
Comprehensive Identification of Deleterious TP53 Missense VUS Variants Based on Their Impact on TP53 Structural Stability



Abstract
:
1. Introduction
2. Results
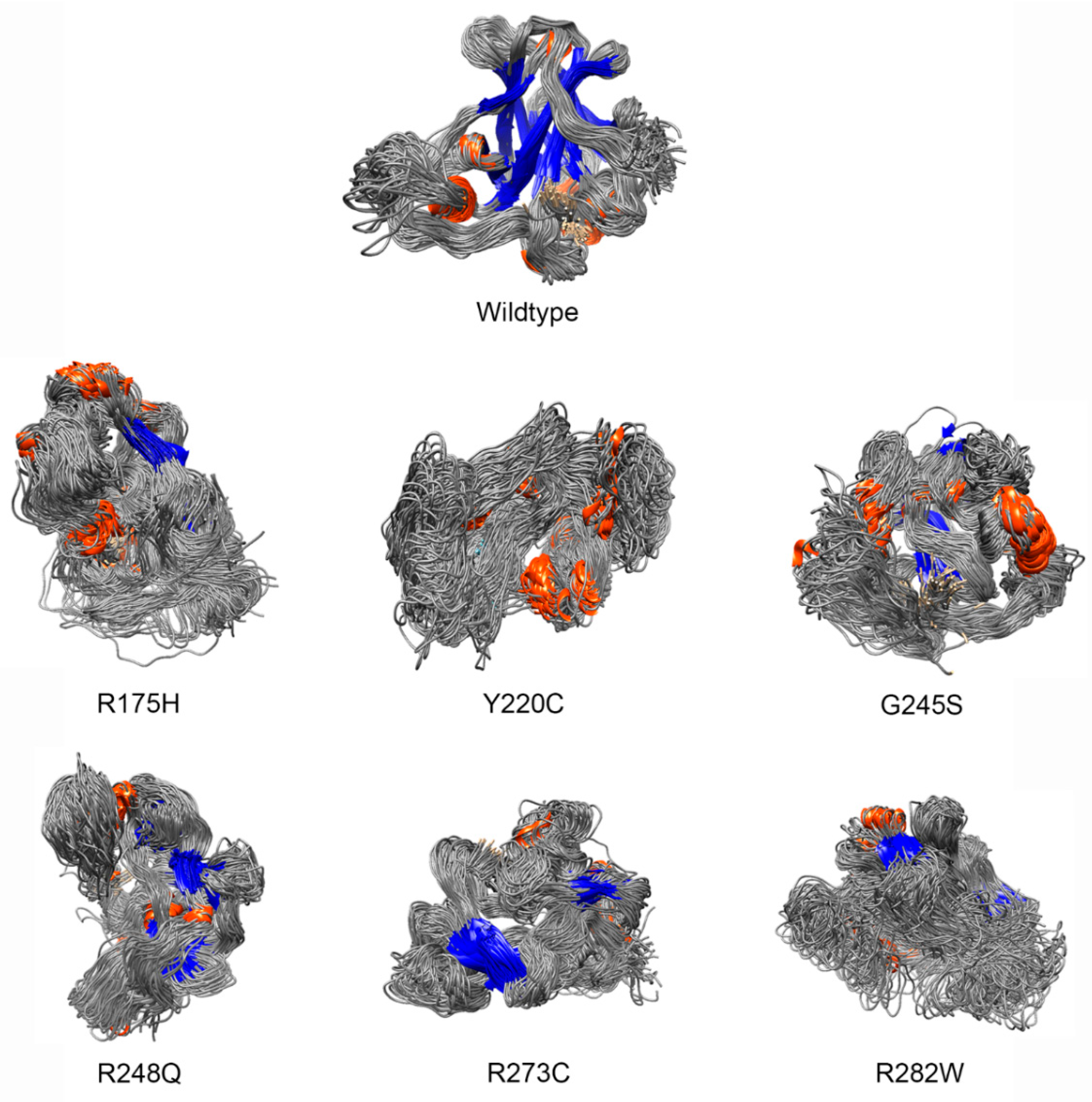
2.1. Construction of Mutant Protein Structures
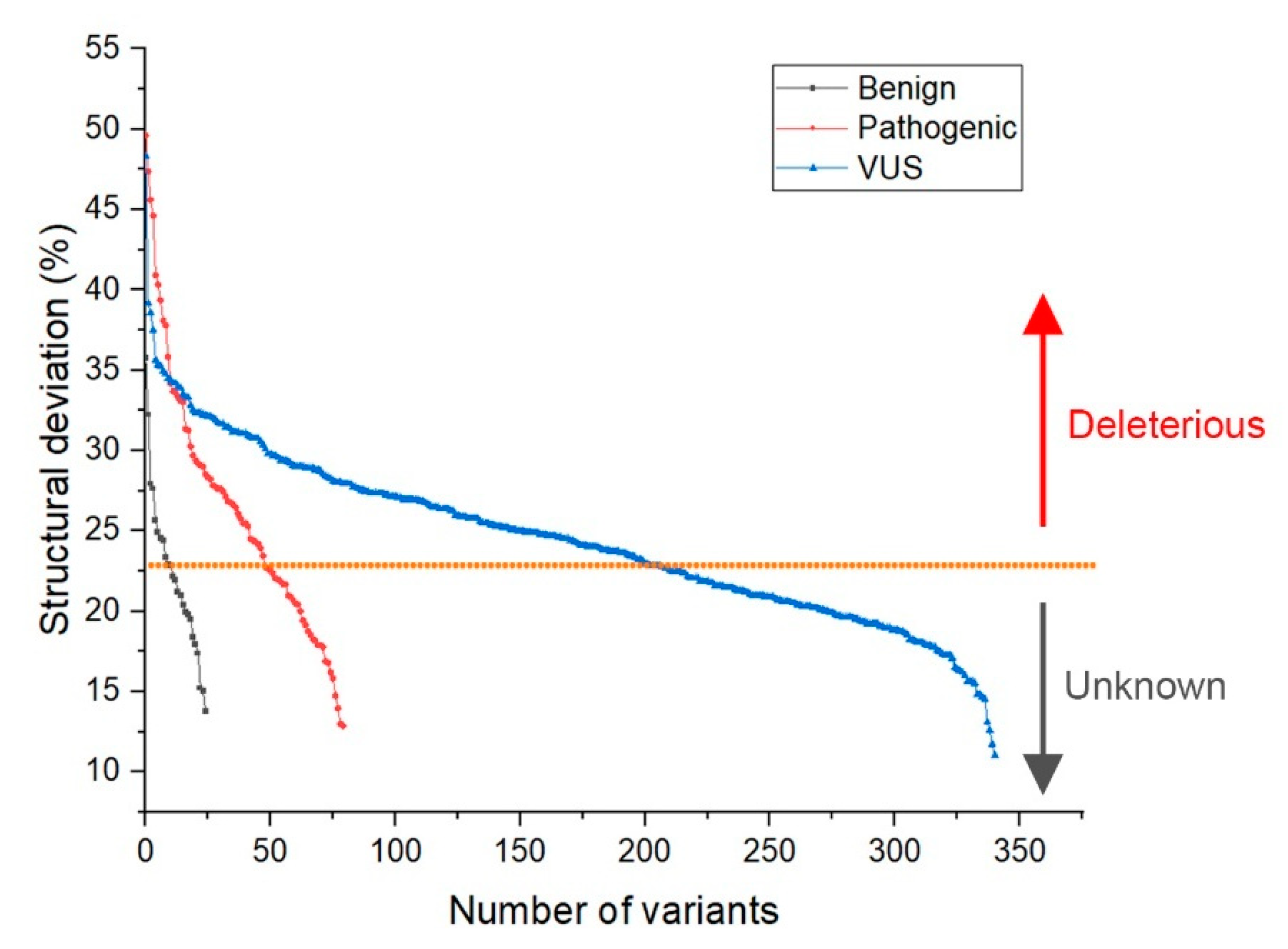
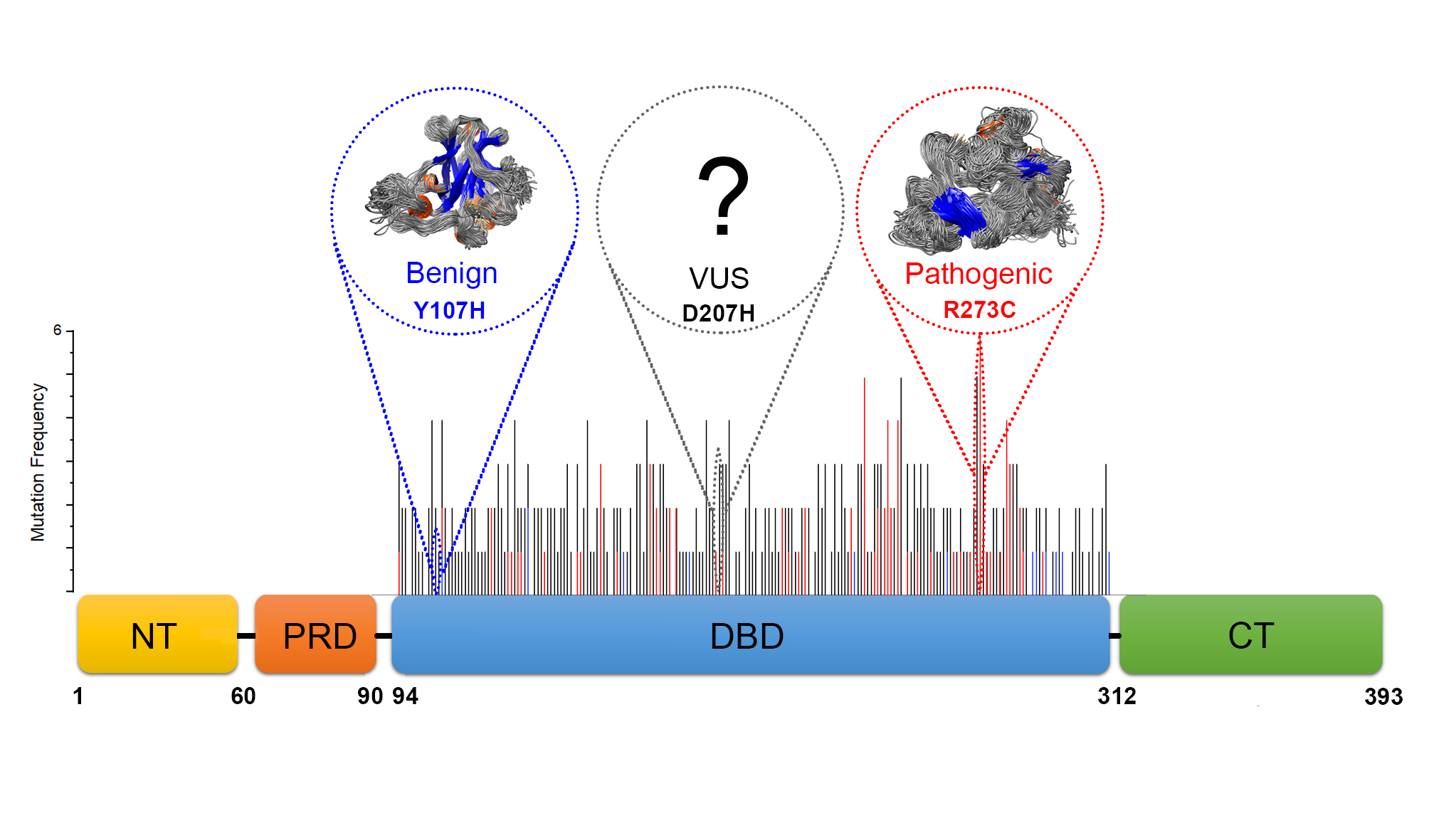
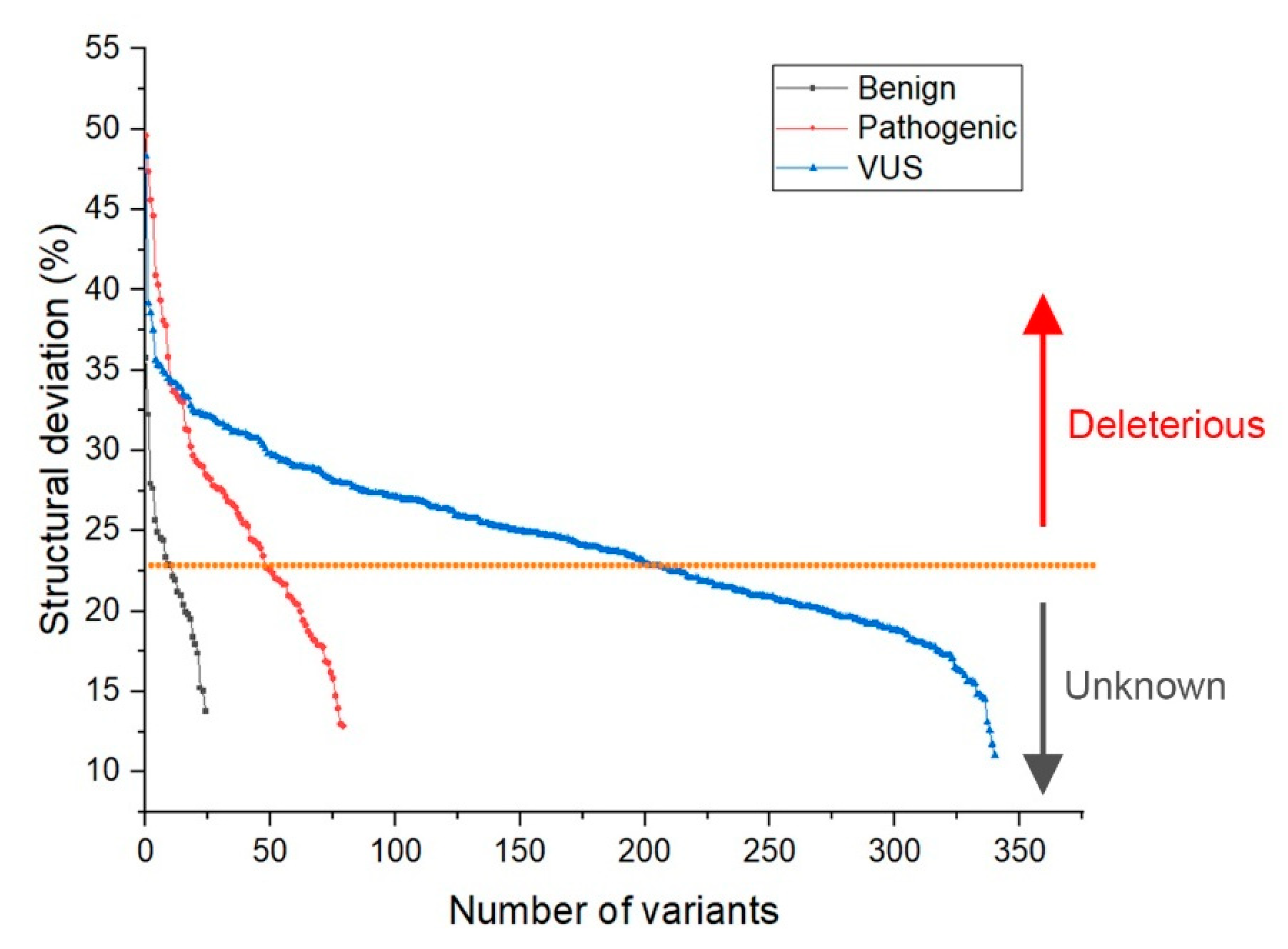
2.2. Identification of Deleterious Variants
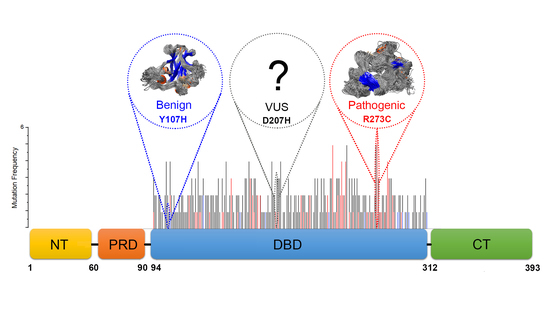
2.3. Features of Deleterious VUS
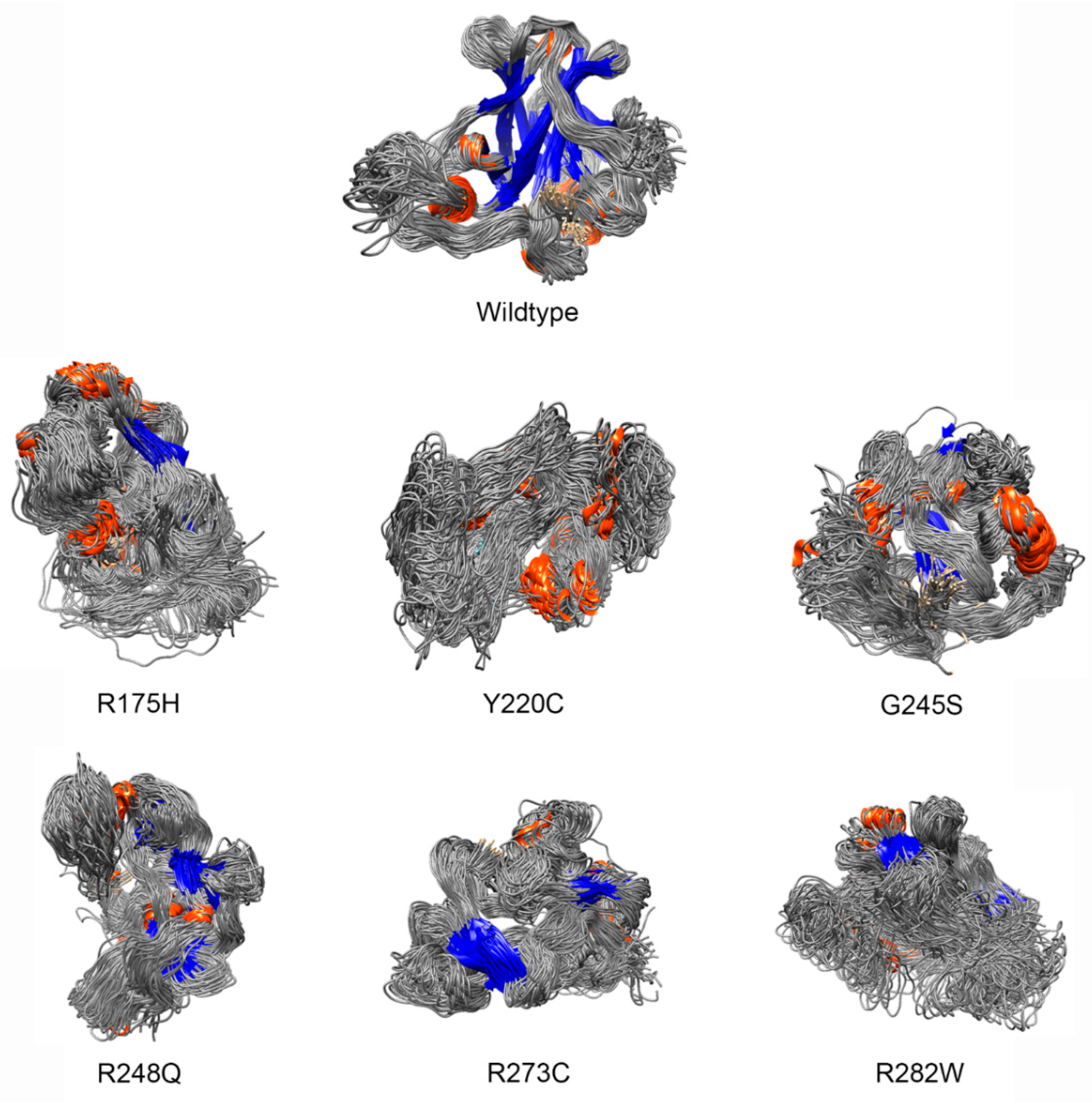
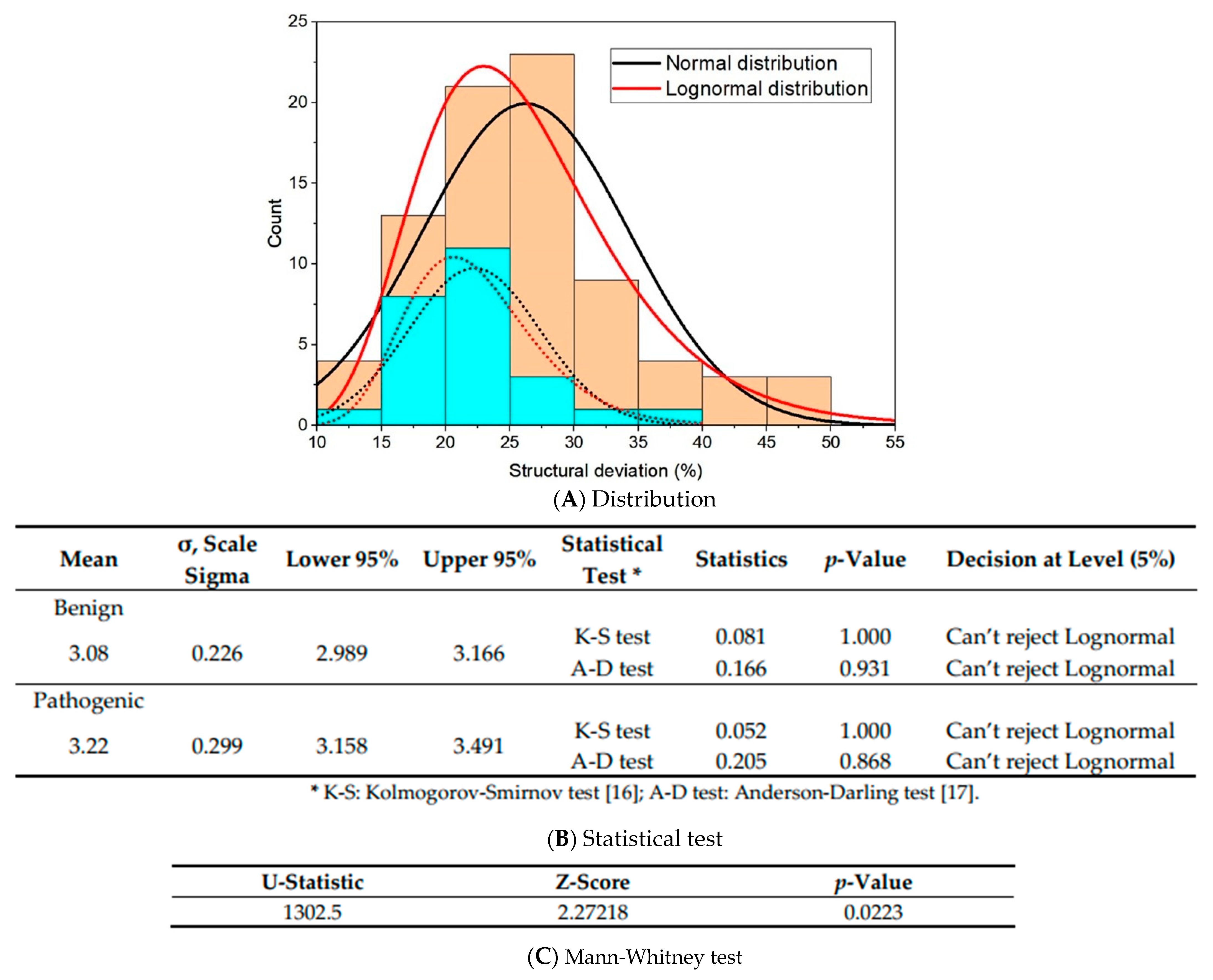
- Deviation distribution. The range of deviation was 3.158–3.878 (23.5–48.3%) (Figure 3). This range was similar to that of known pathogenic variants, except for the top 3—R273C, Y220C, R175H (49.6, 47.4, 45.6%)—as they were highly destructive for the TP53 structure (Figure 2). For benign variants, 6 out of 23 benign variants were above the deleterious structure limits (>23.5%). Here, the Q165K had a structure deviation of 35.7% and was an outlier within the benign variants. The variants below the cutoff line were classified as “unknown”, as certain pathogenic variants may have minimal impact on structure stability [19]; therefore, their overall structure scaffolds can be comparable to benign variants. Overall, the results justified the use of the deviation from known pathogenic variants as the reference to classify the missense VUS.
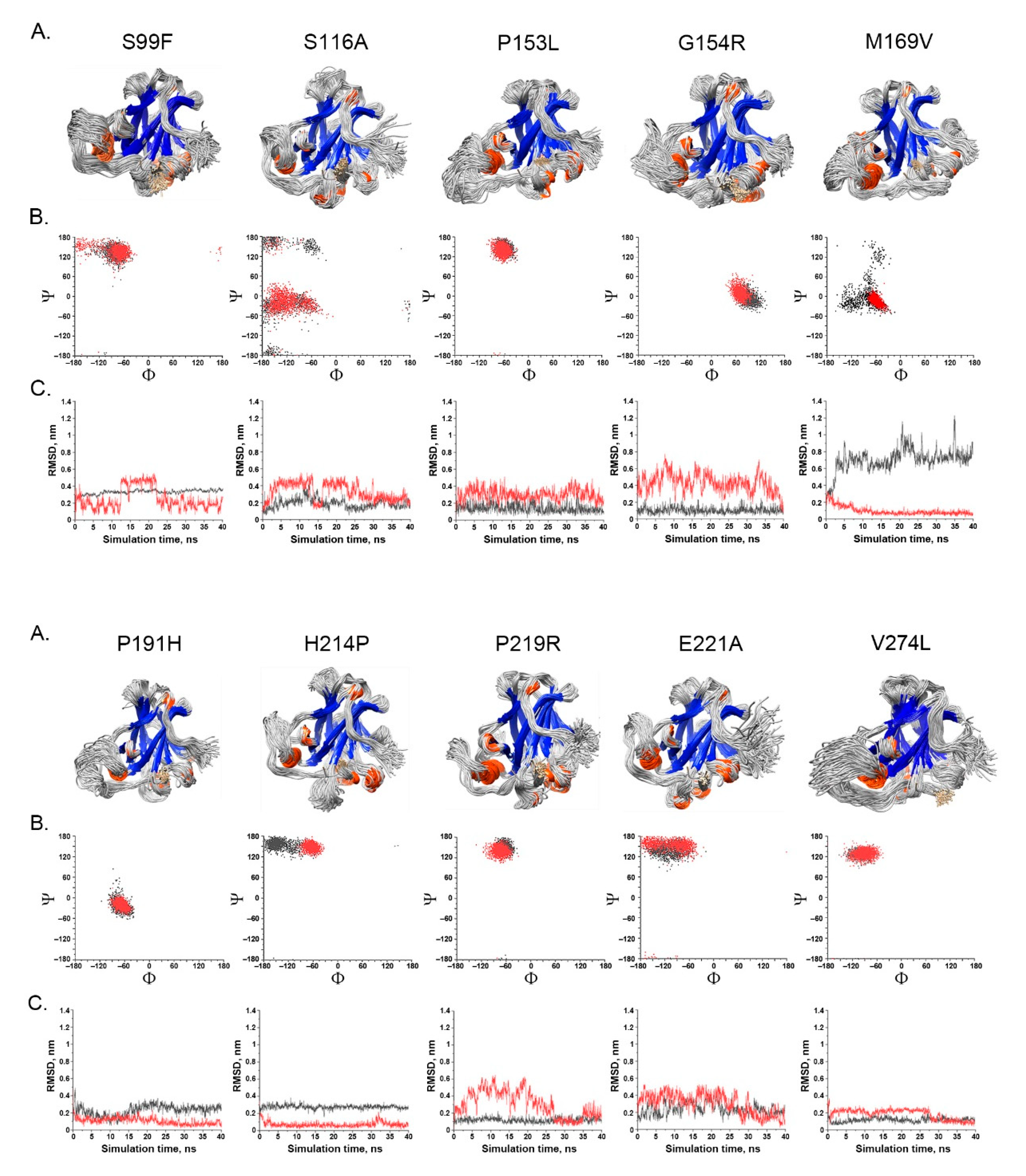
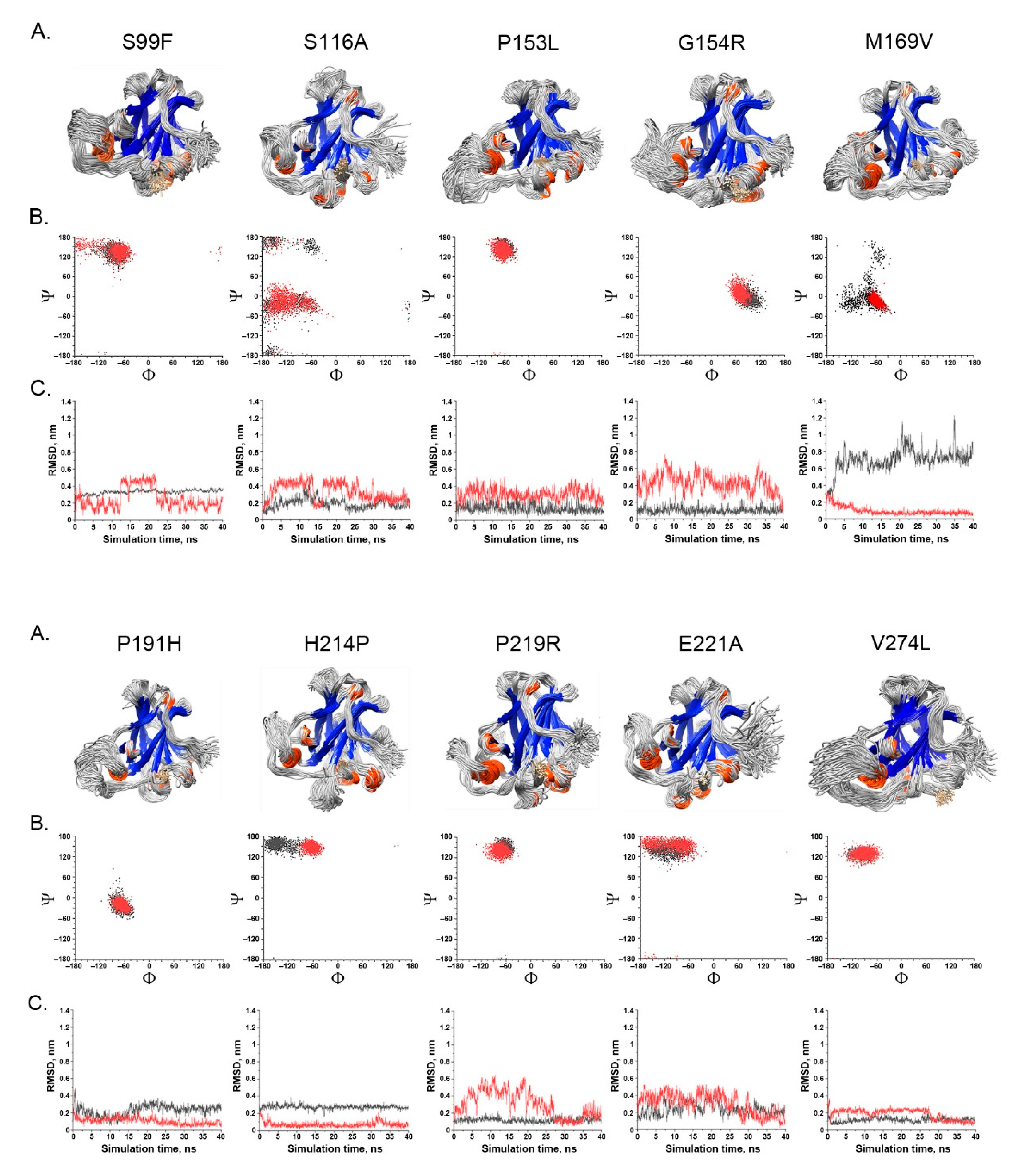
- Spatial change of the substituted residues. The Ramachandran plot (RP) showed the spatial differences of the substituted residues from the wildtype residues, and the root-mean-square-deviation (RMSD) plot also showed the altered position of the substituted residues from the wildtype residues in the global TP53 structure (Figure 4A–C). For example, in S99F, F in RP showed its torsional angle in reflecting the rigidity of the fluctuation, and F in RMSD also showed its larger fluctuation, revealing its instability in the local environment; in G154R, R in RP showed its torsional angle fluctuation deviated from the wildtype G, and R in RMSD showed its large fluctuation, reflecting its instability in its local environment; in H214P, P in RP showed its torsional angle substantially fluctuated from the wildtype residue H, and the lower RMSD showed its high stability in TP53.
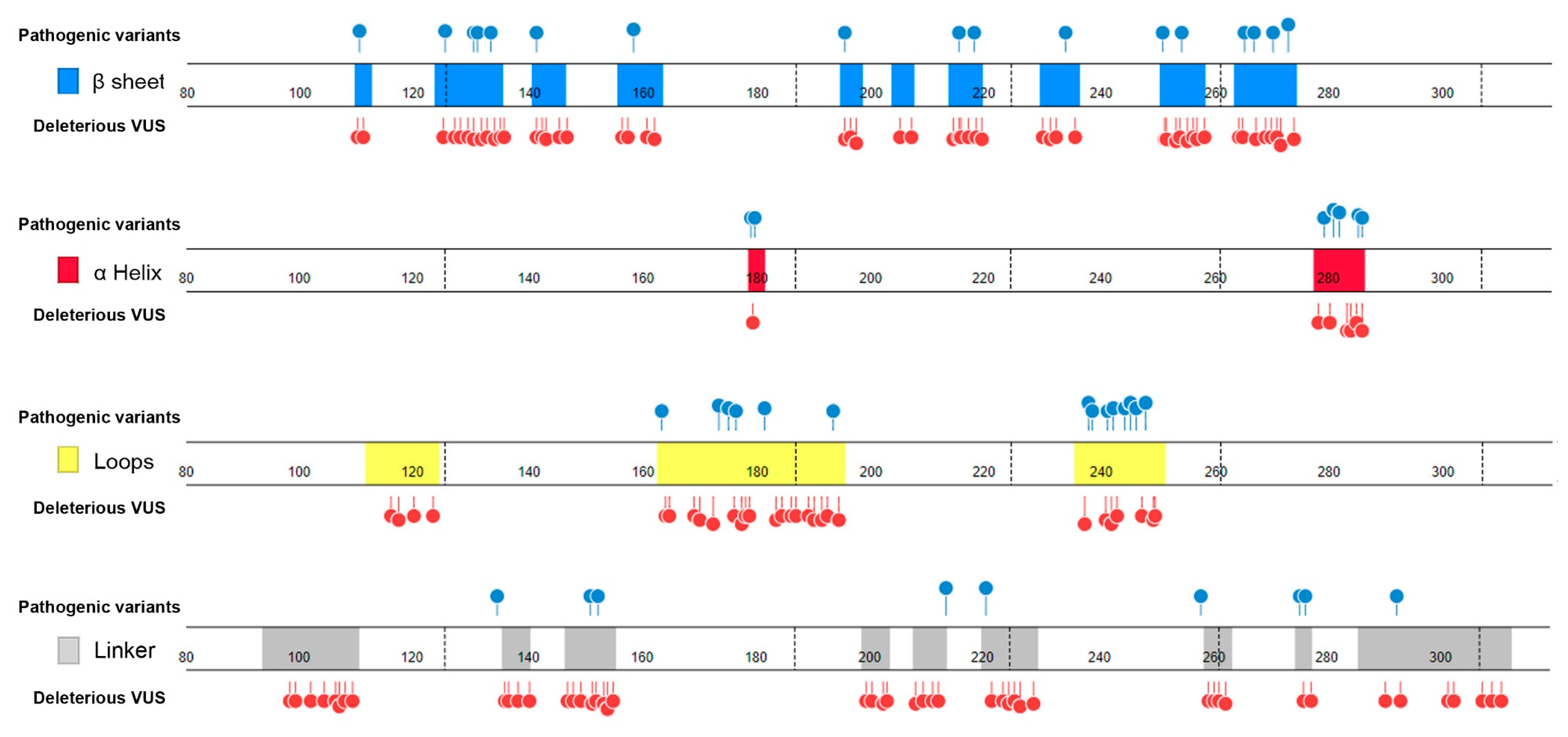
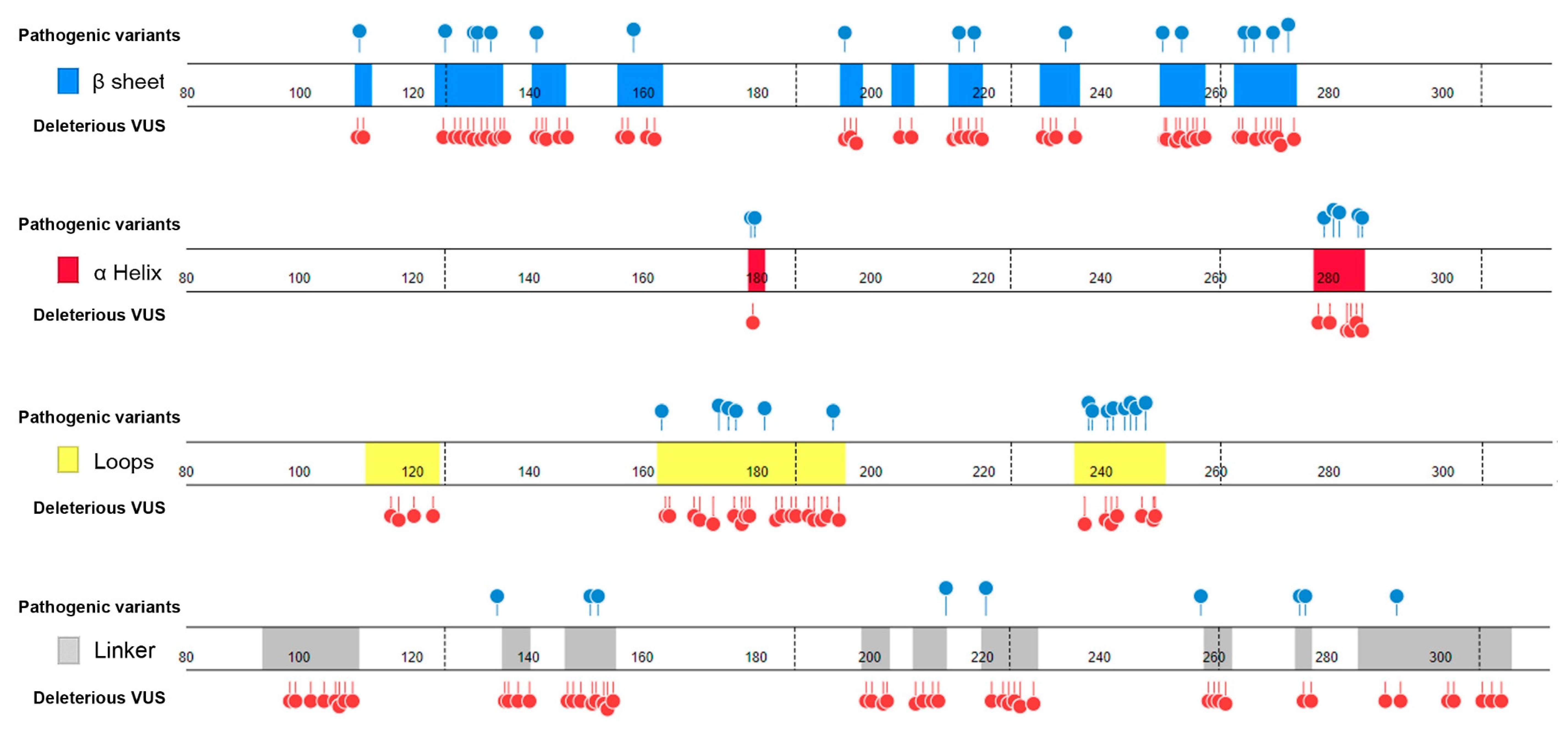
- Distribution in TP53 secondary structure. The deleterious VUS variants were distributed across the entire DNA binding domain, of which 44 were in the regions with few known pathogenic variants, including entire β sheets, loop 1 and loop 2, and all linkers; for the regions overlapped with the known pathogenic variants, the deleterious VUS variants were distributed more widely than the known pathogenic variants (Figure 5). The results indicate that RPMDS provides high sensitivity to detect deleterious missense VUS variants.
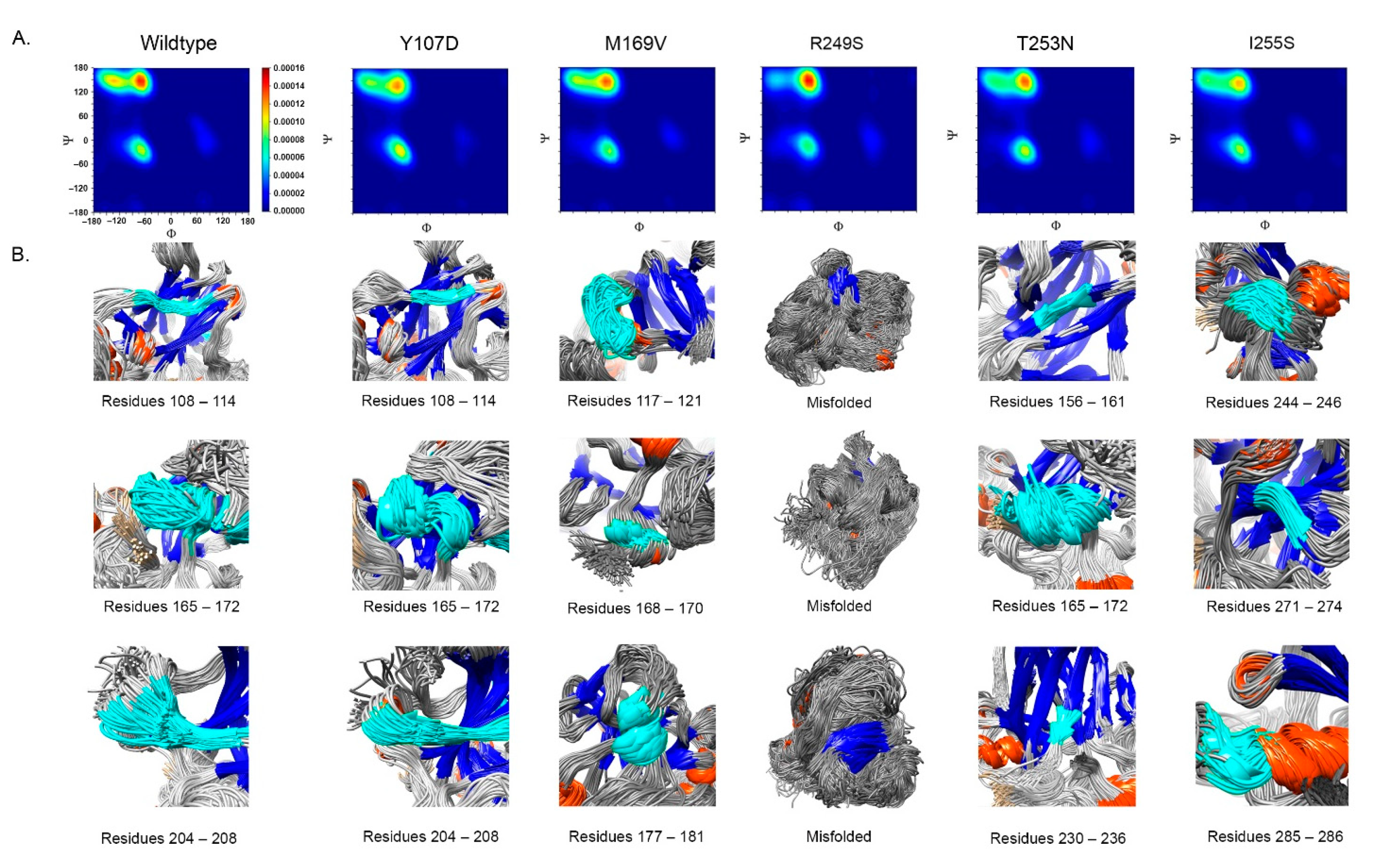
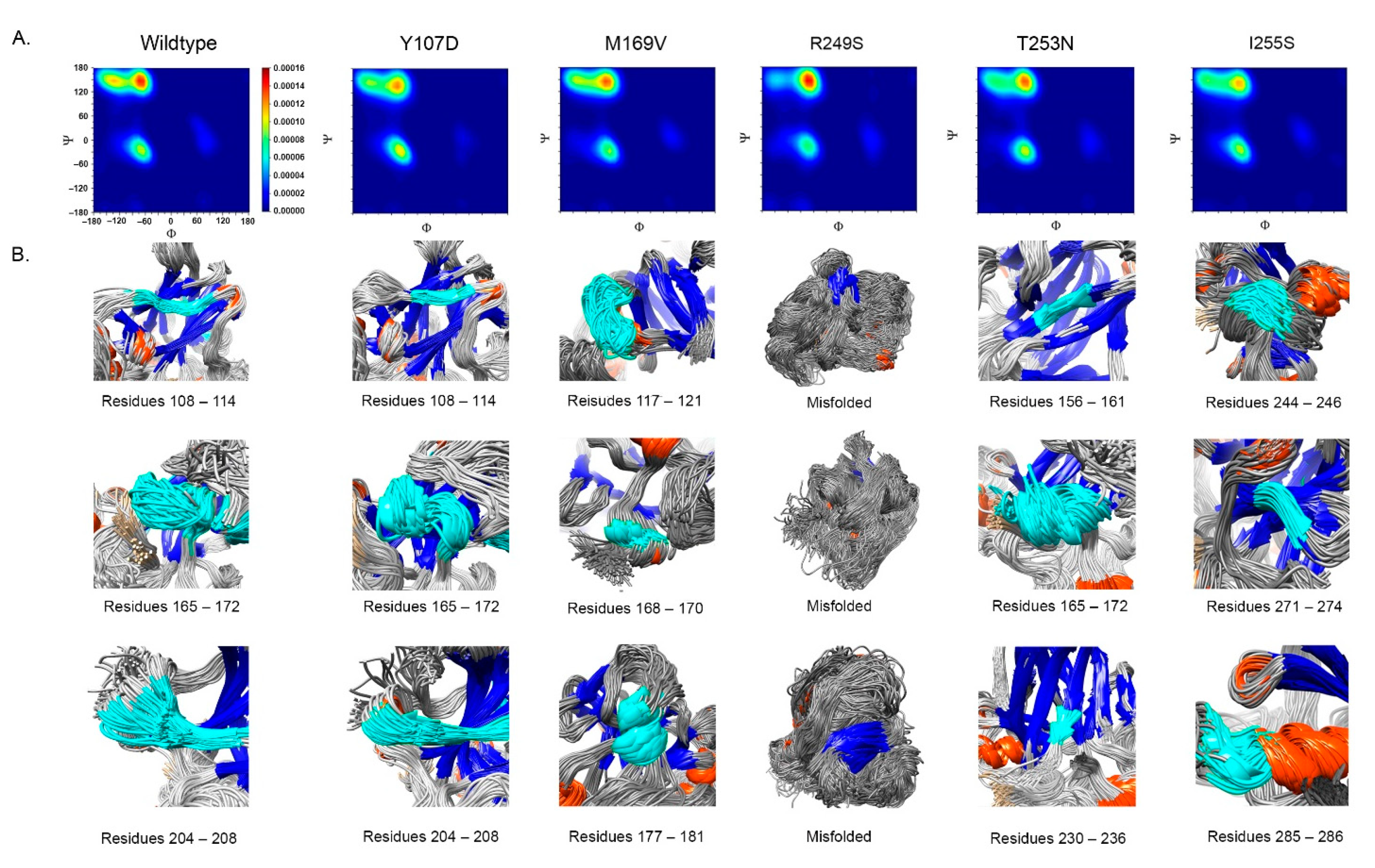
- Impact on TP53 local structure. The Ramachandran density plots showed that the deleterious VUS variants caused more local structural change, whereas known pathogenic variants caused more global structural change (Figure 6A). Taking Y107D, M169V, R249S, T253N, and I255S as examples (Figure 6B): in Y107D, the change of residue caused greater flexibility in the β strand (residues 108–114 (β1) and 204–208 (β6)); this was reflected by the diminishing peak in the Ramachandran density plot: an α helix loop was formed between residues 165–172, and this was not observed in WT; in M169V, the flexible structure was reflected by the diminished peaks at P-II and the β sheet region in the Ramachandran density plot. The extra α-helix bend was formed at residues 117–121 and 168–170, and α-helix 1 (177–181) was more structurally stable than the WT α-helix 1 (H1); with its high deviation of 3.770 (43.4%), R249S misfolded the TP53 structure globally, and the Ramachandran plot revealed a spike at the P-II region, while the β sheet region was diminished; in T253N, greater flexibility of β strand S4 and shortened β strand S8 (residues 156–161 and 230–236, respectively) reflected the diminished peak of the Ramachandran density plot; the residues 165–172 showed a stable structural formation, and this was not detected in the WT; in I255S, the Ramachandran density plot revealed the dissipated peak at the β sheet and P-II spiral regions; higher flexibility was present at the linker residues (244–246) and α-helix 2 (H2) (285–286), and S10 β strand (271–274) was extended by 2 residues.
2.4. Comparison with Other In Silico Methods
3. Discussion
4. Materials and Methods
4.1. Source of Missense VUS Variants
4.2. Molecular Dynamics Simulation (MDS)
4.3. Ramachandran Plot (RP)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Levine, A.J. p53, the Cellular Gatekeeper for Growth and Division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef] [Green Version]
- Fridman, J.S.; Lowe, S.W. Control of apoptosis by p53. Oncogene 2003, 22, 9030–9040. [Google Scholar] [CrossRef] [Green Version]
- Haupt, S.; Berger, M.; Goldberg, Z.; Haupt, Y. Apoptosis—The p53 network. J. Cell Sci. 2003, 116, 4077–4085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petitjean, A.; Mathe, E.; Kato, S.; Ishioka, C.; Tavtigian, S.V.; Hainaut, P.; Olivier, M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum. Mutat. 2007, 28, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Li, F.P.; Fraumeni, J.F., Jr.; Mulvihill, J.J.; Blattner, W.A.; Dreyfus, M.G.; Tucker, M.A.; Miller, R.W. A cancer family syndrome in twenty-four kindreds. Cancer Res. 1988, 48, 5358–5362. [Google Scholar] [PubMed]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vos, J.; Otten, W.; van Asperen, C.; Jansen, A.; Menko, F.; Tibben, A. The counsellees’ view of an unclassified variant in BRCA1/2: Recall, interpretation, and impact on life. Psychooncology 2008, 17, 822–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macklin, S.; Durand, N.; Atwal, P.; Hines, S. Observed frequency and challenges of variant reclassification in a hereditary cancer clinic. Genet. Med. 2018, 20, 346–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tung, N.; Lin, N.U.; Kidd, J.; Allen, B.A.; Singh, N.; Wenstrup, R.J.; Hartman, A.R.; Winer, E.P.; Garber, J.E. Frequency of Germline Mutations in 25 Cancer Susceptibility Genes in a Sequential Series of Patients With Breast Cancer. J. Clin. Oncol. 2016, 34, 1460–1468. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.; Richards, F.M. The interpretation of protein structures: Estimation of static accessibility. J. Mol. Biol. 1971, 55, 379–400. [Google Scholar] [CrossRef]
- McDonald, I.K.; Thornton, J.M. Satisfying Hydrogen Bonding Potential in Proteins. J. Mol. Biol. 1994, 238, 777–793. [Google Scholar] [CrossRef]
- Martin, A.C.; Facchiano, A.M.; Cuff, A.L.; Hernandez-Boussard, T.; Olivier, M.; Hainaut, P.; Thornton, J.M. Integrating mutation data and structural analysis of the TP53 tumor-suppressor protein. Hum. Mutat. 2002, 19, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Tam, B.; Sinha, S.; Wang, S.M. Combining Ramachandran plot and molecular dynamics simulation for structural-based variant classification: Using TP53 variants as model. Comput. Struct. Biotechnol 2020, 18, 4033–4039. [Google Scholar] [CrossRef] [PubMed]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Rosengarth, A.; Luecke, H. Structure of the human p53 core domain in the absence of DNA. Acta Crystallogr. Sect. D 2007, 63, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Massey, F.J. The Kolmogorov-Smirnov Test for Goodness of Fit. J. Am. Stat. Assoc. 1951, 46, 68–78. [Google Scholar] [CrossRef]
- Anderson, T.W.; Darling, D.A. Asymptotic Theory of Certain “Goodness of Fit” Criteria Based on Stochastic Processes. Ann. Math. Statist. 1952, 23, 193–212. [Google Scholar] [CrossRef]
- Mann, H.B.; Whitney, D.R. On a Test of Whether one of Two Random Variables is Stochastically Larger than the Other. Ann. Math. Stat. 1947, 18, 50–60. [Google Scholar] [CrossRef]
- Joerger, A.C.; Fersht, A.R. Structure–function–rescue: The diverse nature of common p53 cancer mutants. Oncogene 2007, 26, 2226–2242. [Google Scholar] [CrossRef] [Green Version]
- Findlay, G.M.; Boyle, E.A.; Hause, R.J.; Klein, J.C.; Shendure, J. Saturation editing of genomic regions by multiplex homology-directed repair. Nature 2014, 513, 120–123. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Li, G.; Wu, J.; Liang, J.; Wang, X. Identification of pathogenic variants in cancer genes using base editing screens with editing efficiency correction. Genome Biol. 2021, 22, 80. [Google Scholar] [CrossRef]
- Starita, L.M.; Islam, M.M.; Banerjee, T.; Adamovich, A.I.; Gullingsrud, J.; Fields, S.; Shendure, J.; Parvin, J.D. A Multiplex Homology-Directed DNA Repair Assay Reveals the Impact of More Than 1,000 BRCA1 Missense Substitution Variants on Protein Function. Am. J. Hum. Genet. 2018, 103, 498–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammadi, L.; Vreeswijk, M.P.; Oldenburg, R.; van den Ouweland, A.; Oosterwijk, J.C.; van der Hout, A.H.; Hoogerbrugge, N.; Ligtenberg, M.; Ausems, M.G.; van der Luijt, R.B.; et al. A simple method for co-segregation analysis to evaluate the pathogenicity of unclassified variants; BRCA1 and BRCA2 as an example. BMC Cancer 2009, 9, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jasin, M. Homologous repair of DNA damage and tumorigenesis:the BRCA connection. Oncogene 2002, 21, 8981–8993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindor, N.M.; Guidugli, L.; Wang, X.; Vallée, M.P.; Monteiro, A.N.; Tavtigian, S.; Goldgar, D.E.; Couch, F.J. A review of a multifactorial probability-based model for classification of BRCA1 and BRCA2 variants of uncertain significance (VUS). Hum. Mutat. 2012, 33, 8–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCoy, M.D.; Madhavan, S.; Nimmagadda, S.; Klimov, D.; Jafri, M.S. Translational Applications of Protein Structure Simulation: Predicting Phenotype of Missense Variants. Biophys. J. 2019, 116 (Suppl. 1), 13a. [Google Scholar] [CrossRef] [Green Version]
- Woods, N.T.; Baskin, R.; Golubeva, V.; Jhuraney, A.; De-Gregoriis, G.; Vaclova, T.; Goldgar, D.E.; Couch, F.J.; Carvalho, M.A.; Iversen, E.S.; et al. Functional assays provide a robust tool for the clinical annotation of genetic variants of uncertain significance. NPJ Genom. Med. 2016, 1, 16001. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2014, 24, 2125–2137. [Google Scholar] [CrossRef] [Green Version]
- Cooper, G.M.; Stone, E.A.; Asimenos, G.; Green, E.D.; Batzoglou, S.; Sidow, A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005, 15, 901–913. [Google Scholar] [CrossRef] [Green Version]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.A.; Edwards, K.J.; Day, I.N.M.; Gaunt, T.R. Predicting the Functional, Molecular, and Phenotypic Consequences of Amino Acid Substitutions using Hidden Markov Models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jhong, J.-H.; Lee, J.; Koo, J.-Y. Meta-analytic support vector machine for integrating multiple omics data. BioData Min. 2017, 10, 2. [Google Scholar] [CrossRef] [Green Version]
- Tavtigian, S.V.; Greenblatt, M.S.; Lesueur, F.; Byrnes, G.B. In silico analysis of missense substitutions using sequence-alignment based methods. Hum. Mutat. 2008, 29, 1327–1336. [Google Scholar] [CrossRef] [Green Version]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C. Stop explaining black box machine learning models for high stakes decisions and use interpretable models instead. Nat. Mach. Intell. 2019, 1, 206–215. [Google Scholar] [CrossRef] [Green Version]
- Papadimitriou, S.; Gazzo, A.; Versbraegen, N.; Nachtegael, C.; Aerts, J.; Moreau, Y.; Van Dooren, S.; Nowé, A.; Smits, G.; Lenaerts, T. Predicting disease-causing variant combinations. Proc. Natl. Acad. Sci. USA 2019, 116, 11878–11887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Šali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. Int. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Sorin, E.J.; Pande, V.S. Exploring the Helix-Coil Transition via All-Atom Equilibrium Ensemble Simulations. Biophys. J. 2005, 88, 2472–2493. [Google Scholar] [CrossRef] [Green Version]
- Hess, B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) Comparison with Updated ClinVar Classification | ||||
| Variant | ClinVar Classification | RPMDS Classification | ||
| Nucleotide | Amino Acid | Original | New | |
| c.706T>G | Y236D | VUS | Pathogenic | Deleterious |
| c.695T>A | I232N | VUS | Pathogenic | Deleterious |
| c.794T>A | L265Q | VUS | Pathogenic | Deleterious |
| c.413C>T | A138V | VUS | Conflict | Deleterious |
| c.422G>T | C141F | VUS | Conflict | Deleterious |
| c.434T>C | L145P | VUS | Conflict | Deleterious |
| c.526T>A | C176S | VUS | Conflict | Deleterious |
| c.556G>A | D186N | VUS | Conflict | Deleterious |
| c.581T>G | L194R | VUS | Conflict | Deleterious |
| c.626G>A | R209K | VUS | Conflict | Deleterious |
| c.814G>A | V272M | VUS | Conflict | Deleterious |
| c.931A>C | N311H | VUS | Conflict | Deleterious |
| c.431A>T | Q144L | VUS | Conflict | Undefined |
| c.452C>G | P151R | VUS | Conflict | Undefined |
| c.658T>C | Y220H | VUS | Conflict | Undefined |
| c.730G>T | G244C | VUS | Conflict | Undefined |
| c.928A>G | N310D | VUS | Conflict | Undefined |
| (B) Comparison with Different In Silico Methods in Classifying 340 Missense VUS | ||||
| Methods | Classification | |||
| Deleterious * | Rate (%) | Undefined ** | Rate (%) | |
| RPMDS | 193 | 56.8 | 147 | 43.2 |
| Polyphen2_HVAR_pred | 202 | 59.4 | 138 | 40.6 |
| Polyphen2_HDIV_pred | 217 | 62.9 | 123 | 37.1 |
| PROVEAN_pred | 233 | 68.5 | 107 | 31.5 |
| LRT_pred | 242 | 71.2 | 98 | 28.8 |
| SIFT_pred | 268 | 78.8 | 72 | 21.2 |
| MutationTaster_pred | 287 | 84.4 | 53 | 15.6 |
| M-CAP_pred | 335 | 98.5 | 5 | 1.5 |
| FATHMM_pred | 340 | 100 | 0 | 0 |
| MetaSVM_pred | 340 | 100 | 0 | 0 |
| MetaLR_pred | 340 | 100 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tam, B.; Sinha, S.; Qin, Z.; Wang, S.M. Comprehensive Identification of Deleterious TP53 Missense VUS Variants Based on Their Impact on TP53 Structural Stability. Int. J. Mol. Sci. 2021, 22, 11345. https://doi.org/10.3390/ijms222111345
Tam B, Sinha S, Qin Z, Wang SM. Comprehensive Identification of Deleterious TP53 Missense VUS Variants Based on Their Impact on TP53 Structural Stability. International Journal of Molecular Sciences. 2021; 22(21):11345. https://doi.org/10.3390/ijms222111345
Chicago/Turabian StyleTam, Benjamin, Siddharth Sinha, Zixin Qin, and San Ming Wang. 2021. "Comprehensive Identification of Deleterious TP53 Missense VUS Variants Based on Their Impact on TP53 Structural Stability" International Journal of Molecular Sciences 22, no. 21: 11345. https://doi.org/10.3390/ijms222111345
APA StyleTam, B., Sinha, S., Qin, Z., & Wang, S. M. (2021). Comprehensive Identification of Deleterious TP53 Missense VUS Variants Based on Their Impact on TP53 Structural Stability. International Journal of Molecular Sciences, 22(21), 11345. https://doi.org/10.3390/ijms222111345