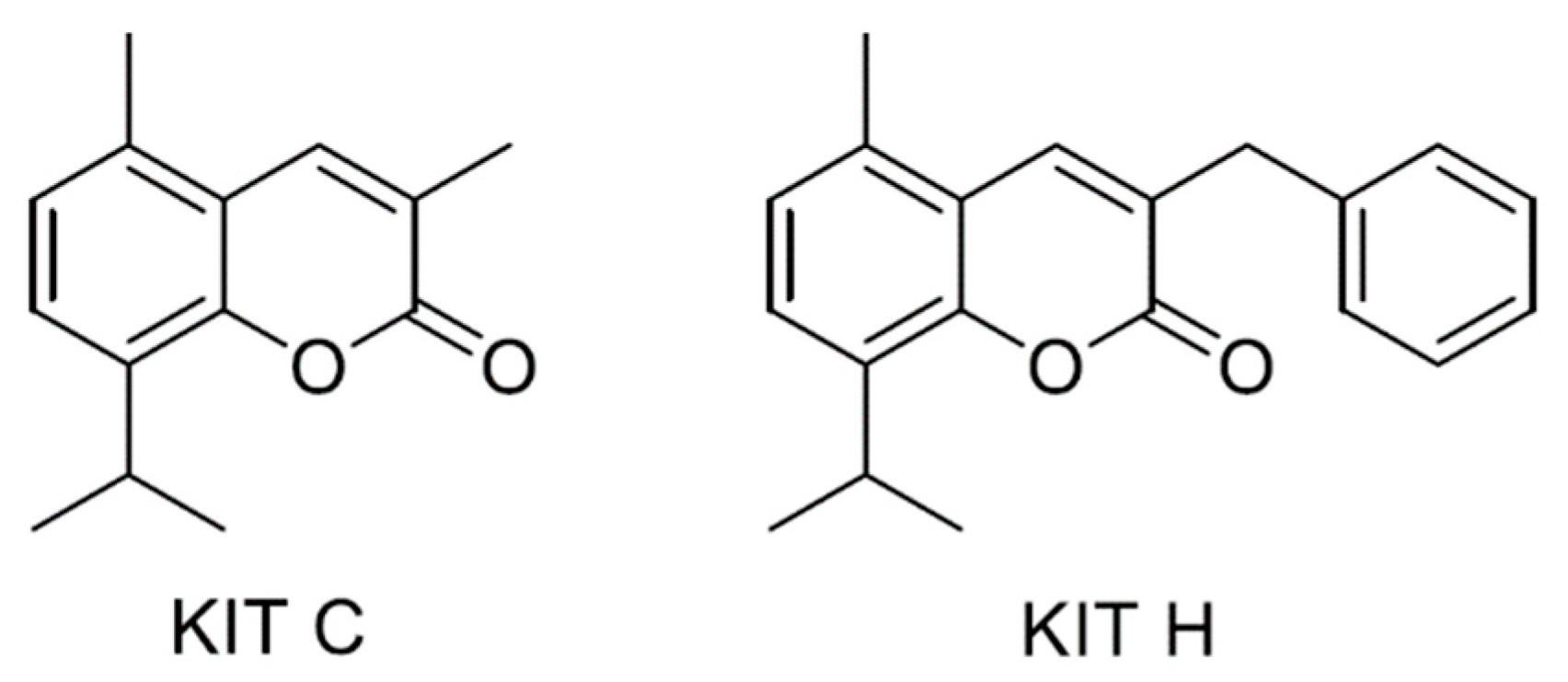
2.1. Cytotoxic Effects of KIT C and KIT H
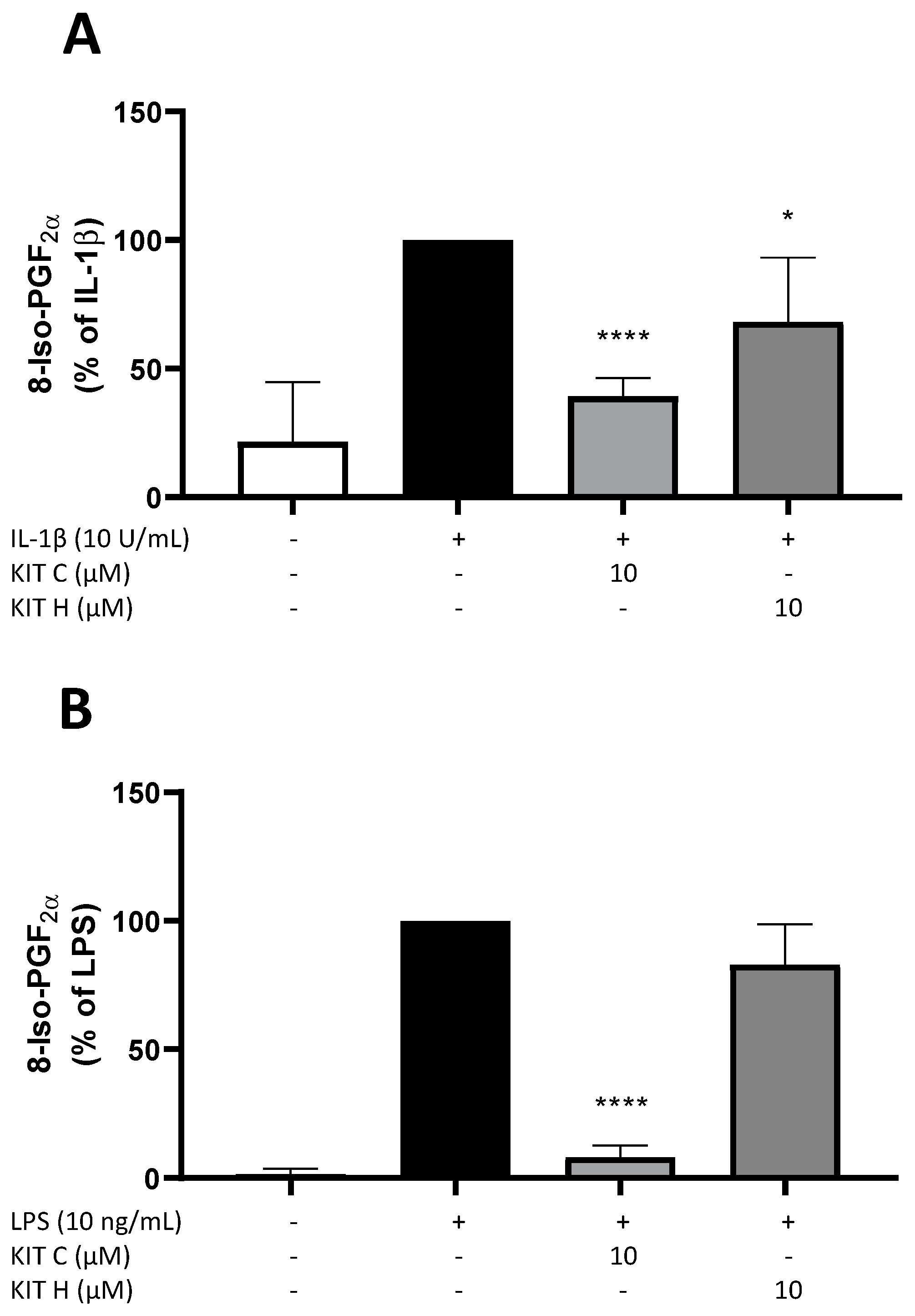
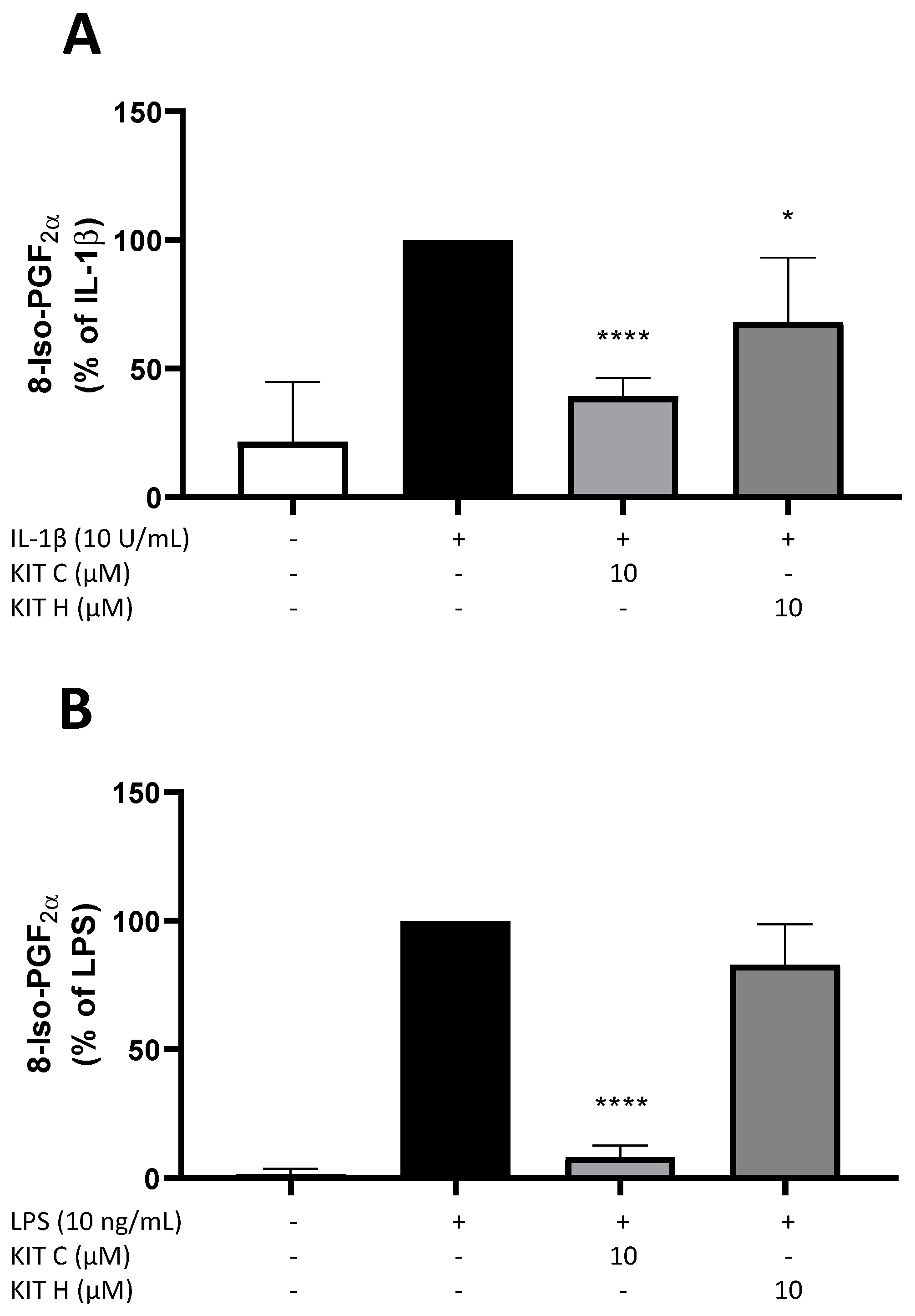
2.2. Effects of KIT C and KIT H on 8-Iso-PGF2α-Release in SK-N-SH and Primary Microglial Cells
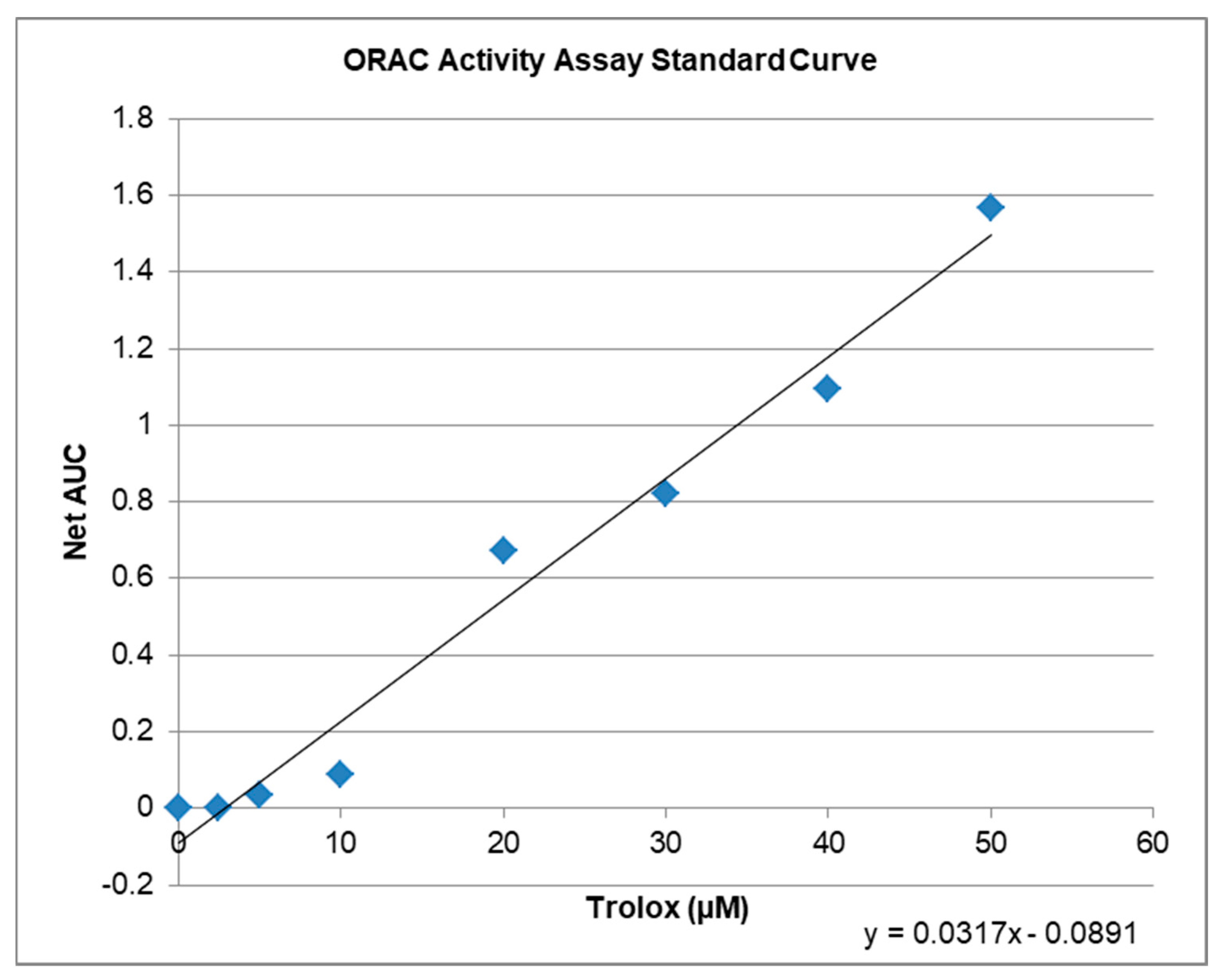
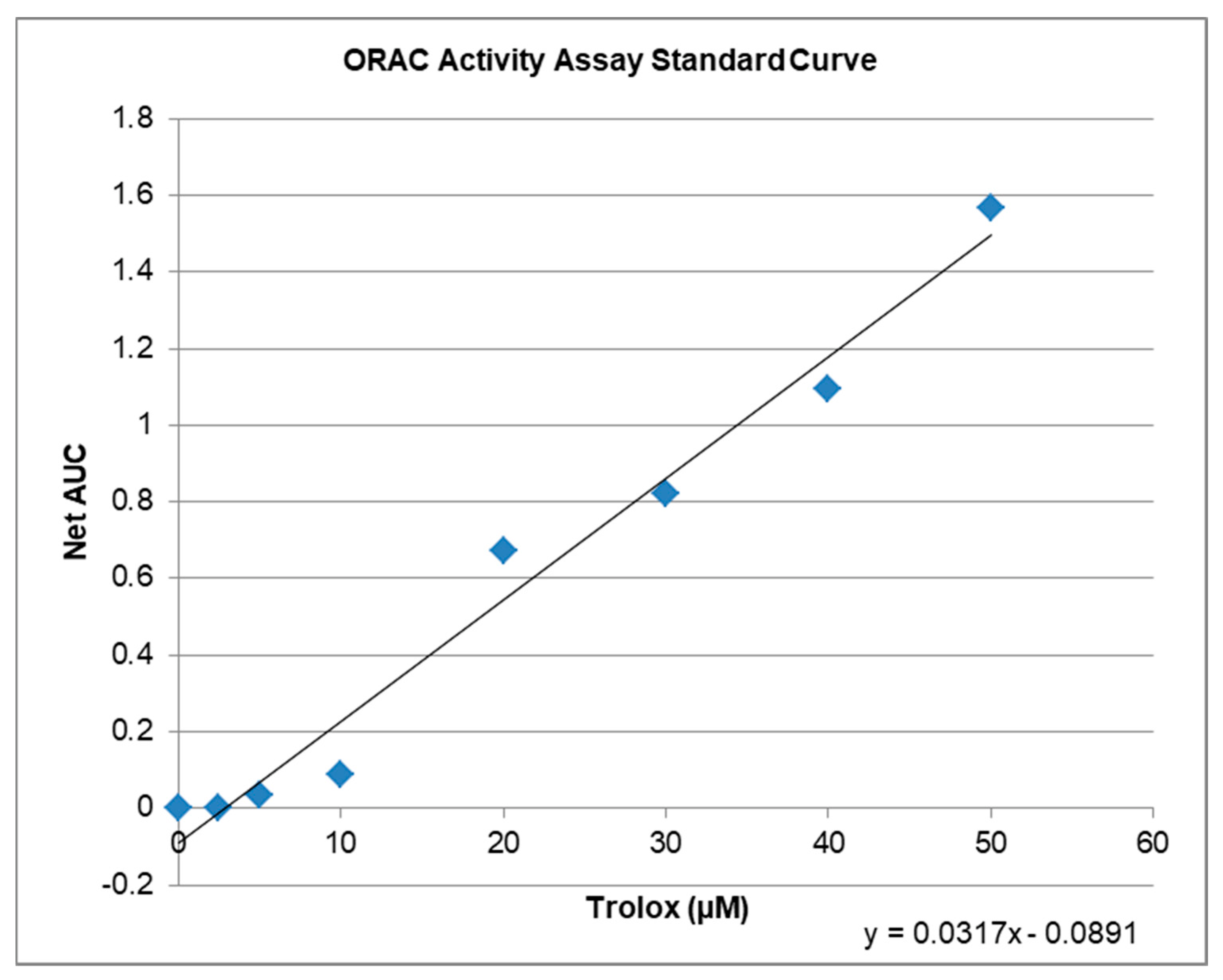
2.3. Antioxidative Capacity of KIT C and H in the ORAC Assay
2.4. GPR55-Dependent Antioxidative Effects
2.5. GPR55-Dependent Antioxidative Effects in GPR55 Knock Out Cells
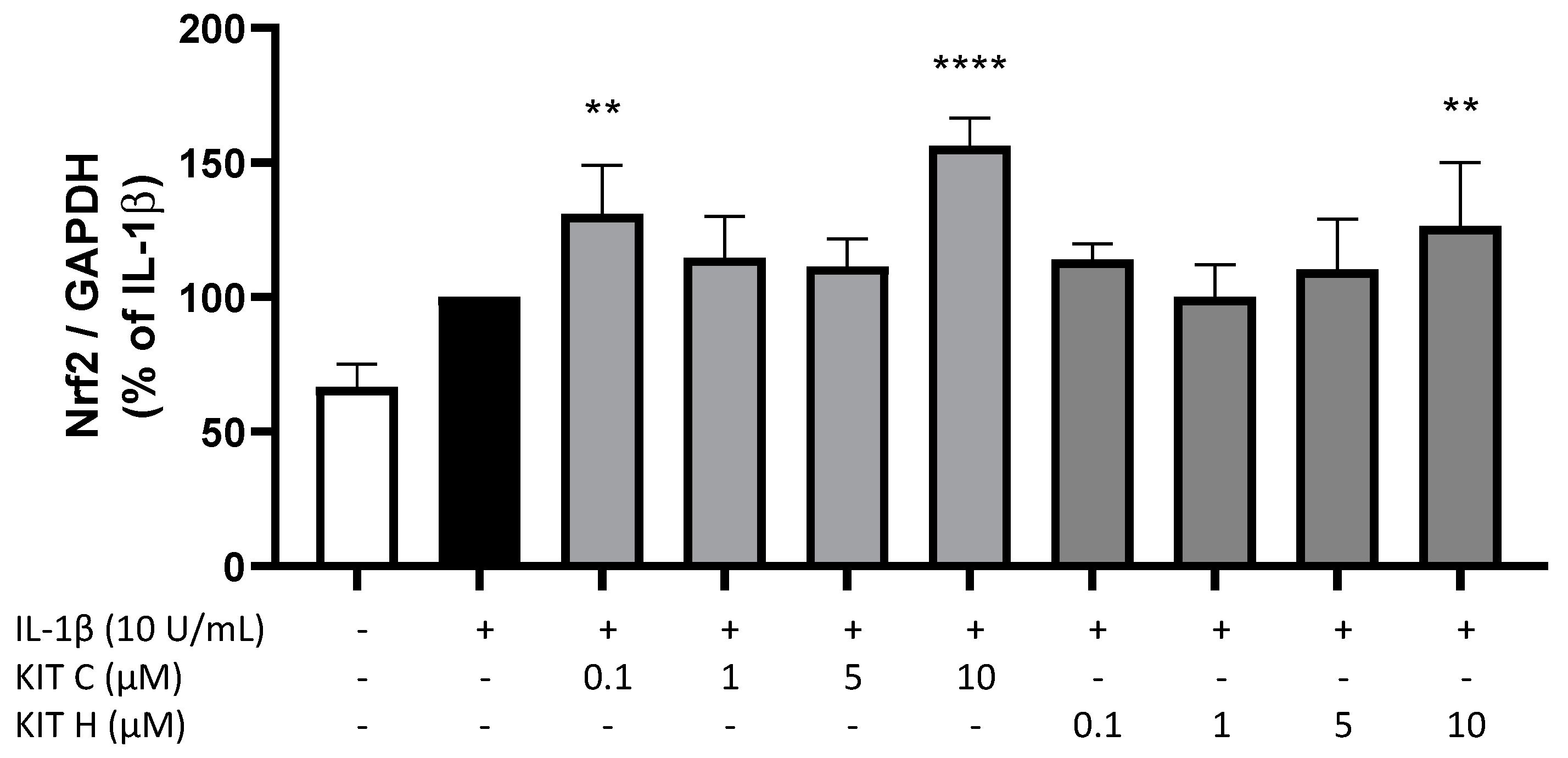
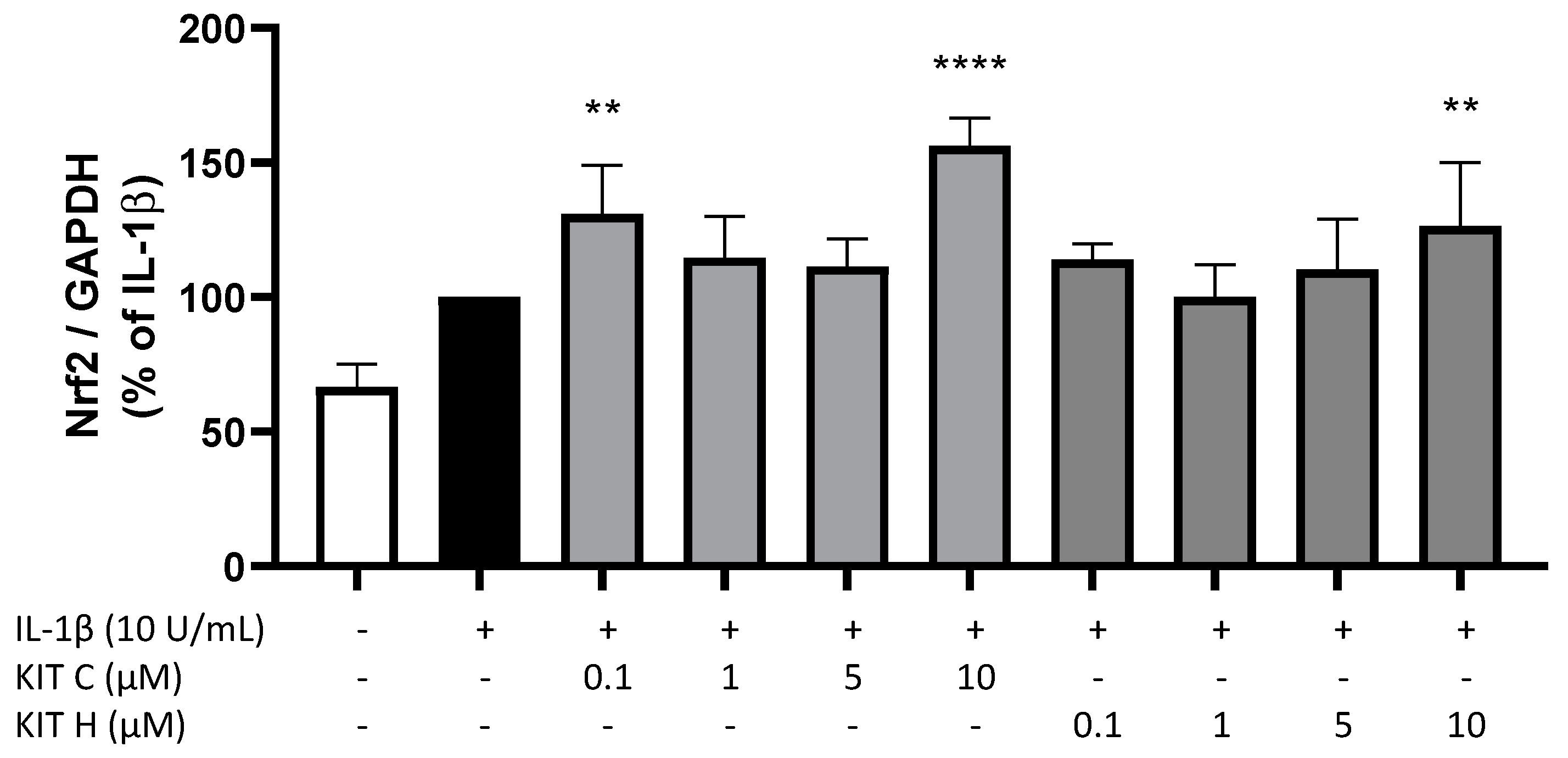
2.6. Nrf-2 Expression in SK-N-SH Cells
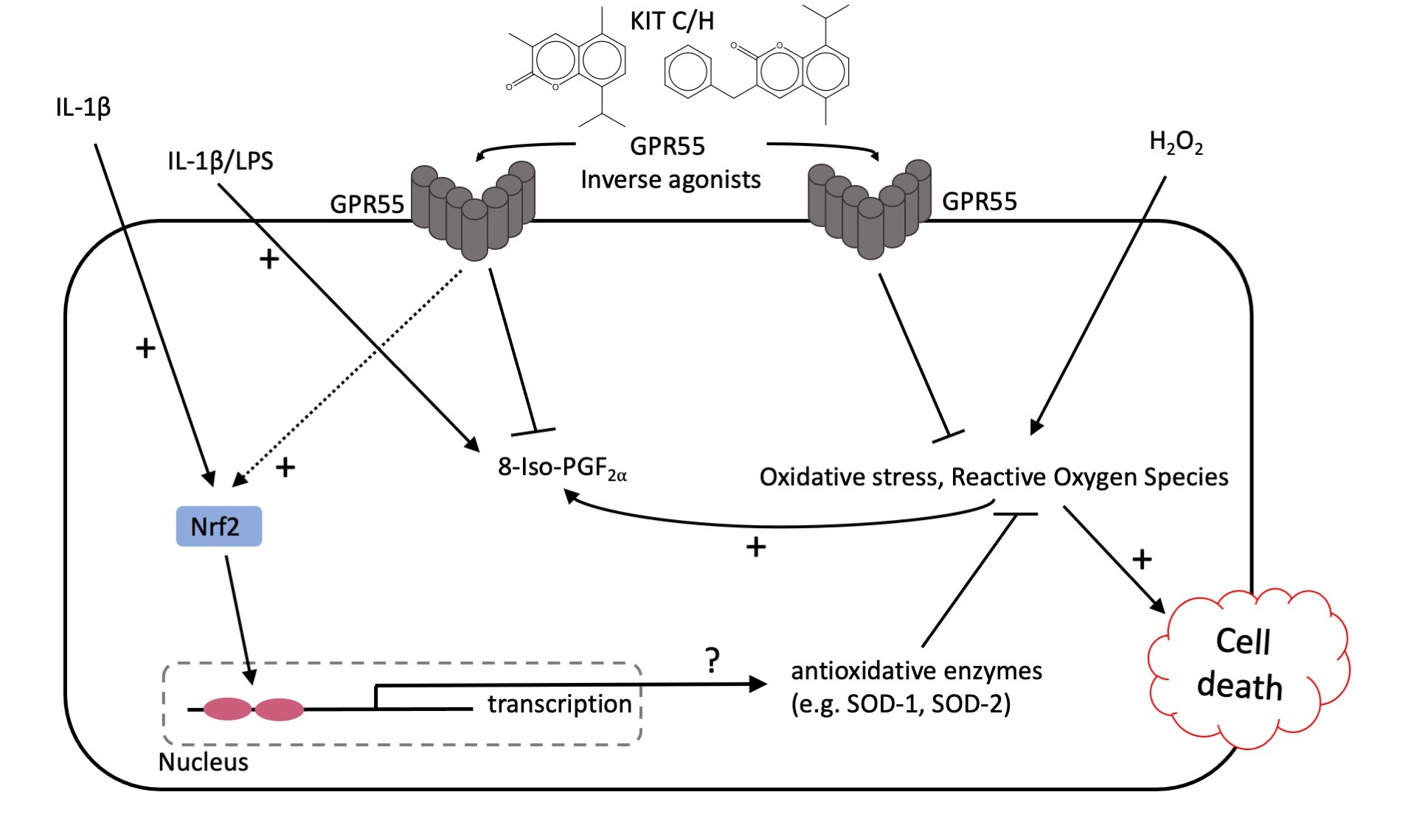
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Other Materials
4.3. Human Neuroblastoma (SK-N-SH) Cell Culture
4.4. GPR55-Knockout with CRISPR/Cas9 System in SK-N-SH Cells
4.5. Primary Mouse Microglial Cultures
4.5.1. Ethics Statement
4.5.2. Primary Mouse Microglia Cultures
4.6. Cell Viability Assay
4.7. Determination of 8-Iso-PGF2α (8-Isoprostane) Release
4.8. ROS-MTT Assay
4.9. RNA Isolation and Quantitative PCR
4.10. ORAC-Assay
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dimopoulos, N.; Piperi, C.; Psarra, V.; Lea, R.W.; Kalofoutis, A. Increased plasma levels of 8-iso-PGF2α and IL-6 in an elderly population with depression. Psychiatry Res. 2008, 161, 59–66. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef]
- Cui, L.; Hou, N.-N.; Wu, H.-M.; Zuo, X.; Lian, Y.-Z.; Zhang, C.-N.; Wang, Z.-F.; Zhang, X.; Zhu, J.-H. Prevalence of Alzheimer’s Disease and Parkinson’s Disease in China: An Updated Systematical Analysis. Front. Aging Neurosci. 2020, 12, 603854. [Google Scholar] [CrossRef] [PubMed]
- Savica, R.; Grossardt, B.R.; Bower, J.H.; Ahlskog, J.E.; Rocca, W.A. Time Trends in the Incidence of Parkinson Disease. JAMA Neurol. 2016, 73, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Sawzdargo, M.; Nguyen, T.; Lee, D.K.; Lynch, K.R.; Cheng, R.; Heng, H.H.Q.; George, S.R.; O’Dowd, B.F. Identification and cloning of three novel human G protein-coupled receptor genes GPR52, ΨGPR53 and GPR55: GPR55 is extensively expressed in human brain. Mol. Brain Res. 1999, 64, 193–198. [Google Scholar] [CrossRef]
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.-O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor: GPR55, a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar] [CrossRef]
- Oka, S.; Nakajima, K.; Yamashita, A.; Kishimoto, S.; Sugiura, T. Identification of GPR55 as a lysophosphatidylinositol receptor. Biochem. Biophys. Res. Commun. 2007, 362, 928–934. [Google Scholar] [CrossRef]
- Shore, D.M.; Reggio, P.H. The therapeutic potential of orphan GPCRs, GPR35 and GPR55. Front. Pharmacol. 2015, 6, 13–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saliba, S.W.; Jauch, H.; Gargouri, B.; Keil, A.; Hurrle, T.; Volz, N.; Mohr, F.; van der Stelt, M.; Bräse, S.; Fiebich, B.L. Anti-neuroinflammatory effects of GPR55 antagonists in LPS-activated primary microglial cells. J. Neuroinflamm. 2018, 15, 322. [Google Scholar] [CrossRef]
- Saliba, S.W.; Gläser, F.; Deckers, A.; Keil, A.; Hurrle, T.; Apweiler, M.; Ferver, F.; Volz, N.; Endres, D.; Bräse, S.; et al. Effects of a Novel GPR55 Antagonist on the Arachidonic Acid Cascade in LPS-Activated Primary Microglial Cells. Int. J. Mol. Sci. 2021, 22, 2503. [Google Scholar] [CrossRef] [PubMed]
- Sato, J.; Makita, N.; Iiri, T. Inverse agonism: The classic concept of GPCRs revisited. Endocr. J. 2016, 63, 507–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wisler, J.W.; Xiao, K.; Thomsen, A.R.; Lefkowitz, R.J. Recent developments in biased agonism. Curr. Opin. Cell Biol. 2014, 27, 18–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strachan, R.T.; Sun, J.; Rominger, D.H.; Violin, J.D.; Ahn, S.; Thomsen, A.; Zhu, X.; Kleist, A.; Costa, T.; Lefkowitz, R.J. Divergent transducer-specific molecular efficacies generate biased agonism at a G protein-coupled receptor (GPCR). J. Biol. Chem. 2014, 289, 14211–14224. [Google Scholar] [CrossRef] [Green Version]
- DeWire, S.M.; Violin, J.D. Biased ligands for better cardiovascular drugs: Dissecting G-protein-coupled receptor pharmacology. Circ. Res. 2011, 109, 205–216. [Google Scholar] [CrossRef]
- Blättermann, S.; Peters, L.; Ottersbach, P.A.; Bock, A.; Konya, V.; Weaver, C.D.; Gonzalez, A.; Schröder, R.; Tyagi, R.; Luschnig, P.; et al. A biased ligand for OXE-R uncouples Gα and Gβγ signaling within a heterotrimer. Nat. Chem. Biol. 2012, 8, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Falasca, M.; Ferro, R. Role of the lysophosphatidylinositol/GPR55 axis in cancer. Adv. Biol. Regul. 2016, 60, 88–93. [Google Scholar] [CrossRef]
- Wang, L.; Chen, Y.; Sternberg, P.; Cai, J. Essential roles of the PI3 kinase/Akt pathway in regulating Nrf2-dependent antioxidant functions in the RPE. Investig. Opthalmology Vis. Sci. 2008, 49, 1671–1678. [Google Scholar] [CrossRef] [Green Version]
- Akimov, M.G.; Gamisonia, A.M.; Dudina, P.V.; Gretskaya, N.M.; Gaydaryova, A.A.; Kuznetsov, A.S.; Zinchenko, G.N.; Bezuglov, V.V. GPR55 Receptor Activation by the N-Acyl Dopamine Family Lipids Induces Apoptosis in Cancer Cells via the Nitric Oxide Synthase (nNOS) Over-Stimulation. Int. J. Mol. Sci. 2021, 22, 622. [Google Scholar] [CrossRef]
- Wang, Y.; Pan, W.; Wang, Y.; Yin, Y. The GPR55 antagonist CID16020046 protects against ox-LDL-induced inflammation in human aortic endothelial cells (HAECs). Arch. Biochem. Biophys. 2020, 681, 108254. [Google Scholar] [CrossRef]
- Milne, G.L.; Yin, H.; Hardy, K.D.; Davies, S.S.; Roberts, L.J. Isoprostane generation and function. Chem. Rev. 2011, 111, 5973–5996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrett, A.R.; Murray, B.K.; Robison, R.A.; O’Neill, K.L. Measuring Antioxidant Capacity Using the ORAC and TOSC Assays. In Advanced Protocols in Oxidative Stress II; Methods in Molecular Biology; Armstrong, D., Ed.; Humana Press: Totowa, NJ, USA, 2010; Volume 594, pp. 251–262. ISBN 9781607614104. [Google Scholar]
- Celorrio, M.; Rojo-Bustamante, E.; Fernández-Suárez, D.; Sáez, E.; de Mendoza, A.E.-H.; Müller, C.E.; Ramírez, M.J.; Oyarzábal, J.; Franco, R.; Aymerich, M.S. GPR55: A therapeutic target for Parkinson’s disease? Neuropharmacology 2017, 125, 319–332. [Google Scholar] [CrossRef]
- García-Gutiérrez, M.S.; Navarrete, F.; Navarro, G.; Reyes-Resina, I.; Franco, R.; Lanciego, J.L.; Giner, S.; Manzanares, J. Alterations in Gene and Protein Expression of Cannabinoid CB2 and GPR55 Receptors in the Dorsolateral Prefrontal Cortex of Suicide Victims. Neurotherapeutics 2018, 15, 796–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acquaviva, A.; Vecchio, D.; Arezzini, B.; Comporti, M.; Gardi, C. Signaling pathways involved in isoprostane-mediated fibrogenic effects in rat hepatic stellate cells. Free Radic. Biol. Med. 2013, 65, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Rempel, V.; Volz, N.; Gläser, F.; Nieger, M.; Bräse, S.; Müller, C.E. Antagonists for the Orphan G-Protein-Coupled Receptor GPR55 Based on a Coumarin Scaffold. J. Med. Chem. 2013, 56, 4798–4810. [Google Scholar] [CrossRef] [PubMed]
- Robertson-Gray, O.J.; Walsh, S.K.; Ryberg, E.; Jönsson-Rylander, A.; Lipina, C.; Wainwright, C.L. l-α-Lysophosphatidylinositol (LPI) aggravates myocardial ischemia/reperfusion injury via a GPR55/ROCK-dependent pathway. Pharmacol. Res. Perspect. 2019, 7, e00487. [Google Scholar] [CrossRef] [PubMed]
- Wróbel, A.; Serefko, A.; Szopa, A.; Ulrich, D.; Poleszak, E.; Rechberger, T. O-1602, an Agonist of Atypical Cannabinoid Receptors GPR55, Reverses the Symptoms of Depression and Detrusor Overactivity in Rats Subjected to Corticosterone Treatment. Front. Pharmacol. 2020, 11, 1002. [Google Scholar] [CrossRef] [PubMed]
- Vomund, S.; Schäfer, A.; Parnham, M.; Brüne, B.; von Knethen, A. Nrf2, the Master Regulator of Anti-Oxidative Responses. Int. J. Mol. Sci. 2017, 18, 2772. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Chemotion: Synthesis of KIT C. Available online: https://dx.doi.org/10.14272/reaction/SA-FUHFF-UHFFFADPSC-LSYXPDGXGO-UHFFFADPSC-NUHFF-NUHFF-NUHFF-ZZZ (accessed on 26 October 2021).
- Chemotion: Analyses of KIT C. Available online: https://dx.doi.org/10.14272/LSYXPDGXGOSFCW-UHFFFAOYSA-N.1 (accessed on 26 October 2021).
- Chemotion: Synthesis of KIT H. Available online: https://www.chemotion-repository.net/pid/9509 (accessed on 26 October 2021).
- Chemotion: Analyses of KIT H. Available online: https://dx.doi.org/10.14272/WNXYNMKVCJSJKO-UHFFFAOYSA-N.1 (accessed on 26 October 2021).
- Saliba, S.W.; Marcotegui, A.R.; Fortwängler, E.; Ditrich, J.; Perazzo, J.C.; Muñoz, E.; de Oliveira, A.C.P.; Fiebich, B.L. AM404, paracetamol metabolite, prevents prostaglandin synthesis in activated microglia by inhibiting COX activity. J. Neuroinflamm. 2017, 14, 246. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Ibeas, P.; Barandalla, M.; Colleoni, S.; Lazzari, G. Pyruvate antioxidant roles in human fibroblasts and embryonic stem cells. Mol. Cell. Biochem. 2017, 429, 137–150. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | µM TE 1 | Net AUC 2 | AUC |
|---|---|---|---|
| Blank | 0.0 | 0.0 | 1.3684 |
| 1 µM KIT C | 5.2 | 0.0743 | 1.4427 |
| 10 µM KIT C | 6.5 | 0.1176 | 1.4860 |
| 1 µM KIT H | 3.4 | 0.0201 | 1.3885 |
| 10 µM KIT H | 4.7 | 0.0607 | 1.4291 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Apweiler, M.; Saliba, S.W.; Streyczek, J.; Hurrle, T.; Gräßle, S.; Bräse, S.; Fiebich, B.L. Targeting Oxidative Stress: Novel Coumarin-Based Inverse Agonists of GPR55. Int. J. Mol. Sci. 2021, 22, 11665. https://doi.org/10.3390/ijms222111665
Apweiler M, Saliba SW, Streyczek J, Hurrle T, Gräßle S, Bräse S, Fiebich BL. Targeting Oxidative Stress: Novel Coumarin-Based Inverse Agonists of GPR55. International Journal of Molecular Sciences. 2021; 22(21):11665. https://doi.org/10.3390/ijms222111665
Chicago/Turabian StyleApweiler, Matthias, Soraya Wilke Saliba, Jana Streyczek, Thomas Hurrle, Simone Gräßle, Stefan Bräse, and Bernd L. Fiebich. 2021. "Targeting Oxidative Stress: Novel Coumarin-Based Inverse Agonists of GPR55" International Journal of Molecular Sciences 22, no. 21: 11665. https://doi.org/10.3390/ijms222111665
APA StyleApweiler, M., Saliba, S. W., Streyczek, J., Hurrle, T., Gräßle, S., Bräse, S., & Fiebich, B. L. (2021). Targeting Oxidative Stress: Novel Coumarin-Based Inverse Agonists of GPR55. International Journal of Molecular Sciences, 22(21), 11665. https://doi.org/10.3390/ijms222111665