
A Combined mRNA- and miRNA-Sequencing Approach Reveals miRNAs as Potential Regulators of the Small Intestinal Transcriptome in Celiac Disease

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
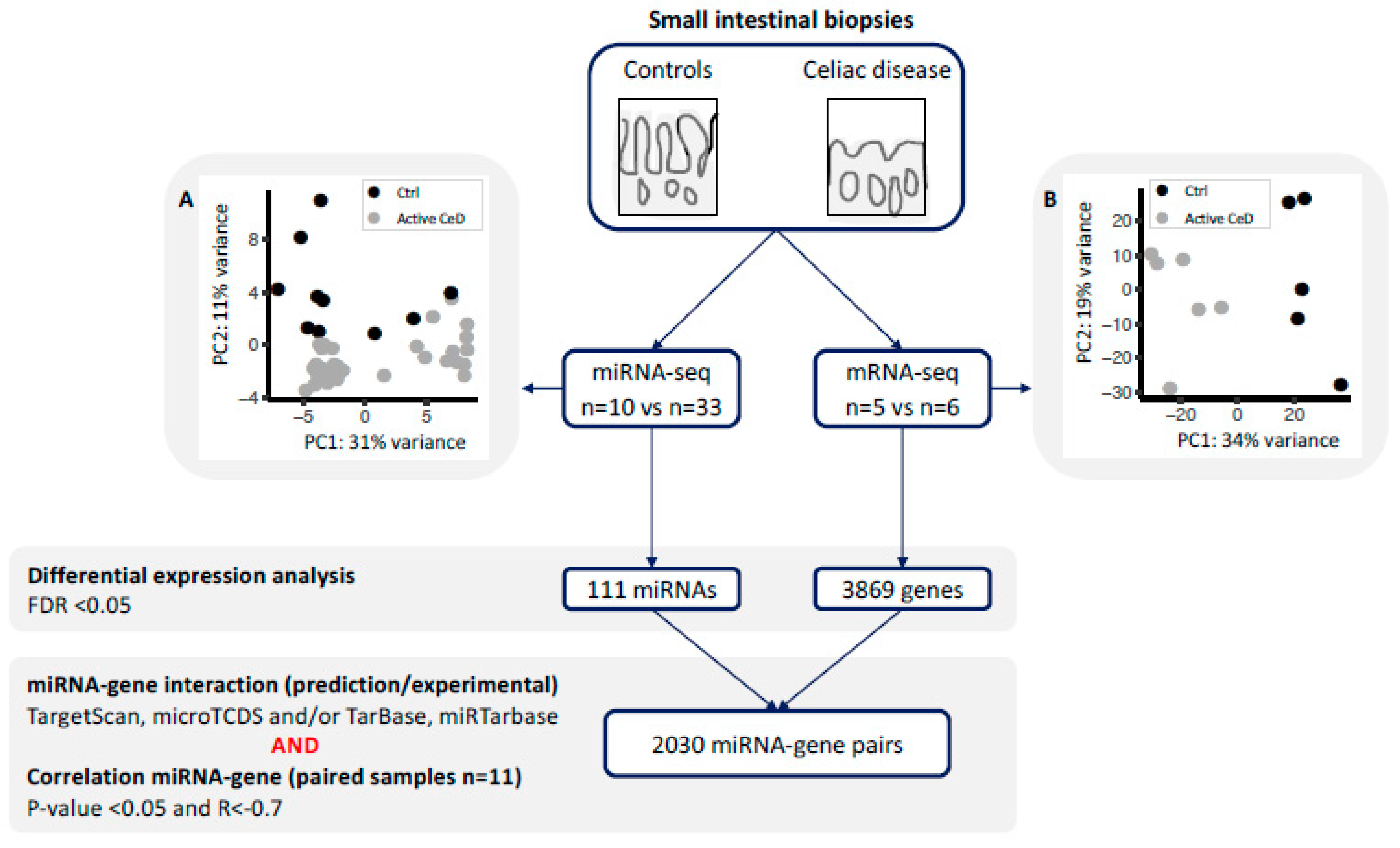
2.1. Differential Expression Analysis in CeD Identifies Immune-Related Genes and miRNAs
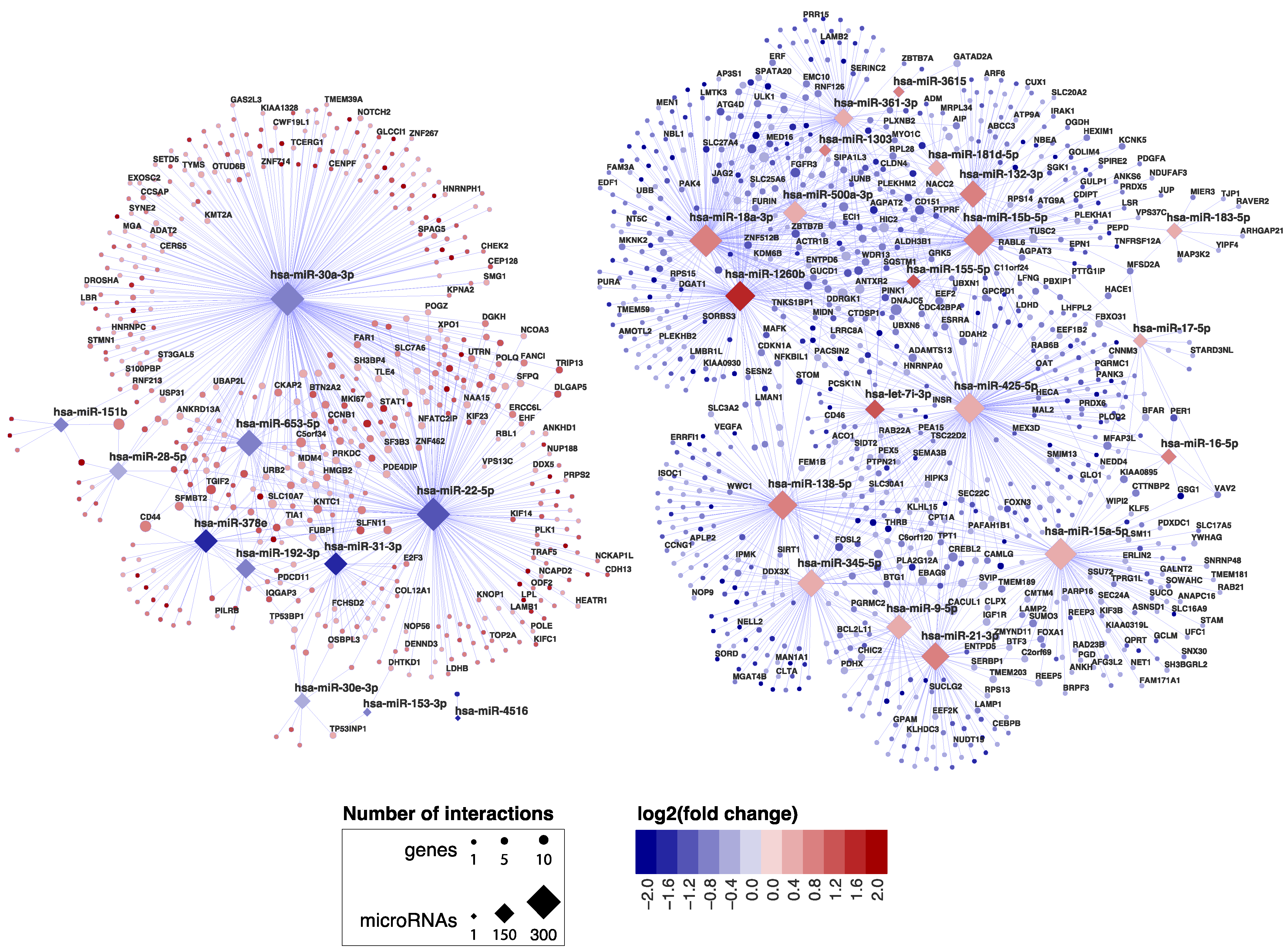
2.2. miRNA‒Target Transcript Interaction Network
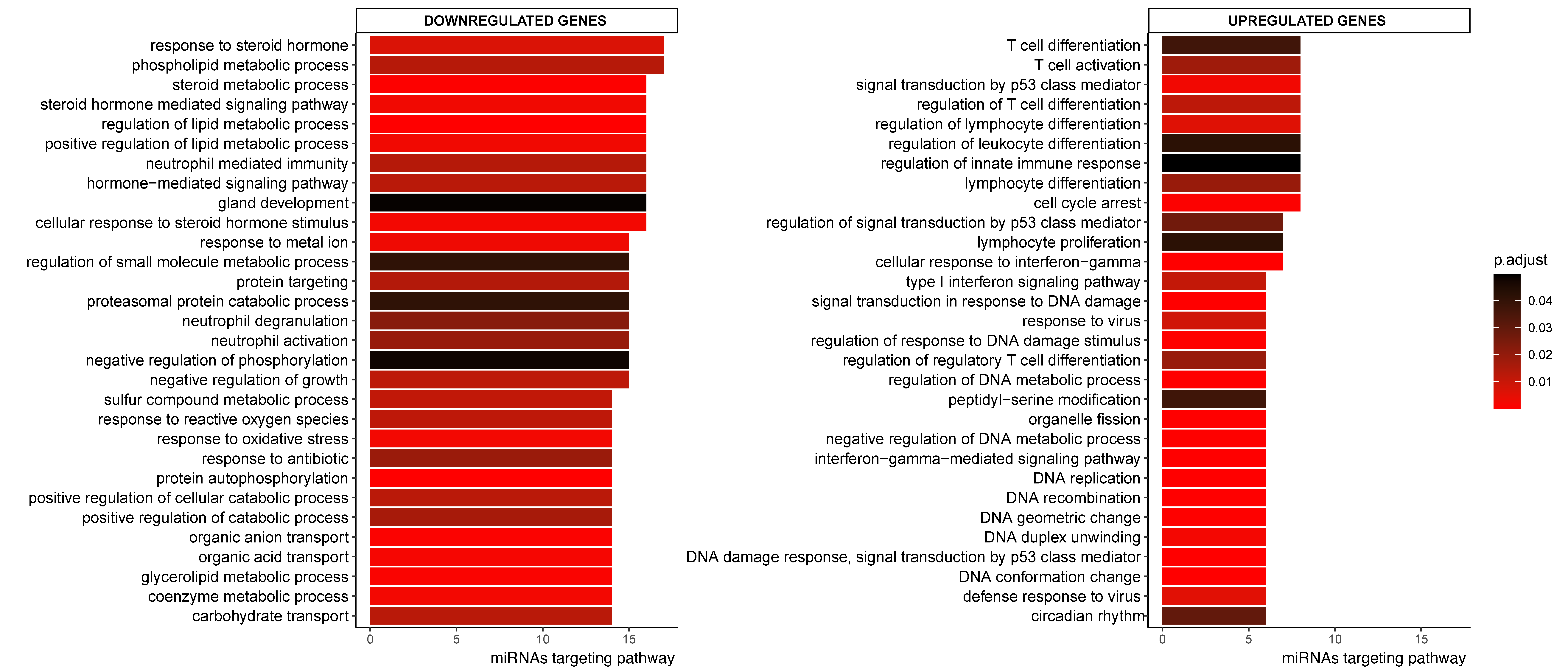
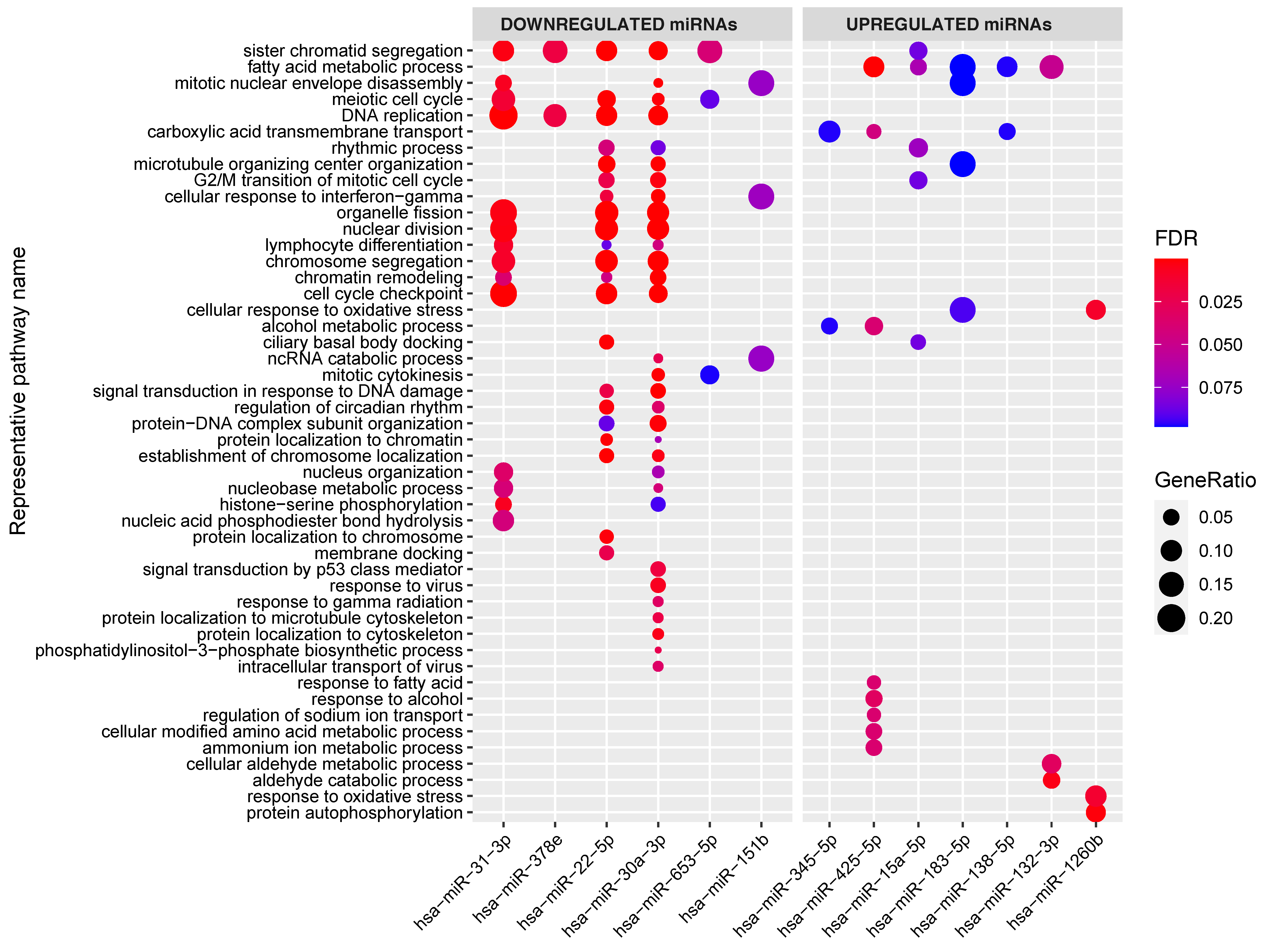
2.3. Pathway Enrichment Analyses
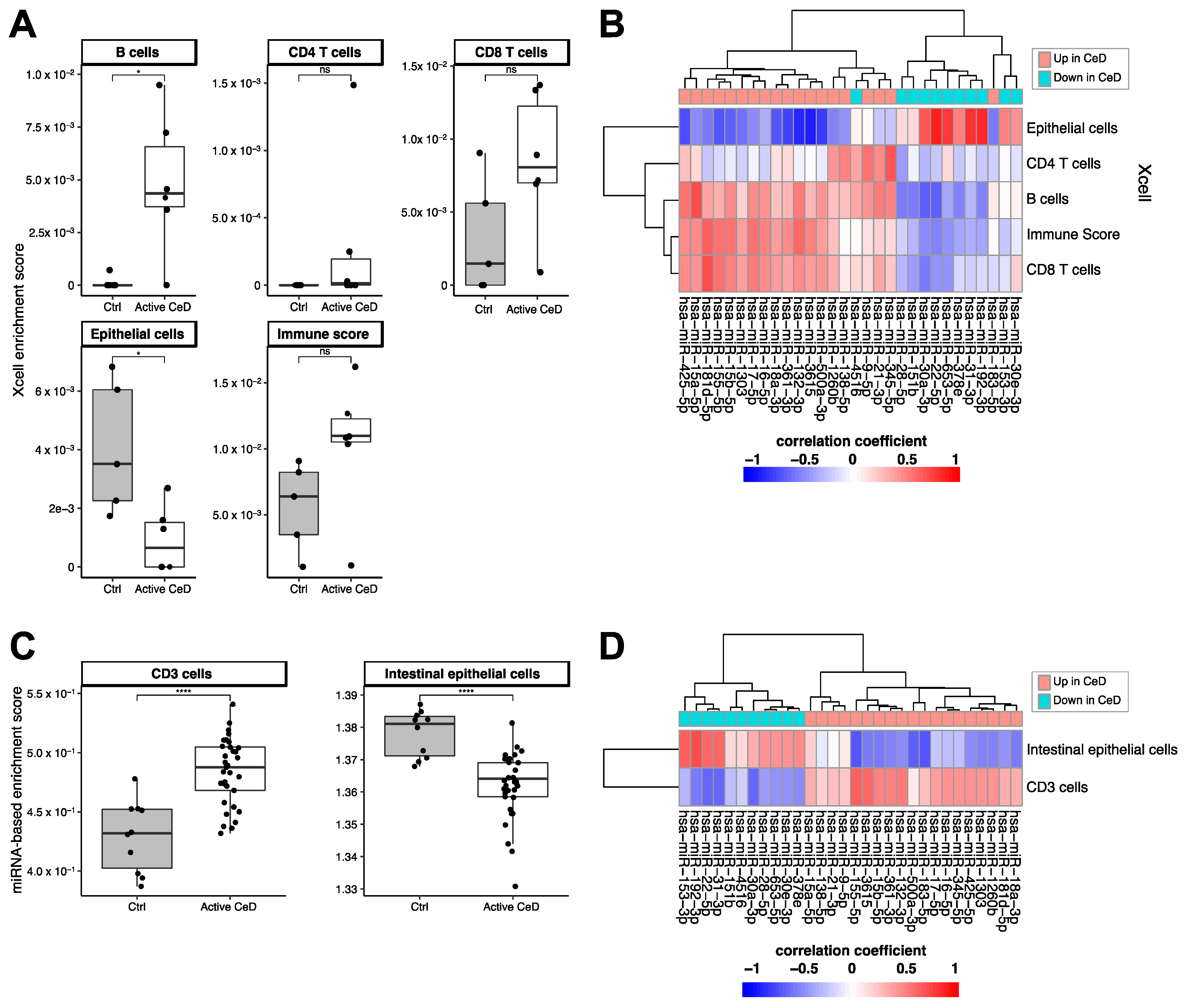
2.4. Cell Type‒Enrichment Analysis
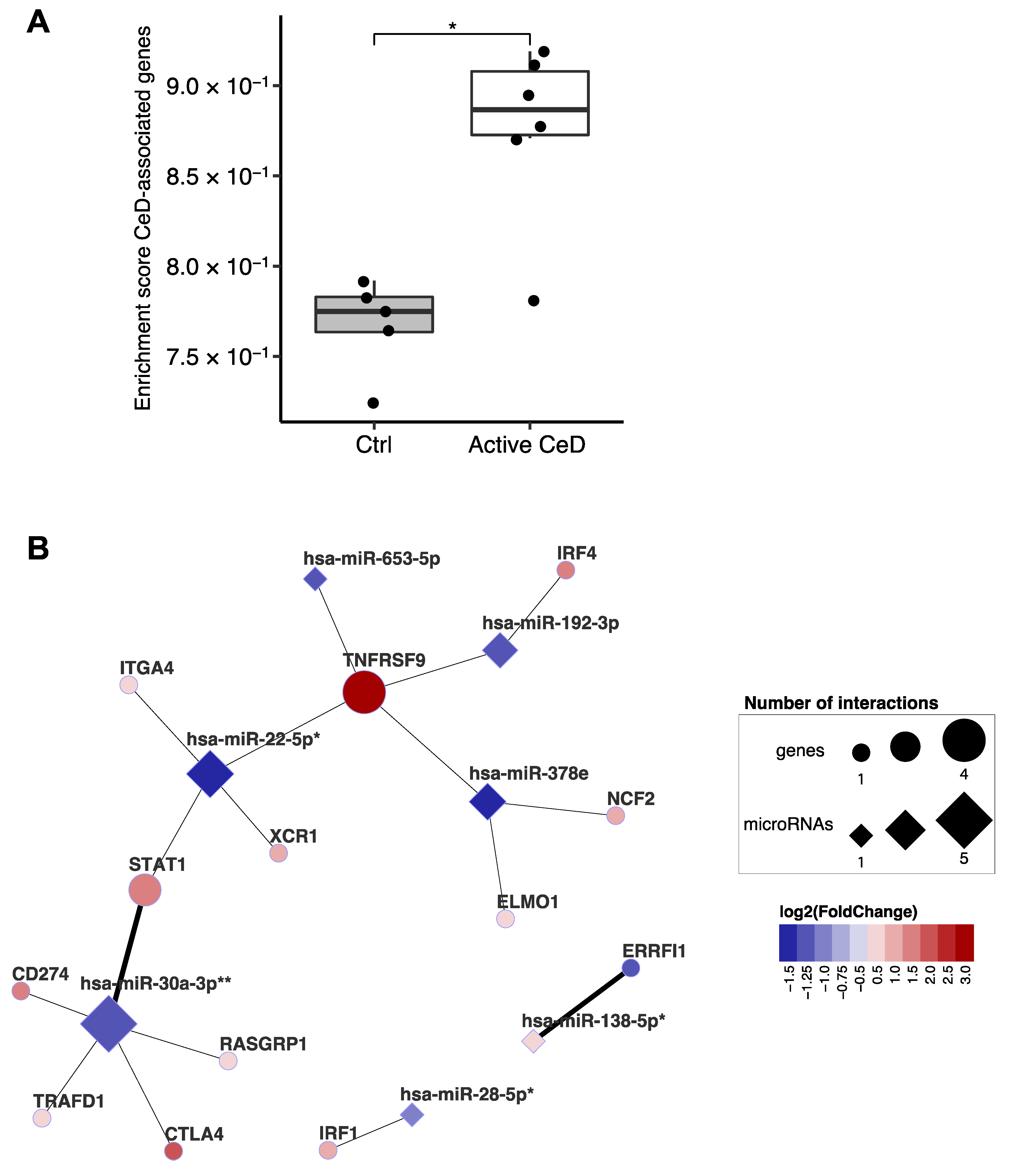
2.5. Linking the miRNA‒Target Transcript Network to Genes in CeD-Associated Genetic Risk Regions
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. RNA Isolation, Small RNA, and mRNA-Sequencing
4.3. Prioritizing Genes Targeted by miRNAs in the Small Intestinal Biopsies
4.3.1. Differential Expression Analyses in miRNA Sequencing and RNA Sequencing
4.3.2. miRNA‒Target Transcript Network
4.4. Enrichment Analyses
4.5. Cell Type‒Enrichment Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lindfors, K.; Ciacci, C.; Kurppa, K.; Lundin, K.E.A.; Makharia, G.K.; Mearin, M.L.; Murray, J.A.; Verdu, E.F.; Kaukinen, K. Coeliac disease. Nat. Rev. Dis. Primers 2019, 5, 1–18. [Google Scholar] [CrossRef]
- Singh, P.; Arora, A.; Strand, T.A.; Leffler, D.A.; Catassi, C.; Green, P.H.; Kelly, C.P.; Ahuja, V.; Makharia, G.K. Global Prevalence of Celiac Disease: Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol. 2018, 16, 823–836.e2. [Google Scholar] [CrossRef]
- Ricaño-Ponce, I.; Gutierrez-Achury, J.; Costa, A.F.; Deelen, P.; Kurilshikov, A.; Zorro, M.M.; Platteel, M.; van der Graaf, A.; Sugai, E.; Moreno, M.L.; et al. Immunochip meta-analysis in European and Argentinian populations identifies two novel genetic loci associated with celiac disease. Eur. J. Hum. Genet. 2020, 28, 313–323. [Google Scholar] [CrossRef]
- Trynka, G.; Hunt, K.A.; Bockett, N.A.; Romanos, J.; Mistry, V.; Szperl, A.; Bakker, S.F.; Bardella, M.T.; Bhaw-Rosun, L.; Castillejo, G.; et al. Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat. Genet. 2011, 43, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Jabri, B.; Abadie, V. IL-15 functions as a danger signal to regulate tissue-resident T cells and tissue destruction. Nat. Rev. Immunol. 2015, 15, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Bragde, H.; Jansson, U.; Jarlsfelt, I.; Söderman, J. Gene expression profiling of duodenal biopsies discriminates celiac disease mucosa from normal mucosa. Pediatr. Res. 2011, 69, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Bragde, H.; Jansson, U.; Fredrikson, M.; Grodzinsky, E.; Söderman, J. Celiac disease biomarkers identified by transcriptome analysis of small intestinal biopsies. Cell. Mol. Life Sci. 2018, 75, 4385–4401. [Google Scholar] [CrossRef]
- Loberman-Nachum, N.; Sosnovski, K.; Di Segni, A.; Efroni, G.; Braun, T.; BenShoshan, M.; Anafi, L.; Avivi, C.; Barshack, I.; Shouval, D.S.; et al. Defining the Celiac Disease Transcriptome using Clinical Pathology Specimens Reveals Biologic Pathways and Supports Diagnosis. Sci. Rep. 2019, 9, 16163. [Google Scholar] [CrossRef]
- van der Graaf, A.; Zorro, M.; Claringbould, A.; Vosa, U.; Aguirre-Gamboa, R.; Li, C.; Mooiweer, J.; Ricano-Ponce, I.; Borek, Z.; Koning, F.; et al. Systematic prioritization of candidate genes in disease loci identifies TRAFD1 as a master regulator of IFNγ signalling in celiac disease. Front. Genet. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Chen, P.; Liao, K.; Xiao, C. MicroRNA says no to mass production. Nat. Immunol. 2018, 19, 1040–1042. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef]
- Ramírez-sánchez, A.D.; Tan, I.L.; Gonera-De Jong, B.C.; Visschedijk, M.C.; Jonkers, I.; Withoff, S. Molecular biomarkers for celiac disease: Past, present and future. Int. J. Mol. Sci. 2020, 21, 8528. [Google Scholar] [CrossRef]
- Capuano, M.; Iaffaldano, L.; Tinto, N.; Montanaro, D.; Capobianco, V.; Izzo, V.; Tucci, F.; Troncone, G.; Greco, L.; Sacchetti, L. MicroRNA-449a overexpression, reduced NOTCH1 signals and scarce goblet cells characterize the small intestine of celiac patients. PLoS ONE 2011, 6, e29094. [Google Scholar] [CrossRef] [PubMed]
- Magni, S.; Comani, G.B.; Elli, L.; Vanessi, S.; Ballarini, E.; Nicolini, G.; Rusconi, M.; Castoldi, M.; Meneveri, R.; Muckenthaler, M.U.; et al. MIRNAs affect the expression of innate and adaptive immunity proteins in celiac disease. Am. J. Gastroenterol. 2014, 109, 1662. [Google Scholar] [CrossRef] [PubMed]
- Buoli Comani, G.; Panceri, R.; Dinelli, M.; Biondi, A.; Mancuso, C.; Meneveri, R.; Barisani, D. miRNA-regulated gene expression differs in celiac disease patients according to the age of presentation. Genes Nutr. 2015, 10, 482. [Google Scholar] [CrossRef]
- Bascuñán, K.A.; Pérez-Bravo, F.; Gaudioso, G.; Vaira, V.; Roncoroni, L.; Elli, L.; Monguzzi, E.; Araya, M. A miRNA-Based Blood and Mucosal Approach for Detecting and Monitoring Celiac Disease. Dig. Dis. Sci. 2019, 65, 1982–1991. [Google Scholar] [CrossRef] [PubMed]
- Comincini, S.; Manai, F.; Meazza, C.; Pagani, S.; Martinelli, C.; Pasqua, N.; Pelizzo, G.; Biggiogera, M.; Bozzola, M. Identification of autophagy-related genes and their regulatory miRNAs associated with celiac disease in children. Int. J. Mol. Sci. 2017, 18, 391. [Google Scholar] [CrossRef] [PubMed]
- Hsin, J.P.; Lu, Y.; Loeb, G.B.; Leslie, C.S.; Rudensky, A.Y. The effect of cellular context on miR-155-mediated gene regulation in four major immune cell types. Nat. Immunol. 2018, 19, 1137–1145. [Google Scholar] [CrossRef]
- Bracken, C.P.; Scott, H.S.; Goodall, G.J. A network-biology perspective of microRNA function and dysfunction in cancer. Nat. Rev. Genet. 2016, 17, 719–732. [Google Scholar] [CrossRef]
- Vaira, V.; Roncoroni, L.; Barisani, D.; Gaudioso, G.; Bosari, S.; Bulfamante, G.; Doneda, L.; Conte, D.; Tomba, C.; Bardella, M.T.; et al. microRNA profiles in coeliac patients distinguish different clinical phenotypes and are modulated by gliadin peptides in primary duodenal fibroblasts. Clin. Sci. 2014, 126, 417–423. [Google Scholar] [CrossRef]
- Huffaker, T.B.; Hu, R.; Runtsch, M.C.; Bake, E.; Chen, X.; Zhao, J.; Round, J.L.; Baltimore, D.; O’Connell, R.M. Epistasis between MicroRNAs 155 and 146a during T Cell-Mediated Antitumor Immunity. Cell Rep. 2012, 2, 1697–1709. [Google Scholar] [CrossRef]
- Torri, A.; Carpi, D.; Bulgheroni, E.; Crosti, M.C.; Moro, M.; Gruarin, P.; Rossi, R.L.; Rossetti, G.; Vizio, D.D.; Hoxha, M.; et al. Extracellular MicroRNA signature of human helper T cell subsets in health and autoimmunity. J. Biol. Chem. 2017, 292, 2903–2915. [Google Scholar] [CrossRef]
- Gracias, D.T.; Stelekati, E.; Hope, J.L.; Boesteanu, A.C.; Doering, T.A.; Norton, J.; Mueller, Y.M.; Fraietta, J.A.; Wherry, E.J.; Turner, M.; et al. The microRNA miR-155 controls CD8+ T cell responses by regulating interferon signaling. Nat. Immunol. 2013, 14, 593–602. [Google Scholar] [CrossRef]
- Singh, U.P.; Murphy, A.E.; Enos, R.T.; Shamran, H.A.; Singh, N.P.; Guan, H.; Hegde, V.L.; Fan, D.; Price, R.L.; Taub, D.D.; et al. miR-155 deficiency protects mice from experimental colitis by reducing T helper type 1/type 17 responses. Immunology 2014, 143, 478–489. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Iny Stein, T.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards suite: From gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinform. 2016, 2016, 1.30.1–1.30.33. [Google Scholar] [CrossRef] [PubMed]
- De Rie, D.; Abugessaisa, I.; Alam, T.; Arner, E.; Arner, P.; Ashoor, H.; Åström, G.; Babina, M.; Bertin, N.; Burroughs, A.M.; et al. An integrated expression atlas of miRNAs and their promoters in human and mouse. Nat. Biotechnol. 2017, 35, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Iversen, R.; Roy, B.; Stamnaes, J.; Høydahl, L.S.; Hnida, K.; Neumann, R.S.; Korponay-Szabó, I.R.; Lundin, K.E.A.; Sollid, L.M. Efficient T cell–B cell collaboration guides autoantibody epitope bias and onset of celiac disease. Proc. Natl. Acad. Sci. USA 2019, 116, 15134–15139. [Google Scholar] [CrossRef] [PubMed]
- Jabri, B.; Sollid, L.M. T Cells in Celiac Disease. J. Immunol. 2017, 198, 3005–3014. [Google Scholar] [CrossRef]
- Abadie, V.; Kim, S.M.; Lejeune, T.; Palanski, B.A.; Ernest, J.D.; Tastet, O.; Voisine, J.; Discepolo, V.; Marietta, E.V.; Hawash, M.B.F.; et al. IL-15, gluten and HLA-DQ8 drive tissue destruction in coeliac disease. Nature 2020, 578, 600–604. [Google Scholar] [CrossRef]
- Briskin, D.; Wang, P.Y.; Bartel, D.P. The biochemical basis for the cooperative action of microRNAs. Proc. Natl. Acad. Sci. USA 2020, 117, 17764–17774. [Google Scholar] [CrossRef]
- Gil-Zamorano, J.; Tomé-Carneiro, J.; Lopez de las Hazas, M.C.; del Pozo-Acebo, L.; Crespo, M.C.; Gómez-Coronado, D.; Chapado, L.A.; Herrera, E.; Latasa, M.J.; Ruiz-Roso, M.B.; et al. Intestinal miRNAs regulated in response to dietary lipids. Sci. Rep. 2020, 10, 18921. [Google Scholar] [CrossRef]
- McKenna, L.B.; Schug, J.; Vourekas, A.; McKenna, J.B.; Bramswig, N.C.; Friedman, J.R.; Kaestner, K.H. MicroRNAs control intestinal epithelial differentiation, architecture, and barrier function. Gastroenterology 2010, 139, 1654–1664.e1. [Google Scholar] [CrossRef] [PubMed]
- Beumer, J.; Clevers, H. Cell fate specification and differentiation in the adult mammalian intestine. Nat. Rev. Mol. Cell Biol. 2021, 22, 39–53. [Google Scholar] [CrossRef]
- Wang, B.; Rong, X.; Palladino, E.N.D.; Wang, J.; Fogelman, A.M.; Martín, M.G.; Alrefai, W.A.; Ford, D.A.; Tontonoz, P. Phospholipid Remodeling and Cholesterol Availability Regulate Intestinal Stemness and Tumorigenesis. Cell Stem Cell 2018, 22, 206–220.e4. [Google Scholar] [CrossRef] [PubMed]
- Stine, R.R.; Sakers, A.P.; TeSlaa, T.; Kissig, M.; Stine, Z.E.; Kwon, C.W.; Cheng, L.; Lim, H.W.; Kaestner, K.H.; Rabinowitz, J.D.; et al. PRDM16 Maintains Homeostasis of the Intestinal Epithelium by Controlling Region-Specific Metabolism. Cell Stem Cell 2019, 25, 830–845.e8. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Ma, X.; Lv, C.; Sheng, X.; Li, X.; Zhao, R.; Song, Y.; Andl, T.; Plikus, M.V.; Sun, J.; et al. Stress responsive miR-31 is a major modulator of mouse intestinal stem cells during regeneration and tumorigenesis. Elife 2017, 6, e29538. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Xu, J.; Li, Y.; Zhao, R.; Du, S.; Lv, C.; Wu, W.; Liu, R.; Sheng, X.; Song, Y.; et al. MicroRNA-31 Reduces Inflammatory Signaling and Promotes Regeneration in Colon Epithelium, and Delivery of Mimics in Microspheres Reduces Colitis in Mice. Gastroenterology 2019, 156, 2281–2296.e6. [Google Scholar] [CrossRef]
- Lukonin, I.; Serra, D.; Challet Meylan, L.; Volkmann, K.; Baaten, J.; Zhao, R.; Meeusen, S.; Colman, K.; Maurer, F.; Stadler, M.B.; et al. Phenotypic landscape of intestinal organoid regeneration. Nature 2020, 586, 275–280. [Google Scholar] [CrossRef]
- Liu, J.Y.; Seno, H.; Miletic, A.V.; Mills, J.C.; Swat, W.; Stappenbeck, T.S. Vav proteins are necessary for correct differentiation of mouse cecal and colonic enterocytes. J. Cell Sci. 2009, 122, 324–334. [Google Scholar] [CrossRef][Green Version]
- Latreille, R.; Servant, R.; Darsigny, M.; Marcoux, S.; Jones, C.; Perreault, N.; Boudreau, F. Transcription factor CUX1 is required for intestinal epithelial wound healing and targets the VAV2-RAC1 Signalling complex. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2347–2355. [Google Scholar] [CrossRef]
- Postema, M.M.; Grega-Larson, N.E.; Meenderink, L.M.; Tyska, M.J. PACSIN2-dependent apical endocytosis regulates the morphology of epithelial microvilli. Mol. Biol. Cell 2019, 30, 2515–2526. [Google Scholar] [CrossRef] [PubMed]
- Moffett, H.F.; Cartwright, A.N.R.; Kim, H.J.; Godec, J.; Pyrdol, J.; Äijö, T.; Martinez, G.J.; Rao, A.; Lu, J.; Golub, T.R.; et al. The microRNA miR-31 inhibits CD8+ T cell function in chronic viral infection. Nat. Immunol. 2017, 18, 791–799. [Google Scholar] [CrossRef]
- Ferguson, B.J.; Mansur, D.S.; Peters, N.E.; Ren, H.; Smith, G.L. DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. eLife 2012, 1, e00047. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Ban, T.; Wang, Z.; Choi, M.K.; Kawamura, T.; Negishi, H.; Nakasato, M.; Lu, Y.; Hangai, S.; Koshiba, R.; et al. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature 2009, 462, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Palone, F.; Vitali, R.; Trovato, C.M.; Montuori, M.; Negroni, A.; Mallardo, S.; Stronati, L. Faecal high mobility group box 1 in children with celiac disease: A pilot study. Dig. Liver Dis. 2018, 50, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Manti, S.; Cuppari, C.; Tardino, L.; Parisi, G.; Spina, M.; Salpietro, C.; Leonardi, S. HMGB1 as a new biomarker of celiac disease in children: A multicenter study. Nutrition 2017, 37, 18–21. [Google Scholar] [CrossRef] [PubMed]
- Boyapati, R.K.; Rossi, A.G.; Satsangi, J.; Ho, G.T. Gut mucosal DAMPs in IBD: From mechanisms to therapeutic implications. Mucosal Immunol. 2016, 9, 567–582. [Google Scholar] [CrossRef]
- Wang, Y.B.; Tan, B.; Mu, R.; Chang, Y.; Wu, M.; Tu, H.Q.; Zhang, Y.C.; Guo, S.S.; Qin, X.H.; Li, T.; et al. Ubiquitin-associated domain-containing ubiquitin regulatory X (UBX) protein UBXN1 is a negative regulator of nuclear factor κB (NF-κB) signaling. J. Biol. Chem. 2015, 290, 10395–10405. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Yang, L.; Cheng, G.; Yang, G.; Xu, Z.; You, F.; Sun, Q.; Lin, R.; Fikrig, E.; Sutton, R.E. UBXN1 Interferes with Rig-I-like Receptor-Mediated Antiviral Immune Response by Targeting MAVS. Cell Rep. 2013, 3, 1057–1070. [Google Scholar] [CrossRef]
- Koizumi, S.I.; Sasaki, D.; Hsieh, T.H.; Taira, N.; Arakaki, N.; Yamasaki, S.; Wang, K.; Sarkar, S.; Shirahata, H.; Miyagi, M.; et al. JunB regulates homeostasis and suppressive functions of effector regulatory T cells. Nat. Commun. 2018, 9, 5344. [Google Scholar] [CrossRef]
- Wheaton, J.D.; Ciofani, M. JunB Controls Intestinal Effector Programs in Regulatory T Cells. Front. Immunol. 2020, 11, 44. [Google Scholar] [CrossRef]
- Polioudakis, D.; Bhinge, A.A.; Killion, P.J.; Lee, B.K.; Abell, N.S.; Iyer, V.R. A Myc-microRNA network promotes exit from quiescence by suppressing the interferon response and cell-cycle arrest genes. Nucleic Acids Res. 2013, 41, 2239–2254. [Google Scholar] [CrossRef]
- Alsaleh, G.; François, A.; Philippe, L.; Gong, Y.Z.; Bahram, S.; Cetin, S.; Pfeffer, S.; Gottenberg, J.E.; Wachsmann, D.; Georgel, P.; et al. MiR-30a-3p negatively regulates BAFF synthesis in systemic sclerosis and rheumatoid arthritis fibroblasts. PLoS ONE 2014, 9, e111266. [Google Scholar] [CrossRef]
- de Almeida, R.C.; Chagas, V.S.; Castro, M.A.A.; Petzl-Erler, M.L. Integrative analysis identifies genetic variants associated with autoimmune diseases affecting putative microRNA binding sites. Front. Genet. 2018, 9, 139. [Google Scholar] [CrossRef]
- Kern, F.; Backes, C.; Hirsch, P.; Fehlmann, T.; Hart, M.; Meese, E.; Keller, A. What’s the target: Understanding two decades of in silico microRNA-target prediction. Brief. Bioinform. 2020, 21, 1999–2010. [Google Scholar] [CrossRef]
- Mittelbrunn, M.; Gutiérrez-Vázquez, C.; Villarroya-Beltri, C.; González, S.; Sánchez-Cabo, F.; González, M.Á.; Bernad, A.; Sánchez-Madrid, F. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat. Commun. 2011, 2, 282. [Google Scholar] [CrossRef]
- Montecalvo, A.; Larregina, A.T.; Shufesky, W.J.; Stolz, D.B.; Sullivan, M.L.G.; Karlsson, J.M.; Baty, C.J.; Gibson, G.A.; Erdos, G.; Wang, Z.; et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood 2012, 119, 756–766. [Google Scholar] [CrossRef] [PubMed]
- Moerkens, R.; Mooiweer, J.; Withoff, S.; Wijmenga, C. Celiac disease-on-chip: Modeling a multifactorial disease in vitro. United Eur. Gastroenterol. J. 2019, 7, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, G.; Carissimo, A.; Chen, A.; Cutillo, L.; Nowakowski, T.J.; Di Bernardo, D.; Blelloch, R. The impact of microRNAs on transcriptional heterogeneity and gene co-expression across single embryonic stem cells. Nat. Commun. 2017, 8, 14126. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zheng, J.; Chen, Z.; Liu, Y.; Dura, B.; Kwak, M.; Xavier-Ferrucio, J.; Lu, Y.C.; Zhang, M.; Roden, C.; et al. Single-cell microRNA-mRNA co-sequencing reveals non-genetic heterogeneity and mechanisms of microRNA regulation. Nat. Commun. 2019, 10, 95. [Google Scholar] [CrossRef] [PubMed]
- Amr, K.S.; Bayoumi, F.S.; Eissa, E.; Abu-Zekry, M. Circulating microRNAs as potential non-invasive biomarkers in pediatric patients with celiac disease. Eur. Ann. Allergy Clin. Immunol. 2019, 51, 159–164. [Google Scholar] [CrossRef]
- Zorro, M.M.; Aguirre-Gamboa, R.; Mayassi, T.; Ciszewski, C.; Barisani, D.; Hu, S.; Weersma, R.K.; Withoff, S.; Li, Y.; Wijmenga, C.; et al. Tissue alarmins and adaptive cytokine induce dynamic and distinct transcriptional responses in tissue-resident intraepithelial cytotoxic T lymphocytes. J. Autoimmun. 2020, 108, 102422. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Paraskevopoulou, M.D.; Georgakilas, G.; Kostoulas, N.; Vlachos, I.S.; Vergoulis, T.; Reczko, M.; Filippidis, C.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-microT web server v5.0: Service integration into miRNA functional analysis workflows. Nucleic Acids Res. 2013, 41, W169–W173. [Google Scholar] [CrossRef] [PubMed]
- Karagkouni, D.; Paraskevopoulou, M.D.; Chatzopoulos, S.; Vlachos, I.S.; Tastsoglou, S.; Kanellos, I.; Papadimitriou, D.; Kavakiotis, I.; Maniou, S.; Skoufos, G.; et al. DIANA-TarBase v8: A decade-long collection of experimentally supported miRNA-gene interactions. Nucleic Acids Res. 2018, 46, D239–D245. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.H.; Chang, N.W.; Shrestha, S.; Hsu, S.D.; Lin, Y.L.; Lee, W.H.; Yang, C.D.; Hong, H.C.; Wei, T.Y.; Tu, S.J.; et al. miRTarBase 2016: Updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2016, 44, D239–D247. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.A.A.; Wang, X.; Fletcher, M.N.C.; Meyer, K.B.; Markowetz, F. RedeR: R/Bioconductor package for representing modular structures, nested networks and multiple levels of hierarchical associations. Genome Biol. 2012, 13, R29. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. Omi. J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. Revigo summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed]
- Aran, D. Cell-type enrichment analysis of bulk transcriptomes using xcell. In Methods in Molecular Biology; Humana: New York, NY, USA, 2020; Volume 2120, pp. 263–276. [Google Scholar]
- Kim-Hellmuth, S.; Aguet, F.; Oliva, M.; Muñoz-Aguirre, M.; Kasela, S.; Wucher, V.; Castel, S.E.; Hamel, A.R.; Viñuela, A.; Roberts, A.L.; et al. Cell type–specific genetic regulation of gene expression across human tissues. Science 2020, 369, eaaz8528. [Google Scholar] [CrossRef] [PubMed]
- Marderstein, A.R.; Uppal, M.; Verma, A.; Bhinder, B.; Tayyebi, Z.; Mezey, J.; Clark, A.G.; Elemento, O. Demographic and genetic factors influence the abundance of infiltrating immune cells in human tissues. Nat. Commun. 2020, 11, 2213–2214. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, I.L.; Barisani, D.; Panceri, R.; Modderman, R.; Visschedijk, M.; Weersma, R.K.; Wijmenga, C.; Jonkers, I.; Coutinho de Almeida, R.; Withoff, S. A Combined mRNA- and miRNA-Sequencing Approach Reveals miRNAs as Potential Regulators of the Small Intestinal Transcriptome in Celiac Disease. Int. J. Mol. Sci. 2021, 22, 11382. https://doi.org/10.3390/ijms222111382
Tan IL, Barisani D, Panceri R, Modderman R, Visschedijk M, Weersma RK, Wijmenga C, Jonkers I, Coutinho de Almeida R, Withoff S. A Combined mRNA- and miRNA-Sequencing Approach Reveals miRNAs as Potential Regulators of the Small Intestinal Transcriptome in Celiac Disease. International Journal of Molecular Sciences. 2021; 22(21):11382. https://doi.org/10.3390/ijms222111382
Chicago/Turabian StyleTan, Ineke Luise, Donatella Barisani, Roberto Panceri, Rutger Modderman, Marijn Visschedijk, Rinse K. Weersma, Cisca Wijmenga, Iris Jonkers, Rodrigo Coutinho de Almeida, and Sebo Withoff. 2021. "A Combined mRNA- and miRNA-Sequencing Approach Reveals miRNAs as Potential Regulators of the Small Intestinal Transcriptome in Celiac Disease" International Journal of Molecular Sciences 22, no. 21: 11382. https://doi.org/10.3390/ijms222111382
APA StyleTan, I. L., Barisani, D., Panceri, R., Modderman, R., Visschedijk, M., Weersma, R. K., Wijmenga, C., Jonkers, I., Coutinho de Almeida, R., & Withoff, S. (2021). A Combined mRNA- and miRNA-Sequencing Approach Reveals miRNAs as Potential Regulators of the Small Intestinal Transcriptome in Celiac Disease. International Journal of Molecular Sciences, 22(21), 11382. https://doi.org/10.3390/ijms222111382