
Genomic Marks Associated with Chromatin Compartments in the CTCF, RNAPII Loop and Genomic Windows


Abstract
:1. Introduction
2. Results
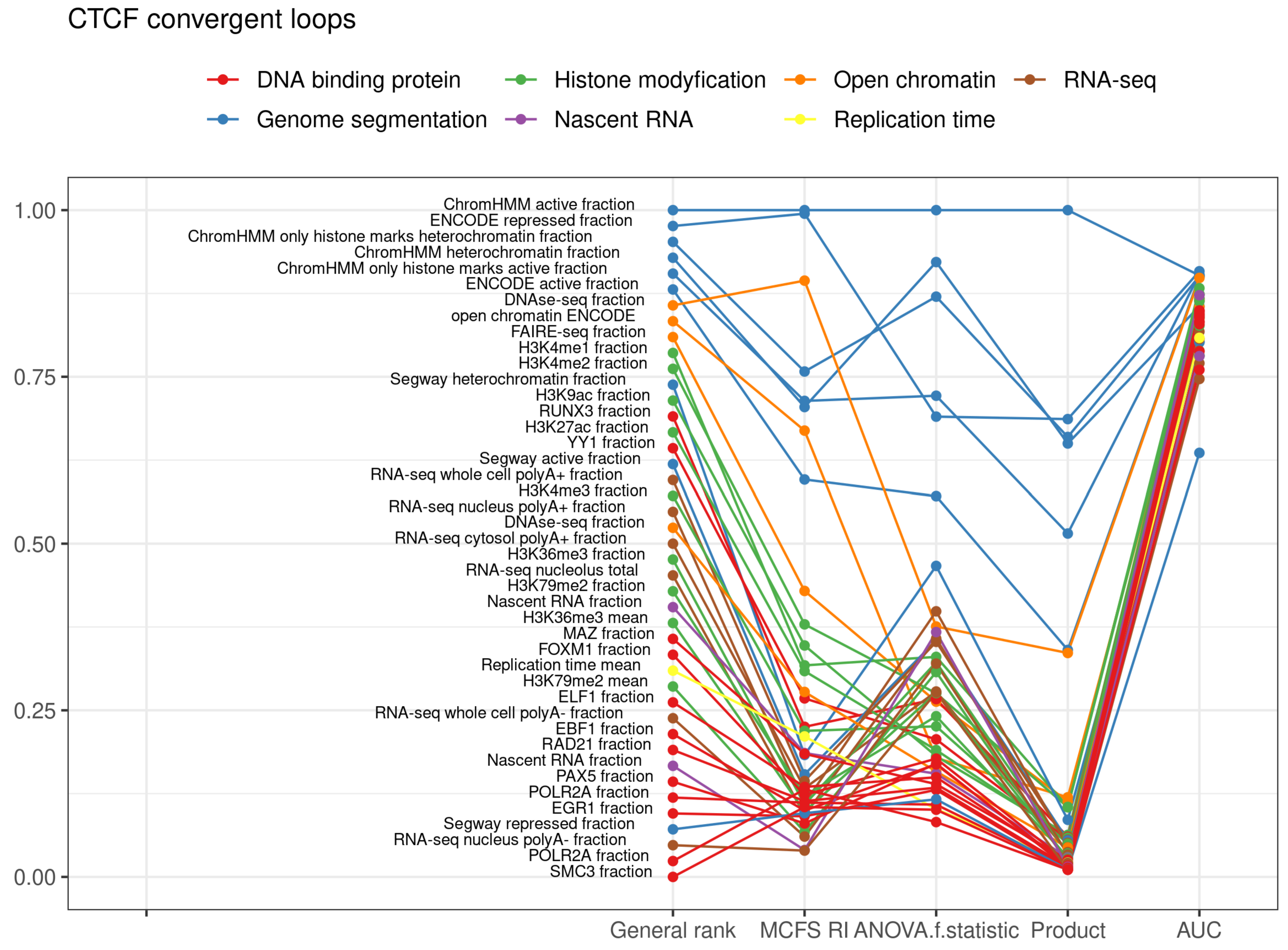
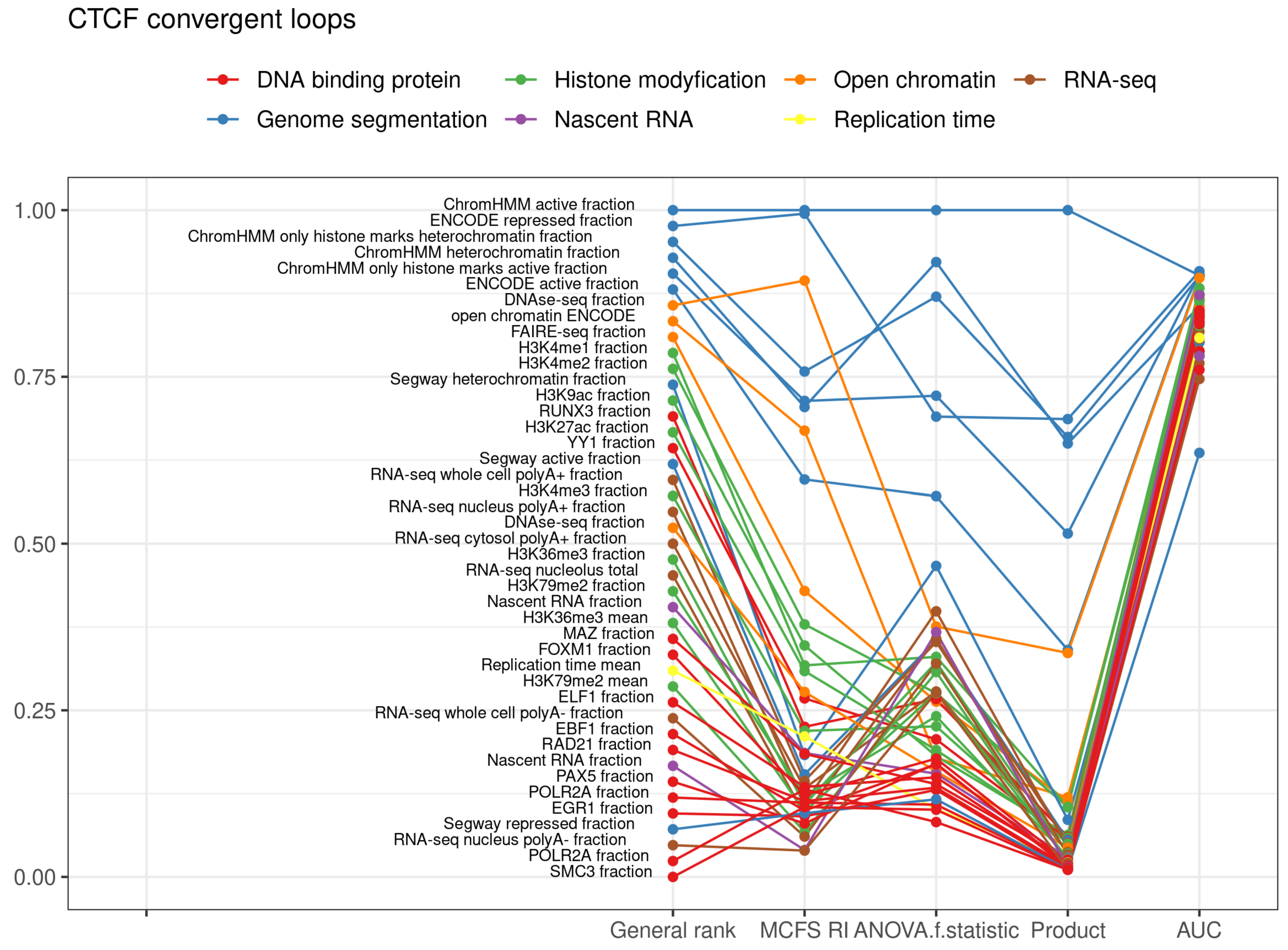
2.1. CTCF Convergent Loops
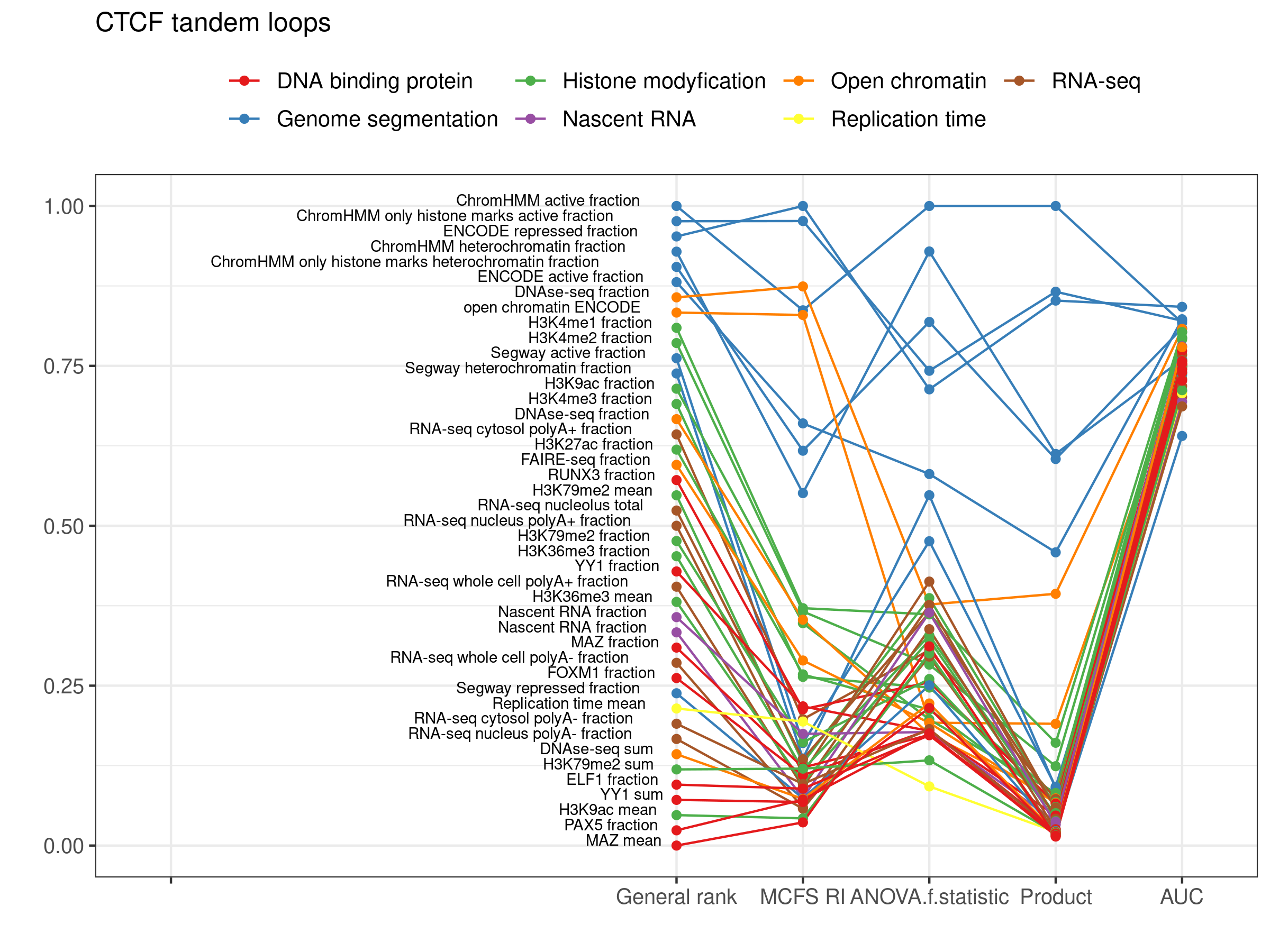
2.2. CTCF Tandem Loops
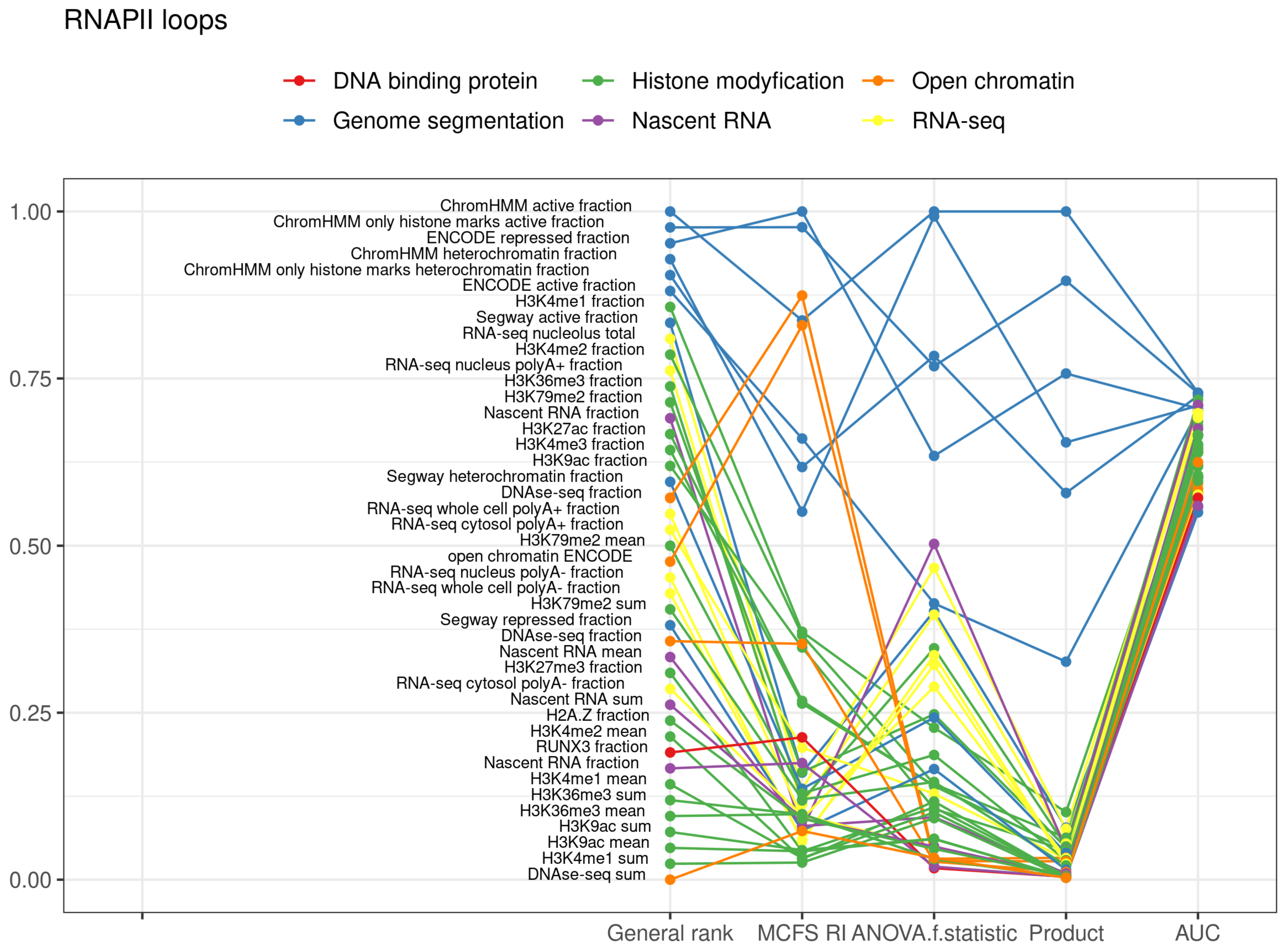
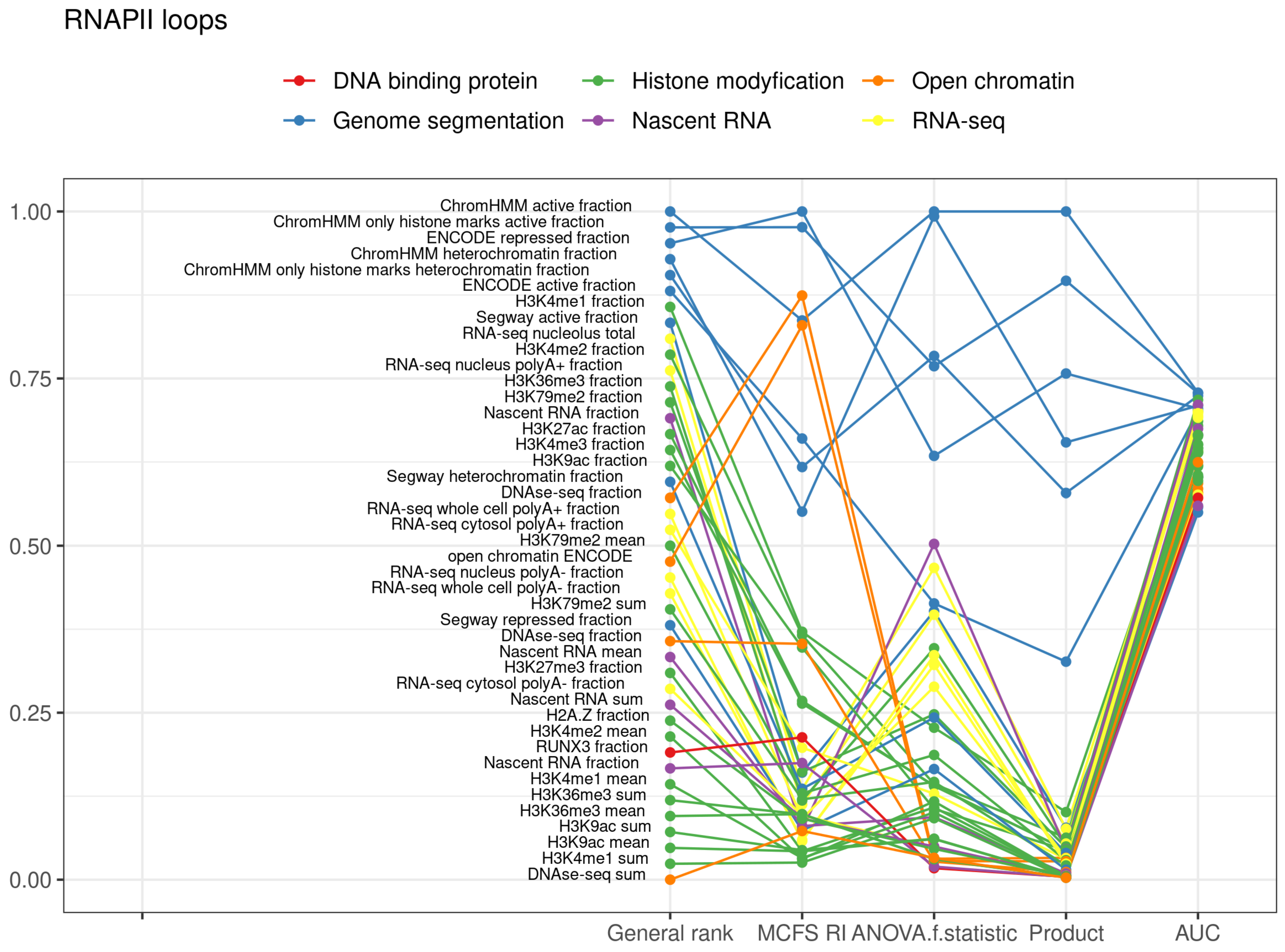
2.3. RNAPII Loops
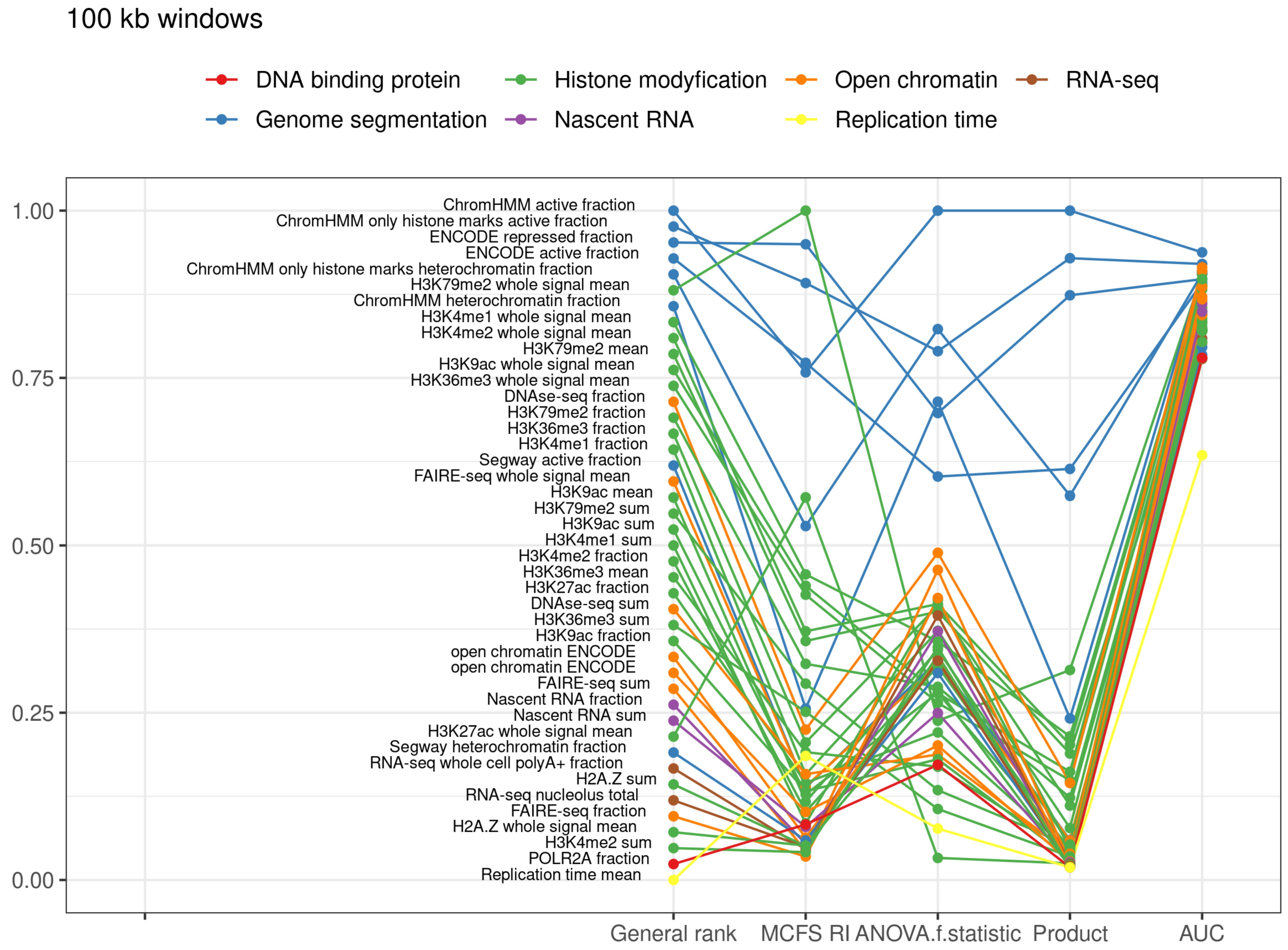
2.4. Genomic Windows
3. Discussion
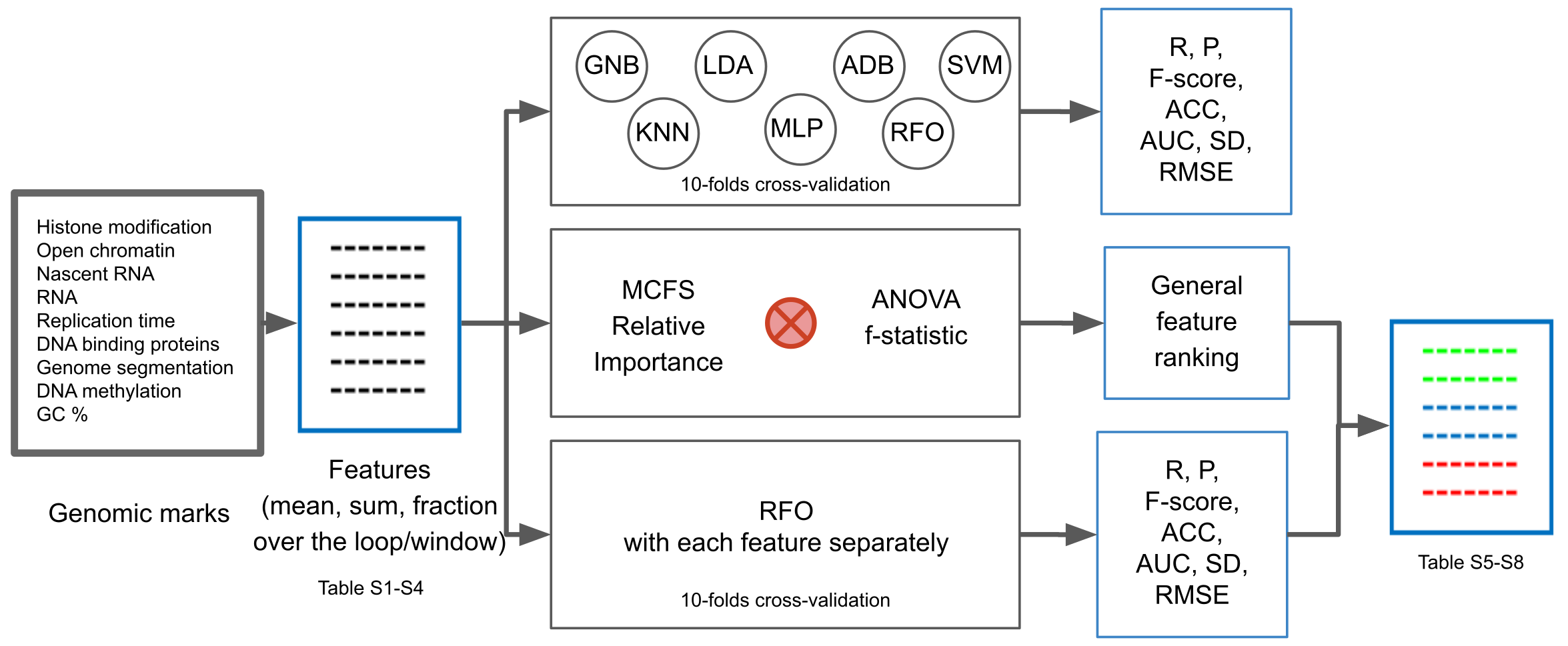
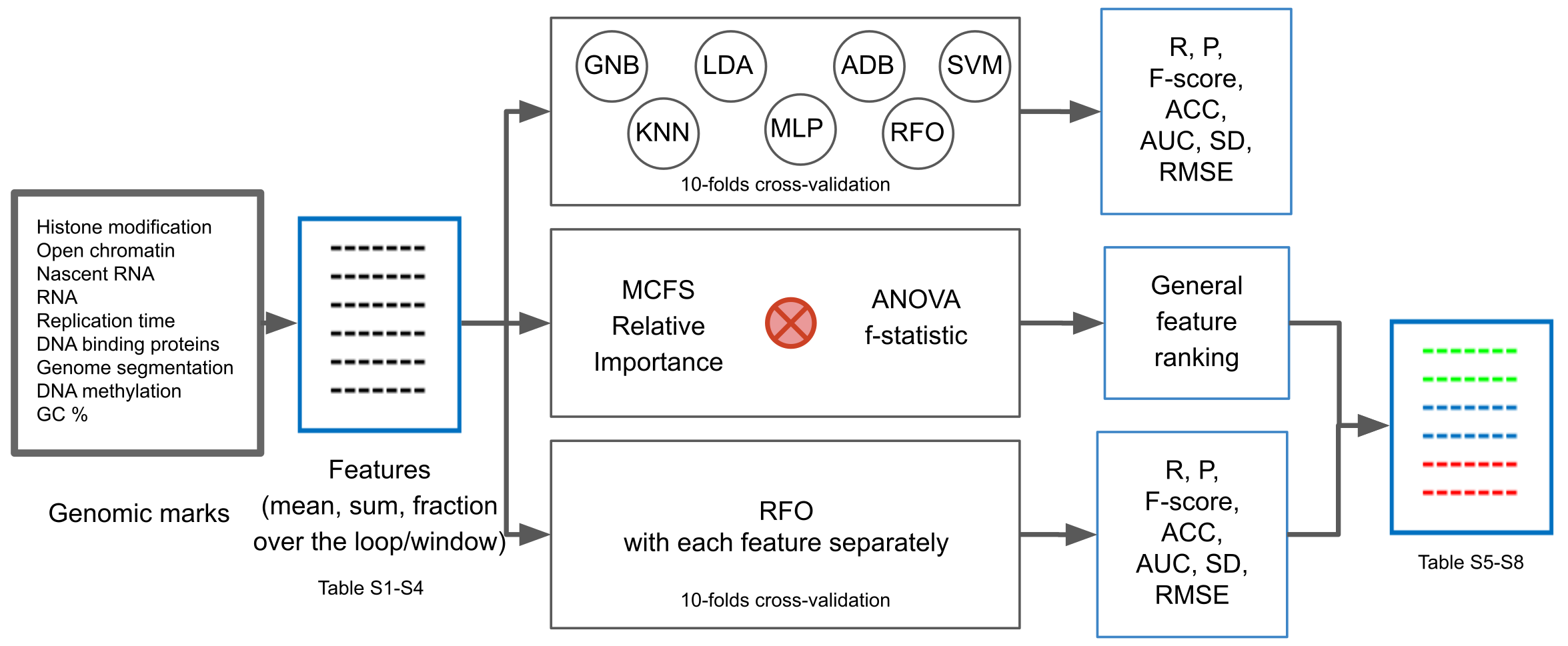
4. Materials and Methods
- DNA methylation at CpG sites Methyl Array data (GEO Accession: GSM999376), Methyl Reduced Representation Bisulfite Sequencing (RRBS) (GSM683906, GSM683927);
- ChIP-seq data for chemical modifications (methylation, acetylation) to the histone proteins, particularly: H3K4me3, H3K27ac, H3K9ac, H4K20me1, H3K4me1, H3K4me2, H3K79me2, H3K36me3, H3K27me3, H3K9me3 and H2AZ (histone alternative variant);
- ChIP-seq data for 87 DNA binding proteins;
- Open chromatin data: DNase-seq, FAIRE-seq, ENCODE synthesis. This is ENCODE synthesis of evidence from different assays: DNase I hypersensitivity (HS), Formaldehyde-Assisted Isolation of Regulatory Elements (FAIRE), and chromatin immunoprecipitation (ChIP) for select regulatory factors (PolII, CTCF, c-Myc). This indicates the regions of the DNA available for direct interaction with non-histone proteins and RNA;
- Genome segmentation: ChromHMM [28], Segway [29], ENCODE synthesis [30], the old ChromHMM method based only on histone modifications [31]. Using two different unsupervised machine learning techniques (ChromHMM and Segway), the genome was automatically segmented into disjoint segments. Each segment belongs to one of a few specific genomic “states” which is assigned an intuitive label (“active” for promoters (also inactive promoters), enhancers, transcription-associated, insulators, “repressed” for Polycomb repressed, “heterochromatin” for heterochromatin, repetitive, copy number variation regions). These methods used ENCODE ChIP-seq, DNase-seq, and FAIRE-seq data. Regions that were not assigned to “heterochromatin” not “repressed” classes by the segmentation methods went to the “active” class;
- RNA-seq transcription data.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bolzer, A.; Kreth, G.; Solovei, I.; Koehler, D.; Saracoglu, K.; Fauth, C.; Müller, S.; Eils, R.; Cremer, C.; Speicher, M.; et al. Three-dimensional maps of all chromosomes in human male fibroblast nuclei and prometaphase rosettes. PLoS Biol. 2005, 3, e157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nora, E.; Lajoie, B.R.; Schulz, E.G.; Giorgetti, L.; Okamoto, I.; Servant, N.; Piolot, T.; Van Berkum, N.L.; Meisig, J.; Sedat, J.; et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 2012, 485, 381–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cremer, T.; Cremer, M.; Hübner, B.; Strickfaden, H.; Smeets, D.; Popken, J.; Sterr, M.; Markaki, Y.; Rippe, K.; Cremer, C. The 4D nucleome: Evidence for a dynamic nuclear landscape based on co-aligned active and inactive nuclear compartments. FEBS Lett. 2015, 589, 2931–2943. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Z.; Luo, O.J.; Li, X.; Zheng, M.; Zhu, J.J.; Szalaj, P.; Trzaskoma, P.; Magalska, A.; Wlodarczyk, J.; Ruszczycki, B.; et al. CTCF-Mediated Human 3D Genome Architecture Reveals Chromatin Topology for Transcription. Cell 2015, 163, 1611–1627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieberman-Aiden, E.; Van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falk, M.; Feodorova, Y.; Naumova, N.; Imakaev, M.; Lajoie, B.R.; Leonhardt, H.; Joffe, B.; Dekker, J.; Fudenberg, G.; Solovei, I.; et al. Heterochromatin drives compartmentalization of inverted and conventional nuclei. Nature 2019, 570, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Draminski, M.; Rada-Iglesias, A.; Enroth, S.; Wadelius, C.; Koronacki, J.; Komorowski, J. Monte Carlo feature selection for supervised classification. Bioinformatics 2008, 24, 110–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Dramiński, M.; Koronacki, J. rmcfs: An R Package for Monte Carlo Feature Selection and Interdependency Discovery. J. Stat. Softw. 2018, 85, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Ernst, J.; Kellis, M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol. 2010, 28, 817–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, C.; Andrews, R.M.; Flicek, P.; Dillon, S.C.; Karaöz, U.; Clelland, G.K.; Wilcox, S.; Beare, D.M.; Fowler, J.C.; Couttet, P.; et al. The landscape of histone modifications across 1% of the human genome in five human cell lines. Genome Res. 2007, 17, 691–707. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Wang, X.; He, K.; Charron, J.-B.F.; Elling, A.A.; Deng, X.W. Genome-wide profiling of histone H3 lysine 9 acetylation and dimethylation in Arabidopsis reveals correlation between multiple histone marks and gene expression. Plant Mol. Biol. 2010, 72, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Peters, A.H.F.M.; Kubicek, S.; Mechtler, K.; O’Sullivan, R.J.; Derijck, A.A.H.A.; Perez-Burgos, L.; Kohlmaier, A.; Opravil, S.; Tachibana, M.; Shinkai, Y.; et al. Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol. Cell. 2003, 12, 1577–1589. [Google Scholar] [CrossRef]
- Nishioka, K.; Rice, J.C.; Sarma, K.; Erdjument-Bromage, H.; Werner, J.; Wang, Y.; Chuikov, S.; Valenzuela, P.; Tempst, P.; Steward, R.; et al. PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Mol. Cell. 2002, 9, 1201–1213. [Google Scholar] [CrossRef]
- Puig-Kröger, A.; Corbí, A. RUNX3: A new player in myeloid gene expression and immune response. J. Cell Biochem. 2006, 98, 744–756. [Google Scholar] [CrossRef]
- Yokomizo-Nakano, T.; Sashida, G. Two faces of RUNX3 in myeloid transformation. Exp. Hematol. 2021, 97, 14–20. [Google Scholar] [CrossRef]
- Weintraub, A.S.; Li, C.; Zamudio, A.V.; Sigova, A.A.; Hannett, N.M.; Day, D.S.; Abraham, B.J.; Cohen, M.A.; Nabet, B.; Buckley, D.L.; et al. YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell 2017, 171, 1573–1588.e28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farooq, Z.; Banday, S.; Pandita, T.K.; Altaf, M. The many faces of histone H3K79 methylation. Mutat. Res. Rev. Mutat. Res. 2016, 768, 46–52. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Chen, S.-A.; Local, A.; Liu, T.; Qiu, Y.; Dorighi, K.M.; Preissl, S.; Rivera, C.M.; Wang, C.; Ye, Z.; et al. Histone H3 lysine 4 monomethylation modulates long-range chromatin interactions at enhancers. Cell Res. 2018, 28, 387. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, X.; Hu, H. H3K4me2 reliably defines transcription factor binding regions in different cells. Genomics 2014, 103, 222–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinz, S.; Romanoski, C.E.; Benner, C.; Glass, C.K. The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol. 2015, 16, 144–154. [Google Scholar] [CrossRef] [Green Version]
- Pekowska, A.; Benoukraf, T.; Ferrier, P.; Spicuglia, S. A unique H3K4me2 profile marks tissue-specific gene regulation. Genome Res. 2010, 20, 1493–1502. [Google Scholar] [CrossRef] [Green Version]
- Fu, S.; Wang, Q.; Moore, J.E.; Purcaro, M.; Pratt, H.E.; Fan, K.; Gu, C.; Jiang, C.; Zhu, R.; Kundaje, A.; et al. Differential analysis of chromatin accessibility and histone modifications for predicting mouse developmental enhancers. Nucleic Acids Res. 2018, 46, 11184–11201. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The ENCODE Project Consortium. A User’s Guide to the Encyclopedia of DNA Elements (ENCODE). PLoS Biol. 2011, 9, e1001046. [Google Scholar] [CrossRef]
- Ernst, J.; Kellis, M. Interplay between chromatin state, regulator binding, and regulatory motifs in six human cell types. Genome Res. 2013, 23, 1142–1154. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, M.M.; Buske, O.J.; Wang, J.; Weng, Z.; Bilmes, J.A.; Noble, W.S. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat. Methods 2012, 9, 473–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, M.; Ernst, J.; Wilder, S.; Kundaje, A.; Harris, R.S.; Libbrecht, M.; Giardine, B.; Ellenbogen, P.M.; Bilmes, J.A.; Birney, E.; et al. Integrative annotation of chromatin elements from ENCODE data. Nucleic Acids Res. 2013, 41, 827–841. [Google Scholar] [CrossRef] [Green Version]
- Ernst, J.; Kheradpour, P.; Mikkelsen, T.S.; Shoresh, N.; Ward, L.; Epstein, C.B.; Zhang, X.; Wang, L.; Issner, R.; Coyne, M.; et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 2011, 473, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef]
- Core, L.J.; Martins, A.L.; Danko, C.G.; Waters, C.T.; Siepel, A.; Lis, J.T. Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat. Genet. 2014, 46, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.E.; Arlt, M.F.; Park, S.H.; Rajendran, S.; Paulsen, M.; Ljungman, M.; Glover, T.W. Large transcription units unify copy number variants and common fragile sites arising under replication stress. Genome Res. 2015, 25, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Pope, B.; Ryba, T.; Dileep, V.; Yue, F.; Wu, W.; Denas, O.; Vera, D.; Wang, Y.; Hansen, R.S.; Canfield, T.K.; et al. Topologically associating domains are stable units of replication-timing regulation. Nature 2014, 515, 402–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classifier | R | P | F-Score | ACC | AUC | SD | RMSE |
|---|---|---|---|---|---|---|---|
| GNB | 0.8139 | 0.8510 | 0.8179 | 0.8139 | 0.9138 | 0.0301 | 0.4296 |
| KNN | 0.8876 | 0.8912 | 0.8881 | 0.8876 | 0.9357 | 0.0234 | 0.2924 |
| LDA | 0.9175 | 0.9185 | 0.9166 | 0.9175 | 0.9677 | 0.0172 | 0.2509 |
| ADB | 0.9142 | 0.9158 | 0.9134 | 0.9142 | 0.9703 | 0.0123 | 0.4697 |
| SVM | 0.9264 | 0.9276 | 0.9253 | 0.9264 | 0.9689 | 0.0218 | 0.2353 |
| MLP | 0.9253 | 0.9262 | 0.9249 | 0.9253 | 0.9728 | 0.0167 | 0.2515 |
| RFO | 0.9209 | 0.9231 | 0.9195 | 0.9209 | 0.9732 | 0.0147 | 0.2419 |
| Mean | 0.9008 | 0.9076 | 0.9008 | 0.9008 | 0.9575 | 0.0195 | 0.3102 |
| The Highest Place in the Ranking | ||||
|---|---|---|---|---|
| Assay Type | CTCF Convergent | CTCF Tandem | RNAPII | 100 kb Windows |
| Genome segmentation | 1 | 1 | 1 | 1 |
| Open chromatin | 7 | 7 | 19 | 13 |
| H3K4me1 | 10 | 9 | 7 | 8 |
| H3K4me2 | 11 | 10 | 10 | 9 |
| H3K9ac | 13 | 13 | 17 | 11 |
| DNA binding protein | 14 | 19 | 35 | 42 |
| H3K27ac | 15 | 17 | 14 | 25 |
| RNA-seq | 18 | 16 | 9 | 36 |
| H3K4me3 | 19 | 14 | 16 | 45 |
| H3K36me3 | 23 | 24 | 12 | 12 |
| H3K79me2 | 25 | 20 | 13 | 6 |
| Nascent RNA | 26 | 28 | 14 | 32 |
| Replication time | 30 | 34 | 120 | 43 |
| H2A.Z | 54 | 55 | 33 | 37 |
| DNA methylation at CpG sites | 60 | 53 | 70 | 151 |
| H3K27me3 | 104 | 56 | 30 | 345 |
| H4K20me1 | 119 | 102 | 45 | 194 |
| H3K9me3 | 336 | 327 | 203 | 270 |
| GC% | 409 | 384 | 51 | 84 |
| Classifier | R | P | F-Score | ACC | AUC | SD | RMSE |
|---|---|---|---|---|---|---|---|
| GNB | 0.7122 | 0.8259 | 0.7318 | 0.7122 | 0.8665 | 0.0331 | 0.5354 |
| KNN | 0.8613 | 0.8648 | 0.8622 | 0.8613 | 0.8942 | 0.0292 | 0.3226 |
| LDA | 0.8912 | 0.8897 | 0.8887 | 0.8912 | 0.9365 | 0.0246 | 0.2846 |
| ADB | 0.8946 | 0.8942 | 0.8927 | 0.8946 | 0.9409 | 0.0201 | 0.4778 |
| SVM | 0.8976 | 0.8962 | 0.8938 | 0.8976 | 0.9287 | 0.036 | 0.2736 |
| MLP | 0.8930 | 0.8928 | 0.8919 | 0.893 | 0.9379 | 0.0216 | 0.3038 |
| RFO | 0.9004 | 0.8997 | 0.8962 | 0.9004 | 0.9411 | 0.0235 | 0.2736 |
| Mean | 0.8643 | 0.8805 | 0.8653 | 0.8643 | 0.9208 | 0.0269 | 0.3531 |
| Classifier | R | P | F-Score | ACC | AUC | SD | RMSE |
|---|---|---|---|---|---|---|---|
| GNB | 0.4544 | 0.7952 | 0.4963 | 0.4544 | 0.7736 | 0.0692 | 0.7372 |
| KNN | 0.8744 | 0.8639 | 0.8629 | 0.8744 | 0.7944 | 0.0648 | 0.3253 |
| LDA | 0.8801 | 0.8735 | 0.8713 | 0.8801 | 0.8657 | 0.0567 | 0.3108 |
| ADB | 0.8824 | 0.8754 | 0.8719 | 0.8824 | 0.8665 | 0.0563 | 0.4890 |
| SVM | 0.8785 | 0.7861 | 0.8243 | 0.8785 | 0.8737 | 0.0514 | 0.2730 |
| MLP | 0.8642 | 0.8606 | 0.8596 | 0.8642 | 0.8206 | 0.0607 | 0.3370 |
| RFO | 0.8910 | 0.8855 | 0.8771 | 0.8910 | 0.8676 | 0.0587 | 0.2930 |
| Mean | 0.8179 | 0.8486 | 0.8091 | 0.8179 | 0.8374 | 0.0597 | 0.3950 |
| Classifier | R | P | F-Score | ACC | AUC | SD | RMSE |
|---|---|---|---|---|---|---|---|
| GNB | 0.8450 | 0.8476 | 0.8424 | 0.8450 | 0.9189 | 0.0180 | 0.3930 |
| KNN | 0.8693 | 0.8720 | 0.8672 | 0.8693 | 0.9255 | 0.0094 | 0.3141 |
| LDA | 0.8983 | 0.9011 | 0.8980 | 0.8983 | 0.9655 | 0.0116 | 0.2796 |
| ADB | 0.8970 | 0.9009 | 0.8973 | 0.8970 | 0.9654 | 0.0107 | 0.4779 |
| SVM | 0.8991 | 0.9036 | 0.8992 | 0.8991 | 0.9633 | 0.0167 | 0.2703 |
| MLP | 0.8907 | 0.8943 | 0.8908 | 0.8907 | 0.9566 | 0.0114 | 0.3116 |
| RFO | 0.8960 | 0.8999 | 0.8961 | 0.8960 | 0.9645 | 0.0108 | 0.2738 |
| Mean | 0.8851 | 0.8885 | 0.8844 | 0.8851 | 0.9514 | 0.0127 | 0.3315 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szczepińska, T.; Mollah, A.F.; Plewczynski, D. Genomic Marks Associated with Chromatin Compartments in the CTCF, RNAPII Loop and Genomic Windows. Int. J. Mol. Sci. 2021, 22, 11591. https://doi.org/10.3390/ijms222111591
Szczepińska T, Mollah AF, Plewczynski D. Genomic Marks Associated with Chromatin Compartments in the CTCF, RNAPII Loop and Genomic Windows. International Journal of Molecular Sciences. 2021; 22(21):11591. https://doi.org/10.3390/ijms222111591
Chicago/Turabian StyleSzczepińska, Teresa, Ayatullah Faruk Mollah, and Dariusz Plewczynski. 2021. "Genomic Marks Associated with Chromatin Compartments in the CTCF, RNAPII Loop and Genomic Windows" International Journal of Molecular Sciences 22, no. 21: 11591. https://doi.org/10.3390/ijms222111591
APA StyleSzczepińska, T., Mollah, A. F., & Plewczynski, D. (2021). Genomic Marks Associated with Chromatin Compartments in the CTCF, RNAPII Loop and Genomic Windows. International Journal of Molecular Sciences, 22(21), 11591. https://doi.org/10.3390/ijms222111591