Abstract
Ulcerative colitis (UC) is a chronic autoimmune disorder affecting the colonic mucosa. UC is a subtype of inflammatory bowel disease along with Crohn’s disease and presents with varying extraintestinal manifestations. No single etiology for UC has been found, but a combination of genetic and environmental factors is suspected. Research has focused on the role of intestinal dysbiosis in the pathogenesis of UC, including the effects of dysbiosis on the integrity of the colonic mucosal barrier, priming and regulation of the host immune system, chronic inflammation, and progression to tumorigenesis. Characterization of key microbial taxa and their implications in the pathogenesis of UC and colitis-associated cancer (CAC) may present opportunities for modulating intestinal inflammation through microbial-targeted therapies. In this review, we discuss the microbiota-immune crosstalk in UC and CAC, as well as the evolution of microbiota-based therapies.
1. Background
Inflammatory bowel disease (IBD) is a chronic autoimmune condition affecting the gastrointestinal (GI) tract. It comprises Crohn’s disease (CD) and ulcerative colitis (UC), and generally presents as a progressive inflammatory condition. UC is characterized by inflammation of colonic mucosa and submucosa starting at the rectum and extending through the colon. Typical symptoms of UC flares include abdominal pain, hematochezia, tenesmus, and loose stools. Extraintestinal manifestations may also present, including ocular pathologies, arthropathies, liver disease such as primary sclerosing cholangitis, and dermatological manifestations [1].
Various genetic and environmental factors have been implicated in UC susceptibility [2]. To date, over 200 single nucleotide polymorphisms (SNPs) have been associated with the risk of developing UC [3]. Epidemiological studies have shown a higher incidence of UC among populations adopting Western diets rich in refined sugars, dairy, protein, and animal fat, and low in dietary fiber including wholegrains, fruits, and vegetables [4]. The role of environmental influences aligns with the hygiene hypothesis, which states that limited exposure to microorganisms during infancy and childhood may impair appropriate priming and development of the immune system, thus promoting autoimmunity [5]. Exposure to antibiotics during gestation and childhood, psychological stress, and family history also affect the risk of developing UC [6]. These factors profoundly alter the intestinal microbiome but may also provide opportunities for new treatment options.
The extent and duration of UC disease activity is associated with an increased risk of neoplasia [7]. The risk of developing colitis-associated cancer (CAC) begins increasing 8 to 10 years after UC diagnosis [8]. Previous studies have estimated a risk of 2% by 10 years, 8% by 20 years, and 18% by 30 years [7]. Other studies have shown that while sporadic colorectal cancer (CRC) affects 1–2% of the general population, over 13% of patients with UC will develop CAC [9]. This corresponds to a 4 to 10-fold increased incidence as compared with sporadic CRC [8]. Sex differences have also been reported, with higher CAC prevalence and mortality rates observed among male patients [10]. The relationship between UC and CAC has influenced the development of clinical practice guidelines, with increased endoscopic surveillance recommended among UC patients starting 8 years after initial UC diagnosis. These recommendations have led to successful reductions in CAC morbidity and mortality [11].
The mechanisms underpinning UC pathogenesis remain unclear, but the dominant hypothesis suggests that environmental factors, including alterations in intestinal microbiota, contribute to an exaggerated immune response and chronic inflammation in genetically susceptible individuals [12]. Conventional treatments for UC have largely relied on dampening the immune response to induce disease remission and promote mucosal healing [13]. Pharmacotherapies such as corticosteroids and disease modifying anti-rheumatic drugs remain the dominant treatment paradigm; however, these medications have significant side effect profiles and may induce immune tolerance with long term use. These medications are also associated with significant healthcare costs, particularly newer biological therapies which require ongoing dosing. Emerging therapies have focused on the potential benefits of microbiota-targeted alternatives, including prebiotics, probiotics, synbiotics, antibiotics, and fecal microbiota transplantation (FMT). This review will discuss key changes in intestinal microbiota associated with UC pathogenesis and immune dysfunction, as well as the role of microbiota-based therapies in affecting intestinal inflammation and progression to neoplasia.
2. Microbiome-Immune Interactions in UC
2.1. Immune System Perturbations in UC
Perturbations in intestinal microbiota and immune dysregulation are key features of UC pathogenesis (Figure 1). Colonization is largely believed to commence during parturition, although limited evidence suggests that some microbial cells might be present in utero during the prenatal period [14]. The largest contributors to intestinal microbiota composition constitute mode of childbirth and feeding during infancy. Subsequent expansion and diversification of the intestinal microbiome continues throughout childhood and adolescence until a relatively stable composition is achieved in adulthood [15].
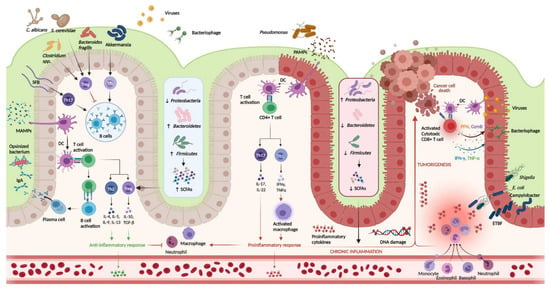
Figure 1.
Host-immune interactions in ulcerative colitis. Intestinal microbiota interact with the immune system through various pathways. In the healthy colon, DCs sample MAMPs and present antigens on major histocompatibility complex class II to naive CD4+ T cells. Naive CD4+ T cells become activated and differentiate into various T cell subtypes depending on the presence of specific cytokines within the local microenvironment. Anti-inflammatory Th subtypes comprise Th2 and Treg cells. CD4+ T cells also activate plasma cells which secrete immunoglobulin A (IgA) which is essential for microbial opsonization. Proinflammatory Th subtypes consist of Th1 cells and Th17 cells, which are upregulated in the diseased colon via interactions between DCs and PAMPs. Chronic inflammation contributes to DNA damage and tumorigenesis. Invading viruses stimulate CD8+ cytotoxic T cell activation via antigen-MHC I interactions. However, CD8+ T cells can also assist in cancer cell death. Disruptions in the mucosal barrier provides avenues for microbial translocation, including ETBF, which has been implicated in colitis-associated cancer. Finally, the production of SCFA is increased in the healthy colon (mediated by increased density of Firmicutes and Bacteroidetes phyla), while increased density of the Proteobacterium phylum is associated with lower concentrations of SCFA and colonic inflammation. DC, dendritic cell; DNA, deoxyribonucleic acid; ETBF, enterotoxigenic Bacteroides fragilis; IFN-γ, interferon-gamma; IgA, immunoglobulin A; MAMPs, microbe-associated molecular patterns; PAMPs, pathogen-associated molecular patterns; SCFAs, short chain fatty acids; SFB, segmented filamentous bacteria; Th, T helper; Treg, T regulatory; TNF-α, tumor necrosis factor-alpha. Created in Biorender.com (accessed: 1 August 2021) [16].
Early life may be considered a common denominator between intestinal microbiota development and susceptibility to UC, as perturbations in early microbial colonization such as caesarean section delivery, dietary changes, exposure to antibiotics, systemic stressors, and infection constitute the same environmental factors associated with the risk of developing UC [17,18,19].
The microbiome plays an important role in maintaining intestinal homeostasis by training the innate and adaptive immune systems to tolerate commensal microbes, while offering protection against harmful pathogens [20,21,22]. Tolerance towards commensal microorganisms is mediated via: (1) reducing contact between luminal microbes and the intestinal mucosa through physical barriers [23], and (2) development of immune hyporesponsiveness [24].
The intestinal mucosal barrier serves as the first line of defense against bacterial translocation into systemic circulation and is composed of physical and immunological elements working together to maintain intestinal health. Alterations in the physiological composition of gut microbes in early life disrupt tolerance to commensals, permit translocation of pathogens, and result in dysregulation of host immune function through various signaling cascades [25]. Microbial dysbiosis, intestinal barrier defects, and alterations in mucin secretion may occur even in the absence of active inflammation, including outside of the colon in UC. This suggests that disruptions to normal intestinal physiology are primary contributors to UC pathogenesis and likely predate inflammation [26].
2.1.1. Physical Barrier
A mucus blanket composed of heavily glycosylated mucins serves as the first physical element of the intestinal mucosal barrier. Mucins may be membrane tethered, secretory, or non-gel forming. Their production and secretion are principally mediated by goblet cells and may be influenced by nonspecific factors such as immune system interactions with microbiota and dietary factors, and specific modulators including epigenetics and transcriptional factors [27]. Among the various pathogen recognition receptor (PRR) ligands, toll-like receptor (TLR) ligands serve as particularly powerful stimuli for goblet cell production of mucins [28]. Intestinal microorganisms synthesize a variety of conserved structural components which act as ligands for PRRs termed microbe-associated molecular patterns (MAMPs), which are expressed by commensals and enteropathogens. In the context of pathobionts, MAMPs are typically referred to as pathogen-associated molecular patterns (PAMPs) [29]. Gram-negative bacteria such as Escherichia coli and Pseudomonas aeruginosa produce PAMPs, lipopolysaccharide (LPS) and flagellin, which bind TLR4 and TLR5, respectively, to alter mucin production and activate inflammatory pathways such as the nuclear factor-κB (NF-κB) cascade. While goblet cells are found throughout the GI tract, they are most concentrated in the colon and rectum where they form a thick mucin bilayer [24,30]. Notably, this increasing density gradient of goblet cells correlates with the density and diversity of gut microbes from proximal to distal aspects of the GI tract [31].
The mucous bilayer in the colon consists of a loosely arranged outer layer (ranging in thickness from 100 to 400 µm in the small bowel, to ~700 µm in the colon) which interacts with microbes, and a dense, impenetrable inner layer (ranging in thickness from 15 to 30 µm in the small bowel, to ∼100 µm in the colon) rich in antimicrobial peptides [27,32]. This mucin meshwork allows for selective diffusion of nutrients and oxygen while limiting microbial contact with the underlying epithelium. Glycosylation of mucins is essential for maintaining intestinal homeostasis and involves either O-glycosylation or N-glycosylation. O-glycans act as important food sources for intestinal microbiota, while N-glycans maintain the mucosal barrier. Together, these carbohydrate moieties influence the composition of the intestinal microbiota and protect against intestinal inflammation and disease susceptibility [27]. For example, increased glycosylation of N-glycans via overexpression of the enzyme β-1,4-galactosyltransferase I (βGalT1) results in a higher Firmicutes to Bacteroidetes ratio, protection against tumor necrosis factor-α (TNF-α) induced inflammation, and decreased susceptibility to dextran sulfate sodium (DSS)-induced colitis [33]. In contrast, reductions in goblet cell densities [34], alterations in mucin production, and discontinuity of the mucous blanket layer have been implicated in UC pathophysiology. Specifically, reduced expression of MUC9 and MUC20, and increases in MUCH16 have been reported across UC patients irrespective of disease activity, while increases in MUC1 and decreases in MUC2 expression appear to be limited to regions of ulceration [1,35,36]. Decreases in mucin glycosylation and sulphation and increases in sialylation impair barrier function and are well described features of UC [37].
Below the mucin layer, the GI tract is lined by a monolayer of intestinal epithelial cells (IECs) connected via junctional complexes, forming villi and crypts. The IECs form the largest physical barrier of the GI tract and are the strongest determinants of protection against the external environment. They physically separate the products of the intestinal lumen from the underlying lamina propria, thereby maintaining intestinal homeostasis. The junctional complexes which connect the IECs are vital in regulating selective transportation of water and nutrients and preventing penetration of the intestinal mucosa by commensals and enteropathogens [38]. These protein complexes are composed of tight junctions, adherens junctions, and desmosomes. The IECs comprise five distinct cell types, including enterocytes, enteroendocrine cells, tuft cells, Paneth cells, and microfold (M) cells [28], which are regenerated by pluripotent stem cells residing within the intestinal crypts [39]. While IECs exhibit primarily protective functions, defects in this barrier layer have been associated with increased susceptibility to gastrointestinal disease. For example, alterations in deoxyribonucleic acid (DNA) methylation and transcriptome patterns have been implicated in UC pathogenesis. Several of the affected pathways include innate immune system function including cytokine signaling and complement activation, as well as extracellular matrix composition including collagen, laminin, and fibril synthesis and degradation [40]. Many of these epigenetic alterations in methylation patterns appear to be independent of microscopic mucosal inflammation and remain stable over time in UC patients. IECs harvested from inflamed mucosa of UC patients exhibit alterations in molecular signaling cascades, including enhanced Notch signaling and TNF-α induced NF-κB signaling [41]. Furthermore, IECs harvested from patients with active UC exhibit higher apoptotic indices which contributes to impaired barrier function and permits translocation of commensal and enteropathogenic microorganisms, resulting in higher levels of proinflammatory cytokines including TNF-α [42]. Increases in TNF-α result in impairment of the mucosal barrier by inducing caspase-dependent apoptosis and caspase-independent necroptosis of multiple IECs [43]. This, in part, explains the therapeutic success of antibodies targeting TNF-α in select patients. However, a subgroup of patients demonstrates little to no response despite adequate dosing and duration of anti-TNF-α treatment, suggesting that intestinal inflammation independent of TNF-α signaling may be involved in certain subgroups of UC patients [44].
2.1.2. Immunoglobulin A
Within the mucus layer reside additional components of the host defense system including antibacterial peptides and secretory immunoglobulin-A (IgA). The gut mucosa harbors the largest concentration of IgA in the human body, which can be produced in a T-cell dependent or T-cell independent manner [45]. Plasma cells within the lamina propria produce dimeric IgA which is shuttled from the basolateral membrane to the apical surface of IECs via the polymeric immunoglobulin receptor (pIgR) [46]. At the apical surface of IECs, the pIgR-Ig complex is cleaved to produce secretory IgA. Once secreted, IgA can mediate its physiological functions including neutralizing bacterial toxins, inhibiting epithelial translocation of PAMPs such as Shigella LPS, coating microorganisms to reduce their immunogenicity, and facilitating the uptake of organisms (such as non-invasive Salmonella) to stimulate stronger adaptive immune responses [47]. Secretory IgA is essential for protecting against microbial invasion, influencing the composition of intestinal microbiota and protecting against intestinal inflammation [46].
The expression of IgA and pIgR can be altered by the intestinal microbiota. Upregulation can be achieved via activation of the NF-κB signaling cascade through commensal bacteria including Bacteroides thetaiotaomicron and certain strains belonging to the Enterobacteriaceae family [48,49]. This upregulation is presumably mediated via direct interactions between commensal MAMPs and TLRs, which stimulate myeloid differentiation factor 88 (MyD88) signaling and increase transcription of pIgR [50]. While proinflammatory cytokines such as interferon (IFN)-γ, TNF-α, interleukin (IL)-1, and IL-4 induce pIgR transcription, paradoxically, intestinal inflammation associated with UC causes downregulation of pIgR expression by IECs [51]. In addition to downregulating pIgR expression, UC is associated with lower concentrations of secretory IgA in the intestinal lumen, higher concentrations of IgA in the serum, decreased transcytosis of dimeric IgA across IECs, and accumulation of IgA within the lamina propria [51].
Crosslinking of IgA with its cognate transmembrane receptor on neutrophils, i.e., FcαRI, stimulates neutrophil recruitment to inflamed tissues and stimulates the release of leukotriene B4 (LTB4), a potent neutrophil chemoattractant [52]. In this manner, a sustained inflammatory loop can be maintained leading to excessive tissue damage. In addition to increased IgA–FcαRI interactions, UC disease activity is also associated with increased neutrophil uptake of IgA-opsonized bacteria within the intestinal mucosa [52]. This contributes to lower concentrations of IgA within the intestinal lumen, diminished immune protection against enteropathogenic invasion, increasing patient susceptibility to inflammation mediated by microbes, and worsened disease activity. Downregulation of pIgR and somatic mutations in IL-17 signaling have been reported in sporadic CRC, which may be driven by particular members of colonic microbiota [53,54]. The influence of microbiota on tumorigenesis is discussed further below.
2.1.3. Innate and Adaptive Immunity
Within the lamina propria are additional bacterial defenses belonging to innate and adaptive immunity. Innate immunity comprises antibacterial peptides, lysozymes, macrophages, and dendritic cells, while adaptive immunity includes T and B cells, which are concentrated within highly organized lymphoid follicles known as Peyer’s patches [55]. Dendritic cells extend their cytoplasmic projections into the intestinal lumen, where they sample intestinal contents and present antigens to T cells within the Peyer’s patches [56]. These dendritic cells are a heterogenous group of antigen-presenting cells with unique biological function that are primarily focused on maintaining a balance between proinflammatory and tolerogenic responses [57].
Genome-wide association studies have identified over 200 loci specifically associated with increased risk of developing UC [3]. Many of these genes have been implicated in innate and adaptive immune system function and impaired autophagy, including specific defects in extracellular matrix protein 1 (ECM1), IL-10, and IL-23R [58].This impaired clearance of microbes causes persistent stimulation of the innate immunity system, prolonged stimulation of the adaptive immune system and chronic inflammation [59]. Inflamed mucosa exhibits upregulation of TLR2 and TLR4 in dendritic cells, which contributes to increased expression of proinflammatory cytokine IL-12 and alterations in microbial interactions [60]. Activated dendritic cells initiate and perpetuate inflammation alone or in combination with adaptive immune cells [57]. Upregulation of IL-13 receptor subunit α-2 (IL-13Rα2) has also been described in intestinal epithelial cells during active UC, which appears to impair goblet cell function, inhibit mucosal regeneration, and alter IL-13 signaling [61]. While low levels of IL-13 are secreted by natural killer cells and macrophages in non-inflamed colonic mucosa, increased release of IL-13 by mononuclear cells in active UC has been implicated in epithelial cell apoptosis and impairment of tight junctions, subsequently producing conduits for microbial translocation and perpetuation of intestinal inflammation [62].
Commensal microorganisms also produce an abundance of PRR ligands which shape homeostatic immune function. IL-17-producing CD4+ Th17 cells are concentrated within the lamina propria and their immunomodulatory role is highly influenced by commensal bacteria, such as segmented filamentous bacteria (SFB) and Bifidobacterium adolescentis [63]. Bacteroides fragilis, which is another commensal bacterium, synthesizes a capsular polysaccharide A (PSA) with potent immunomodulatory roles. This PSA contributes to the activation of the phosphoinositide 3-kinase (PI3K) pathway and downstream cAMP response element-binding protein (CREB)-dependent transcription of anti-inflammatory genes [64]. This supports the priming of CD4+ regulatory T (Treg) cells, production of anti-inflammatory IL-10, immune system maturation, and maintenance of Th1/Th2 balance [65]. These host–microbial interactions underscore how early life exposure to microorganisms is critical for shaping host immune interactions, establishing immunoregulatory networks, and influencing susceptibility to inflammatory diseases in later life.
2.2. Intestinal Microbiota Composition in Ulcerative Colitis
The vast majority of commensal microbiota are found within the GI tract [66]. Alterations in the structure or function of one or multiple classes of microbes, a condition called microbial dysbiosis, may significantly impact host health and has been implicated in various acute and chronic intestinal disorders such as UC [67].
Gut microbes are uniquely distributed across the GI tract with abundance and composition reflecting varying physiologic conditions. Factors such as pH, luminal transit time, nutritional substrates, and mucus layer composition impact microbial colonization and proliferation [20]. Intestinal microbiota are also fundamental for nutrient extraction, complementing host metabolism, supporting host nutrition and growth, and promoting intestinal cell proliferation by providing a unique enzymatic pool to digest macromolecules derived from dietary sources. Among these, the generation of key metabolites such as short-chain fatty acids (SCFAs), vitamins (i.e., vitamin K, B12), folate and bile acids rely on bacterial metabolism [20]. Several gut microbes possess enzymatic machinery to synthesize or modify host neurotransmitters and hormones [68].
The intestinal epithelium represents a key host-microbe interface in UC. Several studies have demonstrated that the inflammatory processes triggering UC are caused by direct contact of dysbiotic microbes with the intestinal mucosa [69]. To better understand the role of the intestinal microbiota in driving inflammatory processes in UC, the bacterial taxonomic profiles and fungi of stool samples and mucosal biopsies of UC patients have been sequenced [70]. While this phylogenetic analysis presents some limitations due to the intra- and interindividual variability of intestinal microbial communities, multiple studies have reported consistent alterations in the intestinal microbiota of UC patients as compared with healthy controls (Table 1). For example, the microbiome in UC is characterized by reduced bacterial α-diversity, reflecting species richness and evenness, and β-diversity (variability) in community composition between UC and healthy subjects [71,72]. UC is associated with a decrease in the number of bacterial taxa from the Firmicutes and Bacteroidetes phyla and a significant increase in bacterial communities from the Proteobacteria phylum [71,72,73,74,75]. These changes are collectively described as a state of bacterial dysbiosis. This dysbiosis could explain the presence of inflammation in the colon of UC patients, as the increased abundance of Gram-negative taxa such as Escherichia-Shigella, Fusobacterium, Actinobacillus, Streptococcus, and Campylobacter shift the host-microbe equilibrium towards a proinflammatory phenotype, supported by evidence of altered expression of several TLRs in subjects with UC [76,77,78]. TLR4 recognizes molecular profiles derived from Gram-negative bacteria (i.e., lipopolysaccharide), thus, playing a key role in limiting their invasion when the intestinal barrier is disrupted during inflammation [75]. Conversely, the depletion of members from the Clostridiaceae family (phylum Firmicutes), such as Faecalibacterium prausnitzii and other species from the genera Clostridium, Ruminococcus, Eubacterium, Roseburia, and Akkermansia significantly lower production of butyrate, propionate, and acetate, and thus impair epithelial barrier function by reducing colonocyte proliferation and affecting Treg cells’ maturation through abnormal production of proinflammatory markers [20,71,72,79,80,81].

Table 1.
Intestinal microbiota alterations in ulcerative colitis and impacts on host immune, intestinal function.
Enterobacteriaceae (phylum Proteobacteria) uptake carbohydrates from the mucus layer, expanding their colonization and abundance while impairing mucosal integrity [20]. Increased Enterobacteriaceae and a lower concentration of Bacteroides observed in colonic or rectal UC-biopsies have been associated with inflammation severity and outcomes of relapse and remission [86]. Bacteroides suppress inflammation mediated by Th1 and Th2 immune cell activity, whereas the abnormal interaction between Enterobacteriaceae or their metabolites with the colonic epithelial cells stimulates the production of proinflammatory cytokines and induces the immune response [86]. Pathogen-induced acute enteritis has also been associated with risk of developing UC. For instance, it has been shown that specific strains of Campylobacter jejuni can cause the translocation of non-pathogenic commensal microbes across the intestinal epithelium by disrupting the integrity of the tight junctions. The passage of commensals through the intestinal barrier can increase the number of interactions between such microbes and host immune receptors, including TLRs, resulting in chronic inflammation [92].
In addition to bacterial dysbiosis, UC has also been associated with its own microbiome changes, highlighting the complexity of untangling microbial crosstalk in the pathogenesis of the disease [70,89]. This also extends to the intestinal (fungal) mycobiome. UC patients during active disease show an increase in the Basidiomycota/Ascomycota ratio as compared with those in remission and healthy controls. Sokol et al. found changes in the abundance of Saccharomyces cerevisiae and Candida albicans in stool samples from UC subjects. The authors also described the ability of Saccharomyces cerevisiae to produce anti-inflammatory IL-10, suggesting a role for this yeast in the pathogenesis of gut inflammation. Interestingly, this study reported the presence of strong correlations between fungi and bacteria only in UC and not in CD subjects, highlighting how such interkingdom interactions can enhance and contribute to the inflammatory phenotype of UC [70]. Subsequently, Qiu et al. showed an increase of Aspergillus in colonic mucosa specimens from UC subjects. Although this study did not find the same changes in the fungal population observed by Sokol et al., it reported positive correlations between Wickerhamomyces, Penicillium, and proinflammatory markers. Our knowledge of the host-fungi relationship in inflammation continues to develop [88].
A metagenomic analysis may provide more reliable information regarding the functional role of the intestinal microbiota in UC than taxonomic profiling, as the functional potential of the microbial genome is more stable and conserved [78,83]. Shotgun metagenomics have identified more than 20,000 gene families and up to 15 metabolic pathways altered in UC subjects (Table 1) [78]. UC is associated with a significant increase in protease and peptidase activity, suggesting a bacterial proteolytic signature involved in driving inflammation. Hence, elastase activity negatively correlates with beneficial bacteria such as Adlercreutzia and Akkermansia, but positively correlates with Bacteroides vulgatus, a bacterial species known for its proteolytic functional profile. These findings suggest that fecal proteolytic activity might be predictive of disease outcomes in UC [78].
Recent advances that have allowed sequencing of whole DNA of intestinal microorganisms have also facilitated the exploration of the virus kingdom within the human microbiome. In line with previous findings, UC is associated with compositional and functional changes of the mucosal virobiota [90,93]. In healthy conditions, the intestinal mucosal layer has a relatively low viral load, composed of a diverse viral population that is relatively stable over time. In contrast, UC-colonic biopsies show an expansion of viral abundance and reduced α-diversity of the viral population, which is mainly enriched by Gram-negative bacteriophages, mostly from the Caudovirales order [90]. The parallel viral and bacterial dysbiosis in UC suggest the presence of functional inter-kingdom crosstalk in sustaining inflammatory processes. The enrichment of Gram-negative bacterial taxa observed in UC could potentially stimulate the expansion of bacteriophages against such bacteria, resulting in bacteriolysis and subsequent release of PAMPs that could trigger inflammatory responses [20,90,91].
Despite recent advances in sequencing technologies, further studies are needed to elucidate the causal role of the intestinal microbiota in modulating the inflammatory processes in UC. This may occur by integrating microbiome sciences with metabolomics and epigenetics [83,94]. Understanding the contribution of each microbial kingdom to host–microbe interactions could significantly improve the management of UC and support opportunities for personalized medicine [95].
3. Therapeutic Implications of Modifying the Intestinal Microbiome in the Treatment of Ulcerative Colitis
3.1. Prebiotics
Prebiotics are defined as nonviable, nondigestive food ingredients which can increase the composition, viability, and growth of beneficial microorganisms (Figure 2) [96]. Prebiotics most commonly comprise inulin or oligosaccharides such as fructans, fructooligosaccharides, galactooligosaccharides, and trans-galactooligosaccharides. Fermentation of prebiotics by intestinal microorganisms generates SCFAs such as butyrate, acetate, and propionate, which are primary nutritional substrates for colonocytes [97]. These SCFAs also have multiple beneficial effects on immune system function and intestinal homeostasis and can act as ligands to G-protein coupled receptors which regulate diverse intestinal functions [98]. Prebiotics may also exert additional metabolic effects on metal ion absorption and fatty acid metabolism and enhance host immunity through upregulation of secretory IgA and cytokine production.
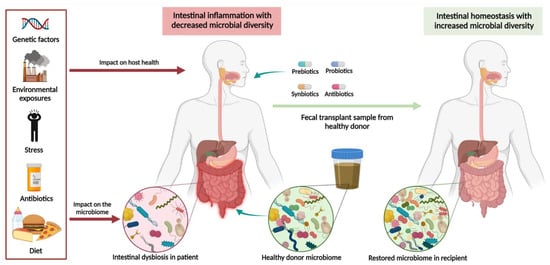
Figure 2.
Microbiota-based therapeutic approaches in ulcerative colitis. Various factors have been implicated in contributing to intestinal dysbiosis including a high-fat or low-fiber diet, exposure to antibiotics in early life, psychological stress, environmental pollutants, and genetic factors. Profound disruptions to intestinal microbiota increase an individual’s susceptibility to developing autoimmune disease, including UC. Increasing research is focusing on the role of prebiotics, probiotics, synbiotics, antibiotics, and fecal microbiota transplantation in restoring intestinal homeostasis. While antibiotic exposure in early life increases the risk of developing UC, certain classes of antibiotics may also be used as a therapy once disease is established. Created in Biorender.com (accessed: 1 August 2021) [16].
Several studies have described the role of prebiotic preparations in the management of UC. Germinated barley foodstuff high in glutamine and hemicellulose is metabolized by Eubacterium and Bifidobacterium into butyrate [99]. Butyrate, along with other SCFA, has been shown to play an important role in promoting remission in active UC and is found in significantly lower concentrations in patients with active IBD [100]. Germinated barley foodstuff has also been implicated in inhibiting inflammation mediated by cytokines IL-6, IL-8, and TNF-α, reducing C-reactive protein, and promoting mucosal regeneration [99,101]. Intake of oligofructose-enriched inulin has also been associated with reductions in fecal calprotectin [102].
3.2. Probiotics
Probiotics comprise live microorganisms that may confer important health benefits to the host when consumed [103]. Commensal bacteria found throughout the GI tract help protect against disease-causing pathogen invasion, synthesize and secrete vitamin B12 and vitamin K, promote immune system priming and maturation, and support the production of SCFA [104]. Probiotics also demonstrate antimicrobial properties, mediated through the reduction of intestinal pH via SCFA production, and downregulation of NF-κB signaling in macrophages by butyrate in UC [100,105]. Collectively, these actions reduce the expression of downstream inflammatory mediators such as TNF-α, IL-6, and IL-12. Probiotics such as Lactobacillus rhamnosus GG also appear to promote epithelial cell survival and growth by stimulating protein kinase B (PKB) and inhibiting TNF-α mediated apoptosis [106].
While several RCTs have explored the role of probiotics on inducing and maintaining remission in UC, these findings are limited by small sample size and study design. A recent meta-analysis involving 11 RCTs showed that while probiotics pose no serious adverse events for patients as compared with a placebo, there is low certainty of evidence to support their role in maintaining disease remission for UC [107].
VSL#3 and E. coli Nissle 1917 have shown the greatest promise for treating UC. VSL#3 is a probiotic cocktail which confers anti-inflammatory benefits via upregulation of IL-10, which inhibits IL-12, IFN-γ, and TNF-α [108]. VSL#3 has also been shown to promote intestinal tight junction integrity and repair of zonula occludens 1 (ZO-1) and occludin post injury via upregulation of the tyrosine-protein phosphatase non-receptor type 2 (PTPN2) gene previously shown to confer protection against IBD [109]. E. coli Nissle 1917 reduces colonic inflammation mediated by TNF-α, IL-6, IL-1β, and IL-17, and strengthens the tight junctions which connect IECs [110,111]. Recent studies have suggested that this probiotic may have the potential for toxicity, even at low doses. E. coli Nissle 1917 carries the pks pathogenicity island in its genome, which codes for a genotoxin suspected to play a role in colorectal cancer development (colibactin). While human data showing detectable levels of colibactin in recipients of E. coli Nissle 1917 are lacking, the possibility of long-term adverse effects should be considered [110].
3.3. Synbiotics
Synbiotics are defined as products containing both probiotics and prebiotics, carefully selected to enhance the viability and growth of beneficial microorganisms within the host. The synergistic actions of ingesting both products simultaneously may carry greater therapeutic potential than either product alone [13]. Synbiotic combinations most commonly include Bifidobacterium longum and Lactobacillus rhamnosus with inulin, fructooligosaccharide, and psyllium [104]. Synbiotics appear to exhibit superior abilities in promoting commensal survival and increased production of SCFAs [111].
A small number of studies have investigated the therapeutic efficacy of synbiotics in the treatment of UC [13]. Among the published data, synbiotics have been shown to decrease CRP [112,113], reduce levels of TNF-α, IL-1β, and IL-8 [114,115], and decrease symptom severity, frequency of short-term disease relapse, and increase duration of remission [115]. While these findings are encouraging, caution should be taken in the interpretation of these results due to small sample size, inconsistent dosing across various studies, and limited availability of placebo-controlled trials. Further randomized controlled trials (RCT) with larger sample sizes are required to assess the impact of synbiotics more effectively in UC treatment.
3.4. Antibiotics
Antibiotics have been included in UC therapy as adjuvants, both in the presence of active bacterial infection, and for their ability to suppress the abnormal proliferation of pathogens and stabilize the luminal and mucosal microbial load in favor of the growth of beneficial bacteria [116]. The most commonly prescribed antibiotic agents include inhibitors of cell wall biosynthesis (amoxicillin, vancomycin, and fosfomycin), and inhibitors of nucleic acids (metronidazole and rifaximin) or protein synthesis (tobramycin and vancomycin) [117,118]. Recent studies have shown that combinations of antibiotics, orally administered from 7 days to 3 months, are more effective than single agents alone for improving clinical outcomes in patients with mild to moderate UC [117,119,120,121]. When used in combinations, these drugs display a broad spectrum of action against both Gram-positive and Gram-negative bacteria, effectively targeting the majority of intestinal pathogens that have been associated with UC and modulating bacterial enzymatic activities [122].
Antibiotics also possess potent anti-inflammatory and immunomodulatory properties [116,123]. Recent studies have shown that antibiotics can prevent tissue invasion and bacterial translocation, thus limiting systemic inflammation [116,124]. This approach has been used in the treatment of pediatric acute severe colitis [120,121].
Nevertheless, the therapeutic efficacy of antibiotics in UC is controversial due to its long-term impact on commensal microbes. Antibiotic treatments have been shown to significantly deplete microbial populations from colonic mucosae of IBD patients, and following cessation of therapy, commensal microbes undergo substantial structural and functional changes which may persist years after termination of the therapy [125]. Long-term exposure to antibiotic treatments impairs commensal bacterial diversity, leading to the abnormal proliferation of fungi, facilitating the growth of antibiotic-resistant species including methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Enterococci (VRE), and increasing susceptibility to secondary infections common in UC including Clostridium difficile colitis [117,125]. The effect of antibiotics on the structure and function of the commensal microbiome seems to be more significant when the therapy is administered during critical windows of early life development. Studies have also shown that children exposed to antibiotic therapies in early life are more susceptible to develop UC or CD in adulthood, implicating antibiotics as risk factors for autoimmune disease [126,127].
3.5. Fecal Microbiota Transplantation
Fecal microbiota transplantation (FMT) involves the transfer of prescreened intestinal bacteria from a healthy donor to an unwell recipient to restore the recipient microbiome to a healthier milieu and reduce symptoms associated with inflammation. The first use of FMT in human history dates to fourth century China for treatment of food poisoning [128]. Subsequent records show use during World War II for the treatment of bacterial dysentery [128]. Despite its long history, it was only in the past decade that FMT has gained recognition for its role in treating recurrent or refractory Clostridioides difficile infection (rCDI), with proven efficacy, safety, tolerability, and patient acceptance [128,129]. The effectiveness of FMT for rCDI is high, with several studies reporting >90% response after two administrations [130,131].
In response to its success for the treatment of rCDI, FMT has also received attention for its therapeutic potential in the treatment of UC. To date, one RCT in pediatric UC [132], and four RCTs in adult UC have assessed the role of FMT in treating chronic inflammation (Table 2) [133,134,135,136]. While methodologies across these studies are mixed, overall, FMT appears to show promise in inducing short-term remission [137]. In two qualitative studies exploring patient experiences with FMT in UC, patients reported positive experiences with treatment and an interest in receiving FMT in the future [138,139]. This high level of patient acceptance may further encourage research on the role of FMT in IBD therapy, leveraged by patient support groups and private foundations. FMT may also support cost-effectiveness as compared with conventional long-term UC therapies, as has been demonstrated in the treatment of rCDI [140].
Table 2.
Summary of methods, outcomes, and results in adult, pediatric fecal microbiota transplant studies.
Microbiome changes in adult and pediatric UC patients who received donor FMT suggests increased bacterial diversity, within 4–6 weeks post-transplant (Table 3) [132,133,135,141]. Paramsothy et al. and Costello et al. both reported an increase in donor-derived species from the Prevotella genus after 8 weeks [135,141] and Anaerofilum pentosovorans and Bacteroides coprophilus species were associated with disease improvement following FMT [141]. A decrease in Bacteroides genus, at 4 and 8 weeks, post FMT, as well as an increase in Clostridium cluster XVIII and Ruminococcus spp was associated with disease remission in recipients [145]. An increase in taxa typically found in the oral cavity, such as Streptococcus spp and Fusobacterium spp, was associated with lack of UC remission. Further, patients in remission after FMT had increased synthesis of SCFAs and secondary bile acids [145].
Table 3.
Summary of microbial changes in adult, pediatric fecal microbiota transplant studies.
In the pediatric population, three of four studies reported some degree of clinical response post FMT [132,142,143]. Only one study reported adverse events such as worsening colitis requiring hospitalization for intravenous corticosteroid administration; this study by Pai et al. was also the only RCT to systematically assess the role of a FMT in pediatric UC patients using a placebo-controlled, blinded study design [132]. Among adult UC trials, 3 of 4 RCTs reported a statistically significant rate of achieving primary and secondary outcomes in the FMT group compared to control arms [133,135,141]. These four studies also employed the use of larger samples as compared with previous FMT studies and used pooled fecal matter from multiple donors to increase bacterial richness at baseline, during transplantation, and after treatment [132,133,135,141].
Various methods of FMT administration have been trialed. These include targeting upper GI routes via naso-gastric, naso-duodenal, and naso-jejunal tubes, as well as lower GI routes including colonoscopy infusions and enema-based therapies. More recently, oral capsules containing lyophilized or liquid FMT product have attracted interest for their ease of administration, convenient at-home use, and simple storage requirements [146,147]. Emerging evidence is showing impressive efficacy of capsule FMT for treatment of rCDI, with recent studies demonstrating equivalent clinical benefits and side effect profiles for capsule FMT as with traditional enema formulations [128,148,149]. To date, no study has been published assessing the efficacy of FMT capsules in the treatment of IBD. However, numerous studies using oral FMT capsular therapy are actively recruiting patients. The results of these studies will provide important information on the future of capsule FMT as a minimally invasive route of delivery, which will cater to patients’ growing interest for convenient at-home administration methods [138].
4. Microbial Influence on Progression to Colitis-Associated Cancer
4.1. Gut Dysbiosis and CAC
Longstanding UC correlates with an increased risk of developing CAC through cumulative inflammatory burden [150]. Complex interactions between various genetic and epigenetic factors, a Western diet high in refined sugars and animal fat, and low in dietary fiber and intestinal dysbiosis have been hypothesized to play key roles in tumorigenesis [4,151,152,153,154].
Gut dysbiosis may contribute to CAC through direct and indirect interactions with the host, such as bacterial metabolites and secreted molecules (e.g., genotoxins and virulence factors), attachment, invasion and translocation, and host defense modulation, leading to direct cell damage and chronic inflammation [155,156]. Among the different microbial-induced colon tumorigenesis theories, the alpha-bug hypothesis [157], driver-passenger hypothesis [158], and common ground hypothesis are most common [159].
In the alpha-bug hypothesis, a single pro-oncogenic microbe termed “alpha-bug” (particularly Enterotoxigenic Bacteroides fragilis (ETBF)) is thought to directly cause epithelial damage, modify colonic microbiota to further promote CAC development, and displace taxa that may protect against metaplasia. The driver-passenger model suggests that although a “driver bacteria” (with the same role as the alpha-bug) initially causes DNA damage, this results in microbial alterations that promote growth of opportunistic bacteria (i.e., bacterial passengers) which contribute to tumorigenesis. More recently, the common ground hypothesis has proposed that exogenous and endogenous factor (e.g., unhealthy diet, exogenous contaminants, and chronic inflammation) initially form a “leaky gut,” which results in transcellular hyperpermeability and bacterial internalization of pathobionts resulting in chronic inflammation and morphological changes in genetically predisposed individuals [157].
Bacteria such as ETBF, Fusobacterium nucleatum, Escherichia coli, and Peptostreptococcus anaerobius have been associated with colon cancer in human and animal models [109,160]. ETBF, through its zinc-metalloprotease toxin (Bacteroides fragilis toxin [BFT]), can trigger a carcinogenic inflammatory cascade by inducing E-cadherin cleavage, leading to increased intestinal permeability and Wnt/β-catenin and NF-κB signaling pathway activation, resulting in myeloid cell activation and increased levels of IL-17. This leads to a downstream series of immunological events that results in uncontrolled proliferation of colonic epithelial cells [161]. In addition, ETBF as well as polyketide synthase (pks)-positive Escherichia coli, have been associated with the creation of biofilms that coat adenomas, which may further promote tumorigenesis by altering the cancer metabolome (upregulation of N1,N12-diacetylspermine) and trigger IL-17-associated inflammation [162,163,164,165].
Fusobacterium nucleatum, a Gram-negative bacterium that resides in the oral cavity, has attracted interest over the past decade given its association with CAC. Fusobacterium nucleatum possesses several mechanisms that may contribute to CAC development [166]. Fusobacterium nucleatum adhesion protein A (fadA) facilitates attachment and invasion by binding to E-cadherin present in epithelial and malignant cells, resulting in expression of inflammatory molecules such as NF-κB, IL-6, IL-8, and IL-18, and TLR2/TLR4 activation [167,168]. Fusobacterium nucleatum has also been shown to accelerate DNA methylation in cancer-specific genes in patients with UC and appears to inhibit natural killer cell cytotoxicity via the Fap2 protein [169]. Colibactin-producing Escherichia coli (Pks+) has been shown to mediate cell damage through DNA alkylation and DNA double-strand breaks, contributing to tumorigenesis [110,170]. Colonic inflammation has been shown to further promote the genotoxic effects of Pks+ Escherichia coli [171]. Peptostreptococcus anaerobius has been shown to accelerate CAC in ApcMin/+ mice, and to attach to malignant cells via integrin α2/β1, a collagen receptor widely expressed on intestinal epithelial cells. This leads to downstream activation of the NF-κB pathway, a key regulator of intestinal inflammation and cancer development and progression [172].
A recent study in mouse CAC models found that α-diversity was decreased during the development of UC to CAC, and that the composition of the intestinal microbiome differed between three groups: control groups exhibited higher levels of Firmicutes, Verrucomicrobia, and Actinobacteria, and UC and CAC groups had higher levels of Proteobacteria, Firmicutes, and Verrucomicrobia [173]. Moreover, several metabolites were correlated with these microbial changes seen in the UC and CAC groups, specifically 12–hydroxy–8,10-octadecadienoic acid and linoleic acid positively correlated with Enterobacteriaceae, Escherichia-Shigella, and Proteobacteria. Thus, these metabolites could act as biomarkers for CAC.
Another animal study using azoxymethane/dextran sulfate sodium (AOM/DSS)-induced CAC murine models, showed that sucralose, a widely used caloric-free sweetener, led to an increase in the number and size of colonic tumors, inflammatory cytokines, and changes in the intestinal microbiota as compared with controls [174]. This highlights the importance of diet on the intestinal microbiota and CAC development.
In a recent study of patients with CAC, the CAC group was found to have decreased α-diversity, higher Proteobacteria, and decreased Firmicutes and Bacteroidetes as compared with healthy controls [175]. Significant differences were also found between the sporadic CRC group and the CAC group, with the latter having higher Proteobacteria, with Bradyrhizobiaceae and Enterobacteriaceae being the two overrepresented families. In addition, the levels of Fusobacterium were higher in the sporadic cancer group as compared with the CAC group. Further, there is evidence to suggest that the composition of the intestinal microbiota can change across different stages of CAC. In later-stage CAC, whereas Akkermansia, Fusobacterium, Peptostreptococcus, Streptococcus, and Ruminococcus were significantly higher, Granulicatella and Lactobacillus were significantly decreased as compared with non-CAC controls [176].
UC and CAC share similar microbial alterations that could potentially contribute to their shared pathogenesis. Whether these microbial alterations are the cause or consequence of chronic inflammation remains to be elucidated.
4.2. Intestinal Microbiota as an Emerging Target for the Treatment of Colitis-Associated Cancer
Given the potential role of the intestinal microbiome in the pathogenesis of CAC, gut bacteria-targeted therapies including probiotics, prebiotics, synbiotics, antibiotics, and FMT may hold promise [177,178,179]. This theory has strong biological plausibility. As we have discussed, mechanisms through which intestinal microbiota modulation occurs in CAC are similar to those seen in UC.
To the best of our knowledge, only two studies have been conducted on the use of FMT in murine models with CAC. Wang et al. used FMT to treat mice with AOM/DSS-induced CAC, which led to an increase in α-diversity as compared with the pre-FMT microbiota [173]. In addition, FMT led to an increase in colonic length, reduction in number of tumors and inflammation, as well as inhibition of proinflammatory molecules (IL-1β, IL-6, and TNF-α) and increase in anti-inflammatory cytokines (IL-10 and TGF-β). Furthermore, FMT-treated mice were found to have increased levels of CD3/CD4 in the lamina propria.
In another study of murine models with implanted colorectal adenoma cells and chemotherapy-induced mucosal injury, the authors found that FMT led to a reduction in diarrhea and intestinal mucositis, as well as suppression of IL-6 [180]. No significant differences were found in α-diversity between the groups.
Taken together, these findings suggest that FMT may be a promising therapy in modulating the intestinal microbiome of murine models with CAC. Larger studies are required to better understand the mechanisms and benefits of FMT in CAC.
5. Concluding Remarks
The intestinal microbiome exerts a major influence on the development and progression of UC and CAC. Our understanding of fungal and viral influences in the GI tract is steadily growing. With the support of culture-based sequencing, advanced metagenomics, and bioinformatics technologies, we are constructing a clearer picture of host–microbial dynamics. This provides more opportunities to understand disease pathogenesis at an individual level and may target treatments more effectively to individual patients’ UC and CAC biology.
While the cause of UC and CAC remains unclear, there is a clear role for the microbiome in regulating host inflammatory response and maintaining intestinal homeostasis. Our existing treatment paradigm of simply dampening immune activation through life-long, systemically acting immune suppression needs to keep pace with intestinal microbiome research. Multiple taxa have been implicated in triggering intestinal immune activation, and this is increasingly established through both structural and functional sequencing techniques. The metabolic contributions of key bacterial taxa play clear roles in epithelial cell function. The development of microbiota-based therapies will continue to have enormous potential. Exciting early data supports the role of FMT, prebiotics, probiotics, synbiotics, and select antibiotics in UC care.
Associations between microbial dysbiosis, chronic inflammation, autoimmunity, and tumorigenesis are well established. The future of GI pharmacotherapy will involve treatments that can halt this progression at its onset.
Author Contributions
J.P. and V.C. contributed equally to this work and performed the majority of the writing; N.N., D.A.R., and N.P. contributed to the writing of the manuscript; N.P. supervised and approved the work. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors disclose no relevant conflict of interest.
References
- Yamamoto-Furusho, J.K.; Martínez-Benítez, B.; Sánchez-Morales, G.E. Histopathologic Parameters at Diagnosis as Early Predictors of Histologic Remission along the Course of Ulcerative Colitis. Gastroenterol. Res. Pract. 2020, 2020, 1–5. [Google Scholar] [CrossRef]
- Jairath, V.; Feagan, B.G. Global Burden of Inflammatory Bowel Disease. Lancet Gastroenterol. Hepatol. 2020, 5, 2–3. [Google Scholar] [CrossRef]
- Ye, B.D.; McGovern, D.P.B. Genetic Variation in IBD: Progress, Clues to Pathogenesis and Possible Clinical Utility. Expert Rev. Clin. Immunol. 2016, 12, 1091–1107. [Google Scholar] [CrossRef] [PubMed]
- Pigneur, B.; Ruemmele, F.M. Nutritional Interventions for the Treatment of IBD: Current Evidence and Controversies. Ther. Adv. Gastroenterol. 2019, 12, 175628481989053. [Google Scholar] [CrossRef]
- Saidel-Odes, L.; Odes, S. Hygiene Hypothesis in Inflammatory Bowel Disease. Ann. Gastroenterol. 2014, 27, 189–190. [Google Scholar]
- Troelsen, F.S.; Jick, S. Antibiotic Use in Childhood and Adolescence and Risk of Inflammatory Bowel Disease: A Case-Control Study in the UK Clinical Practice Research Datalink. Inflamm. Bowel Dis. 2020, 26, 440–447. [Google Scholar] [CrossRef]
- Eaden, J.A.; Abrams, K.R.; Mayberry, J.F. The Risk of Colorectal Cancer in Ulcerative Colitis: A Meta-Analysis. Gut 2001, 48, 526–535. [Google Scholar] [CrossRef]
- Yashiro, M. Ulcerative Colitis-Associated Colorectal Cancer. World J. Gastroenterol. 2014, 20, 16389–16397. [Google Scholar] [CrossRef]
- Kinugasa, T.; Akagi, Y. Status of Colitis-Associated Cancer in Ulcerative Colitis. World J. Gastrointest. Oncol. 2016, 8, 351. [Google Scholar] [CrossRef]
- Greuter, T.; Rieder, F.; Kucharzik, T.; Peyrin-Biroulet, L.; Schoepfer, A.M.; Rubin, D.T.; Vavricka, S.R. Emerging Treatment Options for Extraintestinal Manifestations in IBD. Gut 2021, 70, 796–802. [Google Scholar] [CrossRef]
- Olén, O.; Erichsen, R.; Sachs, M.C.; Pedersen, L.; Halfvarson, J.; Askling, J.; Ekbom, A.; Sørensen, H.T.; Ludvigsson, J.F. Colorectal Cancer in Ulcerative Colitis: A Scandinavian Population-Based Cohort Study. Lancet 2020, 395, 123–131. [Google Scholar] [CrossRef]
- Khan, I.; Ullah, N.; Zha, L.; Bai, Y.; Khan, A.; Zhao, T.; Che, T.; Zhang, C. Alteration of Gut Microbiota in Inflammatory Bowel Disease (IBD): Cause or Consequence? IBD Treatment Targeting the Gut Microbiome. Pathogens 2019, 8, 126. [Google Scholar] [CrossRef]
- Roselli, M.; Finamore, A. Use of Synbiotics for Ulcerative Colitis Treatment. Curr. Clin. Pharmacol. 2019, 15, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Fricke, W.F.; Ravel, J. Microbiome or No Microbiome: Are We Looking at the Prenatal Environment through the Right Lens? Microbiome 2021, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Wopereis, H.; Oozeer, R.; Knipping, K.; Belzer, C.; Knol, J. The First Thousand Days - Intestinal Microbiology of Early Life: Establishing a Symbiosis. Pediatr. Allergy Immunol. 2014, 25, 428–438. [Google Scholar] [CrossRef] [PubMed]
- BioRender. Available online: www.biorender.com (accessed on 1 August 2021).
- Ananthakrishnan, A.N.; Bernstein, C.N.; Iliopoulos, D.; Macpherson, A.; Neurath, M.F.; Ali, R.A.R.; Vavricka, S.R.; Fiocchi, C. Environmental Triggers in IBD: A Review of Progress and Evidence. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, M.; Sabino, J.; Frias-Gomes, C.; Hillenbrand, C.M.; Soudant, C.; Axelrad, J.E.; Shah, S.C.; Ribeiro-Mourão, F.; Lambin, T.; Peter, I.; et al. Early Life Exposures and the Risk of Inflammatory Bowel Disease: Systematic Review and Meta-Analyses. EClinicalMedicine 2021, 36, 100884. [Google Scholar] [CrossRef]
- Torun, A.; Hupalowska, A.; Trzonkowski, P.; Kierkus, J.; Pyrzynska, B. Intestinal Microbiota in Common Chronic Inflammatory Disorders Affecting Children. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Pickard, J.M.; Zeng, M.Y.; Caruso, R.; Núñez, G. Gut Microbiota: Role in Pathogen Colonization, Immune Responses, and Inflammatory Disease. Immunol. Rev. 2017, 279, 70–89. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Hand, T.W. Role of the Microbiota in Immunity and Inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, Stability and Resilience of the Human Gut Microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef]
- MacPherson, A.J.; Slack, E.; Geuking, M.B.; McCoy, K.D. The Mucosal Firewalls against Commensal Intestinal Microbes. Semin. Immunopathol. 2009, 31, 145–149. [Google Scholar] [CrossRef]
- Johansson, M.E.V.; Holmén Larsson, J.M.; Hansson, G.C. The Two Mucus Layers of Colon Are Organized by the MUC2 Mucin, Whereas the Outer Layer Is a Legislator of Host-Microbial Interactions. Proc. Natl. Acad. Sci. USA 2011, 108, 4659–4665. [Google Scholar] [CrossRef]
- Selma-Royo, M.; Calatayud Arroyo, M.; García-Mantrana, I.; Parra-Llorca, A.; Escuriet, R.; Martínez-Costa, C.; Collado, M.C. Perinatal Environment Shapes Microbiota Colonization and Infant Growth: Impact on Host Response and Intestinal Function. Microbiome 2020, 8. [Google Scholar] [CrossRef]
- Alipour, M.; Zaidi, D.; Valcheva, R.; Jovel, J.; Martínez, I.; Sergi, C.; Walter, J.; Mason, A.L.; Ka-Shu Wong, G.; Dieleman, L.A.; et al. Mucosal Barrier Depletion and Loss of Bacterial Diversity Are Primary Abnormalities in Paediatric Ulcerative Colitis. J. Crohn’s Colitis 2016, 10, 462–471. [Google Scholar] [CrossRef]
- Pothuraju, R.; Krishn, S.R.; Gautam, S.K.; Pai, P.; Ganguly, K.; Chaudhary, S.; Rachagani, S.; Kaur, S.; Batra, S.K. Mechanistic and Functional Shades of Mucins and Associated Glycans in Colon Cancer. Cancers 2020, 12, 649. [Google Scholar] [CrossRef] [PubMed]
- Martens, E.C.; Neumann, M.; Desai, M.S. Interactions of Commensal and Pathogenic Microorganisms with the Intestinal Mucosal Barrier. Nat. Rev. Microbiol. 2018, 16, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Altenbach, D.; Robatzek, S. Pattern Recognition Receptors: From the Cell Surface to Intracellular Dynamics. Mol. Plant-Microbe Interact. 2007, 20, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Specian, R.D.; Oliver, M.G. Functional Biology of Intestinal Goblet Cells. Am. J. Physiol.-Cell Physiol. 1991, 260. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut Biogeography of the Bacterial Microbiota. Nat. Rev. Microbiol. 2015, 14, 20–32. [Google Scholar] [CrossRef] [PubMed]
- McGuckin, M.A.; Lindén, S.K.; Sutton, P.; Florin, T.H. Mucin Dynamics and Enteric Pathogens. Nat. Rev. Microbiol. 2011, 9, 265–278. [Google Scholar] [CrossRef]
- Vanhooren, V.; Vandenbroucke, R.E.; Dewaele, S.; Van Hamme, E.; Haigh, J.J.; Hochepied, T.; Libert, C. Mice Overexpressing β-1,4-Galactosyltransferase i Are Resistant to TNF-Induced Inflammation and DSS-Induced Colitis. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Dorofeyev, A.E.; Vasilenko, I.V.; Rassokhina, O.A.; Kondratiuk, R.B. Mucosal Barrier in Ulcerative Colitis and Crohn’s Disease. Gastroenterol. Res. Pract. 2013, 2013, 431231. [Google Scholar] [CrossRef]
- Yamamoto-Furusho, J.K.; Ascaño-Gutiérrez, I.; Furuzawa-Carballeda, J.; Fonseca-Camarillo, G. Differential Expression of MUC12, MUC16, and MUC20 in Patients with Active and Remission Ulcerative Colitis. Mediat. Inflamm. 2015, 2015, 659018. [Google Scholar] [CrossRef]
- Longman, R.J.; Poulsom, R.; Corfield, A.P.; Warren, B.F.; Wright, N.A.; Thomas, M.G. Alterations in the Composition of the Supramucosal Defense Barrier in Relation to Disease Severity of Ulcerative Colitis. J. Histochem. Cytochem. 2006, 54, 1335–1348. [Google Scholar] [CrossRef]
- Grondin, J.A.; Kwon, Y.H.; Far, P.M.; Haq, S.; Khan, W.I. Mucins in Intestinal Mucosal Defense and Inflammation: Learning From Clinical and Experimental Studies. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Groschwitz, K.R.; Hogan, S.P. Intestinal Barrier Function: Molecular Regulation and Disease Pathogenesis. J. Allergy Clin. Immunol. 2009, 124, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Vancamelbeke, M.; Vermeire, S. The Intestinal Barrier: A Fundamental Role in Health and Disease. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 821–834. [Google Scholar] [CrossRef]
- Howell, K.J.; Kraiczy, J.; Nayak, K.M.; Gasparetto, M.; Ross, A.; Lee, C.; Mak, T.N.; Koo, B.K.; Kumar, N.; Lawley, T.; et al. DNA Methylation and Transcription Patterns in Intestinal Epithelial Cells from Pediatric Patients with Inflammatory Bowel Diseases Differentiate Disease Subtypes and Associate With Outcome. Gastroenterology 2018, 154, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, A.; Nagata, S.; Anzai, S.; Takahashi, J.; Kawai, M.; Hama, M.; Nogawa, D.; Yamamoto, K.; Kuno, R.; Suzuki, K.; et al. Ubiquitin D Is Upregulated by Synergy of Notch Signalling and TNF-α in the Inflamed Intestinal Epithelia of IBD Patients. J. Crohn’s Colitis 2019, 13, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Blander, J.M. Death in the Intestinal Epithelium—Basic Biology and Implications for Inflammatory Bowel Disease. FEBS J. 2016, 2720–2730. [Google Scholar] [CrossRef] [PubMed]
- Ruder, B.; Atreya, R.; Becker, C. Tumour Necrosis Factor Alpha in Intestinal Homeostasis and Gut Related Diseases. Int. J. Mol. Sci. 2019, 20, 1887. [Google Scholar] [CrossRef] [PubMed]
- Gaujoux, R.; Starosvetsky, E.; Maimon, N.; Vallania, F.; Bar-Yoseph, H.; Pressman, S.; Weisshof, R.; Goren, I.; Rabinowitz, K.; Waterman, M.; et al. Cell-Centred Meta-Analysis Reveals Baseline Predictors of Anti-TNFα Non-Response in Biopsy and Blood of Patients with IBD. Gut 2019, 68, 604–614. [Google Scholar] [CrossRef]
- Brandtzaeg, P.; Johansen, F.E. Mucosal B Cells: Phenotypic Characteristics, Transcriptional Regulation, and Homing Properties. Immunol. Rev. 2005, 206, 32–63. [Google Scholar] [CrossRef]
- Gommerman, J.L.; Rojas, O.L.; Fritz, J.H. Re-Thinking the Functions of IgA+plasma Cells. Gut Microbes 2015, 5, 652–662. [Google Scholar] [CrossRef]
- Pabst, O.; Cerovic, V.; Hornef, M. Secretory IgA in the Coordination of Establishment and Maintenance of the Microbiota. Trends Immunol. 2016, 37, 287–296. [Google Scholar] [CrossRef]
- Hooper, L.V.; Wong, M.H.; Thelin, A.; Hansson, L.; Falk, P.G.; Gordon, J.I. Molecular Analysis of Commensal Host-Microbial Relationships in the Intestine. Science 2001, 291, 881–884. [Google Scholar] [CrossRef]
- Bruno, M.E.C.; Rogier, E.W.; Frantz, A.L.; Stefka, A.T.; Thompson, S.N.; Kaetzel, C.S. Regulation of the Polymeric Immunoglobulin Receptor in Intestinal Epithelial Cells by Enterobacteriaceae: Implications for Mucosal Homeostasis. Immunol. Investig. 2010, 39, 356–382. [Google Scholar] [CrossRef]
- Wei, H.; Wang, J.Y. Role of Polymeric Immunoglobulin Receptor in Iga and Igm Transcytosis. Int. J. Mol. Sci. 2021, 22, 2284. [Google Scholar] [CrossRef] [PubMed]
- Johansen, F.E.; Kaetzel, C.S. Regulation of the Polymeric Immunoglobulin Receptor and IgA Transport: New Advances in Environmental Factors That Stimulate PIgR Expression and Its Role in Mucosal Immunity. Mucosal Immunol. 2011, 4, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Van der Steen, L.; Tuk, C.W.; Bakema, J.E.; Kooij, G.; Reijerkerk, A.; Vidarsson, G.; Bouma, G.; Kraal, G.; de Vries, H.E.; Beelen, R.H.J.; et al. Immunoglobulin A: FcαRI Interactions Induce Neutrophil Migration Through Release of Leukotriene B4. Gastroenterology 2009, 137. [Google Scholar] [CrossRef]
- Traicoff, J.L.; De Marchis, L.; Ginsburg, B.L.; Zamora, R.E.; Khattar, N.H.; Blanch, V.J.; Plummer, S.; Bargo, S.A.; Templeton, D.J.; Casey, G.; et al. Characterization of the Human Polymeric Immunoglobulin Receptor (PIGR) 3′UTR and Differential Expression of PIGR MRNA during Colon Tumorigenesis. J. Biomed. Sci. 2003, 10, 792–804. [Google Scholar] [CrossRef]
- Hurtado, C.G.; Wan, F.; Housseau, F.; Sears, C.L. Roles for Interleukin 17 and Adaptive Immunity in Pathogenesis of Colorectal Cancer. Gastroenterology 2018, 155, 1706–1715. [Google Scholar] [CrossRef]
- Caputi, V.; Popov, J.; Giron, M.C.; O’Mahony, S. Gut Microbiota as a Mediator of Host Neuro-Immune Interactions: Implications in Neuroinflammatory Disorders. Mod. Trends Psychiatry 2021, 32, 40–57. [Google Scholar] [CrossRef]
- Rescigno, M. CCR6+ Dendritic Cells: The Gut Tactical-Response Unit. Immunity 2006, 24, 508–510. [Google Scholar] [CrossRef][Green Version]
- Sun, T.; Nguyen, A.; Gommerman, J.L. Dendritic Cell Subsets in Intestinal Immunity and Inflammation. J. Immunol. 2020, 204, 1075–1083. [Google Scholar] [CrossRef]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Philip Schumm, L.; Sharma, Y.; Anderson, C.A.; et al. Host-Microbe Interactions Have Shaped the Genetic Architecture of Inflammatory Bowel Disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Steinbach, E.C.; Plevy, S.E. The Role of Macrophages and Dendritic Cells in the Initiation of Inflammation in IBD. Inflamm. Bowel Dis. 2014, 20, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Hart, A.L.; Al-Hassi, H.O.; Rigby, R.J.; Bell, S.J.; Emmanuel, A.V.; Knight, S.C.; Kamm, M.A.; Stagg, A.J. Characteristics of Intestinal Dendritic Cells in Inflammatory Bowel Diseases. Gastroenterology 2005, 129, 50–65. [Google Scholar] [CrossRef] [PubMed]
- Verstockt, B.; Ferrante, M.; Vermeire, S.; Van Assche, G. New Treatment Options for Inflammatory Bowel Diseases. J. Gastroenterol. 2018, 53, 585–590. [Google Scholar] [CrossRef]
- Heller, F.; Fromm, A.; Gitter, A.H.; Mankertz, J.; Schulzke, J.D. Epithelial Apoptosis Is a Prominent Feature of the Epithelial Barrier Disturbance in Intestinal Inflammation: Effect of pro-Inflammatory Interleukin-13 on Epithelial Cell Function. Mucosal Immunol. 2008, 1, 58–61. [Google Scholar] [CrossRef]
- Tan, T.G.; Sefik, E.; Geva-Zatorsky, N.; Kua, L.; Naskar, D.; Teng, F.; Pasman, L.; Ortiz-Lopez, A.; Jupp, R.; Wu, H.J.J.; et al. Identifying Species of Symbiont Bacteria from the Human Gut That, Alone, Can Induce Intestinal Th17 Cells in Mice. Proc. Natl. Acad. Sci. USA 2016, 113, E8141–E8150. [Google Scholar] [CrossRef]
- Erturk-Hasdemir, D.; Oh, S.F.; Okan, N.A.; Stefanetti, G.; Gazzaniga, F.S.; Seeberger, P.H.; Plevy, S.E.; Kasper, D.L. Symbionts Exploit Complex Signaling to Educate the Immune System. Proc. Natl. Acad. Sci. USA 2019, 116, 26157–26166. [Google Scholar] [CrossRef]
- Zhao, F.; Qu, J.; Wang, W.; Li, S.; Xu, S. The Imbalance of Th1/Th2 Triggers an Inflammatory Response in Chicken Spleens after Ammonia Exposure. Poult. Sci. 2020, 99, 3817–3822. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14. [Google Scholar] [CrossRef] [PubMed]
- Ananthakrishnan, A.N. Epidemiology and Risk Factors for IBD. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 205–217. [Google Scholar] [CrossRef]
- Lyte, J.M. Eating for 3.8 × 1013: Examining the Impact of Diet and Nutrition on the Microbiota-Gut-Brain Axis through the Lens of Microbial Endocrinology. Front. Endocrinol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Pei, L.Y.; Ke, Y.S.; Zhao, H.H.; Wang, L.; Jia, C.; Liu, W.Z.; Fu, Q.H.; Shi, M.N.; Cui, J.; Li, S.C. Role of Colonic Microbiota in the Pathogenesis of Ulcerative Colitis. BMC Gastroenterol. 2019, 19. [Google Scholar] [CrossRef]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal Microbiota Dysbiosis in IBD. Gut 2017, 66, 1039–1048. [Google Scholar] [CrossRef]
- Zou, J.; Liu, C.; Jiang, S.; Qian, D.; Duan, J. Cross Talk between Gut Microbiota and Intestinal Mucosal Immunity in the Development of Ulcerative Colitis. Infect. Immun. 2021, 89. [Google Scholar] [CrossRef]
- Pavel, F.M.; Vesa, C.M.; Gheorghe, G.; Diaconu, C.C.; Stoicescu, M.; Munteanu, M.A.; Babes, E.E.; Tit, D.M.; Toma, M.M.; Bungau, S. Highlighting the Relevance of Gut Microbiota Manipulation in Inflammatory Bowel Disease. Diagnostics 2021, 11, 1090. [Google Scholar] [CrossRef]
- Frank, D.N.; St., Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-Phylogenetic Characterization of Microbial Community Imbalances in Human Inflammatory Bowel Diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef]
- Chen, D.L.; Dai, Y.C.; Zheng, L.; Chen, Y.L.; Zhang, Y.L.; Tang, Z.P. Features of the Gut Microbiota in Ulcerative Colitis Patients with Depression: A Pilot Study. Medicine 2021, 100, e24845. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Bai, X.; Cao, X.; Yue, W.; Jiang, W.; Yu, Z. Changes in Intestinal Microbiota and Correlation with TLRs in Ulcerative Colitis in the Coastal Area of Northern China. Microb. Pathog. 2021, 150. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, P.; Macsharry, J.; Darby, T.; Fanning, A.; Shanahan, F.; Houston, A.; Brint, E. Differential Expression of Key Regulators of Toll-like Receptors in Ulcerative Colitis and Crohn’s Disease: A Role for Tollip and Peroxisome Proliferator-Activated Receptor Gamma? Clin. Exp. Immunol. 2016, 183, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Franchimont, D.; Vermeire, S.; El Housni, H.; Pierik, M.; Van Steen, K.; Gustot, T.; Quertinmont, E.; Abramowicz, M.; Van Gossum, A.; Devière, J.; et al. Deficient Host-Bacteria Interactions in Inflammatory Bowel Disease? The Toll-like Receptor (TLR)-4 Asp299gly Polymorphism Is Associated with Crohn’s Disease and Ulcerative Colitis. Gut 2004, 53, 987–992. [Google Scholar] [CrossRef] [PubMed]
- Galipeau, H.J.; Caminero, A.; Turpin, W.; Bermudez-Brito, M.; Santiago, A.; Libertucci, J.; Constante, M.; Raygoza Garay, J.A.; Rueda, G.; Armstrong, S.; et al. Novel Fecal Biomarkers That Precede Clinical Diagnosis of Ulcerative Colitis. Gastroenterology 2021, 160, 1532–1545. [Google Scholar] [CrossRef]
- Wiechers, C.; Zou, M.; Galvez, E.; Beckstette, M.; Ebel, M.; Strowig, T.; Huehn, J.; Pezoldt, J. The Microbiota Is Dispensable for the Early Stages of Peripheral Regulatory T Cell Induction within Mesenteric Lymph Nodes. Cell. Mol. Immunol. 2021, 18, 1211–1221. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Oshima, K.; Suda, W.; Nagano, Y.; Nishikawa, H.; Fukuda, S.; Saito, T.; Narushima, S.; Hase, K.; et al. Treg Induction by a Rationally Selected Mixture of Clostridia Strains from the Human Microbiota. Nature 2013, 500, 232–236. [Google Scholar] [CrossRef]
- Machiels, K.; Joossens, M.; Sabino, J.; De Preter, V.; Arijs, I.; Eeckhaut, V.; Ballet, V.; Claes, K.; Van Immerseel, F.; Verbeke, K.; et al. A Decrease of the Butyrate-Producing Species Roseburia Hominis and Faecalibacterium Prausnitzii Defines Dysbiosis in Patients with Ulcerative Colitis. Gut 2014, 63, 1275–1283. [Google Scholar] [CrossRef]
- Jiang, P.; Wu, S.; Luo, Q.; Zhao, X.; Chen, W.-H. Metagenomic Analysis of Common Intestinal Diseases Reveals Relationships among Microbial Signatures and Powers Multidisease Diagnostic Models. mSystems 2021, 6. [Google Scholar] [CrossRef]
- Ryan, F.J.; Ahern, A.M.; Fitzgerald, R.S.; Laserna-Mendieta, E.J.; Power, E.M.; Clooney, A.G.; O’Donoghue, K.W.; McMurdie, P.J.; Iwai, S.; Crits-Christoph, A.; et al. Colonic Microbiota Is Associated with Inflammation and Host Epigenomic Alterations in Inflammatory Bowel Disease. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- Ohkusa, T.; Yoshida, T.; Sato, N.; Watanabe, S.; Tajiri, H.; Okayasu, I. Commensal Bacteria Can Enter Colonic Epithelial Cells and Induce Proinflammatory Cytokine Secretion: A Possible Pathogenic Mechanism of Ulcerative Colitis. J. Med. Microbiol. 2009, 58, 535–545. [Google Scholar] [CrossRef]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermúdez-Humarán, L.G.; Gratadoux, J.-J.J.J.; Blugeon, S.; Bridonneau, C.; Furet, J.P.J.-P.; Corthier, G.; et al. Faecalibacterium Prausnitzii Is an Anti-Inflammatory Commensal Bacterium Identified by Gut Microbiota Analysis of Crohn Disease Patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef]
- Nishihara, Y.; Ogino, H.; Tanaka, M.; Ihara, E.; Fukaura, K.; Nishioka, K.; Chinen, T.; Tanaka, Y.; Nakayama, J.; Kang, D.; et al. Mucosa-Associated Gut Microbiota Reflects Clinical Course of Ulcerative Colitis. Sci. Rep. 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Kedia, S.; Ghosh, T.S.; Jain, S.; Desigamani, A.; Kumar, A.; Gupta, V.; Bopanna, S.; Yadav, D.P.; Goyal, S.; Makharia, G.; et al. Gut Microbiome Diversity in Acute Severe Colitis Is Distinct from Mild to Moderate Ulcerative Colitis. J. Gastroenterol. Hepatol. 2021, 36, 731–739. [Google Scholar] [CrossRef]
- Qiu, X.; Ma, J.; Jiao, C.; Mao, X.; Zhao, X.; Lu, M.; Wang, K.; Zhang, H. Alterations in the Mucosa-Associated Fungal Microbiota in Patients with Ulcerative Colitis. Oncotarget 2017, 8, 107577–107588. [Google Scholar] [CrossRef] [PubMed]
- Hoarau, G.; Mukherjee, P.K.; Gower-Rousseau, C.; Hager, C.; Chandra, J.; Retuerto, M.A.; Neut, C.; Vermeire, S.; Clemente, J.; Colombel, J.F.; et al. Bacteriome and Mycobiome Interactions Underscore Microbial Dysbiosis in Familial Crohn’s Disease. MBio 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Lu, X.J.; Zhang, Y.; Cheung, C.P.; Lam, S.; Zhang, F.; Tang, W.; Ching, J.Y.L.; Zhao, R.; Chan, P.K.S.; et al. Gut Mucosal Virome Alterations in Ulcerative Colitis. Gut 2019, 68, 1169–1179. [Google Scholar] [CrossRef]
- Gogokhia, L.; Buhrke, K.; Bell, R.; Hoffman, B.; Brown, D.G.; Hanke-Gogokhia, C.; Ajami, N.J.; Wong, M.C.; Ghazaryan, A.; Valentine, J.F.; et al. Expansion of Bacteriophages Is Linked to Aggravated Intestinal Inflammation and Colitis. Cell Host Microbe 2019, 25, 285–299.e8. [Google Scholar] [CrossRef] [PubMed]
- Friswell, M.; Campbell, B.; Rhodes, J. The Role of Bacteria in the Pathogenesis of Inflammatory Bowel Disease. Gut Liver 2010, 4, 295–306. [Google Scholar] [CrossRef]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-Specific Alterations in the Enteric Virome in Inflammatory Bowel Disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef]
- Lavelle, A.; Sokol, H. Gut Microbiota-Derived Metabolites as Key Actors in Inflammatory Bowel Disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 223–237. [Google Scholar] [CrossRef]
- Sinopoulou, V.; Gordon, M.; Dovey, T.M.; Akobeng, A.K. Interventions for the Management of Abdominal Pain in Ulcerative Colitis. Cochrane Database Syst. Rev. 2021, 2021. [Google Scholar] [CrossRef]
- Gyawali, R.; Nwamaioha, N.; Fiagbor, R.; Zimmerman, T.; Newman, R.H.; Ibrahim, S.A. The Role of Prebiotics in Disease Prevention and Health Promotion. In Dietary Interventions in Gastrointestinal Diseases: Foods, Nutrients, and Dietary Supplements; Elsevier: Amsterdam, The Netherlands, 2019; pp. 151–167. ISBN 9780128144695. [Google Scholar]
- Davani-Davari, D.; Negahdaripour, M.; Karimzadeh, I.; Seifan, M.; Mohkam, M.; Masoumi, S.J.; Berenjian, A.; Ghasemi, Y. Prebiotics: Definition, Types, Sources, Mechanisms, and Clinical Applications. Foods 2019, 8, 92. [Google Scholar] [CrossRef] [PubMed]
- Priyadarshini, M.; Kotlo, K.U.; Dudeja, P.K.; Layden, B.T. Role of Short Chain Fatty Acid Receptors in Intestinal Physiology and Pathophysiology. Compr. Physiol. 2018, 8, 1065–1090. [Google Scholar] [CrossRef]
- Kanauchi, O.; Matsumoto, Y.; Matsumura, M.; Fukuoka, M.; Bamba, T. The Beneficial Effects of Microflora, Especially Obligate Anaerobes, and Their Products on the Colonic Environment in Inflammatory Bowel Disease. Curr. Pharm. Des. 2005, 11, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Venegas, D.P.; De La Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.M.; Faber, K.N.; Hermoso, M.A.; Parada Venegas, D.; et al. Short Chain Fatty Acids (SCFAs)Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef]
- Faghfoori, Z.; Navai, L.; Shakerhosseini, R.; Somi, M.H.; Nikniaz, Z.; Norouzi, M.F. Effects of an Oral Supplementation of Germinated Barley Foodstuff on Serum Tumour Necrosis Factor-α, Interleukin-6 and -8 in Patients with Ulcerative Colitis. Ann. Clin. Biochem. 2011, 48, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Casellas, F.; Borruel, N.; Torrejón, A.; Varela, E.; Antolin, M.; Guarner, F.; Malagelada, J.R. Oral Oligofructose-Enriched Inulin Supplementation in Acute Ulcerative Colitis Is Well Tolerated and Associated with Lowered Faecal Calprotectin. Aliment. Pharmacol. Ther. 2007, 25, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.; Guarner, F.; Reid, G.; Gibson, G.R.; Merenstein, D.J.; Pot, B.; Morelli, L.; Canani, R.B.; Flint, H.J.; Salminen, S.; et al. Expert Consensus Document: The International Scientific Association for Probiotics and Prebiotics Consensus Statement on the Scope and Appropriate Use of the Term Probiotic. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 506–514. [Google Scholar] [CrossRef]
- Iannotti, F.A.; Di Marzo, V. The Gut Microbiome, Endocannabinoids and Metabolic Disorders. J. Endocrinol. 2021, 248, R83–R97. [Google Scholar] [CrossRef] [PubMed]
- Chibbar, R.; Dieleman, L.A. The Gut Microbiota in Celiac Disease and Probiotics. Nutrients 2019, 11, 2375. [Google Scholar] [CrossRef] [PubMed]
- Bellavia, M.; Tomasello, G.; Romeo, M.; Damiani, P.; Lo Monte, A.I.; Lozio, L.; Campanella, C.; Gammazza, A.M.; Rappa, F.; Zummo, G.; et al. Gut Microbiota Imbalance and Chaperoning System Malfunction Are Central to Ulcerative Colitis Pathogenesis and Can Be Counteracted with Specifically Designed Probiotics: A Working Hypothesis. Med. Microbiol. Immunol. 2013, 202, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Iheozor-Ejiofor, Z.; Kaur, L.; Gordon, M.; Baines, P.A.; Sinopoulou, V.; Akobeng, A.K. Probiotics for Maintenance of Remission in Ulcerative Colitis. Cochrane Database Syst. Rev. 2020, 2020, CD007443. [Google Scholar] [CrossRef] [PubMed]
- Plaza-Diaz, J.; Gomez-Llorente, C.; Abadia-Molina, F.; Saez-Lara, M.J.; Campaña-Martin, L.; Muñoz-Quezada, S.; Romero, F.; Gil, A.; Fontana, L. Effects of Lactobacillus Paracasei CNCM I-4034, Bifidobacterium Breve CNCM I-4035 and Lactobacillus Rhamnosus CNCM I-4036 on Hepatic Steatosis in Zucker Rats. PLoS ONE 2014, 9, e98401. [Google Scholar] [CrossRef]
- Hering, L.; Katkeviciute, E.; Schwarzfischer, M.; Busenhart, P.; Gottier, C.; Mrdjen, D.; Komuczki, J.; Wawrzyniak, M.; Lang, S.; Atrott, K.; et al. Protein Tyrosine Phosphatase Non-Receptor Type 2 Function in Dendritic Cells Is Crucial to Maintain Tissue Tolerance. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Nougayrède, J.P.; Homburg, S.; Taieb, F.; Boury, M.; Brzuszkiewicz, E.; Gottschalk, G.; Buchrieser, C.; Hacker, J.; Dobrindt, U.; Oswald, E. Escherichia Coli Induces DNA Double-Strand Breaks in Eukaryotic Cells. Science 2006, 313, 848–851. [Google Scholar] [CrossRef]
- De Vrese, M.; Marteau, P.R. Probiotics and Prebiotics: Effects on Diarrhea. J. Nutr. 2007, 137. [Google Scholar] [CrossRef]
- Fujimori, S.; Gudis, K.; Mitsui, K.; Seo, T.; Yonezawa, M.; Tanaka, S.; Tatsuguchi, A.; Sakamoto, C. A Randomized Controlled Trial on the Efficacy of Synbiotic versus Probiotic or Prebiotic Treatment to Improve the Quality of Life in Patients with Ulcerative Colitis. Nutrition 2009, 25, 520–525. [Google Scholar] [CrossRef]
- Mawdsley, J.E.D. Synbiotic Therapy for Ulcerative Colitis. Gut 2005, 54, 1346. [Google Scholar] [CrossRef][Green Version]
- Federico, A.; Tuccillo, C.; Grossi, E.; Abbiati, R.; Garbagna, N.; Romano, M.; Tiso, A.; Del Vecchio Blanco, C.; Loguercio, C. The Effect of a New Symbiotic Formulation on Plasma Levels and Peripheral Blood Mononuclear Cell Expression of Some Pro-Inflammatory Cytokines in Patients with Ulcerative Colitis: A Pilot Study. Eur. Rev. Med. Pharmacol. Sci. 2009, 13, 285–293. [Google Scholar]
- Malathi, K.; Nandini, R.; Kr, D.; Bn, S. Journal of Hepatology and A Randomized Open Label Study to Evaluate the Efficacy and Tolerability of Synbiotic in the Treatment of Ulcerative Colitis. J. Hepatol. Gastrointest. Disord. 2019, 5, 1–4. [Google Scholar]
- Ledder, O. Antibiotics in Inflammatory Bowel Diseases: Do We Know What We’re Doing? Transl. Pediatr. 2019, 8, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Abraham, B.; Quigley, E.M.M. Antibiotics and Probiotics in Inflammatory Bowel Disease: When to Use Them? Frontline Gastroenterol. 2020, 11, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, Y.; Sato, N.; Tsukinaga, S.; Uchiyama, K.; Koido, S.; Ishikawa, D.; Ohkusa, T. Long-Term Outcomes of Antibiotic Combination Therapy for Ulcerative Colitis. Ther. Adv. Chronic Dis. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.; Levine, A.; Escher, J.C.; Griffiths, A.M.; Russell, R.K.; Dignass, A.; Dias, J.A.; Bronsky, J.; Braegger, C.P.; Cucchiara, S.; et al. Management of Pediatric Ulcerative Colitis: Joint ECCO and ESPGHAN Evidence-Based Consensus Guidelines. J. Pediatr. Gastroenterol. Nutr. 2012, 55, 340–361. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.; Bishai, J.; Reshef, L.; Abitbol, G.; Focht, G.; Marcus, D.; Ledder, O.; Lev-Tzion, R.; Orlanski-Meyer, E.; Yerushalmi, B.; et al. Antibiotic Cocktail for Pediatric Acute Severe Colitis and the Microbiome: The PRASCO Randomized Controlled Trial. Inflamm. Bowel Dis. 2020, 26, 1733–1742. [Google Scholar] [CrossRef]
- Turner, D.; Levine, A.; Kolho, K.L.; Shaoul, R.; Ledder, O. Combination of Oral Antibiotics May Be Effective in Severe Pediatric Ulcerative Colitis: A Preliminary Report. J. Crohn’s Colitis 2014, 8, 1464–1470. [Google Scholar] [CrossRef]
- Rafii, F.; Ruseler-van Embden, J.G.H.; Van Lieshout, L.M.C. Changes in Bacterial Enzymes and PCR Profiles of Fecal Bacteria from a Patient with Ulcerative Colitis before and after Antimicrobial Treatments. Dig. Dis. Sci. 1999, 44, 637–642. [Google Scholar] [CrossRef]
- Esposito, G.; Nobile, N.; Gigli, S.; Seguella, L.; Pesce, M.; d’Alessandro, A.; Bruzzese, E.; Capoccia, E.; Steardo, L.; Cuomo, R.; et al. Rifaximin Improves Clostridium Difficile Toxin A-Induced Toxicity in Caco-2 Cells by the PXR-Dependent TLR4/MyD88/NF-ΚB Pathway. Front. Pharmacol. 2016, 7. [Google Scholar] [CrossRef]
- Jia, L.; Chen, H.; Yang, J.; Fang, X.; Niu, W.; Zhang, M.; Li, J.; Pan, X.; Ren, Z.; Sun, J.; et al. Combinatory Antibiotic Treatment Protects against Experimental Acute Pancreatitis by Suppressing Gut Bacterial Translocation to Pancreas and Inhibiting NLRP3 Inflammasome Pathway. Innate Immun. 2020, 26, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Becattini, S.; Taur, Y.; Pamer, E.G. Antibiotic-Induced Changes in the Intestinal Microbiota and Disease. Trends Mol. Med. 2016, 22, 458–478. [Google Scholar] [CrossRef] [PubMed]
- Hviid, A.; Svanström, H.; Frisch, M. Antibiotic Use and Inflammatory Bowel Diseases in Childhood. Gut 2011, 60, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Kronman, M.P.; Zaoutis, T.E.; Haynes, K.; Feng, R.; Coffin, S.E. Antibiotic Exposure and IBD Development Among Children: A Population-Based Cohort Study. Pediatrics 2012, 130, e794–e803. [Google Scholar] [CrossRef]
- Jiang, Z.-D.; Jenq, R.R.; Ajami, N.J.; Petrosino, J.F.; Alexander, A.A.; Ke, S.; Iqbal, T.; DuPont, A.W.; Muldrew, K.; Shi, Y.; et al. Safety and Preliminary Efficacy of Orally Administered Lyophilized Fecal Microbiota Product Compared with Frozen Product given by Enema for Recurrent Clostridium Difficile Infection: A Randomized Clinical Trial. PLoS ONE 2018, 13, e0205064. [Google Scholar] [CrossRef] [PubMed]
- Zellmer, C.; De Wolfe, T.J.; Van Hoof, S.; Blakney, R.; Safdar, N. Patient Perspectives on Fecal Microbiota Transplantation for Clostridium Difficile Infection. Infect. Dis. Ther. 2016, 5, 155–164. [Google Scholar] [CrossRef]
- Kassam, Z.; Lee, C.H.; Yuan, Y.; Hunt, R.H. Fecal Microbiota Transplantation for Clostridium Difficile Infection: Systematic Review and Meta-Analysis. Am. J. Gastroenterol. 2013, 108, 500–508. [Google Scholar] [CrossRef]
- Moayyedi, P.; Yuan, Y.; Baharith, H.; Ford, A.C. Faecal Microbiota Transplantation for Clostridium Difficile -associated Diarrhoea: A Systematic Review of Randomised Controlled Trials. Med. J. Aust. 2017, 207, 166–172. [Google Scholar] [CrossRef]
- Pai, N.; Popov, J.; Hill, L.; Hartung, E.; Grzywacz, K.; Moayyedi, P. Results of the First Pilot Randomized Controlled Trial of Fecal Microbiota Transplant in Pediatric Ulcerative Colitis: Lessons, Limitations, and Future Prospects. Gastroenterology 2021. [Google Scholar] [CrossRef]
- Moayyedi, P.; Surette, M.G.; Kim, P.T.; Libertucci, J.; Wolfe, M.; Onischi, C.; Armstrong, D.; Marshall, J.K.; Kassam, Z.; Reinisch, W.; et al. Fecal Microbiota Transplantation Induces Remission in Patients with Active Ulcerative Colitis in a Randomized Controlled Trial. Gastroenterology 2015, 149, 102–109.e6. [Google Scholar] [CrossRef] [PubMed]
- Rossen, N.G.; Fuentes, S.; van der Spek, M.J.; Tijssen, J.G.; Hartman, J.H.A.; Duflou, A.; Löwenberg, M.; van den Brink, G.R.; Mathus-Vliegen, E.M.H.; de Vos, W.M.; et al. Findings From a Randomized Controlled Trial of Fecal Transplantation for Patients with Ulcerative Colitis. Gastroenterology 2015, 149, 110–118.e4. [Google Scholar] [CrossRef] [PubMed]
- Paramsothy, S.; Kamm, M.A.; Kaakoush, N.O.; Walsh, A.J.; van den Bogaerde, J.; Samuel, D.; Leong, R.W.L.L.; Connor, S.; Ng, W.; Paramsothy, R.; et al. Multidonor Intensive Faecal Microbiota Transplantation for Active Ulcerative Colitis: A Randomised Placebo-Controlled Trial. Lancet 2017, 389, 1218–1228. [Google Scholar] [CrossRef]
- Costello, S.P.; Soo, W.; Bryant, R.V.; Jairath, V.; Hart, A.L.; Andrews, J.M. Systematic Review with Meta-Analysis: Faecal Microbiota Transplantation for the Induction of Remission for Active Ulcerative Colitis. Aliment. Pharmacol. Ther. 2017, 46, 213–224. [Google Scholar] [CrossRef]
- Narula, N.; Kassam, Z.; Yuan, Y.; Colombel, J.-F.; Ponsioen, C.; Reinisch, W.; Moayyedi, P. Systematic Review and Meta-Analysis: Fecal Microbiota Transplantation for Treatment of Active Ulcerative Colitis. Inflamm. Bowel Dis. 2017, 23, 1702–1709. [Google Scholar] [CrossRef]
- Popov, J.; Hartung, E.; Hill, L.; Chauhan, U.; Pai, N. Pediatric Patient and Parent Perceptions of Fecal Microbiota Transplantation for the Treatment of Ulcerative Colitis. J. Pediatr. Gastroenterol. Nutr. 2020. [Google Scholar] [CrossRef]
- Chauhan, U.; Popov, J.; Farbod, Y.; Kalantar, M.; Wolfe, M.; Moayyedi, P.; Marshall, J.K.; Halder, S.; Kaasalainen, S. Fecal Microbiota Transplantation for the Treatment of Ulcerative Colitis: A Qualitative Assessment of Patient Perceptions and Experiences. J. Can. Assoc. Gastroenterol. 2021. [Google Scholar] [CrossRef]
- Abdali, Z.I.; Roberts, T.E.; Barton, P.; Hawkey, P.M. Economic Evaluation of Faecal Microbiota Transplantation Compared to Antibiotics for the Treatment of Recurrent Clostridioides Difficile Infection. EClinicalMedicine 2020, 24, 100420. [Google Scholar] [CrossRef] [PubMed]
- Costello, S.P.; Hughes, P.A.; Waters, O.; Bryant, R.V.; Vincent, A.D.; Blatchford, P.; Katsikeros, R.; Makanyanga, J.; Campaniello, M.A.; Mavrangelos, C.; et al. Effect of Fecal Microbiota Transplantation on 8-Week Remission in Patients with Ulcerative Colitis. JAMA 2019, 321, 156. [Google Scholar] [CrossRef]
- Kellermayer, R.; Nagy-Szakal, D.; Harris, R.A.; Luna, R.A.; Pitashny, M.; Schady, D.; Mir, S.A.V.; Lopez, M.E.; Gilger, M.A.; Belmont, J.; et al. Serial Fecal Microbiota Transplantation Alters Mucosal Gene Expression in Pediatric Ulcerative Colitis. Am. J. Gastroenterol. 2015, 110, 604–606. [Google Scholar] [CrossRef] [PubMed]
- Kunde, S.; Pham, A.; Bonczyk, S.; Crumb, T.; Duba, M.; Conrad, H.; Cloney, D.; Kugathasan, S. Safety, Tolerability, and Clinical Response after Fecal Transplantation in Children and Young Adults with Ulcerative Colitis. J. Pediatr. Gastroenterol. Nutr. 2013, 56, 597–601. [Google Scholar] [CrossRef]
- Suskind, D.L.; Singh, N.; Nielson, H.; Wahbeh, G. Fecal Microbial Transplant via Nasogastric Tube for Active Pediatric Ulcerative Colitis. J. Pediatr. Gastroenterol. Nutr. 2015, 60, 27–29. [Google Scholar] [CrossRef]
- Paramsothy, S.; Nielsen, S.; Kamm, M.A.; Deshpande, N.P.; Faith, J.J.; Clemente, J.C.; Paramsothy, R.; Walsh, A.J.; van den Bogaerde, J.; Samuel, D.; et al. Specific Bacteria and Metabolites Associated with Response to Fecal Microbiota Transplantation in Patients with Ulcerative Colitis. Gastroenterology 2019, 156, 1440–1454.e2. [Google Scholar] [CrossRef]
- Pai, N.; Popov, J.; Hill, L.; Hartung, E. Protocol for a Double-Blind, Randomised, Placebo-Controlled Pilot Study for Assessing the Feasibility and Efficacy of Faecal Microbiota Transplant in a Paediatric Crohn’s Disease Population: PediCRaFT Trial. BMJ Open 2019, 9, e030120. [Google Scholar] [CrossRef] [PubMed]
- Verdier, C.; Denis, S.; Gasc, C.; Boucinha, L.; Uriot, O.; Delmas, D.; Dore, J.; Le Camus, C.; Schwintner, C.; Blanquet-diot, S. An Oral Fmt Capsule as Efficient as an Enema for Microbiota Reconstruction Following Disruption by Antibiotics, as Assessed in an in Vitro Human Gut Model. Microorganisms 2021, 9, 358. [Google Scholar] [CrossRef] [PubMed]
- Youngster, I.; Mahabamunuge, J.; Systrom, H.K.; Sauk, J.; Khalili, H.; Levin, J.; Kaplan, J.L.; Hohmann, E.L. Oral, Frozen Fecal Microbiota Transplant (FMT) Capsules for Recurrent Clostridium Difficile Infection. BMC Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kao, D.; Roach, B.; Silva, M.; Beck, P.; Rioux, K.; Kaplan, G.G.; Chang, H.-J.; Coward, S.; Goodman, K.J.; Xu, H.; et al. Effect of Oral Capsule– vs. Colonoscopy-Delivered Fecal Microbiota Transplantation on Recurrent Clostridium Difficile Infection. JAMA 2017, 318, 1985. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.H.R.; Al Bakir, I.; Ding, N.S.J.; Lee, G.H.; Askari, A.; Warusavitarne, J.; Moorghen, M.; Humphries, A.; Ignjatovic-Wilson, A.; Thomas-Gibson, S.; et al. Cumulative Burden of Inflammation Predicts Colorectal Neoplasia Risk in Ulcerative Colitis: A Large Single-Centre Study. Gut 2019, 68, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Malki, A.; Elruz, R.A.; Gupta, I.; Allouch, A.; Vranic, S.; Al Moustafa, A.E. Molecular Mechanisms of Colon Cancer Progression and Metastasis: Recent Insights and Advancements. Int. J. Mol. Sci. 2021, 22, 130. [Google Scholar] [CrossRef]
- Veettil, S.K.; Wong, T.Y.; Loo, Y.S.; Playdon, M.C.; Lai, N.M.; Giovannucci, E.L.; Chaiyakunapruk, N. Role of Diet in Colorectal Cancer Incidence: Umbrella Review of Meta-Analyses of Prospective Observational Studies. JAMA Netw. Open 2021, 4, e2037341. [Google Scholar] [CrossRef]
- Al Bakir, I.; Curtius, K.; Graham, T.A. From Colitis to Cancer: An Evolutionary Trajectory That Merges Maths and Biology. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Kang, M.; Martin, A. Microbiome and Colorectal Cancer: Unraveling Host-Microbiota Interactions in Colitis-Associated Colorectal Cancer Development. Semin. Immunol. 2017, 32, 3–13. [Google Scholar] [CrossRef]
- Ternes, D.; Karta, J.; Tsenkova, M.; Wilmes, P.; Haan, S.; Letellier, E. Microbiome in Colorectal Cancer: How to Get from Meta-Omics to Mechanism? Trends Microbiol. 2020, 28, 401–423. [Google Scholar] [CrossRef]
- Schmitt, M.; Greten, F.R. The Inflammatory Pathogenesis of Colorectal Cancer. Nat. Rev. Immunol. 2021. [Google Scholar] [CrossRef]
- Sears, C.L.; Pardoll, D.M. Perspective: Alpha-Bugs, Their Microbial Partners, and the Link to Colon Cancer. J. Infect. Dis. 2011, 203, 306–311. [Google Scholar] [CrossRef]
- Tjalsma, H.; Boleij, A.; Marchesi, J.R.; Dutilh, B.E. A Bacterial Driver-Passenger Model for Colorectal Cancer: Beyond the Usual Suspects. Nat. Rev. Microbiol. 2012, 10, 575–582. [Google Scholar] [CrossRef]
- Yu, L.C.-H.H. Microbiota Dysbiosis and Barrier Dysfunction in Inflammatory Bowel Disease and Colorectal Cancers: Exploring a Common Ground Hypothesis. J. Biomed. Sci. 2018, 25, 79. [Google Scholar] [CrossRef]
- Hernández-Luna, M.A.; López-Briones, S.; Luria-Pérez, R. The Four Horsemen in Colon Cancer. J. Oncol. 2019, 2019. [Google Scholar] [CrossRef]
- Chung, L.; Thiele Orberg, E.; Geis, A.L.; Chan, J.L.; Fu, K.; DeStefano Shields, C.E.; Dejea, C.M.; Fathi, P.; Chen, J.; Finard, B.B.; et al. Bacteroides Fragilis Toxin Coordinates a Pro-Carcinogenic Inflammatory Cascade via Targeting of Colonic Epithelial Cells. Cell Host Microbe 2018, 23, 203–214.e5. [Google Scholar] [CrossRef] [PubMed]
- Dejea, C.M.; Fathi, P.; Craig, J.M.; Boleij, A.; Taddese, R.; Geis, A.L.; Wu, X.; DeStefano Shields, C.E.; Hechenbleikner, E.M.; Huso, D.L.; et al. Patients with Familial Adenomatous Polyposis Harbor Colonic Biofilms Containing Tumorigenic Bacteria. Science 2018, 359, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.H.; Dejea, C.M.; Edler, D.; Hoang, L.T.; Santidrian, A.F.; Felding, B.H.; Ivanisevic, J.; Cho, K.; Wick, E.C.; Hechenbleikner, E.M.; et al. Metabolism Links Bacterial Biofilms and Colon Carcinogenesis. Cell Metab. 2015, 21, 891–897. [Google Scholar] [CrossRef]
- Tomkovich, S.; Dejea, C.M.; Winglee, K.; Drewes, J.L.; Chung, L.; Housseau, F.; Pope, J.L.; Gauthier, J.; Sun, X.; Mühlbauer, M.; et al. Human Colon Mucosal Biofilms from Healthy or Colon Cancer Hosts Are Carcinogenic. J. Clin. Investig. 2019, 129, 1699–1712. [Google Scholar] [CrossRef]
- Chew, S.S.; Tan, L.T.H.; Law, J.W.F.; Pusparajah, P.; Goh, B.H.; Ab Mutalib, N.S.; Lee, L.H. Targeting Gut Microbial Biofilms—A Key to Hinder Colon Carcinogenesis? Cancers 2020, 12, 2272. [Google Scholar] [CrossRef] [PubMed]
- Ranjbar, M.; Salehi, R.; Haghjooy Javanmard, S.; Rafiee, L.; Faraji, H.; Jafarpor, S.; Ferns, G.A.; Ghayour-Mobarhan, M.; Manian, M.; Nedaeinia, R. The Dysbiosis Signature of Fusobacterium Nucleatum in Colorectal Cancer-Cause or Consequences? A Systematic Review. Cancer Cell Int. 2021, 21, 194. [Google Scholar] [CrossRef]
- Proença, M.A.; Biselli, J.M.; Succi, M.; Severino, F.E.; Berardinelli, G.N.; Caetano, A.; Reis, R.M.; Hughes, D.J.; Silva, A.E. Relationship between Fusobacterium Nucleatum, Inflammatory Mediators and MicroRNAs in Colorectal Carcinogenesis. World J. Gastroenterol. 2018, 24, 5351–5365. [Google Scholar] [CrossRef]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium Nucleatum Promotes Colorectal Carcinogenesis by Modulating E-Cadherin/β-Catenin Signaling via Its FadA Adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Tahara, T.; Hirata, I.; Nakano, N.; Tahara, S.; Horiguchi, N.; Kawamura, T.; Okubo, M.; Ishizuka, T.; Yamada, H.; Yoshida, D.; et al. Potential Link between Fusobacterium Enrichment and DNA Methylation Accumulation in the Inflammatory Colonic Mucosa in Ulcerative Colitis. Oncotarget 2017, 8, 61917–61926. [Google Scholar] [CrossRef]
- Chiloeches, M.L.; Bergonzini, A.; Frisan, T. Bacterial Toxins Are a Never-Ending Source of Surprises: From Natural Born Killers to Negotiators. Toxins 2021, 13, 426. [Google Scholar] [CrossRef]
- Arthur, J.C.; Perez-Chanona, E.; Mühlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal Inflammation Targets Cancer-Inducing Activity of the Microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef]
- Long, X.; Wong, C.C.; Tong, L.; Chu, E.S.H.; Ho Szeto, C.; Go, M.Y.Y.; Coker, O.O.; Chan, A.W.H.; Chan, F.K.L.; Sung, J.J.Y.; et al. Peptostreptococcus Anaerobius Promotes Colorectal Carcinogenesis and Modulates Tumour Immunity. Nat. Microbiol. 2019, 4, 2319–2330. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Hua, W.; Li, C.; Chang, H.; Liu, R.; Ni, Y.; Sun, H.; Li, Y.; Wang, X.; Hou, M.; et al. Protective Role of Fecal Microbiota Transplantation on Colitis and Colitis-Associated Colon Cancer in Mice Is Associated with Treg Cells. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef]
- Li, X.; Liu, Y.; Wang, Y.; Li, X.; Liu, X.; Guo, M.; Tan, Y.; Qin, X.; Wang, X.; Jiang, M. Sucralose Promotes Colitis-Associated Colorectal Cancer Risk in a Murine Model Along with Changes in Microbiota. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Richard, M.L.; Liguori, G.; Lamas, B.; Brandi, G.; da Costa, G.; Hoffmann, T.W.; Pierluigi Di Simone, M.; Calabrese, C.; Poggioli, G.; Langella, P.; et al. Mucosa-Associated Microbiota Dysbiosis in Colitis Associated Cancer. Gut Microbes 2018, 9, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.W.; Du, L.T.; Li, W.; Yang, Y.M.; Zhang, Y.; Wang, C.X. Biodiversity and Richness Shifts of Mucosa-Associated Gut Microbiota with Progression of Colorectal Cancer. Res. Microbiol. 2020, 171, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Cleynen, I.; Boucher, G.; Jostins, L.; Schumm, L.P.; Zeissig, S.; Ahmad, T.; Andersen, V.; Andrews, J.M.; Annese, V.; Brand, S.; et al. Inherited Determinants of Crohn’s Disease and Ulcerative Colitis Phenotypes: A Genetic Association Study. Lancet 2016, 387, 156–167. [Google Scholar] [CrossRef]
- Perillo, F.; Amoroso, C.; Strati, F.; Giuffrè, M.R.; Díaz-Basabe, A.; Lattanzi, G.; Facciotti, F. Gut Microbiota Manipulation as a Tool for Colorectal Cancer Management: Recent Advances in Its Use for Therapeutic Purposes. Int. J. Mol. Sci. 2020, 21, 5389. [Google Scholar] [CrossRef]
- Fong, W.; Li, Q.; Yu, J. Gut Microbiota Modulation: A Novel Strategy for Prevention and Treatment of Colorectal Cancer. Oncogene 2020, 39, 4925–4943. [Google Scholar] [CrossRef]
- Chang, C.W.; Lee, H.C.; Li, L.H.; Chiau, J.S.C.; Wang, T.E.; Chuang, W.H.; Chen, M.J.; Wang, H.Y.; Shih, S.C.; Liu, C.Y.; et al. Fecal Microbiota Transplantation Prevents Intestinal Injury, Upregulation of Toll-like Receptors, and 5-Fluorouracil/Oxaliplatin-Induced Toxicity in Colorectal Cancer. Int. J. Mol. Sci. 2020, 21, 386. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).