Full-Spectrum Targeted Mutagenesis in Plant and Animal Cells


Abstract
1. Introduction
2. Directed Mutagenesis Using CRISPR Nucleases
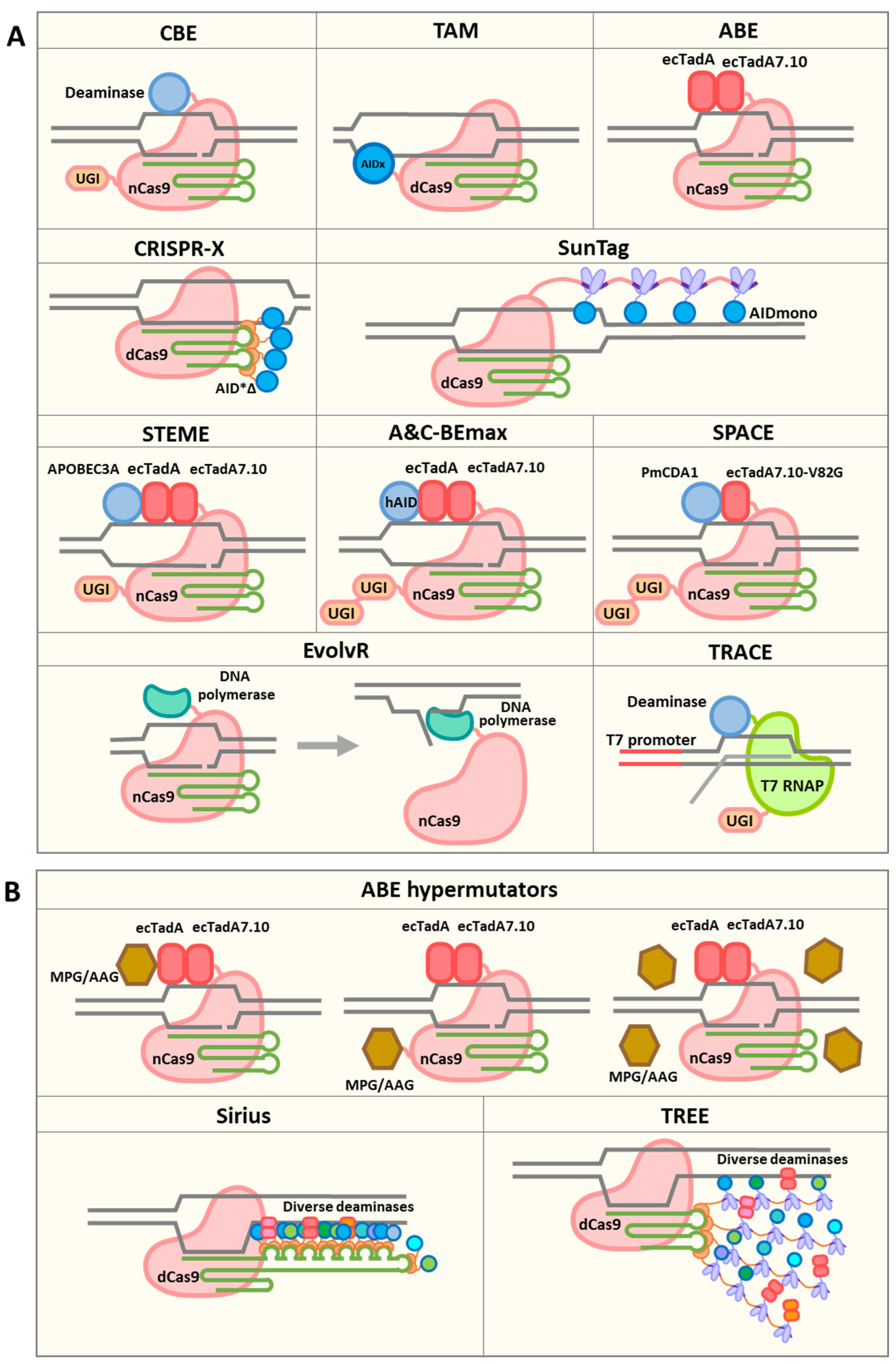
3. Diversifying Base Editors
4. EvolvR
5. Other Approaches for Targeted Mutagenesis
6. In Situ and Ex Vivo Directed Evolution Using Targeted Mutagenesis
7. Limitations and Future Prospects
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAG | Alkyl adenine DNA glycosylase |
| ABE | Adenine base editor |
| ACC | Acetyl-coenzyme A carboxylase |
| AID | Activation-induced cytidine deaminase |
| AIDmono | Monomeric AID |
| AP site | Apurinic/apyrimidinic site |
| APOBEC3A | Apolipoprotein B mRNA editing enzyme catalytic polypeptide 3A |
| APOBEC3B | Apolipoprotein B mRNA editing enzyme catalytic polypeptide 3B |
| BE | Base editor |
| BER | Base excision repair |
| BFP | Blue fluorescent protein |
| Cas | CRISPR-associated protein |
| CBE | Cytosine base editor |
| CGBE | C-to-G base editor |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| dCas9 | Catalytically dead Cas9 |
| DSB | Double-strand break |
| GESTALT | Genome editing of synthetic target arrays for lineage tracing |
| GFP | Green fluorescent protein |
| gRNA | Guide RNA |
| HDR | Homology-directed repair |
| Indels | Insertions and deletions |
| MEK1 kinase | Mitogen-activated protein kinase kinase 1 |
| MEMOIR | Memory by engineered mutagenesis with optical in situ readout |
| MPG | N-methylpurine glycosylase |
| mSCRIBE | Mammalian synthetic cellular recorders integrating biological events |
| MSH2 | MutS homolog 2 |
| nCas9 | Cas9 nickase |
| NHEJ | Non-homologous end joining |
| ORF | Open reading frame |
| PAM | Protospacer adjacent motif |
| PmCDA1 | Petromyzon marinus cytidine deaminase 1 |
| rAPOBEC1 | Rat apolipoprotein B mRNA editing enzyme catalytic polypeptide 1 |
| scRNA-seq | Single-cell RNA sequencing |
| sgRNA | Single guide RNA |
| SPACE | Synchronous programmable adenine and cytosine editor |
| ssDNA | Single-stranded DNA |
| STEME | Saturated targeted endogenous mutagenesis editor |
| TAM | Targeted AID-mediated mutagenesis |
| TLS | Translesion synthesis |
| TRACE | T7 polymerase-driven continuous editing |
| UGI | Uracil glycosylase inhibitor |
| UNG | Uracil-DNA glycosylase |
| XRCC1 | X-ray repair cross-complementing protein 1 |
References
- Simon, A.J.; D’Oelsnitz, S.; Ellington, A.D. Synthetic evolution. Nat. Biotechnol. 2019, 37, 730–743. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Mccarty, N.S.; Graham, A.E.; Studená, L.; Ledesma-Amaro, R. Multiplexed CRISPR Technologies for Gene Editing and Transcriptional Regulation. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; Dicarlo, J.E.; Norville, J.E.; Church, G.M. RNA-Guided Human Genome Engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Chavez, A.; Tung, A.; Chan, Y.; Kaas, C.; Yin, Y.; Cecchi, R.; Lopez-Garnier, S.; Kelsic, E.D.; Schubert, M.; et al. High-Throughput Creation and Functional Profiling of DNA Sequence Variant Libraries Using CRISPR–Cas9 in Yeast. Nat. Biotechnol. 2018, 36, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Donovan, K.F.; Hegde, M.; Sullender, M.; Vaimberg, E.W.; Johannessen, C.M.; Root, D.E.; Doench, J.G. Creation of Novel Protein Variants with CRISPR/Cas9-Mediated Mutagenesis: Turning a Screening By-Product into a Discovery Tool. PLoS ONE 2017, 12, e0170445. [Google Scholar] [CrossRef]
- Butt, H.; Eid, A.; Momin, A.A.; Bazin, J.; Crespi, M.; Arold, S.T.; Mahfouz, M.M. CRISPR Directed Evolution of the Spliceosome for Resistance to Splicing Inhibitors. Genome Biol. 2019, 20, 1–9. [Google Scholar] [CrossRef]
- Mason, D.M.; Weber, C.R.; Parola, C.; Meng, S.M.; Greiff, V.; Kelton, W.J.; Reddy, S.T. High-Throughput Antibody Engineering in Mammalian Cells by CRISPR/Cas9-Mediated Homology-Directed Mutagenesis. Nucleic Acids Res. 2018, 46, 7436–7449. [Google Scholar] [CrossRef]
- Erdogan, M.; Fabritius, A.; Basquin, J.; Griesbeck, O. Targeted In Situ Protein Diversification and Intra-organelle Validation in Mammalian Cells. Cell Chem. Biol. 2020, 27, 610–621.e5. [Google Scholar] [CrossRef]
- Van Vu, T.; Sung, Y.W.; Kim, J.; Doan, D.T.H.; Tran, M.T.; Kim, J.-Y. Challenges and Perspectives in Homology-Directed Gene Targeting in Monocot Plants. Rice 2019, 12, 1–29. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable Editing of a Target Base in Genomic DNA Without Double-Stranded DNA Cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Kunz, C.; Saito, Y.; Schar, P. DNA Repair in Mammalian Cells. Cell. Mol. Life Sci. 2009, 66, 1021–1038. [Google Scholar] [CrossRef] [PubMed]
- Rees, H.A.; Liu, D.R. Base Editing: Precision Chemistry on the Genome and Transcriptome of Living Cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Arazoe, T.; Yachie, N.; Banno, S.; Kakimoto, M.; Tabata, M.; Mochizuki, M.; Miyabe, A.; Araki, M.; Hara, K.Y.; et al. Targeted Nucleotide Editing Using Hybrid Prokaryotic and Vertebrate Adaptive Immune Systems. Science 2016, 353, aaf8729. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.D.; Huang, M.; Dai, P.; Liu, T.; Fan, S.; Cheng, X.; Zhao, Y.; Yeap, L.-S.; Meng, F.-L. Intrinsic Nucleotide Preference of Diversifying Base Editors Guides Antibody Ex Vivo Affinity Maturation. Cell Rep. 2018, 25, 884–892.e3. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.B.; Komor, A.C.; Levy, J.M.; Packer, M.S.; Zhao, K.T.; Liu, D.R. Increasing the Genome-Targeting Scope and Precision of Base Editing with Engineered Cas9-Cytidine Deaminase Fusions. Nat. Biotechnol. 2017, 35, 371–376. [Google Scholar] [CrossRef]
- Komor, A.C.; Zhao, K.T.; Packer, M.S.; Gaudelli, N.M.; Waterbury, A.L.; Koblan, L.W.; Kim, Y.B.; Badran, A.H.; Liu, D.R. Improved Base Excision Repair Inhibition and Bacteriophage Mu Gam Protein Yields C:G-to-T:A Base Editors with Higher Efficiency and Product Purity. Sci. Adv. 2017, 3, eaao4774. [Google Scholar] [CrossRef]
- Li, X.; Wang, Y.; Liu, Y.; Yang, B.; Wang, X.; Wei, J.; Lu, Z.; Zhang, Y.; Wu, J.; Huang, X.; et al. Base Editing with a Cpf1–Cytidine Deaminase Fusion. Nat. Biotechnol. 2018, 36, 324–327. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable Base Editing of A•T to G•C in Genomic DNA without DNA Cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Hua, K.; Tao, X.; Zhu, J.-K. Expanding the Base Editing Scope in Rice by Using Cas9 Variants. Plant Biotechnol. J. 2018, 17, 499–504. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, X.; Wang, L.; Yin, S.; Zhu, B.; Xie, L.; Duan, Q.; Hu, H.; Zheng, R.; Wei, Y.; et al. Increasing Targeting Scope of Adenosine Base Editors in Mouse and Rat Embryos Through Fusion of TadA Deaminase with Cas9 Variants. Protein Cell 2018, 9, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Gaudelli, N.M.; Lam, D.K.; Rees, H.A.; Solá-Esteves, N.M.; Barrera, L.A.; Born, D.A.; Edwards, A.; Gehrke, J.M.; Lee, S.-J.; Liquori, A.J.; et al. Directed Evolution of Adenine Base Editors with Increased Activity and Therapeutic Application. Nat. Biotechnol. 2020, 38, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhang, J.; Yin, W.; Zhang, Z.; Song, Y.; Chang, X. Targeted AID-Mediated Mutagenesis (TAM) Enables Efficient Genomic Diversification in Mammalian Cells. Nat. Methods 2016, 13, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Sale, J.E.; Petersen-Mahrt, S.K.; Neuberger, M.S. AID Is Essential for Immunoglobulin V Gene Conversion in a Cultured B Cell Line. Curr. Biol. 2002, 12, 435–438. [Google Scholar] [CrossRef]
- Arakawa, H.; Hauschild, J.; Buerstedde, J.-M. Requirement of the Activation-Induced Deaminase (AID) Gene for Immunoglobulin Gene Conversion. Science 2002, 295, 1301–1306. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Hess, G.T.; Frésard, L.; Han, K.; Lee, C.H.; Li, A.; Cimprich, K.A.; Montgomery, L.F.S.B.; Bassik, M.C. Directed Evolution Using dCas9-Targeted Somatic Hypermutation in Mammalian Cells. Nat. Methods 2016, 13, 1036–1042. [Google Scholar] [CrossRef]
- Zhang, Y.; Malzahn, A.A.; Sretenovic, S.; Qi, Y. The Emerging and Uncultivated Potential of CRISPR Technology in Plant Science. Nat. Plants 2019, 5, 778–794. [Google Scholar] [CrossRef]
- Joung, J.; Konermann, S.M.; Gootenberg, J.S.; Abudayyeh, O.O.; Platt, R.J.; Brigham, M.D.; Sanjana, N.E.; Zhang, F. Genome-scale CRISPR-Cas9 Knockout and Transcriptional Activation Screening. Nat. Protoc. 2017, 12, 828–863. [Google Scholar] [CrossRef]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved Vectors and Genome-Wide Libraries for CRISPR Screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef]
- Halperin, S.O.; Tou, C.J.; Wong, E.B.; Modavi, C.; Schaffer, D.V.; Dueber, J.E. CRISPR-Guided DNA Polymerases Enable Diversification of all Nucleotides in a Tunable Window. Nat. Cell Biol. 2018, 560, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Liu, S.; Padula, S.; Lesman, D.; Griswold, K.; Lin, A.; Zhao, T.; Marshall, J.L.; Chen, F. Efficient, Continuous Mutagenesis in Human Cells Using a Pseudo-Random DNA Editor. Nat. Biotechnol. 2019, 38, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.L.; Papa, L.J.; Shoulders, M.D. A Processive Protein Chimera Introduces Mutations Across Defined DNA Regions In Vivo. J. Am. Chem. Soc. 2018, 140, 11560–11564. [Google Scholar] [CrossRef] [PubMed]
- DeVilder, M.-C.; Moyon, M.; Gautreau-Rolland, L.; Navet, B.; Perroteau, J.; Delbos, F.; Gesnel, M.-C.; Breathnach, R.; Saulquin, X. Ex vivo evolution of human antibodies by CRISPR-X: From a Naive B Cell Repertoire to Affinity Matured Antibodies. BMC Biotechnol. 2019, 19, 1–13. [Google Scholar] [CrossRef]
- Li, C.; Zhang, R.; Meng, X.; Chen, S.; Zong, Y.; Lu, C.; Qiu, J.-L.; Chen, Y.-H.; Li, J.; Gao, C. Targeted, Random Mutagenesis of Plant Genes with Dual Cytosine and Adenine Base Editors. Nat. Biotechnol. 2020, 38, 875–882. [Google Scholar] [CrossRef]
- Gasiunas, G.; Young, J.K.; Karvelis, T.; Kazlauskas, D.; Urbaitis, T.; Jasnauskaite, M.; Grusyte, M.M.; Paulraj, S.; Wang, P.-H.; Hou, Z.; et al. A Catalogue of Biochemically Diverse CRISPR-Cas9 Orthologs. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Fedorova, I.; Vasileva, A.; Selkova, P.; Abramova, M.; Arseniev, A.; Pobegalov, G.; Kazalov, M.; Musharova, O.; Goryanin, I.; Artamonova, D.; et al. PpCas9 from Pasteurella pneumotropica—a Compact Type II-C Cas9 Ortholog Active in Human Cells. Nucleic Acids Res. 2020, 48, 12297–12309. [Google Scholar] [CrossRef]
- Jacobsen, T.; Ttofali, F.; Liao, C.; Manchalu, S.; Gray, B.N.; Beisel, C.L. Characterization of Cas12a Nucleases Reveals Diverse PAM Profiles Between Closely-Related Orthologs. Nucleic Acids Res. 2020, 48, 5624–5638. [Google Scholar] [CrossRef]
- Hu, J.H.; Miller, S.M.; Geurts, M.H.; Tang, W.; Chen, L.; Sun, N.; Zeina, C.M.; Gao, X.; Rees, H.A.; Lin, Z.; et al. Evolved Cas9 Variants with Broad PAM Compatibility and High DNA Specificity. Nat. Cell Biol. 2018, 556, 57–63. [Google Scholar] [CrossRef]
- Nishimasu, H.; Shi, X.; Ishiguro, S.; Gao, L.Y.; Hirano, S.; Okazaki, S.; Noda, T.; Abudayyeh, O.O.; Gootenberg, J.S.; Mori, H.; et al. Engineered CRISPR-Cas9 Nuclease with Expanded Targeting Space. Science 2018, 361, 1259–1262. [Google Scholar] [CrossRef]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In Vivo Genome Editing using Staphylococcus aureus Cas9. Nat. Cell Biol. 2015, 520, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Topkar, V.V.; Zheng, Z.; Joung, J.K. Broadening the Targeting Range of Staphylococcus aureus CRISPR-Cas9 by Modifying PAM Recognition. Nat. Biotechnol. 2015, 33, 1293–1298. [Google Scholar] [CrossRef] [PubMed]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; Van Der Oost, J.; Regev, A.; et al. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Shmakov, S.; Abudayyeh, O.O.; Makarova, K.S.; Wolf, Y.I.; Gootenberg, J.S.; Semenova, E.; Minakhin, L.; Joung, J.; Konermann, S.M.; Severinov, K.; et al. Discovery and Functional Characterization of Diverse Class 2 CRISPR-Cas Systems. Mol. Cell 2015, 60, 385–397. [Google Scholar] [CrossRef]
- Walton, R.T.; Christie, K.A.; Whittaker, M.N.; Kleinstiver, B.P. Unconstrained Genome Targeting with Near-PAMless Engineered CRISPR-Cas9 Variants. Science 2020, 368, 290–296. [Google Scholar] [CrossRef]
- Najm, F.J.; Strand, C.; Donovan, K.F.; Hegde, M.; Sanson, K.R.; Vaimberg, E.W.; Sullender, M.E.; Hartenian, E.; Kalani, Z.; Fusi, N.; et al. Orthologous CRISPR–Cas9 Enzymes for Combinatorial Genetic Screens. Nat. Biotechnol. 2018, 36, 179–189. [Google Scholar] [CrossRef]
- Ma, H.; Tu, L.-C.; Naseri, A.; Chung, Y.-C.; Grünwald, D.; Zhang, S.; Pederson, T. CRISPR-Sirius: RNA Scaffolds for Signal Amplification in Genome Imaging. Nat. Methods 2018, 15, 928–931. [Google Scholar] [CrossRef]
- Kunii, A.; Hara, Y.; Takenaga, M.; Hattori, N.; Fukazawa, T.; Ushijima, T.; Yamamoto, T.; Sakuma, T. Three-Component Repurposed Technology for Enhanced Expression: Highly Accumulable Transcriptional Activators via Branched Tag Arrays. CRISPR J. 2018, 1, 337–347. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, B.; Chen, L.; Xie, L.; Yu, W.; Wang, Y.; Li, L.; Yin, S.; Yang, L.; Hu, H.; et al. Dual Base Editor Catalyzes Both Cytosine and Adenine Base Conversions in Human Cells. Nat. Biotechnol. 2020, 38, 856–860. [Google Scholar] [CrossRef]
- Grünewald, J.; Zhou, R.; Lareau, C.A.; Garcia, S.P.; Iyer, S.; Miller, B.R.; Langner, L.M.; Hsu, J.Y.; Aryee, M.J.; Joung, J.K. A Dual-Deaminase CRISPR Base Editor Enables Concurrent Adenine and Cytosine Editing. Nat. Biotechnol. 2020, 38, 861–864. [Google Scholar] [CrossRef]
- Zhao, D.; Li, J.; Li, S.; Xin, X.; Hu, M.; Price, M.A.; Rosser, S.J.; Bi, C.; Zhang, X. Glycosylase Base Editors Enable C-to-A and C-to-G Base Changes. Nat. Biotechnol. 2020, 2020, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kurt, I.C.; Zhou, R.; Iyer, S.; Garcia, S.P.; Miller, B.R.; Langner, L.M.; Grünewald, J.; Joung, J.K. CRISPR C-to-G Base Editors for Inducing Targeted DNA Transversions in Human Cells. Nat. Biotechnol. 2020, 2020, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Park, J.E.; Paa, P.; Rajakumar, P.D.; Chew, Y.T.; Manivannan, S.N.; Chew, W.L. Precise and Programmable C:G to G:C Base Editing in Genomic DNA. bioRxiv 2020. [Google Scholar] [CrossRef]
- McKenna, A.; Findlay, G.M.; Gagnon, J.A.; Horwitz, M.S.; Schier, A.F.; Shendure, J. Whole-Organism Lineage Tracing by Combinatorial and Cumulative Genome Editing. Science 2016, 353, aaf7907. [Google Scholar] [CrossRef] [PubMed]
- Frieda, K.L.; Linton, J.M.; Hormoz, S.; Choi, J.; Chow, K.-H.K.; Singer, Z.S.; Budde, M.W.; Elowitz, M.B.; Cai, J.C.L. Synthetic Recording and In Situ Readout of Lineage Information in Single Cells. Nat. Cell Biol. 2017, 541, 107–111. [Google Scholar] [CrossRef]
- Spanjaard, B.; Hu, B.; Mitic, N.; Olivares-Chauvet, P.; Janjuha, S.; Ninov, N.; Junker, J.P. Simultaneous Lineage Tracing and Cell-Type Identification Using CRISPR–Cas9-Induced Genetic Scars. Nat. Biotechnol. 2018, 36, 469–473. [Google Scholar] [CrossRef]
- Alemany, A.; Florescu, M.; Baron, C.S.; Peterson-Maduro, J.; Van Oudenaarden, A. Whole-Organism Clone Tracing Using Single-Cell Sequencing. Nat. Cell Biol. 2018, 556, 108–112. [Google Scholar] [CrossRef]
- Bowling, S.; Sritharan, D.; Osorio, F.G.; Nguyen, M.; Cheung, P.; Rodriguez-Fraticelli, A.; Patel, S.; Yuan, W.-C.; Fujiwara, Y.; Li, B.E.; et al. An Engineered CRISPR-Cas9 Mouse Line for Simultaneous Readout of Lineage Histories and Gene Expression Profiles in Single Cells. Cell 2020, 181, 1410–1422.e27. [Google Scholar] [CrossRef]
- Hwang, B.; Lee, W.; Yum, S.-Y.; Jeon, Y.; Cho, N.; Jang, G.; Bang, D. Lineage Tracing Using a Cas9-Deaminase Barcoding System Targeting Endogenous L1 Elements. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Perli, S.D.; Cui, C.H.; Lu, T.K. Continuous Genetic Recording with Self-Targeting CRISPR-Cas in Human Cells. Science 2016, 353, aag0511. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome Editing With CRISPR–Cas Nucleases, Base Editors, Transposases and Prime Editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Deaminase | Hotspot 1 | Editing Profile | Editing Profile (With UGI or in UGN/MSH2 Double Mutant) | Reference |
|---|---|---|---|---|
| hAIDx (P182X) | WGC | C/G to all nucleotides, with a preference for C/G to T/A | C/G to T/A | [15,23] |
| hAID*Δ | WRC | [15] | ||
| AIDmono | WRC, AGCT | |||
| PmCDA1 | WRC | |||
| APOBEC3A (A3A) | TCA, TC, TCC (using nCas9) | |||
| APOBEC3B mutant (A3BAct) | TCA, TC | |||
| rAPOBEC1 | TC | |||
| AID-3C (AIDmono with APOBEC3C substrate-recognition loop) | TTC | |||
| AID-3F (AIDmono with APOBEC3F substrate-recognition loop) | TGC |
| Target Protein | Evolution Goal | Evolution System | Biological System | Identified Major Mutations | Reference |
|---|---|---|---|---|---|
| Anti-hapten 4-hydroxy-3-nitrophenyl acetyl antibody B1-8 | Improve antigen affinity | CRISPR-X with AIDmono/APOBEC3A | HEK293T cells | W33L, T30I, T30S, S31R, T58I, A97G | [15] |
| Human monoclonal antibody A2Ab | Improve antigen affinity | CRISPR-X with AID*Δ | HEK 293 cells | D74H, W102 L, M112I, G121D, R124P | [34] |
| 30S ribosomal proteins S5 and S12 | Spectinomycin and streptomycin resistance | EvolvR | E. coli | Δ17–19, K23N and Δ24, Δ24, Δ26, G27D (rpsE) | [31] |
| BCR-ABL | Imatinib (Gleevec) resistance | TAM | Human K562 cells | T315I, T319I | [23] |
| Mitogen-activated protein kinase kinase 1 (MEK1 kinase) | Selumetinib and trametinib resistance | TRACE | Human A375 cells | E38K, V211D | [32] |
| Acetyl-coenzyme A carboxylase (ACC) | Herbicide (haloxyfop) resistance | STEMES | Rice | W2125C, P1927F, S1866F, A1884P | [35] |
| Blue fluorescent protein (BFP) | Shift fluorescence spectrum to that of green fluorescent protein (GFP) | TRACE | HEK293T cells | H66Y | [32] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iaffaldano, B.; Reiser, J. Full-Spectrum Targeted Mutagenesis in Plant and Animal Cells. Int. J. Mol. Sci. 2021, 22, 857. https://doi.org/10.3390/ijms22020857
Iaffaldano B, Reiser J. Full-Spectrum Targeted Mutagenesis in Plant and Animal Cells. International Journal of Molecular Sciences. 2021; 22(2):857. https://doi.org/10.3390/ijms22020857
Chicago/Turabian StyleIaffaldano, Brian, and Jakob Reiser. 2021. "Full-Spectrum Targeted Mutagenesis in Plant and Animal Cells" International Journal of Molecular Sciences 22, no. 2: 857. https://doi.org/10.3390/ijms22020857
APA StyleIaffaldano, B., & Reiser, J. (2021). Full-Spectrum Targeted Mutagenesis in Plant and Animal Cells. International Journal of Molecular Sciences, 22(2), 857. https://doi.org/10.3390/ijms22020857