
Aberrant Upregulation of Indoleamine 2,3-Dioxygenase 1 Promotes Proliferation and Metastasis of Hepatocellular Carcinoma Cells via Coordinated Activation of AhR and β-Catenin Signaling

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
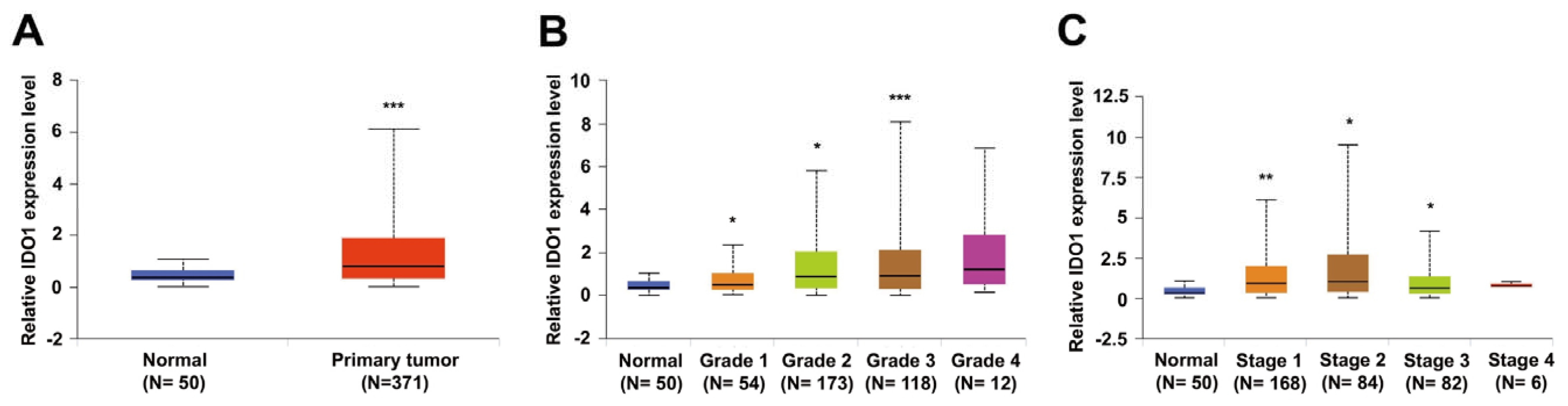
2.1. IDO1 Expression Is Highly Correlated with HCC Progression
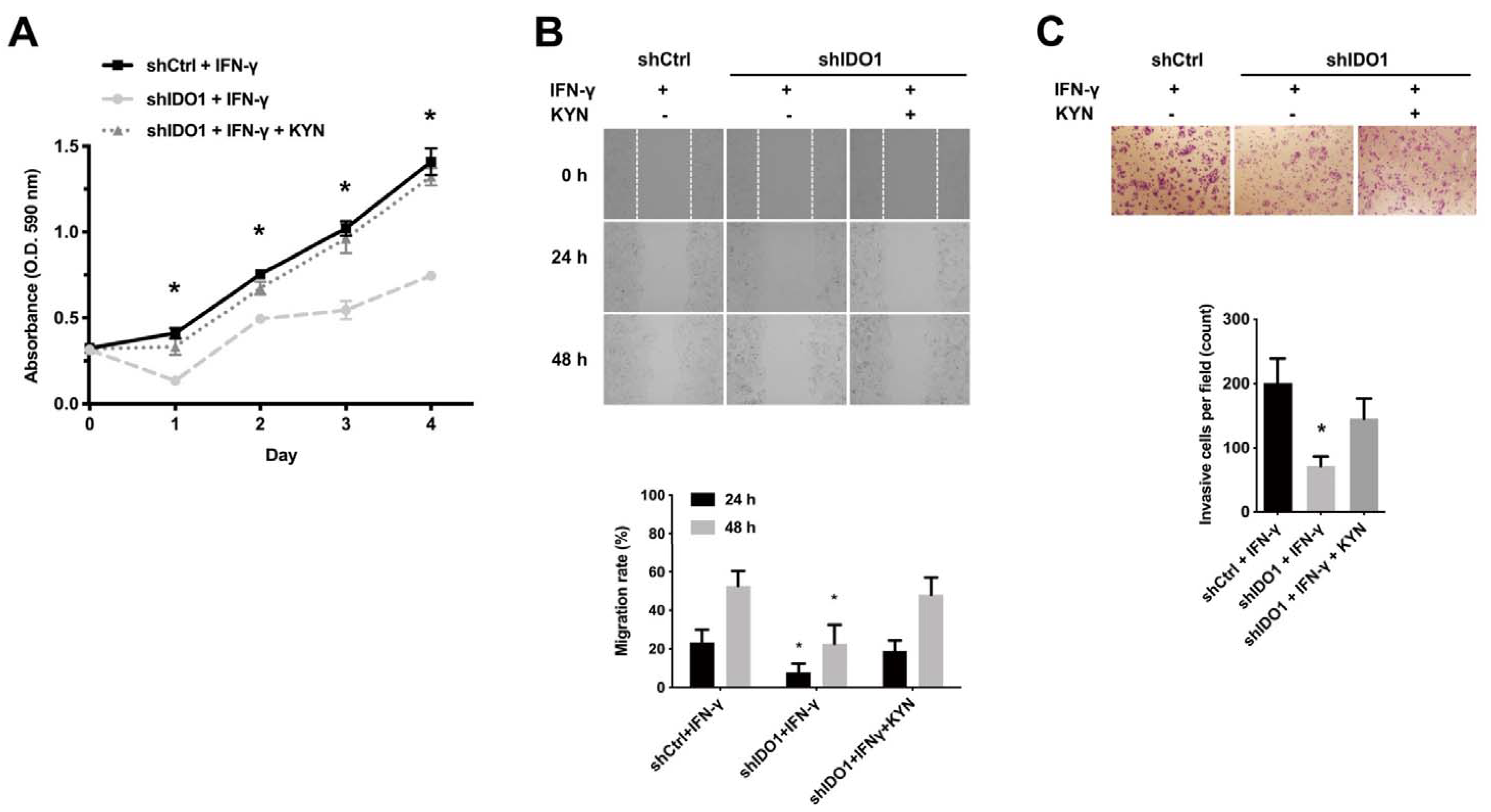
2.2. IDO1 Activity Is Implicated in HCC Proliferation and Metastasis
2.3. AhR and β-Catenin Are Potential Downstream Mediators of IDO1 Tumor-Promoting Activity
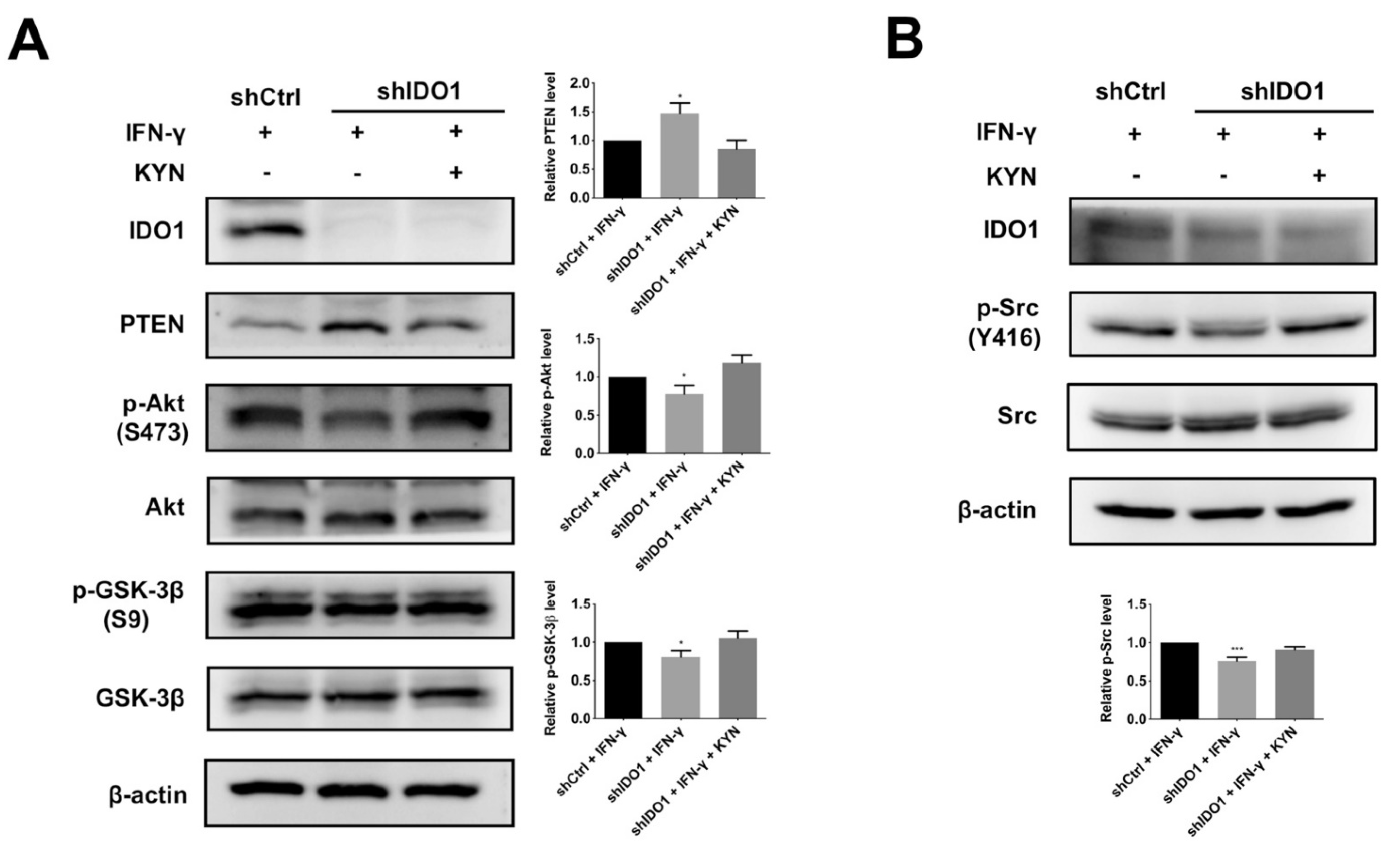
2.4. IDO1 Activates β-Catenin through the Src-PTEN-PI3K/Akt-GSK-3β Axis following AhR Activation
2.5. IDO1 Promotes Proliferation and Metastasis of HCC Cells through Modulating Proliferation- and EMT-Related Molecules
3. Discussion
4. Materials and Methods
4.1. Analysis of HCC Clinical Data from The Cancer Genome Atlas (TCGA) Database
4.2. Cell Culture, IDO1 Knockdown and Functional Assays
4.3. Cell Proliferation, Wound Healing, and Invasion Assays
4.4. Immunofluorescence Staining and Immunoblotting
4.5. Cell Fractionation Assay
4.6. Co-Immunoprecipitation (Co-IP)
4.7. Chromatin Immunoprecipitation (ChIP) Assay
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HCC | Hepatocellular carcinoma |
| EMT | Epithelial–mesenchymal transition |
| IDO1 | Indoleamine 2,3-dioxygenase 1 |
| IFN-γ | Interferon-γ |
| AhR | Aryl hydrocarbon receptor |
| KYN | Kynurenine |
| PI3K | Phosphoinositide 3-kinases |
| Akt | Protein kinase B |
| CYP1A1 | Cytochrome P450 1A1 |
| PTEN | Phosphatidylinositol 3,4,5-trisphosphate 3-phosphatase and dual-specificity protein phosphatase PTEN |
| Src | Proto-oncogene tyrosine-protein kinase Src |
| GSK-3β | Glycogen synthase kinase-3 beta |
| c-myc | Myc proto-oncogene protein |
| cyclin D1 | G1/S-specific cyclin-D1 |
| p53 | Cellular tumor antigen p53 |
| p21WAF/Cip1 | Cyclin-dependent kinase inhibitor 1 |
| p27Kip | Cyclin-dependent kinase inhibitor 1B |
| E-cadherin | Cadherin-1 |
| Snail | Zinc finger protein SNAI1 |
| ZEB-1 | Zinc finger E-box-binding homeobox 1 |
References
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.-W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Ward, E.M.; Johnson, C.J.; Cronin, K.A.; Ma, J.M.; Ryerson, A.B.; Mariotto, A.; Lake, A.J.; Wilson, R.; Sherman, R.L.; et al. Annual Report to the Nation on the Status of Cancer, 1975–2014, Featuring Survival. JNCI J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed]
- Lok, A.S.; Seeff, L.B.; Morgan, T.R.; Di Bisceglie, A.M.; Sterling, R.K.; Curto, T.M.; Everson, G.T.; Lindsay, K.L.; Lee, W.M.; Bonkovsky, H.L.; et al. Incidence of Hepatocellular Carcinoma and Associated Risk Factors in Hepatitis C-Related Advanced Liver Disease. Gastroenterology 2009, 136, 138–148. [Google Scholar] [CrossRef]
- Chang, M.H.; You, S.L.; Chen, C.J.; Liu, C.J.; Lee, C.M.; Lin, S.M.; Chu, H.-C.; Wu, T.-C.; Yang, S.-S.; Kuo, H.-S.; et al. Decreased Incidence of Hepatocellular Carcinoma in Hepatitis B Vaccinees: A 20-Year Follow-up Study. Jnci-J. Natl. Cancer Inst. 2009, 101, 1348–1355. [Google Scholar] [CrossRef]
- Levrero, M.; Zucman-Rossi, J. Mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatol. 2016, 64, S84–S101. [Google Scholar] [CrossRef] [PubMed]
- Estes, C.; Anstee, Q.M.; Arias-Loste, M.T.; Bantel, H.; Bellentani, S.; Caballeria, J.; Colombo, M.; Craxi, A.; Crespo, J.; Day, C.P.; et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016–2030. J. Hepatol. 2018, 69, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Marotta, F.; Vangieri, B.; Cecere, A.; Gattoni, A. The pathogenesis of hepatocellular carcinoma is multifactorial event. Novel immunological treatment in prospect. Clin. Ter. 2004, 155, 187–199. [Google Scholar] [PubMed]
- Mapara, M.Y.; Sykes, M. Tolerance and cancer: Mechanisms of tumor evasion and strategies for breaking tolerance. J. Clin. Oncol. 2004, 22, 1136–1151. [Google Scholar] [CrossRef] [PubMed]
- Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 2005, 5, 263–274. [Google Scholar] [CrossRef]
- Nagano, J.; Shimizu, M.; Hara, T.; Shirakami, Y.; Kochi, T.; Nakamura, N.; Ohtaki, H.; Ito, H.; Tanaka, T.; Tsurumi, H.; et al. Effects of indoleamine 2,3-dioxygenase deficiency on high-fat diet-induced hepatic inflammation. PLoS ONE 2013, 8, e73404. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Hoshi, M.; Ohtaki, H.; Taguchi, A.; Ando, K.; Ishikawa, T.; Osawa, Y.; Hara, A.; Moriwaki, H.; Saito, K.; et al. Ability of IDO to attenuate liver injury in alpha-galactosylceramide-induced hepatitis model. J. Immunol. 2010, 185, 4554–4560. [Google Scholar] [CrossRef]
- Uyttenhove, C.; Pilotte, L.; Theate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; van den Eynde, B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Mellor, A.L.; Munn, D.H. IDO expression by dendritic cells: Tolerance and tryptophan catabolism. Nat. Rev. Immunol. 2004, 4, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Godin-Ethier, J.; Hanafi, L.A.; Piccirillo, C.A.; Lapointe, R. Indoleamine 2,3-dioxygenase expression in human cancers: Clinical and immunologic perspectives. Clin. Cancer Res. 2011, 17, 6985–6991. [Google Scholar] [CrossRef] [PubMed]
- Takikawa, O. Biochemical and medical aspects of the indoleamine 2,3-dioxygenase-initiated L-tryptophan metabolism. Biochem. Biophys. Res. Commun. 2005, 338, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Speeckaert, R.; Vermaelen, K.; van Geel, N.; Autier, P.; Lambert, J.; Haspeslagh, M.; van Gele, M.; Thielemans, K.; Neyns, B.; Roche, N.; et al. Indoleamine 2,3-dioxygenase, a new prognostic marker in sentinel lymph nodes of melanoma patients. Eur. J. Cancer 2012, 48, 2004–2011. [Google Scholar] [CrossRef]
- Zhong, W.; Tong, Y.; Li, Y.; Yuan, J.; Hu, S.; Hu, T.; Song, G. Mesenchymal stem cells in inflammatory microenvironment potently promote metastatic growth of cholangiocarcinoma via activating Akt/NF-kappaB signaling by paracrine CCL5. Oncotarget 2017, 8, 73693–73704. [Google Scholar] [CrossRef] [PubMed]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef]
- Muller, A.J.; DuHadaway, J.B.; Donover, P.S.; Sutanto-Ward, E.; Prendergast, G.C. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat. Med. 2005, 11, 312–319. [Google Scholar] [CrossRef]
- Hou, D.Y.; Muller, A.J.; Sharma, M.D.; DuHadaway, J.; Banerjee, T.; Johnson, M.; Mellor, A.L.; Prendergast, G.C.; Munn, D. Inhibition of indoleamine 2,3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res. 2007, 67, 792–801. [Google Scholar] [CrossRef]
- Pan, K.; Wang, H.; Chen, M.S.; Zhang, H.K.; Weng, D.S.; Zhou, J.; Huang, W.; Li, J.-J.; Song, H.-F.; Xia, J.-C. Expression and prognosis role of indoleamine 2,3-dioxygenase in hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2008, 134, 1247–1253. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Thaker, A.I.; Rao, M.S.; Bishnupuri, K.S.; Kerr, T.A.; Foster, L.; Marinshaw, J.M.; Newberry, R.; Stenson, W.F.; Ciorba, M.A. IDO1 metabolites activate beta-catenin signaling to promote cancer cell proliferation and colon tumorigenesis in mice. Gastroenterology 2013, 145, 416–425. [Google Scholar] [CrossRef]
- Braeuning, A.; Kohle, C.; Buchmann, A.; Schwarz, M. Coordinate regulation of cytochrome P450 1a1 expression in mouse liver by the aryl hydrocarbon receptor and the beta-catenin pathway. Toxicol. Sci. 2011, 122, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Al-Dhfyan, A.; Alhoshani, A.; Korashy, H.M. Aryl hydrocarbon receptor/cytochrome P450 1A1 pathway mediates breast cancer stem cells expansion through PTEN inhibition and beta-Catenin and Akt activation. Mol. Cancer 2017, 16, 14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Jiao, L.Y.; Li, T.J.; Zhu, Y.Y.; Zhou, J.W.; Tian, J. GSK-3beta suppresses HCC cell dissociation in vitro by upregulating epithelial junction proteins and inhibiting Wnt/beta-catenin signaling pathway. J. Cancer 2017, 8, 1598–1608. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Yu, Q.; Liu, J.H.; Zhang, J.; Wang, H.; Koul, D.; McMurray, J.S.; Fang, X.; Yung, W.A.; Siminovitch, K.A.; et al. Src family protein-tyrosine kinases alter the function of PTEN to regulate phosphatidylinositol 3-kinase/AKT cascades. J. Biol. Chem. 2003, 278, 40057–40066. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Peng, Z.; Raufman, J.P. Src-mediated aryl hydrocarbon and epidermal growth factor receptor cross talk stimulates colon cancer cell proliferation. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1006–G1015. [Google Scholar] [CrossRef] [PubMed]
- Shang, S.; Hua, F.; Hu, Z.W. The regulation of beta-catenin activity and function in cancer: Therapeutic opportunities. Oncotarget 2017, 8, 33972–33989. [Google Scholar] [CrossRef]
- Lai, D.W.; Liu, S.H.; Karlsson, A.I.; Lee, W.J.; Wang, K.B.; Chen, Y.C.; Shen, C.-C.; Wu, S.-M.; Liu, C.-Y.; Tien, H.-R.; et al. The novel Aryl hydrocarbon receptor inhibitor biseugenol inhibits gastric tumor growth and peritoneal dissemination. Oncotarget 2014, 5, 7788–7804. [Google Scholar] [CrossRef][Green Version]
- Batlle, E.; Sancho, E.; Franci, C.; Dominguez, D.; Monfar, M.; Baulida, J.; De Herreros, A.G. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell. Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Berasain, C.; Castillo, J.; Perugorria, M.J.; Latasa, M.U.; Prieto, J.; Avila, M.A. Inflammation and liver cancer: New molecular links. Ann. N. Y. Acad. Sci. 2009, 1155, 206–221. [Google Scholar] [CrossRef] [PubMed]
- WHO. Hepatitis Viruses. In International Agency for Research on Cancer (IARC); World Health Organization: Lyon, France, 1994. [Google Scholar]
- Hornyak, L.; Dobos, N.; Koncz, G.; Karanyi, Z.; Pall, D.; Szabo, Z.; Halmos, G.; Székvölgyi, L. The Role of Indoleamine-2,3-Dioxygenase in Cancer Development, Diagnostics, and Therapy. Front. Immunol. 2018, 9, 151. [Google Scholar] [CrossRef] [PubMed]
- Asghar, K.; Farooq, A.; Zulfiqar, B.; Rashid, M.U. Indoleamine 2,3-dioxygenase: As a potential prognostic marker and immunotherapeutic target for hepatocellular carcinoma. World J. Gastroentero 2017, 23, 2286–2293. [Google Scholar] [CrossRef]
- Li, S.L.; Han, X.; Lyu, N.; Xie, Q.K.; Deng, H.J.; Mu, L.W.; Pan, T.; Huang, X.; Wang, X.; Shi, Y.; et al. Mechanism and prognostic value of indoleamine 2,3-dioxygenase 1 expressed in hepatocellular carcinoma. Cancer Sci. 2018, 109, 3726–3736. [Google Scholar] [CrossRef]
- Shibata, Y.; Hara, T.; Nagano, J.; Nakamura, N.; Ohno, T.; Ninomiya, S.; Masahito, S.; Tanaka, T.; Saito, K.; Seishima, M.; et al. The Role of Indoleamine 2,3-Dioxygenase in Diethylnitrosamine-Induced Liver Carcinogenesis. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.P.; Fu, S.F.; Chen, X.; Ye, J.; Ye, Y.; Kong, L.D.; Kong, L.-D.; Zhu, Z. The Clinicopathological and Prognostic Significance of IDO1 Expression in Human Solid Tumors: Evidence from a Systematic Review and Meta-Analysis. Cell Physiol. Biochem. 2018, 49, 134–143. [Google Scholar] [CrossRef]
- Le Naour, J.; Galluzzi, L.; Zitvogel, L.; Kroemer, G.; Vacchelli, E. Trial watch: IDO inhibitors in cancer therapy. Oncoimmunology 2020, 9, 1777625. [Google Scholar] [CrossRef]
- Bishnupuri, K.S.; Alvarado, D.M.; Khouri, A.N.; Shabsovich, M.; Chen, B.; Dieckgraefe, B.K.; Ciorba, M.A. IDO1 and Kynurenine Pathway Metabolites Activate PI3K-Akt Signaling in the Neoplastic Colon Epithelium to Promote Cancer Cell Proliferation and Inhibit Apoptosis. Cancer Res. 2019, 79, 1138–1150. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.; Chang, M.Y.; Parker, K.H.; Beury, D.W.; DuHadaway, J.B.; Flick, H.E.; Boulden, J.; Sutanto-Ward, E.; Soler, A.P.; Laury-Kleintop, L.D.; et al. IDO is a nodal pathogenic driver of lung cancer and metastasis development. Cancer Discov. 2012, 2, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Randi, A.S.; Sanchez, M.S.; Alvarez, L.; Cardozo, J.; Pontillo, C.; de Pisarev, D.L.K. Hexachlorobenzene triggers AhR translocation to the nucleus, c-Src activation and EGFR transactivation in rat liver. Toxicol. Lett. 2008, 177, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Lan, K.H.; Zhou, X.; Tan, M.; Esteva, F.J.; Sahin, A.A.; Klos, K.S.; Li, P.; Monia, B.P.; Nguyen, N.T.; et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004, 6, 117–127. [Google Scholar] [CrossRef]
- Ozaki, T.; Nakagawara, A. Role of p53 in Cell Death and Human Cancers. Cancers 2011, 3, 994–1013. [Google Scholar] [CrossRef] [PubMed]
- Chu, I.M.; Hengst, L.; Slingerland, J.M. The Cdk inhibitor p27 in human cancer: Prognostic potential and relevance to anticancer therapy. Nat. Rev. Cancer 2008, 8, 253–267. [Google Scholar] [CrossRef]
- Paajarvi, G.; Viluksela, M.; Pohjanvirta, R.; Stenius, U.; Hogberg, J. TCDD activates Mdm2 and attenuates the p53 response to DNA damaging agents. Carcinogenesis 2005, 26, 201–208. [Google Scholar] [CrossRef]
- Pollet, M.; Shaik, S.; Mescher, M.; Frauenstein, K.; Tigges, J.; Braun, S.A.; Sondenheimer, K.; Kaveh, M.; Bruhs, A.; Meller, S.; et al. The AHR represses nucleotide excision repair and apoptosis and contributes to UV-induced skin carcinogenesis. Cell Death Differ. 2018, 25, 1823–1836. [Google Scholar] [CrossRef]
- Jin, U.H.; Karki, K.; Cheng, Y.T.; Michelhaugh, S.K.; Mittal, S.; Safe, S. The aryl hydrocarbon receptor is a tumor suppressor-like gene in glioblastoma. J. Biol. Chem. 2019, 294, 11342–11353. [Google Scholar] [CrossRef] [PubMed]
- Schmalhofer, O.; Brabletz, S.; Brabletz, T. E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009, 28, 151–166. [Google Scholar] [CrossRef]
- Chen, J.Y.; Li, C.F.; Kuo, C.C.; Tsai, K.K.; Hou, M.F.; Hung, W.C. Cancer/stroma interplay via cyclooxygenase-2 and indoleamine 2,3-dioxygenase promotes breast cancer progression. Breast Cancer Res. 2014, 16, 410. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, H.T.; Gadalla, R.; El-Husseiny, N.; Hassan, H.; Wang, Z.; Ibrahim, S.A.; El-Shinawi, M.; Sherr, D.H.; Mohamed, M.M. Inflammatory breast cancer: Activation of the aryl hydrocarbon receptor and its target CYP1B1 correlates closely with Wnt5a/b-beta-catenin signalling, the stem cell phenotype and disease progression. J. Adv. Res. 2019, 16, 75–86. [Google Scholar] [CrossRef]
- Guaita, S.; Puig, I.; Franci, C.; Garrido, M.; Dominguez, D.; Batlle, E.; Sancho, E.; Dedhar, S.; de Herreros, A.G.; Baulida, J. Snail induction of epithelial to mesenchymal transition in tumor cells is accompanied by MUC1 repression and ZEB1 expression. J. Biol. Chem. 2002, 277, 39209–39216. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.Y.; Wang, P.P.; Huang, Z.L.; Peng, L.; Lin, C.S.; Gao, Z.L.; Su, S. Tumoral indoleamine 2, 3-dioxygenase 1 is regulated by monocytes and T lymphocytes collaboration in hepatocellular carcinoma. Oncotarget 2016, 7, 14781–14790. [Google Scholar] [CrossRef] [PubMed]
- Friemel, J.; Rechsteiner, M.; Frick, L.; Bohm, F.; Struckmann, K.; Egger, M.; Moch, H.; Heikenwaelder, M.; Weber, A. Intratumor heterogeneity in hepatocellular carcinoma. Clin. Cancer Res. 2015, 21, 1951–1961. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.C.; Hsu, C.H.; Hsu, C.; Cheng, A.L. Tumor Heterogeneity in Hepatocellular Carcinoma: Facing the Challenges. Liver Cancer 2016, 5, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.T.; Liao, L.Z.; Lu, C.H.; Huang, Y.H.; Lin, Y.K.; Lin, J.H.; Chow, L.-P. Quantitative phosphoproteomic analysis identifies the potential therapeutic target EphA2 for overcoming sorafenib resistance in hepatocellular carcinoma cells. Exp. Mol. Med. 2020, 52, 497–513. [Google Scholar] [CrossRef]
- Lecoq, A.L.; Viengchareun, S.; Hage, M.; Bouligand, J.; Young, J.; Boutron, A.; Zizzari, P.; Lombès, M.; Chanson, P.; Kamenický, P. AIP mutations impair AhR signaling in pituitary adenoma patients fibroblasts and in GH3 cells. Endocr. Relat. Cancer 2016, 23, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.D.; Denisenko, O.; Bomsztyk, K. Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat. Protoc. 2006, 1, 179–185. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.-T.; Wu, P.-H.; Hu, C.-C.; Nien, H.-C.; Wang, J.-T.; Sheu, J.-C.; Chow, L.-P. Aberrant Upregulation of Indoleamine 2,3-Dioxygenase 1 Promotes Proliferation and Metastasis of Hepatocellular Carcinoma Cells via Coordinated Activation of AhR and β-Catenin Signaling. Int. J. Mol. Sci. 2021, 22, 11661. https://doi.org/10.3390/ijms222111661
Chen C-T, Wu P-H, Hu C-C, Nien H-C, Wang J-T, Sheu J-C, Chow L-P. Aberrant Upregulation of Indoleamine 2,3-Dioxygenase 1 Promotes Proliferation and Metastasis of Hepatocellular Carcinoma Cells via Coordinated Activation of AhR and β-Catenin Signaling. International Journal of Molecular Sciences. 2021; 22(21):11661. https://doi.org/10.3390/ijms222111661
Chicago/Turabian StyleChen, Chih-Ta, Pei-Hua Wu, Chia-Chi Hu, Hsiao-Ching Nien, Jin-Town Wang, Jin-Chuan Sheu, and Lu-Ping Chow. 2021. "Aberrant Upregulation of Indoleamine 2,3-Dioxygenase 1 Promotes Proliferation and Metastasis of Hepatocellular Carcinoma Cells via Coordinated Activation of AhR and β-Catenin Signaling" International Journal of Molecular Sciences 22, no. 21: 11661. https://doi.org/10.3390/ijms222111661
APA StyleChen, C.-T., Wu, P.-H., Hu, C.-C., Nien, H.-C., Wang, J.-T., Sheu, J.-C., & Chow, L.-P. (2021). Aberrant Upregulation of Indoleamine 2,3-Dioxygenase 1 Promotes Proliferation and Metastasis of Hepatocellular Carcinoma Cells via Coordinated Activation of AhR and β-Catenin Signaling. International Journal of Molecular Sciences, 22(21), 11661. https://doi.org/10.3390/ijms222111661