
Reduced GFAP Expression in Bergmann Glial Cells in the Cerebellum of Sigma-1 Receptor Knockout Mice Determines the Neurobehavioral Outcomes after Traumatic Brain Injury

 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
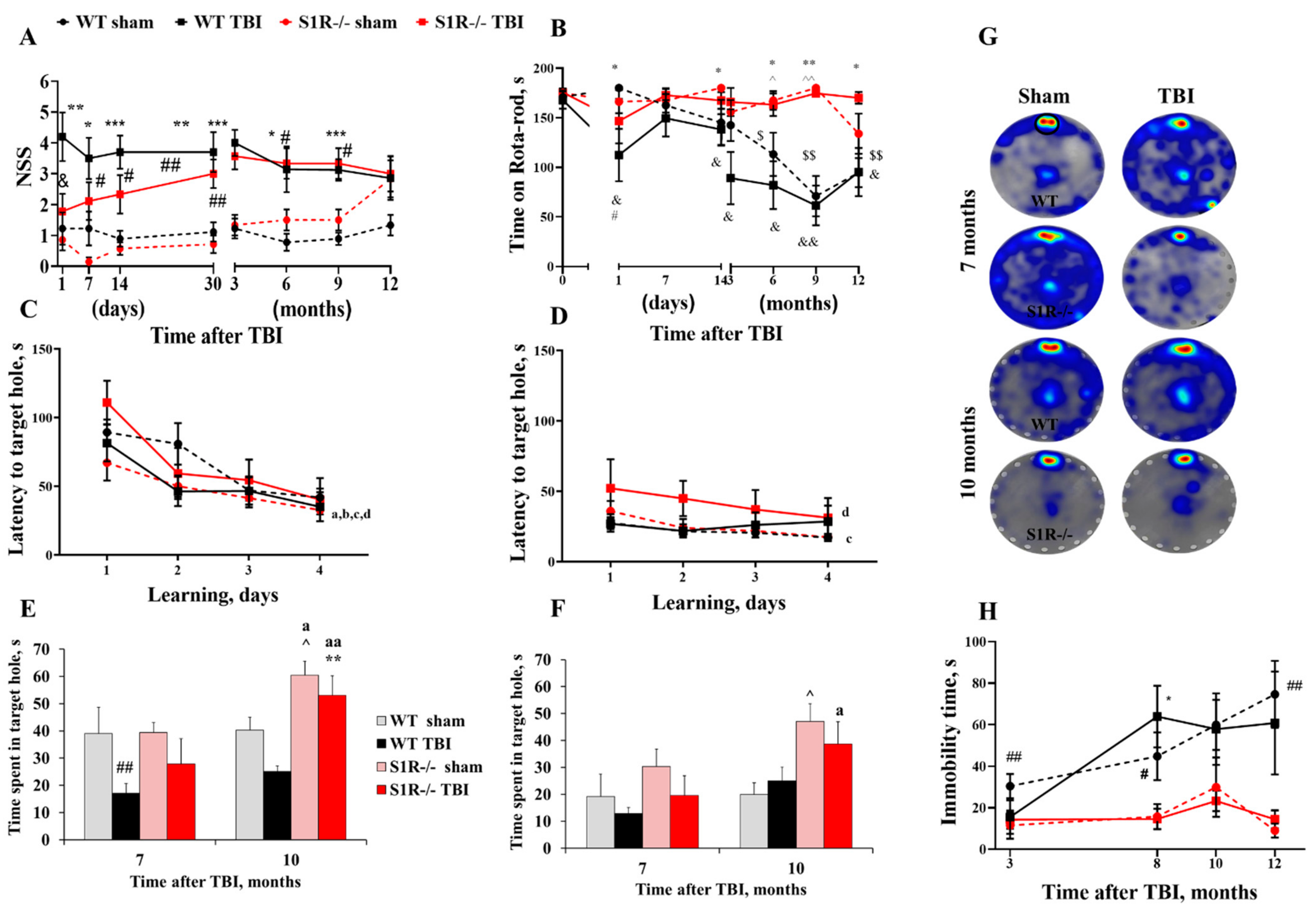
2.1. S1R-/- Mice Exhibit Improved Acute Sensorimotor Ability and Motor Coordination following TBI
2.2. S1R-/- Mice Have Preserved Cognitive Ability after TBI
2.3. S1R-/- Mice Show Reduced Despair-like Behavior and Increased Anxiety, Regardless of Injury Status
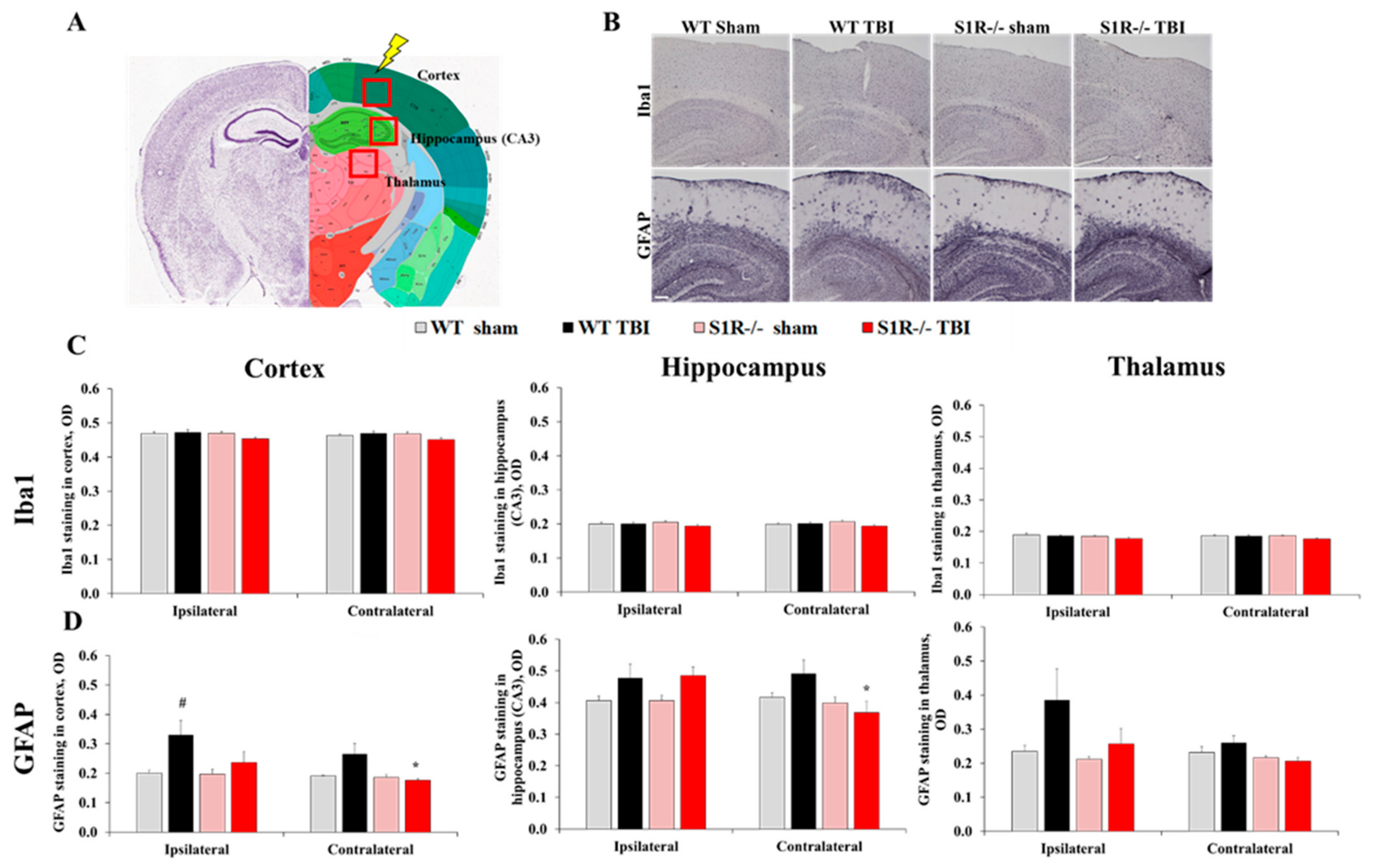
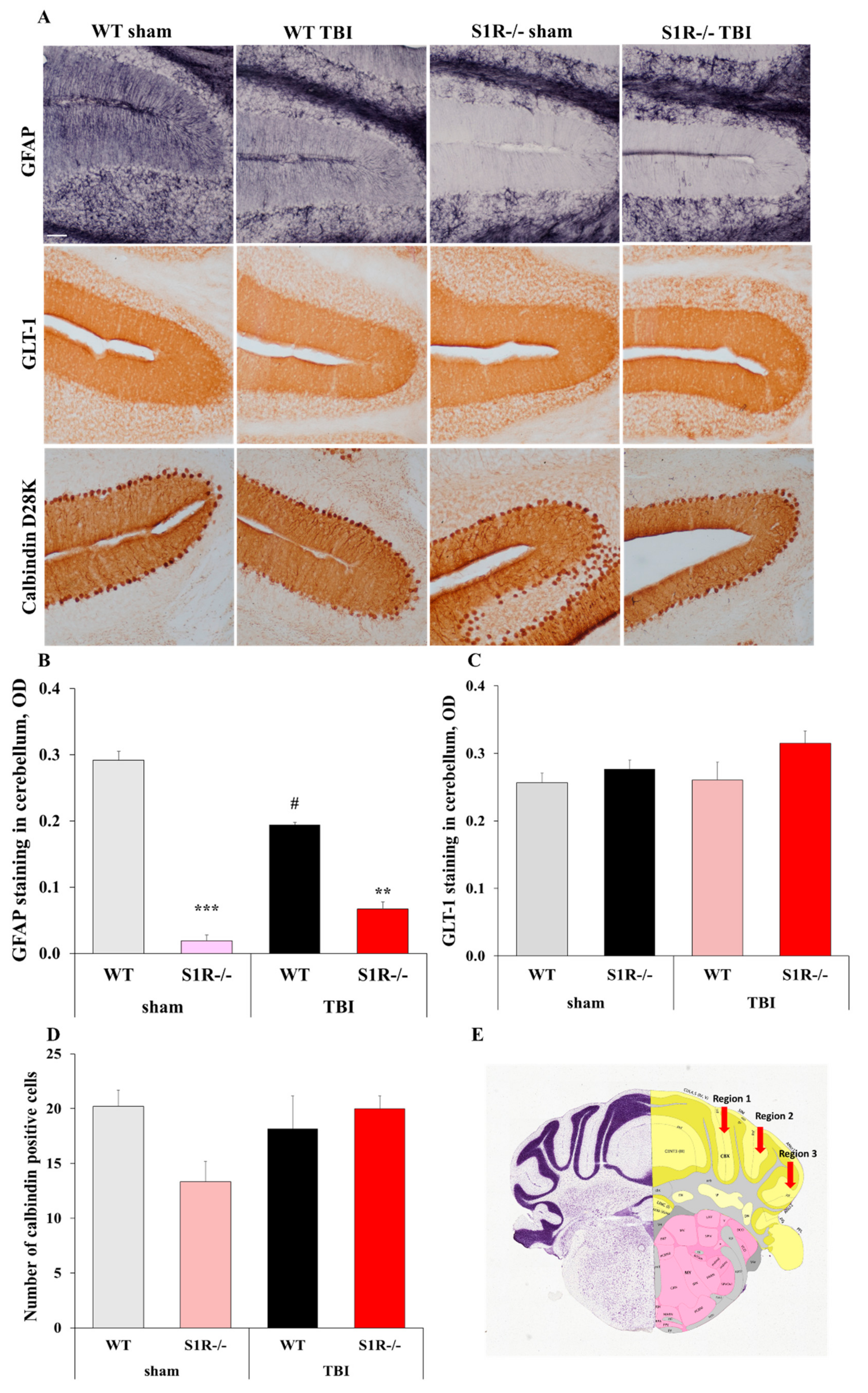
2.4. S1R-/- Mice Exhibit Decreased Glial Fibrillary Acidic Protein (GFAP) Staining in the Molecular Layer of the Cerebellum
2.5. Health Outcome Measures after TBI
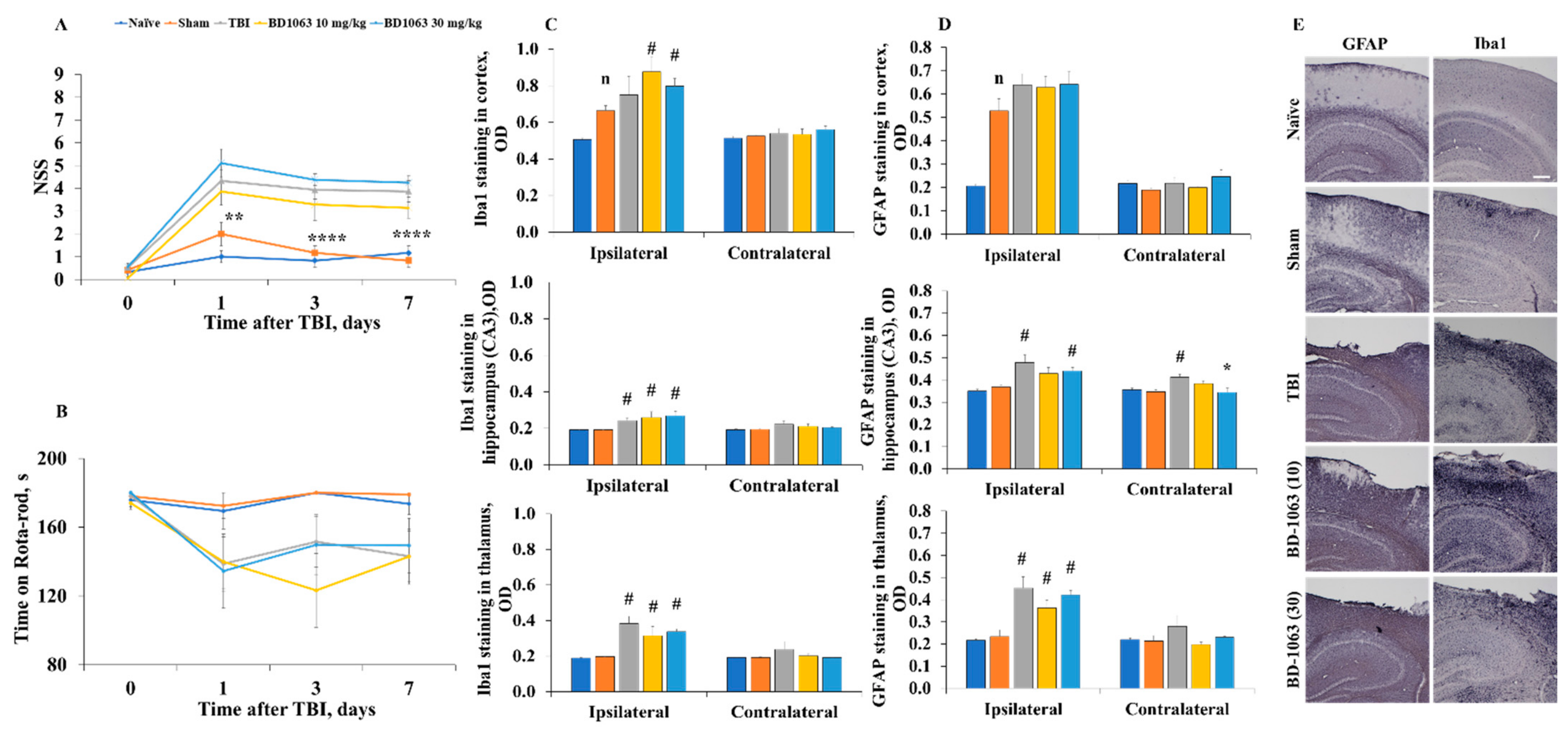
2.6. BD-1063 Does Not Influence Acute Injury Measures or Behavioral and Histological Outcomes Following TBI
3. Discussion
4. Materials and Methods
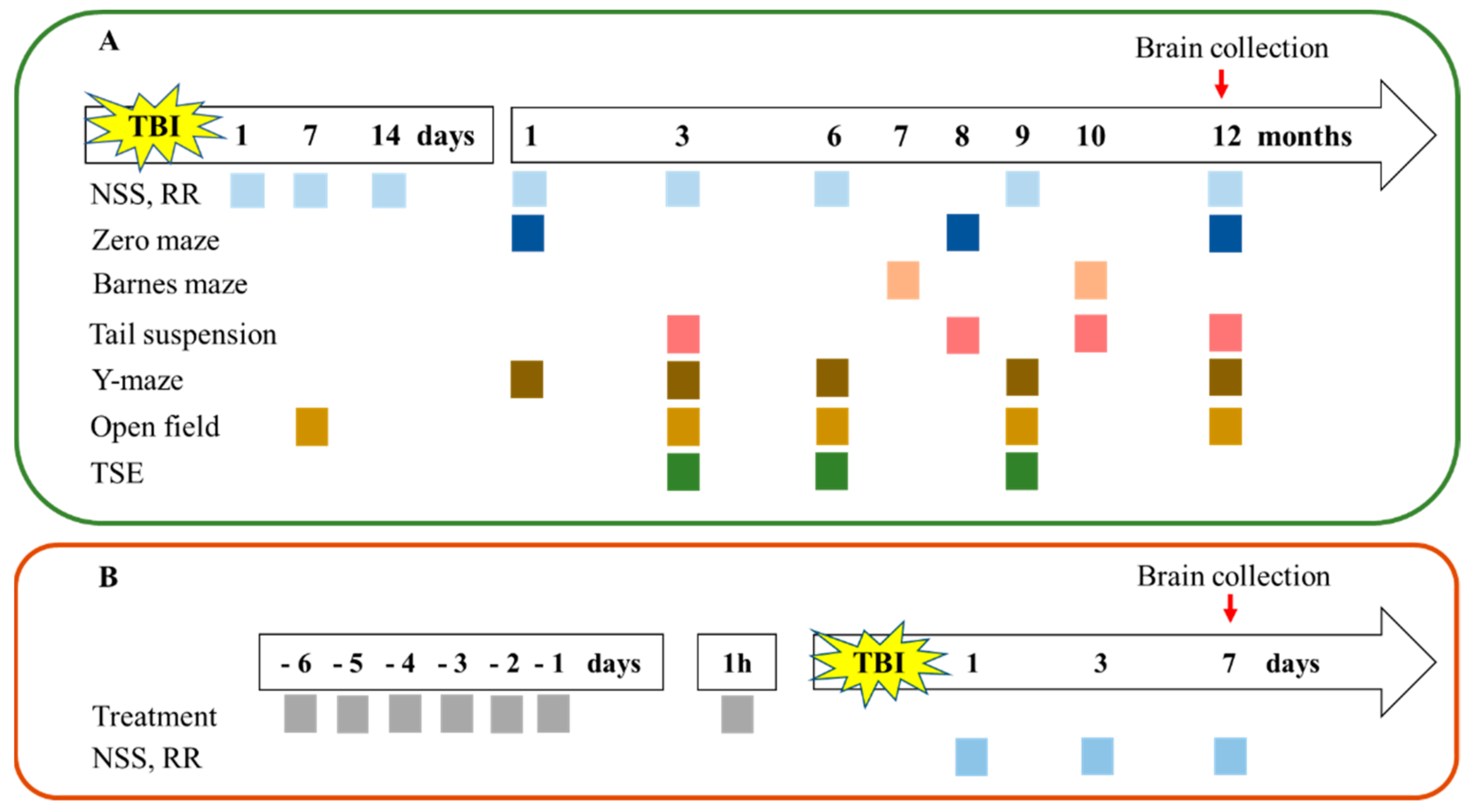
4.1. Animals and Experimental Design
4.2. Lateral Fluid Percussion Injury
4.3. Neurological Severity Score
4.4. Rota-Rod Test
4.5. Y-Maze Test
4.6. Tail Suspension Test
4.7. Open Field Test
4.8. Elevated Zero Maze Test
4.9. Barnes Maze Test
4.10. TSE PhenoMaster System (Indirect Calorimetry)
4.11. Determination of BD-1063 in the Brain Tissue Using UPLC⁄MS
4.12. Quantitative PCR
4.13. Immunohistochemistry
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berkner, J.; Mannix, R.; Qiu, J. Clinical traumatic brain injury in the preclinical setting. Methods Mol. Biol. 2016, 1462, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Maas, A.I.R.; Menon, D.K.; Adelson, P.D.; Andelic, N.; Bell, M.J.; Belli, A.; Bragge, P.; Brazinova, A.; Büki, A.; Chesnut, R.M.; et al. Traumatic brain injury: Integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017, 16, 987–1048. [Google Scholar] [CrossRef] [Green Version]
- Blennow, K.; Brody, D.L.; Kochanek, P.M.; Levin, H.; McKee, A.; Ribbers, G.M.; Yaffe, K.; Zetterberg, H. Traumatic brain injuries. Nat. Rev. Dis. Prim. 2016, 2, 16084. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.; Stewart, W.; Dams-O’Connor, K.; Diaz-Arrastia, R.; Horton, L.; Menon, D.K.; Polinder, S. The chronic and evolving neurological consequences of traumatic brain injury. Lancet Neurol. 2017, 16, 813–825. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.Y.; Lee, A.Y.W. Traumatic brain injuries: Pathophysiology and potential therapeutic targets. Front. Cell. Neurosci. 2019, 13, 528. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.H.; Martens, K.M.; Bashir, A.; Cheung, H.; Stukas, S.; Gibbs, E.; Namjoshi, D.R.; Button, E.B.; Wilkinson, A.; Barron, C.J.; et al. CHIMERA repetitive mild traumatic brain injury induces chronic behavioural and neuropathological phenotypes in wild-type and APP/PS1 mice. Alzheimer Res. Ther. 2019, 11, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucker, L.B.; Velosky, A.G.; Fu, A.H.; McCabe, J.T. Chronic neurobehavioral sex differences in a murine model of repetitive concussive brain injury. Front. Neurol. 2019, 10, 509. [Google Scholar] [CrossRef]
- Riggio, S. Traumatic brain injury and its neurobehavioral sequelae. Neurol. Clin. 2011, 29, 35–47. [Google Scholar] [CrossRef]
- Nguyen, L.; Lucke-Wold, B.P.; Mookerjee, S.A.; Cavendish, J.Z.; Robson, M.J.; Scandinaro, A.L.; Matsumoto, R.R. Role of sigma-1 receptors in neurodegenerative diseases. J. Pharmacol. Sci. 2015, 127, 17–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, T.P.; Junien, J.L. Sigma Receptors in the Central Nervous System and the Periphery; Itzhak, Y., Ed.; London Academic Press: Cambridge, MA, USA, 1994; pp. 21–44. [Google Scholar]
- Vavers, E.; Svalbe, B.; Lauberte, L.; Stonans, I.; Misane, I.; Dambrova, M.; Zvejniece, L. The activity of selective sigma-1 receptor ligands in seizure models in vivo. Behav. Brain Res. 2017, 328, 13–18. [Google Scholar] [CrossRef]
- Nguyen, L.; Lucke-Wold, B.P.; Mookerjee, S.; Kaushal, N.; Matsumoto, R.R. Sigma-1 receptors and neurodegenerative diseases: Towards a hypothesis of Sigma-1 receptors as amplifiers of neurodegeneration and neuroprotection. Adv. Exp. Med. Biol. 2017, 964, 133–152. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Ma, Y.; Ren, Z.; Xu, B.; Zhang, Y.; Chen, J.; Yang, B. Sigma-1 receptor modulates neuroinflammation after traumatic brain injury. Cell. Mol. Neurobiol. 2016, 36, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Wegleiter, K.; Hermann, M.; Posod, A.; Wechselberger, K.; Stanika, R.I.; Obermair, G.J.; Kiechl-Kohlendorfer, U.; Urbanek, M.; Griesmaier, E. The sigma-1 receptor agonist 4-phenyl-1-(4-phenylbutyl) piperidine (PPBP) protects against newborn excitotoxic brain injury by stabilizing the mitochondrial membrane potential in vitro and inhibiting microglial activation in vivo. Exp. Neurol. 2014, 261, 501–509. [Google Scholar] [CrossRef]
- Sánchez-Blázquez, P.; Pozo-Rodrigálvarez, A.; Merlos, M.; Garzón, J. The sigma-1 receptor antagonist, S1RA, reduces stroke damage, ameliorates post-stroke neurological deficits and suppresses the overexpression of MMP-9. Mol. Neurobiol. 2018, 55, 4940–4951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schetz, J.A.; Perez, E.; Liu, R.; Chen, S.; Lee, I.; Simpkins, J.W. A prototypical Sigma-1 receptor antagonist protects against brain ischemia. Brain Res. 2007, 1181, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castany, S.; Gris, G.; Vela, J.M.; Verdú, E.; Boadas-Vaello, P. Critical role of sigma-1 receptors in central neuropathic pain-related behaviours after mild spinal cord injury in mice. Sci. Rep. 2018, 8, 3873. [Google Scholar] [CrossRef]
- Lattard, A.; Poulen, G.; Bartolami, S.; Gerber, Y.N.; Perrin, F.E. Negative impact of Sigma-1 receptor agonist treatment on tissue integrity and motor function following spinal cord injury. Front. Pharmacol. 2021, 12, 614949. [Google Scholar] [CrossRef]
- Semple, B.D.; Zamani, A.; Rayner, G.; Shultz, S.R.; Jones, N.C. Affective, neurocognitive and psychosocial disorders associated with traumatic brain injury and post-traumatic epilepsy. Neurobiol. Dis. 2019, 123, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Roozenbeek, B.; Lingsma, H.F.; Maas, A.I. New considerations in the design of clinical trials for traumatic brain injury. Clin. Investig. 2012, 2, 153–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moritz, C.; Berardi, F.; Abate, C.; Peri, F. Live imaging reveals a new role for the sigma-1 (σ1) receptor in allowing microglia to leave brain injuries. Neurosci. Lett. 2015, 591, 13–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, J.; Sha, S.; Zhou, L.; Wang, C.; Yin, J.; Chen, L. Sigma-1 receptor deficiency reduces MPTP-induced parkinsonism and death of dopaminergic neurons. Cell Death Dis. 2015, 6, e1832. [Google Scholar] [CrossRef]
- Bravo-Caparrós, I.; Ruiz-Cantero, M.C.; Perazzoli, G.; Cronin, S.J.F.; Vela, J.M.; Hamed, M.F.; Penninger, J.M.; Baeyens, J.M.; Cobos, E.J.; Nieto, F.R. Sigma-1 receptors control neuropathic pain and macrophage infiltration into the dorsal root ganglion after peripheral nerve injury. FASEB J. 2020, 34, 5951–5966. [Google Scholar] [CrossRef]
- Chevallier, N.; Keller, E.; Maurice, T. Behavioural phenotyping of knockout mice for the sigma-1 (σ₁) chaperone protein revealed gender-related anxiety, depressive-like and memory alterations. J. Psychopharmacol. 2011, 25, 960–975. [Google Scholar] [CrossRef] [PubMed]
- Maurice, T.; Strehaiano, M.; Duhr, F.; Chevallier, N. Amyloid toxicity is enhanced after pharmacological or genetic invalidation of the σ(1) receptor. Behav. Brain Res. 2018, 339, 1–10. [Google Scholar] [CrossRef]
- Ryskamp, D.A.; Korban, S.; Zhemkov, V.; Kraskovskaya, N.; Bezprozvanny, I. Neuronal Sigma-1 receptors: Signaling functions and protective roles in neurodegenerative diseases. Front. Neurosci. 2019, 13, 862. [Google Scholar] [CrossRef]
- Jia, J.; Cheng, J.; Wang, C.; Zhen, X. Sigma-1 receptor-modulated neuroinflammation in neurological diseases. Front. Cell. Neurosci. 2018, 12, 314. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Loane, D.J. Neuroinflammation after traumatic brain injury: Opportunities for therapeutic intervention. Brain. Behav. Immun. 2012, 26, 1191–1201. [Google Scholar] [CrossRef]
- Maragakis, N.J.; Rothstein, J.D. Mechanisms of disease: Astrocytes in neurodegenerative disease. Nat. Clin. Pract. Neurol. 2006, 2, 679–689. [Google Scholar] [CrossRef]
- Pekny, M.; Wilhelmsson, U.; Pekna, M. The dual role of astrocyte activation and reactive gliosis. Neurosci. Lett. 2014, 565, 30–38. [Google Scholar] [CrossRef]
- Rosa, J.M.; Farre-Alins, V.; Navarrete, M.; Palomino-Antolin, A.; Fernandez-Lopez, E.; Narros-Fernandez, P.; Egea, J. TLR4-pathway impairs synaptic number and cerebrovascular functions through astrocyte activation following traumatic brain injury. Br. J. Pharmacol. 2021, 178, 3395–3413. [Google Scholar] [CrossRef]
- Pischiutta, F.; Micotti, E.; Hay, J.R.; Marongiu, I.; Sammali, E.; Tolomeo, D.; Vegliante, G.; Stocchetti, N.; Forloni, G.; De Simoni, M.-G.; et al. Single severe traumatic brain injury produces progressive pathology with ongoing contralateral white matter damage one year after injury. Exp. Neurol. 2018, 300, 167–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, T.-Y.; Hung, D.T.; Su, T.-P.; Tsai, S.-Y.A. Loss of Sigma-1 receptor chaperone promotes astrocytosis and enhances the Nrf2 antioxidant defense. Oxid. Med. Cell. Longev. 2017, 2017, 4582135. [Google Scholar] [CrossRef] [PubMed]
- Loane, D.J.; Kumar, A.; Stoica, B.A.; Cabatbat, R.; Faden, A.I. Progressive neurodegeneration after experimental brain trauma: Association with chronic microglial activation. J. Neuropathol. Exp. Neurol. 2014, 73, 14–29. [Google Scholar] [CrossRef] [Green Version]
- Carron, S.F.; Alwis, D.S.; Rajan, R. Traumatic brain injury and neuronal functionality changes in sensory cortex. Front. Syst. Neurosci. 2016, 10, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapoport, M.; van Reekum, R.; Mayberg, H. The role of the cerebellum in cognition and behavior. J. Neuropsychiatry Clin. Neurosci. 2000, 12, 193–198. [Google Scholar] [CrossRef]
- Mautes, A.E.; Fukuda, K.; Noble, L.J. Cellular response in the cerebellum after midline traumatic brain injury in the rat. Neurosci. Lett. 1996, 214, 95–98. [Google Scholar] [CrossRef]
- Park, E.; McKnight, S.; Ai, J.; Baker, A.J. Purkinje cell vulnerability to mild and severe forebrain head trauma. J. Neuropathol. Exp. Neurol. 2006, 65, 226–234. [Google Scholar] [CrossRef] [Green Version]
- Cerrato, V. Cerebellar astrocytes: Much more than passive bystanders in ataxia pathophysiology. J. Clin. Med. 2020, 9, 757. [Google Scholar] [CrossRef] [Green Version]
- Nolte, C.; Matyash, M.; Pivneva, T.; Schipke, C.G.; Ohlemeyer, C.; Hanisch, U.K.; Kirchhoff, F.; Kettenmann, H. GFAP promoter-controlled EGFP-expressing transgenic mice: A tool to visualize astrocytes and astrogliosis in living brain tissue. Glia 2001, 33, 72–86. [Google Scholar] [CrossRef]
- Wang, F.; Xu, Q.; Wang, W.; Takano, T.; Nedergaard, M. Bergmann glia modulate cerebellar Purkinje cell bistability via Ca2+-dependent K+ uptake. Proc. Natl. Acad. Sci. USA 2012, 109, 7911–7916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, T.; Beppu, K.; Tanaka, K.F.; Fukazawa, Y.; Shigemoto, R.; Matsui, K. Application of an optogenetic byway for perturbing neuronal activity via glial photostimulation. Proc. Natl. Acad. Sci. USA 2012, 109, 20720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, W.; Allen, N.D.; Skynner, M.; Gusterson, B.; Clark, A.J. Inducible ablation of astrocytes shows that these cells are required for neuronal survival in the adult brain. Glia 2001, 34, 272–282. [Google Scholar] [CrossRef]
- Tyszkiewicz, C.; Pardo, I.D.; Ritenour, H.N.; Liu, C.N.; Somps, C. Increases in GFAP immunoreactive astrocytes in the cerebellar molecular layer of young adult CBA/J mice. Lab. Anim. Res. 2021, 37, 24. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Imura, T.; Sofroniew, M.V.; Fushiki, S. Loss of adenomatous polyposis coli in Bergmann glia disrupts their unique architecture and leads to cell nonautonomous neurodegeneration of cerebellar Purkinje neurons. Glia 2011, 59, 857–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nardai, S.; László, M.; Szabó, A.; Alpár, A.; Hanics, J.; Zahola, P.; Merkely, B.; Frecska, E.; Nagy, Z. N,N-dimethyltryptamine reduces infarct size and improves functional recovery following transient focal brain ischemia in rats. Exp. Neurol. 2020, 327, 113245. [Google Scholar] [CrossRef]
- Katnik, C.; Garcia, A.; Behensky, A.A.; Yasny, I.E.; Shuster, A.M.; Seredenin, S.B.; Petrov, A.V.; Seifu, S.; McAleer, J.; Willing, A.; et al. Treatment with afobazole at delayed time points following ischemic stroke improves long-term functional and histological outcomes. Neurobiol. Dis. 2014, 62, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Gaja-Capdevila, N.; Hernández, N.; Zamanillo, D.; Vela, J.M.; Merlos, M.; Navarro, X.; Herrando-Grabulosa, M. Neuroprotective effects of Sigma 1 receptor ligands on motoneuron death after spinal root injury in mice. Int. J. Mol. Sci. 2021, 22, 6956. [Google Scholar] [CrossRef] [PubMed]
- Langa, F.; Codony, X.; Tovar, V.; Lavado, A.; Giménez, E.; Cozar, P.; Cantero, M.; Dordal, A.; Hernández, E.; Pérez, R.; et al. Generation and phenotypic analysis of sigma receptor type I (sigma 1) knockout mice. Eur. J. Neurosci. 2003, 18, 2188–2196. [Google Scholar] [CrossRef] [PubMed]
- Kilkenny, C.; Browne, W.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Animal research: Reporting in vivo experiments: The ARRIVE guidelines. Br. J. Pharmacol. 2010, 160, 1577–1579. [Google Scholar] [CrossRef]
- McGrath, J.C.; Drummond, G.B.; McLachlan, E.M.; Kilkenny, C.; Wainwright, C.L. Guidelines for reporting experiments involving animals: The ARRIVE guidelines. Br. J. Pharmacol. 2010, 160, 1573–1576. [Google Scholar] [CrossRef] [Green Version]
- Kupats, E.; Stelfa, G.; Zvejniece, B.; Grinberga, S.; Vavers, E.; Makrecka-Kuka, M.; Svalbe, B.; Zvejniece, L.; Dambrova, M. Mitochondrial-protective effects of R-Phenibut after experimental traumatic brain injury. Oxid. Med. Cell. Longev. 2020, 2020, 9364598. [Google Scholar] [CrossRef]
- Zvejniece, L.; Stelfa, G.; Vavers, E.; Kupats, E.; Kuka, J.; Svalbe, B.; Zvejniece, B.; Albert-Weissenberger, C.; Sirén, A.-L.; Plesnila, N.; et al. Skull fractures induce neuroinflammation and worsen outcomes after closed head injury in mice. J. Neurotrauma 2019, 37, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Zvejniece, L.; Svalbe, B.; Veinberg, G.; Grinberga, S.; Vorona, M.; Kalvinsh, I.; Dambrova, M. Investigation into stereoselective pharmacological activity of phenotropil. Basic Clin. Pharmacol. Toxicol. 2011, 109, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Zvejniece, L.; Vavers, E.; Svalbe, B.; Vilskersts, R.; Domracheva, I.; Vorona, M.; Veinberg, G.; Misane, I.; Stonans, I.; Kalvinsh, I.; et al. The cognition-enhancing activity of E1R, a novel positive allosteric modulator of sigma-1 receptors. Br. J. Pharmacol. 2014, 171, 761–771. [Google Scholar] [CrossRef] [Green Version]
- Can, A.; Dao, D.T.; Terrillion, C.E.; Piantadosi, S.C.; Bhat, S.; Gould, T.D. The tail suspension test. J. Vis. Exp. 2012, 59, e3769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gawel, K.; Gibula, E.; Marszalek-Grabska, M.; Filarowska, J.; Kotlinska, J.H. Assessment of spatial learning and memory in the Barnes maze task in rodents-methodological consideration. Naunyn Schmiedebergs Arch. Pharmacol. 2019, 392, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Verollet, R. A major step towards efficient sample preparation with bead-beating. Biotechniques 2008, 44, 832–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vavers, E.; Zvejniece, B.; Stelfa, G.; Svalbe, B.; Vilks, K.; Kupats, E.; Mezapuke, R.; Lauberte, L.; Dambrova, M.; Zvejniece, L. Genetic inactivation of the sigma-1 chaperone protein results in decreased expression of the R2 subunit of the GABA-B receptor and increased susceptibility to seizures. Neurobiol. Dis. 2021, 150, 105244. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stelfa, G.; Vavers, E.; Svalbe, B.; Serzants, R.; Miteniece, A.; Lauberte, L.; Grinberga, S.; Gukalova, B.; Dambrova, M.; Zvejniece, L. Reduced GFAP Expression in Bergmann Glial Cells in the Cerebellum of Sigma-1 Receptor Knockout Mice Determines the Neurobehavioral Outcomes after Traumatic Brain Injury. Int. J. Mol. Sci. 2021, 22, 11611. https://doi.org/10.3390/ijms222111611
Stelfa G, Vavers E, Svalbe B, Serzants R, Miteniece A, Lauberte L, Grinberga S, Gukalova B, Dambrova M, Zvejniece L. Reduced GFAP Expression in Bergmann Glial Cells in the Cerebellum of Sigma-1 Receptor Knockout Mice Determines the Neurobehavioral Outcomes after Traumatic Brain Injury. International Journal of Molecular Sciences. 2021; 22(21):11611. https://doi.org/10.3390/ijms222111611
Chicago/Turabian StyleStelfa, Gundega, Edijs Vavers, Baiba Svalbe, Rinalds Serzants, Anna Miteniece, Lasma Lauberte, Solveiga Grinberga, Baiba Gukalova, Maija Dambrova, and Liga Zvejniece. 2021. "Reduced GFAP Expression in Bergmann Glial Cells in the Cerebellum of Sigma-1 Receptor Knockout Mice Determines the Neurobehavioral Outcomes after Traumatic Brain Injury" International Journal of Molecular Sciences 22, no. 21: 11611. https://doi.org/10.3390/ijms222111611
APA StyleStelfa, G., Vavers, E., Svalbe, B., Serzants, R., Miteniece, A., Lauberte, L., Grinberga, S., Gukalova, B., Dambrova, M., & Zvejniece, L. (2021). Reduced GFAP Expression in Bergmann Glial Cells in the Cerebellum of Sigma-1 Receptor Knockout Mice Determines the Neurobehavioral Outcomes after Traumatic Brain Injury. International Journal of Molecular Sciences, 22(21), 11611. https://doi.org/10.3390/ijms222111611