
Differential Effects of Human P301L Tau Expression in Young versus Aged Mice

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Experimental Design
2.3. Morris Water Maze (MWM)
2.4. Visible Platform Test
2.5. Y-Maze Spontaneous Alternation Test
2.6. Enzyme-Based Microelectrode Arrays (MEAs)
2.7. In Vivo Anesthetized Recordings
2.8. Immunoblotting
2.9. Data Analysis
3. Results
3.1. Tau Expression Is Similar in Young vs. Aged Mice
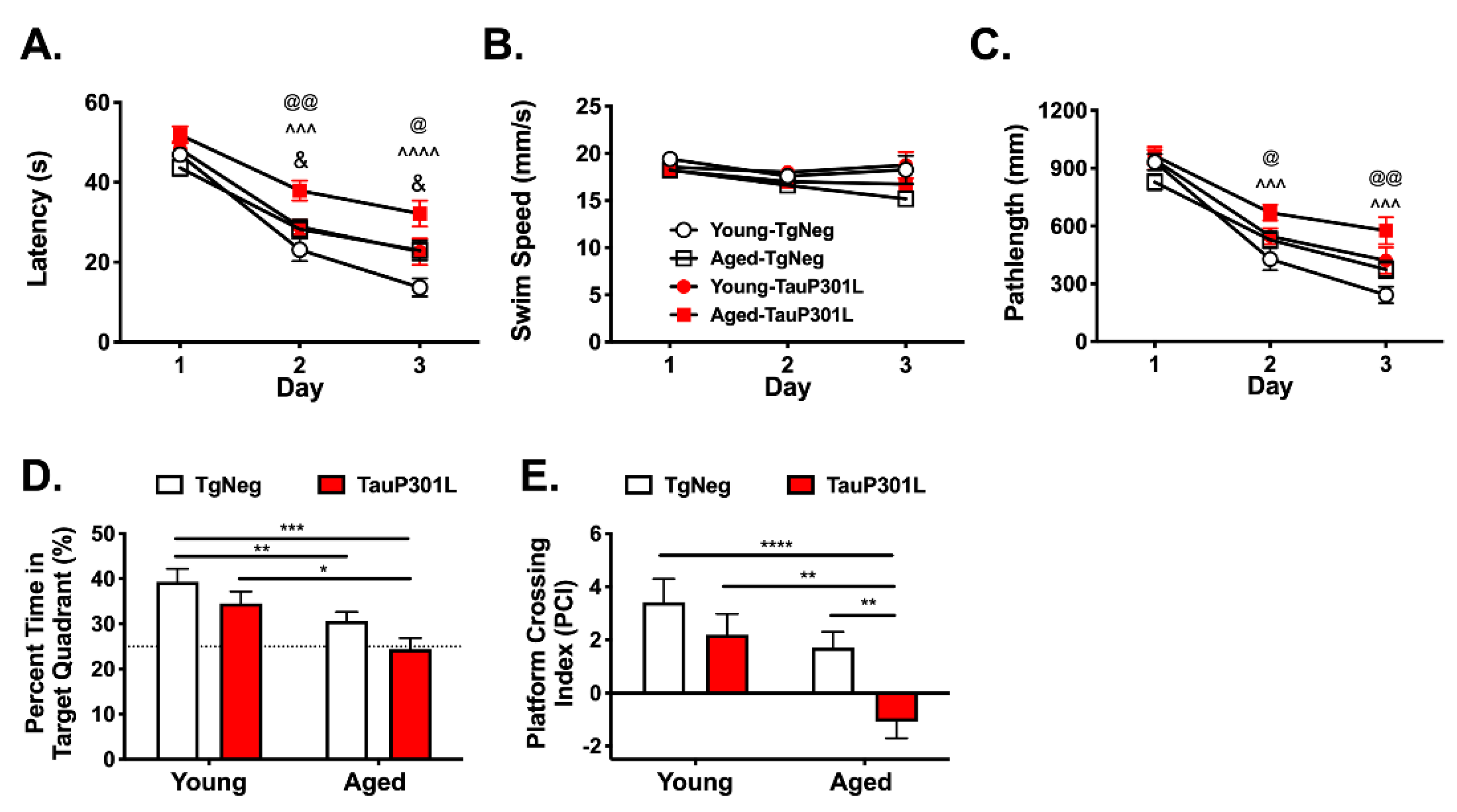
3.2. Age and P301L Tau Expression Alter Learning and Memory in the Morris Water Maze and Y-Maze
3.2.1. Visible Platform Training
3.2.2. Morris Water Maze
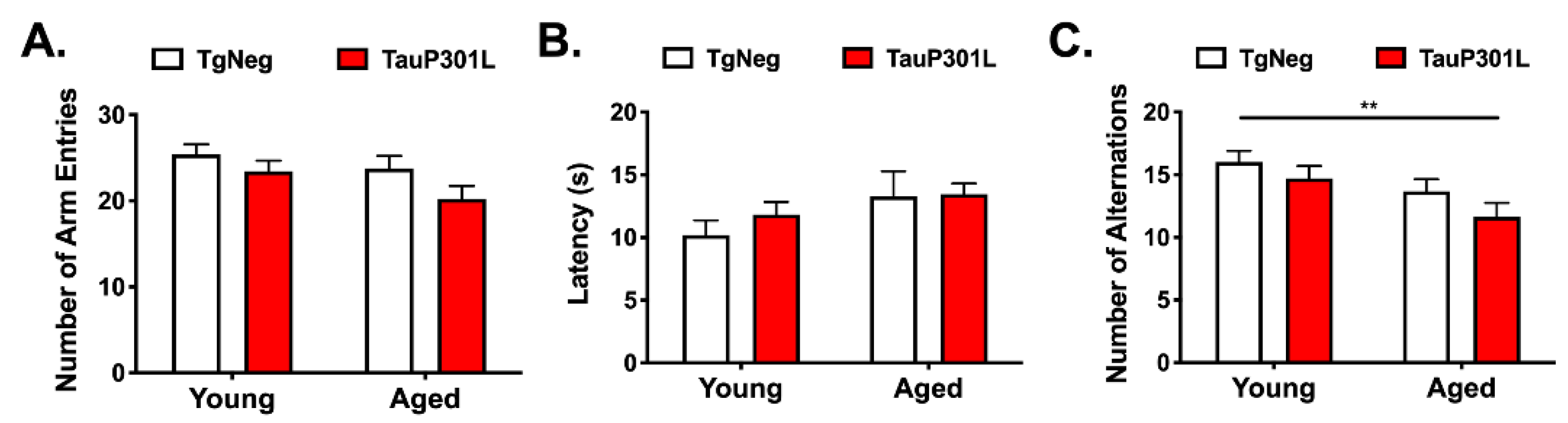
3.2.3. Y-Maze Spontaneous Alternation Test
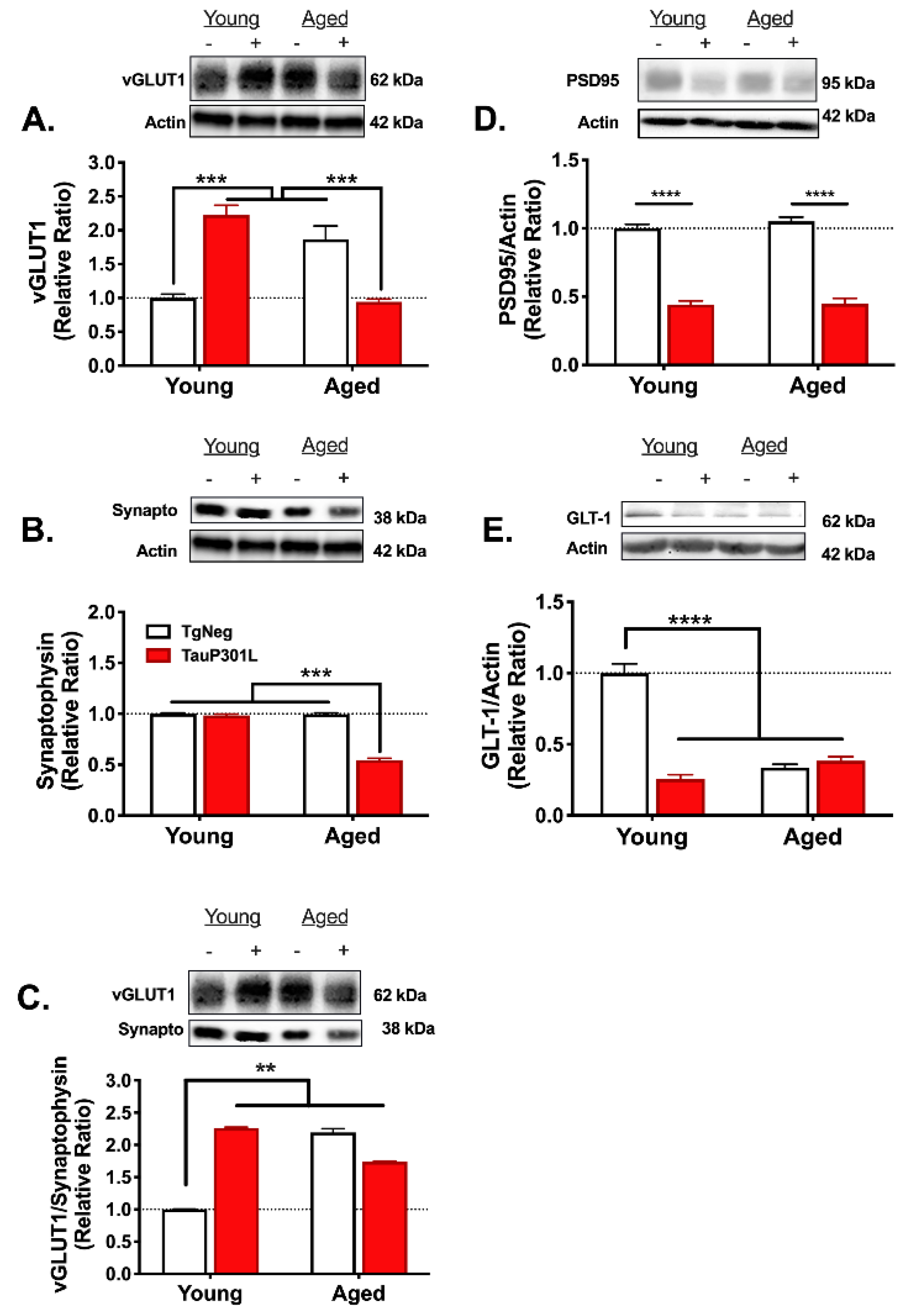
3.3. Age- and Tau-Associated Changes in Glutamate Regulation
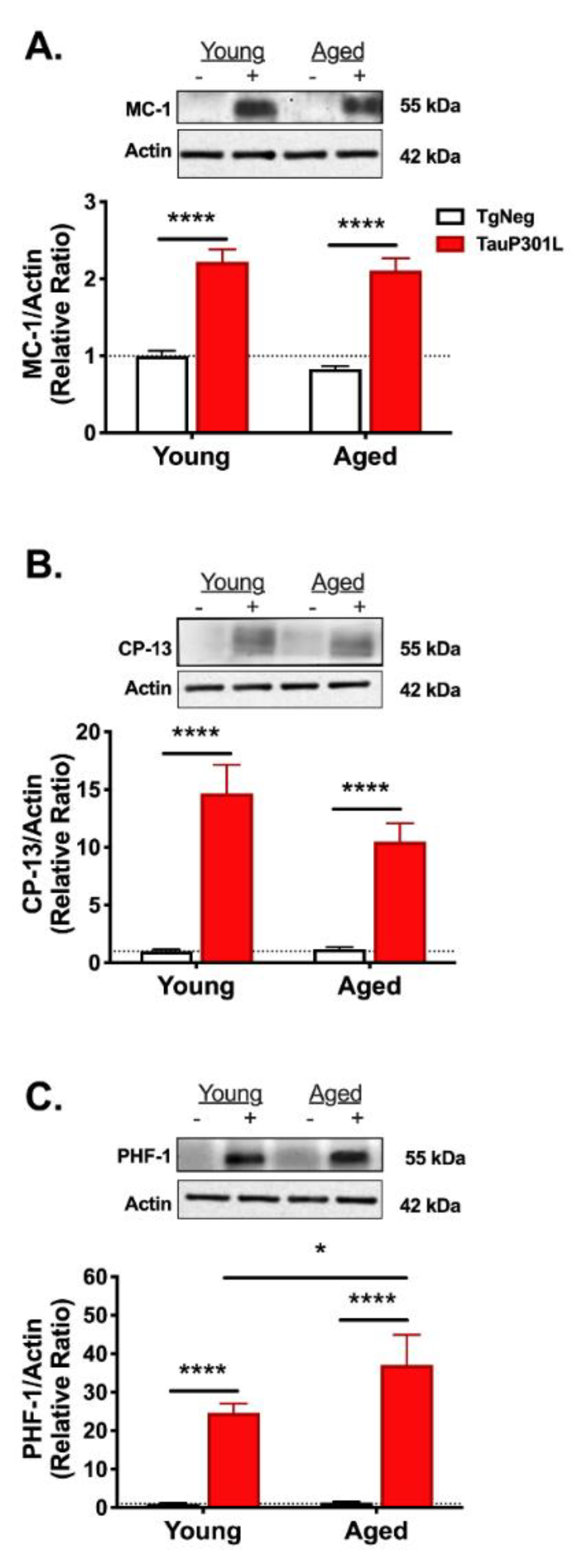
3.4. Age- and Tau-Associated Changes in Protein Expression
3.4.1. Tripartite Synapse
3.4.2. Tau Pathology
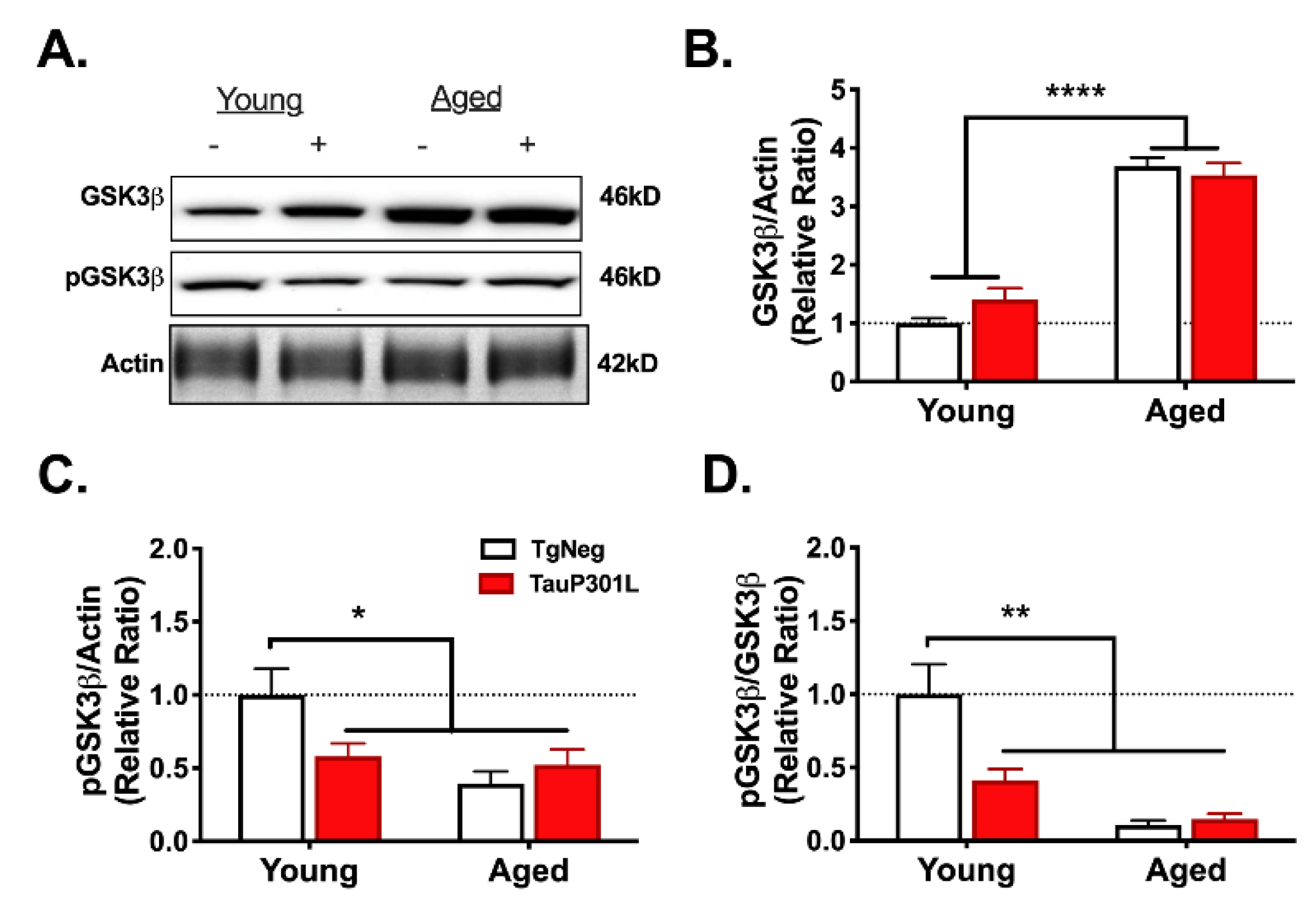
3.4.3. Glycogen Synthase Kinase-3-Beta
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Han, R.Z.; Hu, J.J.; Weng, Y.C.; Li, D.F.; Huang, Y. NMDA receptor antagonist MK-801 reduces neuronal damage and preserves learning and memory in a rat model of traumatic brain injury. Neurosci. Bull. 2009, 25, 367–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saura, C.A.; Valero, J. The role of CREB signaling in Alzheimer’s disease and other cognitive disorders. Rev. Neurosci. 2011, 22, 153–169. [Google Scholar] [CrossRef] [PubMed]
- Aschner, M.; Yao, C.P.; Allen, J.W.; Tan, K.H. Methylmercury alters glutamate transport in astrocytes. Neurochem. Int. 2000, 37, 199–206. [Google Scholar] [CrossRef]
- Hinzman, J.M.; Thomas, T.C.; Quintero, J.E.; Gerhardt, G.A.; Lifshitz, J. Disruptions in the regulation of extracellular glutamate by neurons and glia in the rat striatum two days after diffuse brain injury. J. Neurotrauma 2012, 29, 1197–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuperstein, I.; Broersen, K.; Benilova, I.; Rozenski, J.; Jonckheere, W.; Debulpaep, M.; Vandersteen, A.; Segers-Nolten, I.; Van Der Werf, K.; Subramaniam, V.; et al. Neurotoxicity of Alzheimer’s disease Aβ peptides is induced by small changes in the Aβ42 to Aβ40 ratio. EMBO J. 2010, 29, 3408–3420. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E.; Fukunaga, Y.; Bading, H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 2002, 5, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wong, T.P.; Aarts, M.; Rooyakkers, A.; Liu, L.; Lai, T.W.; Wu, D.C.; Lu, J.; Tymianski, M.; Craig, A.M.; et al. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J. Neurosci. 2007, 27, 2846–2857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meur, K.L.; Galante, M.; Angulo, M.C.; Audinat, E. Tonic activation of NMDA receptors by ambient glutamate of non-synaptic origin in the rat hippocampus. J. Physiol. 2007, 580, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Hunsberger, H.C.; Hickman, J.E.; Reed, M.N. Riluzole rescues alterations in rapid glutamate transients in the hippocampus of rTg4510 mice. Metab. Brain Dis. 2016, 31, 711–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunsberger, H.C.; Rudy, C.C.; Batten, S.R.; Gerhardt, G.A.; Reed, M.N. P301L tau expression affects glutamate release and clearance in the hippocampal trisynaptic pathway. J. Neurochem. 2015, 132, 169–182. [Google Scholar] [CrossRef] [Green Version]
- Hunsberger, H.C.; Weitzner, D.S.; Rudy, C.C.; Hickman, J.E.; Libell, E.M.; Speer, R.R.; Gerhardt, G.A.; Reed, M.N. Riluzole rescues glutamate alterations, cognitive deficits, and tau pathology associated with P301L tau expression. J. Neurochem. 2015, 135, 381–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olney, J.W.; Misra, C.H.; de Gubareff, T. Cysteine-S-sulfate: Brain damaging metabolite in sulfite oxidase deficiency. J. Neuropathol. Exp. Neurol. 1975, 34, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Bassett, S.S.; Yousem, D.M.; Cristinzio, C.; Kusevic, I.; Yassa, M.A.; Caffo, B.S.; Zeger, S.L. Familial risk for Alzheimer’s disease alters fMRI activation patterns. Brain 2006, 129, 1229–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondi, M.W.; Houston, W.S.; Eyler, L.T.; Brown, G.G. fMRI evidence of compensatory mechanisms in older adults at genetic risk for Alzheimer disease. Neurology 2005, 64, 501–508. [Google Scholar] [CrossRef] [Green Version]
- Bookheimer, S.Y.; Strojwas, M.H.; Cohen, M.S.; Saunders, A.M.; Pericak-Vance, M.A.; Mazziotta, J.C.; Small, G.W. Patterns of brain activation in people at risk for Alzheimer’s disease. N. Engl. J. Med. 2000, 343, 450–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farb, N.A.S.; Grady, C.L.; Strother, S.; Tang-Wai, D.F.; Masellis, M.; Black, S.; Freedman, M.; Pollock, B.G.; Campbell, K.L.; Hasher, L.; et al. Abnormal network connectivity in frontotemporal dementia: Evidence for prefrontal isolation. Cortex 2013, 49, 1856–1873. [Google Scholar] [CrossRef] [PubMed]
- Filippini, N.; MacIntosh, B.J.; Hough, M.G.; Goodwin, G.M.; Frisoni, G.B.; Smith, S.M.; Matthews, P.M.; Beckmann, C.F.; Mackay, C.E. Distinct patterns of brain activity in young carriers of the APOE-epsilon4 allele. Proc. Natl. Acad. Sci. USA 2009, 106, 7209–7214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quiroz, Y.T.; Budson, A.E.; Celone, K.; Ruiz, A.; Newmark, R.; Castrillón, G.; Lopera, F.; Stern, C.E. Hippocampal hyperactivation in presymptomatic familial Alzheimer’s disease. Ann. Neurol. 2010, 68, 865–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperling, R.A.; Laviolette, P.S.; O’Keefe, K.; O’Brien, J.; Rentz, D.M.; Pihlajamaki, M.; Marshall, G.; Hyman, B.T.; Selkoe, D.J.; Hedden, T.; et al. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron 2009, 63, 178–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitwell, J.L.; Josephs, K.A.; Avula, R.; Tosakulwong, N.; Weigand, S.D.; Senjem, M.L.; Vemuri, P.; Jones, D.T.; Gunter, J.L.; Baker, M.; et al. Altered functional connectivity in asymptomatic MAPT subjects A comparison to bvFTD. Neurology 2011, 77, 866–874. [Google Scholar] [CrossRef] [PubMed]
- DeVos, S.L.; Goncharoff, D.K.; Chen, G.; Kebodeaux, C.S.; Yamada, K.; Stewart, F.R.; Schuler, D.R.; Maloney, S.E.; Wozniak, D.F.; Rigo, F.; et al. Antisense reduction of tau in adult mice protects against seizures. J. Neurosci. 2013, 33, 12887–12897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holth, J.K.; Bomben, V.C.; Reed, J.G.; Inoue, T.; Younkin, L.; Younkin, S.G.; Pautler, R.G.; Botas, J.; Noebels, J.L. Tau loss attenuates neuronal network hyperexcitability in mouse and Drosophila genetic models of epilepsy. J. Neurosci. 2013, 33, 1651–1659. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-β toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberson, E.D.; Halabisky, B.; Yoo, J.W.; Yao, J.; Chin, J.; Yan, F.; Wu, T.; Hamto, P.; Devidze, N.; Yu, G.-Q.; et al. Amyloid-{beta}/fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J. Neurosci. 2011, 31, 700–711. [Google Scholar] [CrossRef] [Green Version]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najlerahim, A.; Francis, P.T.; Bowen, D.M. Age-related alteration in excitatory amino acid neurotransmission in rat brain. Neurobiol. Aging 1990, 11, 155–158. [Google Scholar] [CrossRef]
- Wheeler, D.D.; Ondo, J.G. Time course of the aging of the high affinity L-glutamate transporter in rat cortical synaptosomes. Exp. Gerontol. 1986, 21, 159–168. [Google Scholar] [CrossRef]
- Nickell, J.; Salvatore, M.F.; Pomerleau, F.; Apparsundaram, S.; Gerhardt, G.A. Reduced plasma membrane surface expression of GLAST mediates decreased glutamate regulation in the aged striatum. Neurobiol. Aging 2007, 28, 1737–1748. [Google Scholar] [CrossRef] [PubMed]
- Simkin, D.; Hattori, S.; Ybarra, N.; Musial, T.F.; Buss, E.W.; Richter, H.; Oh, M.M.; Nicholson, D.A.; Disterhoft, J.F. Aging-Related Hyperexcitability in CA3 Pyramidal Neurons Is Mediated by Enhanced A-Type K+ Channel Function and Expression. J. Neurosci. 2015, 35, 13206–13218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, I.A.; Ikonen, S.; Gallagher, M.; Eichenbaum, H.; Tanila, H. Age-associated alterations of hippocampal place cells are subregion specific. J. Neurosci. 2005, 25, 6877–6886. [Google Scholar] [CrossRef] [PubMed]
- Yassa, M.A.; Lacy, J.W.; Stark, S.M.; Albert, M.S.; Gallagher, M.; Stark, C.E. Pattern separation deficits associated with increased hippocampal CA3 and dentate gyrus activity in nondemented older adults. Hippocampus 2011, 21, 968–979. [Google Scholar] [CrossRef] [Green Version]
- Dutar, P.; Potier, B. Susceptibility to Aβo and TBOA of LTD and Extrasynaptic NMDAR-Dependent Tonic Current in the Aged Rat Hippocampus. Neurochem. Res. 2019, 44, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Potier, B.; Billard, J.-M.; Rivière, S.; Sinet, P.-M.; Denis, I.; Champeil-Potokar, G.; Grintal, B.; Jouvenceau, A.; Kollen, M.; Dutar, P. Reduction in glutamate uptake is associated with extrasynaptic NMDA and metabotropic glutamate receptor activation at the hippocampal CA1 synapse of aged rats. Aging Cell 2010, 9, 722–735. [Google Scholar] [CrossRef]
- Song, M.S.; Rauw, G.; Baker, G.B.; Kar, S. Memantine protects rat cortical cultured neurons against beta-amyloid-induced toxicity by attenuating tau phosphorylation. Eur. J. Neurosci. 2008, 28, 1989–2002. [Google Scholar] [CrossRef]
- Amadoro, G.; Ciotti, M.T.; Costanzi, M.; Cestari, V.; Calissano, P.; Canu, N. NMDA receptor mediates tau-induced neurotoxicity by calpain and ERK/MAPK activation. Proc. Natl. Acad. Sci. USA 2006, 103, 2892–2897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufman, A.M.; Milnerwood, A.J.; Sepers, M.D.; Coquinco, A.; She, K.; Wang, L.; Lee, H.; Craig, A.M.; Cynader, M.; Raymond, L.A. Opposing roles of synaptic and extrasynaptic NMDA receptor signaling in cocultured striatal and cortical neurons. J. Neurosci. 2012, 32, 3992–4003. [Google Scholar] [CrossRef] [Green Version]
- Tackenberg, C.; Grinschgl, S.; Trutzel, A.; Santuccione, A.C.; Frey, M.C.; Konietzko, U.; Grimm, J.; Brandt, R.; Nitsch, R.M. NMDA receptor subunit composition determines beta-amyloid-induced neurodegeneration and synaptic loss. Cell Death Dis. 2013, 4, e608. [Google Scholar] [CrossRef] [Green Version]
- Hoover, B.R.; Reed, M.N.; Su, J.; Penrod, R.D.; Kotilinek, L.A.; Grant, M.K.; Pitstick, R.; Carlson, G.A.; Lanier, L.M.; Yuan, L.-L.; et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 2010, 68, 1067–1081. [Google Scholar] [CrossRef] [Green Version]
- Ramsden, M.; Kotilinek, L.; Forster, C.; Paulson, J.; McGowan, E.; SantaCruz, K.; Guimaraes, A.; Yue, M.; Lewis, J.; Carlson, G.; et al. Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J. Neurosci. 2005, 25, 10637–10647. [Google Scholar] [CrossRef] [PubMed]
- Santacruz, K.; Lewis, J.; Spires, T.; Paulson, J.; Kotilinek, L.; Ingelsson, M.; Guimaraes, A.; DeTure, M.; Ramsden, M.; McGowan, E.; et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science 2005, 309, 476–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulson, J.B.; Ramsden, M.; Forster, C.; Sherman, M.A.; McGowan, E.; Ashe, K.H. Amyloid plaque and neurofibrillary tangle pathology in a regulatable mouse model of Alzheimer’s disease. Am. J. Pathol. 2008, 173, 762–772. [Google Scholar] [CrossRef] [Green Version]
- Hunsberger, H.C.; Rudy, C.C.; Weitzner, D.S.; Zhang, C.; Tosto, D.E.; Knowlan, K.; Xu, Y.; Reed, M.N. Effect size of memory deficits in mice with adult-onset P301L tau expression. Behav. Brain Res. 2014, 272, 181–195. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Wang, S.; Brenner, M.; Paton, J.F.; Kasparov, S. Enhancement of cell-specific transgene expression from a Tet-Off regulatory system using a transcriptional amplification strategy in the rat brain. J. Gene Med. 2008, 10, 583–592. [Google Scholar] [CrossRef] [Green Version]
- Vorhees, C.V.; Williams, M.T. Morris water maze: Procedures for assessing spatial and related forms of learning and memory. Nat. Protoc. 2006, 1, 848–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dellu, F.; Mayo, W.; Cherkaoui, J.; Le Moal, M.; Simon, H. A two-trial memory task with automated recording: Study in young and aged rats. Brain Res. 1992, 588, 132–139. [Google Scholar] [CrossRef]
- Burmeister, J.J.; Gerhardt, G.A. Self-referencing ceramic-based multisite microelectrodes for the detection and elimination of interferences from the measurement of L-glutamate and other analytes. Anal. Chem. 2001, 73, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Hunsberger, H.C.; Setti, S.E.; Heslin, R.T.; Quintero, J.E.; Gerhardt, G.A.; Reed, M.N. Using Enzyme-based Biosensors to Measure Tonic and Phasic Glutamate in Alzheimer’s Mouse Models. J. Vis. Exp. 2017, 55418. [Google Scholar] [CrossRef] [Green Version]
- Paxinos, G.; Franklin, K. Mouse Brain in Stereotaxic Coordinates; Academic Press: Cambridge, MA, USA, 2012. [Google Scholar]
- Hascup, K.N.; Findley, C.A.; Sime, L.N.; Hascup, E.R. Hippocampal alterations in glutamatergic signaling during amyloid progression in AβPP/PS1 mice. Sci. Rep. 2020, 10, 14503. [Google Scholar] [CrossRef] [PubMed]
- Hascup, K.N.; Lynn, M.K.; Fitzgerald, P.J.; Randall, S.; Kopchick, J.J.; Boger, H.A.; Bartke, A.; Hascup, E.R. Enhanced Cognition and Hypoglutamatergic Signaling in a Growth Hormone Receptor Knockout Mouse Model of Successful Aging. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 329–337. [Google Scholar] [CrossRef] [Green Version]
- Hascup, K.N.; Rutherford, E.C.; Quintero, J.E.; Day, B.K.; Nickell, J.R.; Pomerleau, F.; Huettl, P.; Burmeister, J.J.; Gerhardt, G.A. Second-by-Second Measures of L-Glutamate and Other Neurotransmitters Using Enzyme-Based Microelectrode Arrays. In Electrochemical Methods for Neuroscience; Michael, A.C., Borland, L.M., Eds.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2007. [Google Scholar]
- Hinzman, J.M.; Thomas, T.C.; Burmeister, J.J.; Quintero, J.E.; Huettl, P.; Pomerleau, F.; Gerhardt, G.A.; Lifshitz, J. Diffuse brain injury elevates tonic glutamate levels and potassium-evoked glutamate release in discrete brain regions at two days post-injury: An enzyme-based microelectrode array study. J. Neurotrauma 2010, 27, 889–899. [Google Scholar] [CrossRef]
- Day, B.K.; Pomerleau, F.; Burmeister, J.J.; Huettl, P.; Gerhardt, G.A. Microelectrode array studies of basal and potassium-evoked release of L-glutamate in the anesthetized rat brain. J. Neurochem. 2006, 96, 1626–1635. [Google Scholar] [CrossRef] [PubMed]
- O’Kane, R.L.; Martínez-López, I.; DeJoseph, M.R.; Viña, J.R.; Hawkins, R.A. Na(+)-dependent glutamate transporters (EAAT1, EAAT2, and EAAT3) of the blood-brain barrier. A mechanism for glutamate removal. J. Biol. Chem. 1999, 274, 31891–31895. [Google Scholar] [CrossRef] [Green Version]
- Westerman, M.A.; Cooper-Blacketer, D.; Mariash, A.; Kotilinek, L.; Kawarabayashi, T.; Younkin, L.H.; Carlson, G.A.; Younkin, S.G.; Ashe, K.H. The relationship between Abeta and memory in the Tg2576 mouse model of Alzheimer’s disease. J. Neurosci. 2002, 22, 1858–1867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, R.N. The value of spontaneous alternation behavior (SAB) as a test of retention in pharmacological investigations of memory. Neurosci. Biobehav. Rev. 2004, 28, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Costa, V.; Duchatelle, P.; Boulouard, M.; Dauphin, F. Selective 5-HT6 receptor blockade improves spatial recognition memory and reverses age-related deficits in spatial recognition memory in the mouse. Neuropsychopharmacology 2009, 34, 488–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deacon, R.M.J.; Cholerton, L.L.; Talbot, K.; Nair-Roberts, R.G.; Sanderson, D.J.; Romberg, C.; Koros, E.; Bornemann, K.D.; Rawlins, J.N.P. Age-dependent and -independent behavioral deficits in Tg2576 mice. Behav. Brain Res. 2008, 189, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Kennard, J.A.; Harrison, F.E. Intravenous ascorbate improves spatial memory in middle-aged APP/PSEN1 and wild type mice. Behav. Brain Res. 2014, 264, 34–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokkinidis, L. Neurochemical and Neuroanatomical Correlates of Behavioral Habituation and Sensitization: An Overview and Elaboration of Animal Experimentation. In Spontaneous Alternation Behavior; Springer: New York, NY, USA, 1989; pp. 109–130. [Google Scholar]
- Stephens, M.L.; Quintero, J.E.; Pomerleau, F.; Huettl, P.; Gerhardt, G.A. Age-related changes in glutamate release in the CA3 and dentate gyrus of the rat hippocampus. Neurobiol. Aging. 2011, 32, 811–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojcik, S.M.; Rhee, J.S.; Herzog, E.; Sigler, A.; Jahn, R.; Takamori, S.; Brose, N.; Rosenmund, C. An essential role for vesicular glutamate transporter 1 (VGLUT1) in postnatal development and control of quantal size. Proc. Natl. Acad. Sci. USA 2004, 101, 7158–7163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masliah, E.; Alford, M.; Mallory, M.; Rockenstein, E.; Moechars, D.; Van Leuven, F. Abnormal glutamate transport function in mutant amyloid precursor protein transgenic mice. Exp. Neurol. 2000, 163, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Martin, L.J.; Kuncl, R.W. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N. Engl. J. Med. 1992, 326, 1464–1468. [Google Scholar] [CrossRef]
- Deng, Y.; Xu, Z.F.; Liu, W.; Xu, B.; Yang, H.B.; Wei, Y.G. Riluzole-triggered GSH synthesis via activation of glutamate transporters to antagonize methylmercury-induced oxidative stress in rat cerebral cortex. Oxid. Med. Cell Longev. 2012, 2012, 534705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.J.; Chung, Y.H.; Joo, K.M.; Lim, H.C.; Jeon, G.S.; Kim, D.; Lee, W.B.; Kim, Y.S.; Cha, C.I. Age-related changes in glycogen synthase kinase 3beta (GSK3beta) immunoreactivity in the central nervous system of rats. Neurosci. Lett. 2006, 409, 134–139. [Google Scholar] [CrossRef]
- Wang, J.Z.; Wu, Q.; Smith, A.; Grundke-Iqbal, I.; Iqbal, K. Tau is phosphorylated by GSK-3 at several sites found in Alzheimer disease and its biological activity markedly inhibited only after it is prephosphorylated by A-kinase. FEBS Lett. 1998, 436, 28–34. [Google Scholar] [CrossRef] [Green Version]
- Lindsay, J.; Laurin, D.; Verreault, R.; Hébert, R.; Helliwell, B.; Hill, G.B.; McDowell, I. Risk factors for Alzheimer’s disease: A prospective analysis from the Canadian Study of Health and Aging. Am. J. Epidemiol. 2002, 156, 445–453. [Google Scholar] [CrossRef] [Green Version]
- Leem, J.W.; Kim, H.K.; Hulsebosch, C.E.; Gwak, Y.S. Ionotropic glutamate receptors contribute to maintained neuronal hyperexcitability following spinal cord injury in rats. Exp. Neurol. 2010, 224, 321–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neugebauer, V.; Lücke, T.; Schaible, H.G. Requirement of metabotropic glutamate receptors for the generation of inflammation-evoked hyperexcitability in rat spinal cord neurons. Eur. J. Neurosci. 1994, 6, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Van Den Pol, A.N.; Obrietan, K.; Belousov, A. Glutamate hyperexcitability and seizure-like activity throughout the brain and spinal cord upon relief from chronic glutamate receptor blockade in culture. Neuroscience 1996, 74, 653–674. [Google Scholar] [CrossRef]
- Freeman, G.B.; Gibson, G.E. Selective alteration of mouse brain neurotransmitter release with age. Neurobiol. Aging 1987, 8, 147–152. [Google Scholar] [CrossRef]
- Massieu, L.; Tapia, R. Glutamate uptake impairment and neuronal damage in young and aged rats in vivo. J. Neurochem. 1997, 69, 1151–1160. [Google Scholar] [CrossRef]
- Meldrum, M.J.; Glenton, P.; Dawson, R., Jr. [3H]D-aspartic acid release in brain slices of adult and aged Fischer 344 rates. Neurochem. Res. 1992, 17, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Breteler, M.M.; de Groot, R.R.; van Romunde, L.K.; Hofman, A. Risk of dementia in patients with Parkinson’s disease, epilepsy, and severe head trauma: A register-based follow-up study. Am. J. Epidemiol. 1995, 142, 1300–1305. [Google Scholar] [CrossRef] [PubMed]
- Breteler, M.M.; van Duijn, C.M.; Chandra, V.; Fratiglioni, L.; Graves, A.B.; Heyman, A.; Jorm, A.F.; Kokmen, E.; Kondo, K.; Mortimer, J.A.; et al. Medical history and the risk of Alzheimer’s disease: A collaborative re-analysis of case-control studies. EURODEM Risk Factors Research Group. Int. J. Epidemiol. 1991, 20 (Suppl. 2), S36–S42. [Google Scholar] [CrossRef] [PubMed]
- Beagle, A.J.; Darwish, S.M.; Ranasinghe, K.G.; La, A.L.; Karageorgiou, E.; Vossel, K.A. Relative Incidence of Seizures and Myoclonus in Alzheimer’s Disease, Dementia with Lewy Bodies, and Frontotemporal Dementia. J. Alzheimer Dis. 2017, 60, 211–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Born, H.A. Seizures in Alzheimer’s disease. Neuroscience 2015, 286, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Vossel, K.A.; Tartaglia, M.C.; Nygaard, H.B.; Zeman, A.Z.; Miller, B.L. Epileptic activity in Alzheimer’s disease: Causes and clinical relevance. Lancet Neurol. 2017, 16, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Mauri, M.; Sinforiani, E.; Zucchella, C.; Cuzzoni, M.G.; Bono, G. Progression to dementia in a population with amnestic mild cognitive impairment: Clinical variables associated with conversion. Funct. Neurol. 2012, 27, 49–54. [Google Scholar] [PubMed]
- Bakker, A.; Kirwan, C.B.; Miller, M.; Stark, C.E. Pattern separation in the human hippocampal CA3 and dentate gyrus. Science 2008, 319, 1640–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakker, A.; Krauss, G.L.; Albert, M.S.; Speck, C.L.; Jones, L.R.; Stark, C.E.; Yassa, M.A.; Bassett, S.S.; Shelton, A.L.; Gallagher, M. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron 2012, 74, 467–474. [Google Scholar] [CrossRef] [Green Version]
- Gamache, J.; Benzow, K.; Forster, C.; Kemper, L.; Hlynialuk, C.; Furrow, E.; Ashe, K.H.; Koob, M.D. Factors other than hTau overexpression that contribute to tauopathy-like phenotype in rTg4510 mice. Nat. Commun. 2019, 10, 2479. [Google Scholar] [CrossRef]
- Balducci, C.; Santamaria, G.; La Vitola, P.; Brandi, E.; Grandi, F.; Viscomi, A.R.; Beeg, M.; Gobbi, M.; Salmona, M.; Ottonello, S.; et al. Doxycycline counteracts neuroinflammation restoring memory in Alzheimer’s disease mouse models. Neurobiol. Aging 2018, 70, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.C.; Barnes, C.A.; Rao, G.; McNaughton, B.L. Increase in perforant path quantal size in aged F-344 rats. Neurobiol. Aging 1991, 12, 441–448. [Google Scholar] [CrossRef]
- Bell, K.F.; Bennett, D.A.; Cuello, A.C. Paradoxical upregulation of glutamatergic presynaptic boutons during mild cognitive impairment. J. Neurosci. 2007, 27, 10810–10817. [Google Scholar] [CrossRef] [PubMed]
- Kirvell, S.L.; Esiri, M.; Francis, P.T. Down-regulation of vesicular glutamate transporters precedes cell loss and pathology in Alzheimer’s disease. J. Neurochem. 2006, 98, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.; Shai, R.M.; Wu, Y.; Miller, C.A. P3–441: Expression of mRNAs regulating local translation in synaptic terminals of MCI and early ad brain. Alzheimer Dement. 2006, 2, S505–S506. [Google Scholar] [CrossRef]
- Le Duigou, C.; Simonnet, J.; Teleñczuk, M.T.; Fricker, D.; Miles, R. Recurrent synapses and circuits in the CA3 region of the hippocampus: An associative network. Front. Cell Neurosci. 2014, 7, 262. [Google Scholar] [CrossRef]
- Yaguchi, T.; Nishizaki, T. Extracellular high K+ stimulates vesicular glutamate release from astrocytes by activating voltage-dependent calcium channels. J. Cell Physiol. 2010, 225, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Moechars, D.; Weston, M.C.; Leo, S.; Callaerts-Vegh, Z.; Goris, I.; Daneels, G.; Buist, A.; Cik, M.; van der Spek, P.; Kass, S.; et al. Vesicular glutamate transporter VGLUT2 expression levels control quantal size and neuropathic pain. J. Neurosci. 2006, 26, 12055–12066. [Google Scholar] [CrossRef] [PubMed]
- Calhoun, M.E.; Kurth, D.; Phinney, A.L.; Long, J.M.; Hengemihle, J.; Mouton, P.R.; Ingram, D.K.; Jucker, M. Hippocampal neuron and synaptophysin-positive bouton number in aging C57BL/6 mice. Neurobiol. Aging. 1998, 19, 599–606. [Google Scholar] [CrossRef]
- Frick, K.M.; Fernandez, S.M. Enrichment enhances spatial memory and increases synaptophysin levels in aged female mice. Neurobiol. Aging 2003, 24, 615–626. [Google Scholar] [CrossRef]
- Chen, Y.C.; Chen, Q.S.; Lei, J.L.; Wang, S.L. Physical training modifies the age-related decrease of GAP-43 and synaptophysin in the hippocampal formation in C57BL/6J mouse. Brain Res. 1998, 806, 238–245. [Google Scholar] [CrossRef]
- Smith, T.D.; Adams, M.M.; Gallagher, M.; Morrison, J.H.; Rapp, P.R. Circuit-specific alterations in hippocampal synaptophysin immunoreactivity predict spatial learning impairment in aged rats. J. Neurosci. 2000, 20, 6587–6593. [Google Scholar] [CrossRef] [PubMed]
- Counts, S.E.; Alldred, M.J.; Che, S.; Ginsberg, S.D.; Mufson, E.J. Synaptic gene dysregulation within hippocampal CA1 pyramidal neurons in mild cognitive impairment. Neuropharmacology 2014, 79, 172–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyffeler, M.; Zhang, W.N.; Feldon, J.; Knuesel, I. Differential expression of PSD proteins in age-related spatial learning impairments. Neurobiol. Aging 2007, 28, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Preissmann, D.; Leuba, G.; Savary, C.; Vernay, A.; Kraftsik, R.; Riederer, I.M.; Schenk, F.; Riederer, B.M.; Savioz, A. Increased postsynaptic density protein-95 expression in the frontal cortex of aged cognitively impaired rats. Exp. Biol. Med. 2012, 237, 1331–1340. [Google Scholar] [CrossRef]
- Pooler, A.M.; Phillips, E.C.; Lau, D.H.; Noble, W.; Hanger, D.P. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013, 14, 389–394. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Weitzner, D.S.; Engler-Chiurazzi, E.B.; Kotilinek, L.A.; Ashe, K.H.; Reed, M.N. Morris Water Maze Test: Optimization for Mouse Strain and Testing Environment. J. Vis. Exp. 2015, e52706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arriagada, P.V.; Growdon, J.H.; Hedley-Whyte, E.T.; Hyman, B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 1992, 42, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Bancher, C.; Braak, H.; Fischer, P.; Jellinger, K.A. Neuropathological staging of Alzheimer lesions and intellectual status in Alzheimer’s and Parkinson's disease patients. Neurosci. Lett. 1993, 162, 179–182. [Google Scholar] [CrossRef]
- Guillozet, A.L.; Weintraub, S.; Mash, D.C.; Mesulam, M.M. Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Arch. Neurol. 2003, 60, 729–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Amount Protein Loaded | Source |
|---|---|---|
| Tau-5 | 2 ug | Invitrogen |
| HT7 | 15 ug | Thermo Fisher |
| CP13 | 2 ug | Peter Davies |
| MC1 | 2 ug | Peter Davies |
| PHF1 | 2 ug | Peter Davies |
| vGlut1 | 2 ug | Millipore |
| Synaptophysin | 2 ug | Sigma |
| GLT-1 | 20 ug | Millipore |
| PSD-95 | 20 ug | Millipore |
| GSK3β | 20 ug | Cell Signalling |
| pGSK3β | 20 ug | Cell Signalling |
| Actin | 2–20 ug | Santa Cruz |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hunsberger, H.C.; Setti, S.E.; Rudy, C.C.; Weitzner, D.S.; Pfitzer, J.C.; McDonald, K.L.; Hong, H.; Bhattacharya, S.; Suppiramaniam, V.; Reed, M.N. Differential Effects of Human P301L Tau Expression in Young versus Aged Mice. Int. J. Mol. Sci. 2021, 22, 11637. https://doi.org/10.3390/ijms222111637
Hunsberger HC, Setti SE, Rudy CC, Weitzner DS, Pfitzer JC, McDonald KL, Hong H, Bhattacharya S, Suppiramaniam V, Reed MN. Differential Effects of Human P301L Tau Expression in Young versus Aged Mice. International Journal of Molecular Sciences. 2021; 22(21):11637. https://doi.org/10.3390/ijms222111637
Chicago/Turabian StyleHunsberger, Holly C., Sharay E. Setti, Carolyn C. Rudy, Daniel S. Weitzner, Jeremiah C. Pfitzer, Kelli L. McDonald, Hao Hong, Subhrajit Bhattacharya, Vishnu Suppiramaniam, and Miranda N. Reed. 2021. "Differential Effects of Human P301L Tau Expression in Young versus Aged Mice" International Journal of Molecular Sciences 22, no. 21: 11637. https://doi.org/10.3390/ijms222111637
APA StyleHunsberger, H. C., Setti, S. E., Rudy, C. C., Weitzner, D. S., Pfitzer, J. C., McDonald, K. L., Hong, H., Bhattacharya, S., Suppiramaniam, V., & Reed, M. N. (2021). Differential Effects of Human P301L Tau Expression in Young versus Aged Mice. International Journal of Molecular Sciences, 22(21), 11637. https://doi.org/10.3390/ijms222111637