Mitophagy Modulation, a New Player in the Race against ALS




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
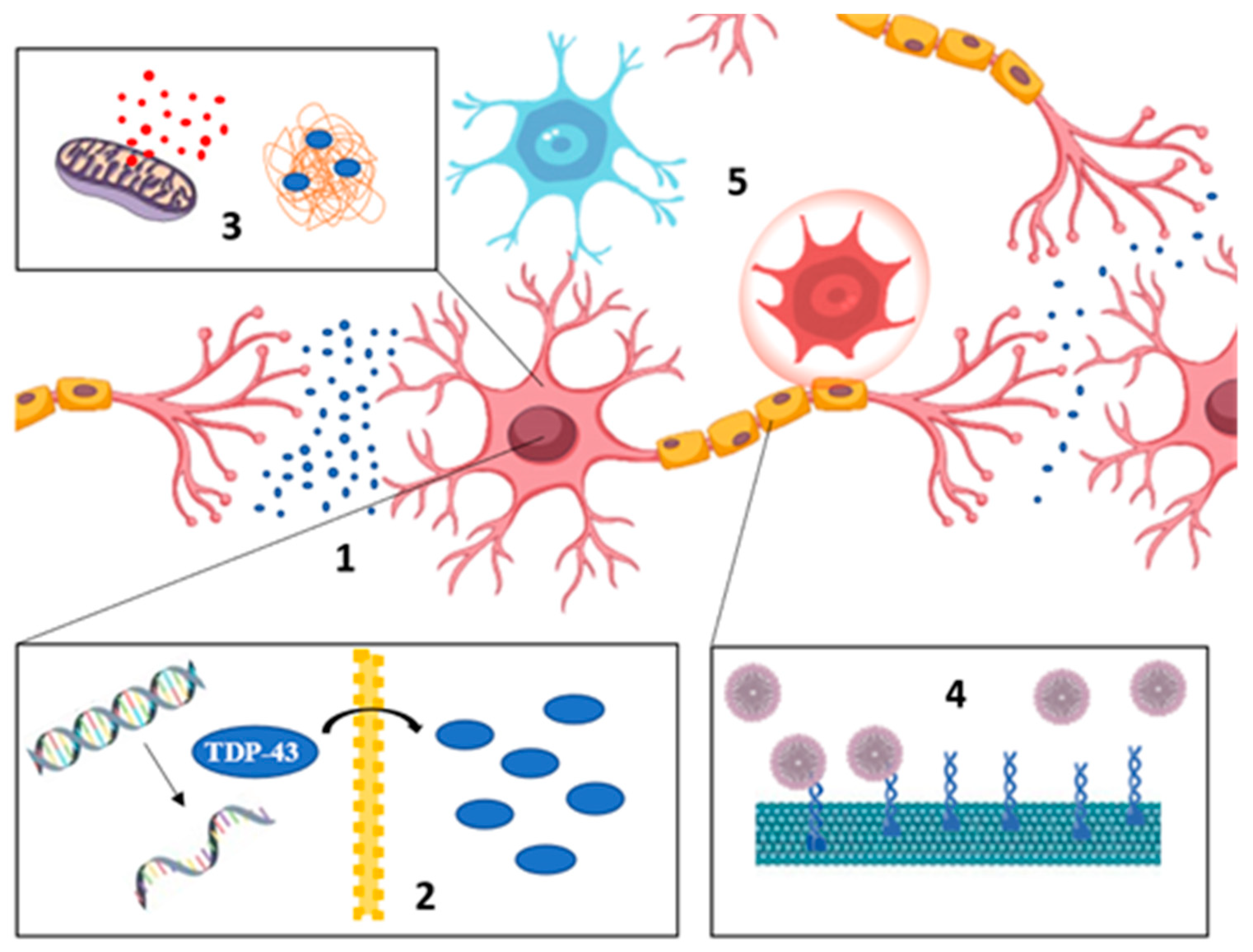
2. Mitochondrial Homeostasis in ALS
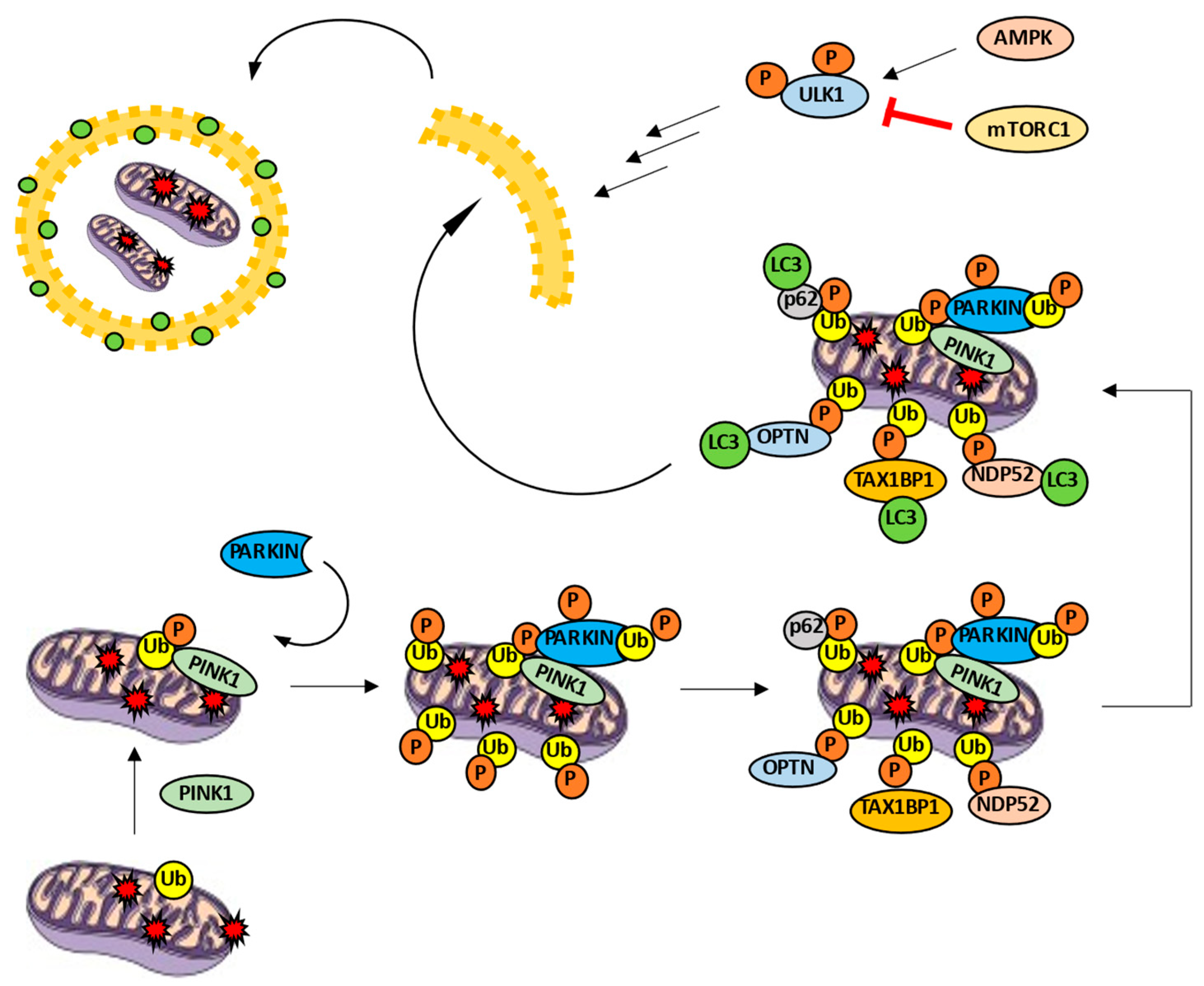
3. Autophagy and Mitophagy
Mitophagy Machinery: A Mitochondrial-Selective Autophagy
4. Mitophagy and ALS
4.1. Mitophagy and Neuroinflammation in ALS
4.2. Mitophagy as Target
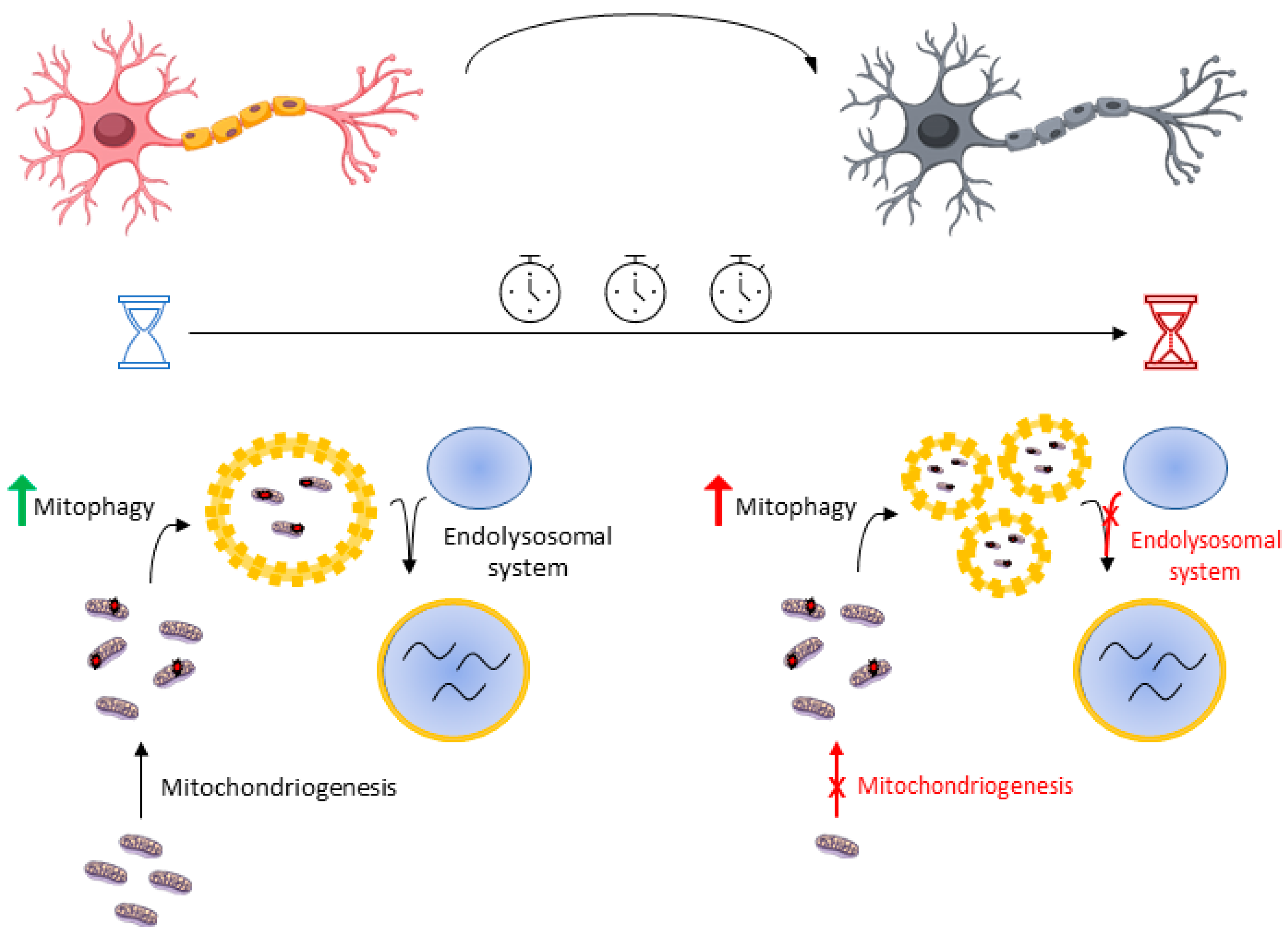
4.2.1. Mitophagy Is a Highly (Temporal) Dynamic Puzzle
4.2.2. Mitophagy Is a Highly (Neuron-Type) Dynamic Puzzle
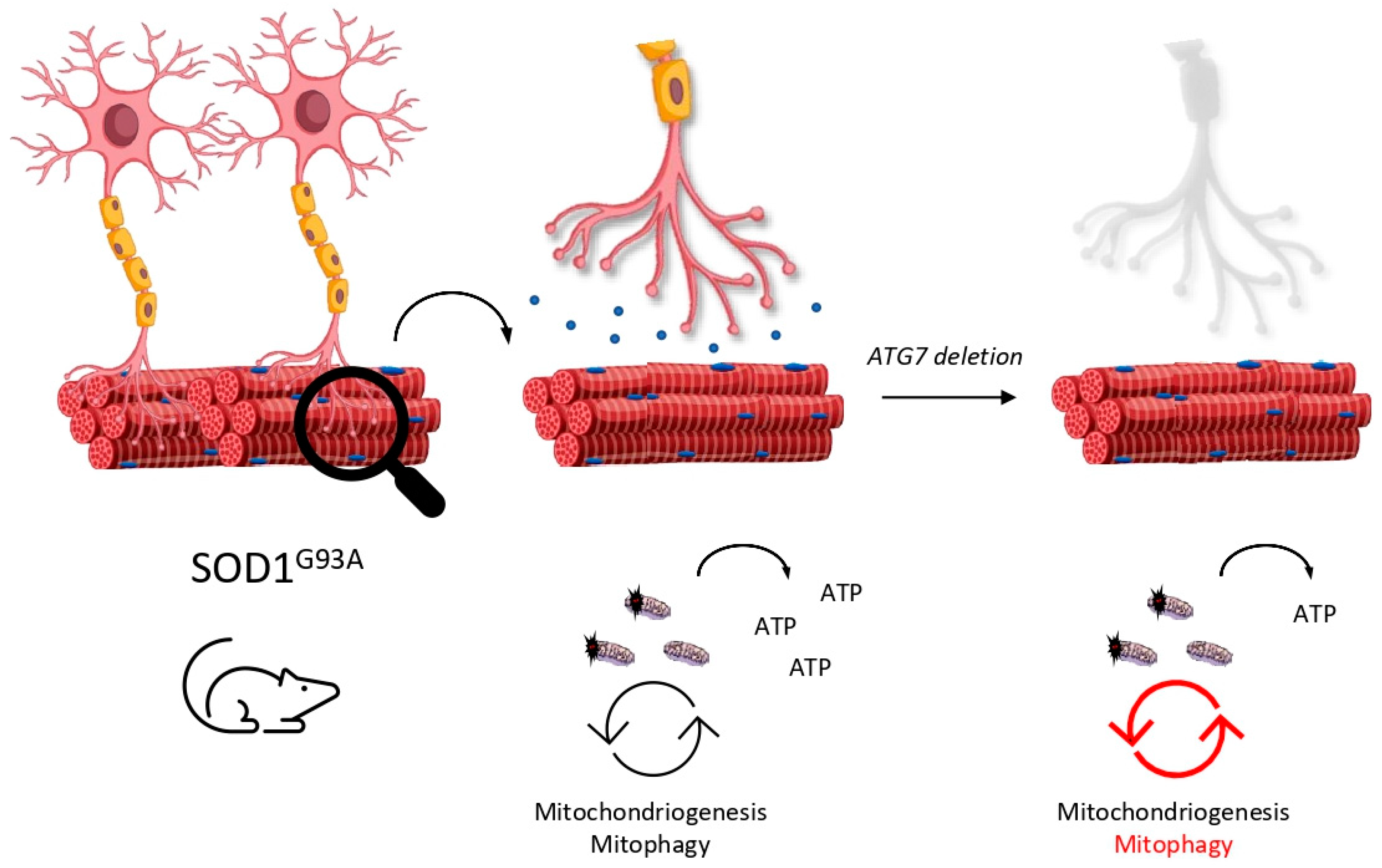
4.2.3. Mitophagy Is a Highly (Cell-Type) Dynamic Puzzle
5. What Is on the Immediate Horizon?
6. Concluding Remarks
Funding
Conflicts of Interest
Abbreviations
| ALS | Amyotrophic lateral sclerosis |
| AMBRA1 | Autophagy and beclin 1 regulator 1 |
| AMPK | AMP-activated protein kinase |
| ATP | Adenosine triphosphate |
| CCCP | Carbonyl cyanide m-chlorophenyl hydrazone |
| CNS | Central nervous system |
| CS | Citrate synthase |
| DAMPs | Damage-associated molecular patterns |
| fALS | Familiar ALS |
| FF motoneuron | Fast fatigable motoneuron |
| FR motoneuron | Fast resistant motoneuron |
| FUNDC1 | FUN14 domain containing protein 1 |
| FUS | Fused in sarcoma |
| GABARAPL1 | Gamma-aminobutyric acid receptor-associated protein-like 1 |
| IFN | Interferon type I |
| ISG | Interferon-stimulated genes |
| IRF3 | Interferon regulatory factor 3 |
| LAP | LC3-associated phagocytosis |
| LC3 | Microtubule-associated proteins 1A/1B light chain 3B |
| LIR | LC3-interaction region |
| MARCH5 | Membrane-associated ring finger 5 ubiquitin-ligase |
| MFN1 | Mitofusin 1 |
| MFN2 | Mitofusin 2 |
| MIRO1 | Mitochondrial Rho GTPase 1 |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| MULAN | Mitochondrial ubiquitin-ligase activator of NF-kB |
| NBR1 | Neighbor of BRCA1 gene 1 |
| NDP52 | Nuclear dot protein 52 |
| NF-kB | Nuclear factor-kappa B |
| NMJ | Neuromuscular junction |
| OPTN | Optineurin |
| P2X7 | P2X purinoceptor 7 |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PINK1 | PTEN-induced kinase 1 |
| RLPK1 | Receptor-like protein kinase 1 |
| ROS | Reactive oxygen species |
| sALS | Sporadic ALS |
| SMCR8 | Smith-Magenis syndrome chromosome region candidate 8 |
| SOD1 | Superoxide dismutase 1 |
| TAX1BP1 | TAX1 binding protein |
| TBK1 | TANK-binding kinase 1 |
| TDP-43 | 43 kDa transactive response DNA binding protein |
| TFAM | Mitochondrial transcription factor A |
| TIM | Translocase of the inner membrane |
| TLR3 | Toll-like receptor 3 |
| TOM | Translocase of the outer membrane |
| ULK1 | Unc-51 like autophagy activating kinase 1 |
| UPS | Ubiquitin-proteasome system |
| VDAC1 | Voltage-dependent anion channel 1 |
References
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [PubMed]
- Arthur, K.C.; Calvo, A.; Price, T.R.; Geiger, J.T.; Chiò, A.; Traynor, B.J. Projected Increase in Amyotrophic Lateral Sclerosis from 2015 to 2040. Nat. Commun. 2016, 7, 12408. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, M.; Brown, R.H. Genetics of Amyotrophic Lateral Sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, a024125. [Google Scholar] [CrossRef]
- Van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic Lateral Sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; Van Den Berg, L.H. Amyotrophic Lateral Sclerosis. Nat. Rev. Dis. Prim. 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar]
- Yellen, G. Fueling Thought: Management of Glycolysis and Oxidative Phosphorylation in Neuronal Metabolism. J. Cell Biol. 2018, 21, 2235–2246. [Google Scholar]
- Carrera-Juliá, S.; Moreno, M.L.; Barrios, C.; de la Rubia Ortí, J.E.; Drehmer, E. Antioxidant Alternatives in the Treatment of Amyotrophic Lateral Sclerosis: A Comprehensive Review. Front. Physiol. 2020, 11, 63. [Google Scholar]
- Carrì, M.T.; D’Ambrosi, N.; Cozzolino, M. Pathways to Mitochondrial Dysfunction in ALS Pathogenesis. Biochem. Biophys. Res. Commun. 2017, 483, 1187–1193. [Google Scholar] [CrossRef]
- Khalil, B.; Liévens, J.C. Mitochondrial Quality Control in Amyotrophic Lateral Sclerosis: Towards a Common Pathway? Neural Regen. Res. 2017, 12, 1052–1061. [Google Scholar] [CrossRef]
- Cook, C.; Petrucelli, L. Genetic Convergence Brings Clarity to the Enigmatic Red Line in ALS. Neuron 2019, 101, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- De Vos, K.J.; Chapman, A.L.; Tennant, M.E.; Manser, C.; Tudor, E.L.; Lau, K.F.; Brownlees, J.; Ackerley, S.; Shaw, P.J.; Mcloughlin, D.M.; et al. Familial Amyotrophic Lateral Sclerosis-Linked SOD1 Mutants Perturb Fast Axonal Transport to Reduce Axonal Mitochondria Content. Hum. Mol. Genet. 2007, 16, 2720–2728. [Google Scholar] [CrossRef] [PubMed]
- Vande Velde, C.; McDonald, K.K.; Boukhedimi, Y.; McAlonis-Downes, M.; Lobsiger, C.S.; Hadj, S.B.; Zandona, A.; Julien, J.P.; Shah, S.B.; Cleveland, D.W. Misfolded SOD1 Associated with Motor Neuron Mitochondria Alters Mitochondrial Shape and Distribution Prior to Clinical Onset. PLoS ONE 2011, 6, e22031. [Google Scholar] [CrossRef] [PubMed]
- Magrané, J.; Cortez, C.; Gan, W.B.; Manfredi, G. Abnormal Mitochondrial Transport and Morphology Are Common Pathological Denominators in SOD1 and TDP43 ALS Mouse Models. Hum. Mol. Genet. 2014, 23, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Gertz, B.; Pan, Y.; Price, A.C.; Molkentin, J.D.; Chang, Q. The Mitochondrial Permeability Transition Pore in Motor Neurons: Involvement in the Pathobiology of ALS Mice. Exp. Neurol. 2009, 218, 333–346. [Google Scholar] [CrossRef]
- Jiang, Z.; Wang, W.; Perry, G.; Zhu, X.; Wang, X. Mitochondrial Dynamic Abnormalities in Amyotrophic Lateral Sclerosis. Transl. Neurodegener. 2015, 4, 14. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn Superoxide Dismutase Gene Are Associated with Familial Amyotrophic Lateral Sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Blokhuis, A.M.; Groen, E.J.N.; Koppers, M.; Van Den Berg, L.H.; Pasterkamp, R.J. Protein Aggregation in Amyotrophic Lateral Sclerosis. Acta Neuropathol. 2013, 125, 777–794. [Google Scholar] [CrossRef]
- Mori, K.; Weng, S.-M.; Arzberger, T.; May, S.; Rentzsch, K.; Kremmer, E.; Schmid, B. The C9orf72 GGGGC Repeat is Translated into Aggregating Dipeptide-Repeat Proteins in FTLD/ALS. Science 2013, 339, 1335–1338. [Google Scholar] [CrossRef]
- Millecamps, S.; Julien, J.P. Axonal Transport Deficits and Neurodegenerative Diseases. Nat. Rev. Neurosci. 2013, 14, 161–176. [Google Scholar] [CrossRef]
- Zhou, J.; Li, A.; Li, X.; Yi, J. Dysregulated Mitochondrial Ca2+ and ROS Signaling in Skeletal Muscle of ALS Mouse Model. Arch. Biochem. Biophys. 2019, 663, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Jemmerson, R.; Dubinsky, J.M.; Brustovetsky, N. Cytochrome c Release from CNS Mitochondria and Potential for Clinical Intervention in Apoptosis-Mediated CNS Diseases. Antioxid. Redox Signal. 2005, 7, 1158–1173. [Google Scholar] [CrossRef] [PubMed]
- Von Bernhardi, R.; Eugenín-von Bernhardi, L.; Eugenín, J. Microglial Cell Dysregulation in Brain Aging and Neurodegeneration. Front. Aging Neurosci. 2015, 7, 124. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The Role of Mitochondria in Amyotrophic Lateral Sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, T.; Maragkakis, M. Amyotrophic Lateral Sclerosis Associated FUS Mutation Shortens Mitochondria and Induces Neurotoxicity. Sci. Rep. 2018, 8, 15575. [Google Scholar] [CrossRef] [PubMed]
- Moller, A.; Bauer, C.S.; Cohen, R.N.; Webster, C.P.; De Vos, K.J. Amyotrophic Lateral Sclerosis-Associated Mutant SOD1 Inhibits Anterograde Axonal Transport of Mitochondria by Reducing Miro1 Levels. Hum. Mol. Genet. 2017, 26, 4668–4679. [Google Scholar] [CrossRef]
- Gautam, M.; Jara, J.H.; Kocak, N.; Rylaarsdam, L.E.; Dong, K.; Bigio, E.H.; Özdinler, P.H. Mitochondria, ER, and Nuclear Membrane Defects Reveal Early Mechanisms for Upper Motor Neuron Vulnerability with Respect to TDP-43 Pathology. Acta Neuropathol. 2019, 137, 47–69. [Google Scholar] [CrossRef]
- Mizushima, N. A Brief History of Autophagy from Cell Biology to Physiology and Disease. Nat. Cell Biol. 2018, 20, 521–527. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, X.; Song, L.; Le, W. Autophagy Dysregulation in Amyotrophic Lateral Sclerosis. Brain Pathol. 2012, 22, 110–116. [Google Scholar] [CrossRef]
- Otomo, A.; Pan, L.; Hadano, S. Dysregulation of the Autophagy-Endolysosomal System in Amyotrophic Lateral Sclerosis and Related Motor Neuron Diseases. Neurol. Res. Int. 2012, 2012, 498428. [Google Scholar] [CrossRef]
- Neuspiel, M.; Schauss, A.C.; Braschi, E.; Zunino, R.; Rippstein, P.; Rachubinski, R.A.; Andrade-Navarro, M.A.; McBride, H.M. Cargo-Selected Transport from the Mitochondria to Peroxisomes Is Mediated by Vesicular Carriers. Curr. Biol. 2008, 18, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.Q.; Ren, C.; Xia, Z.F.; Yao, Y.M. Organelle-Specific Autophagy in Inflammatory Diseases: A Potential Therapeutic Target Underlying the Quality Control of Multiple Organelles. Autophagy 2020, 12, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and MTOR Regulate Autophagy through Direct Phosphorylation of Ulk1 Joungmok. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Shin, J.H.; Lee, J.E.; Choi, E.J. Role of Autophagy in the Pathogenesis of Amyotrophic Lateral Sclerosis. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 2517–2524. [Google Scholar] [CrossRef]
- Fivenson, E.M.; Lautrup, S.; Sun, N.; Scheibye-Knudsen, M.; Stevnsner, T.; Nilsen, H.; Bohr, V.A.; Fang, E.F. Mitophagy in Neurodegeneration and Aging. Neurochem. Int. 2017, 109, 202–209. [Google Scholar] [CrossRef]
- Granatiero, V.; Manfredi, G. Mitochondrial Transport and Turnover in the Pathogenesis of Amyotrophic Lateral Sclerosis. Biology 2019, 8, 36. [Google Scholar] [CrossRef]
- Zhang, C.W.; Hang, L.; Yao, T.P.; Lim, K.L. Parkin Regulation and Neurodegenerative Disorders. Front. Aging Neurosci. 2016, 7, 248. [Google Scholar] [CrossRef]
- Chen, Y.; Dorn, G.W., 2nd. PINK1- Phosphorylated Mitofusin 2 Is a Parkin Receptor for Culling Damaged Mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef]
- Weil, R.; Laplantine, E.; Curic, S.; Génin, P. Role of Optineurin in the Mitochondrial Dysfunction: Potential Implications in Neurodegenerative Diseases and Cancer. Front. Immunol. 2018, 9, 1243. [Google Scholar] [CrossRef]
- Oakes, J.A.; Davies, M.C.; Collins, M.O. TBK1: A New Player in ALS Linking Autophagy and Neuroinflammation. Mol. Brain 2017, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The Ubiquitin Kinase PINK1 Recruits Autophagy Receptors to Induce Mitophagy. Nature 2015, 534, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Perez, F.A.; Palmiter, R.D. Parkin-Deficient Mice Are Not a Robust Model of Parkinsonism. Proc. Natl. Acad. Sci. USA 2005, 102, 2174–2179. [Google Scholar] [CrossRef] [PubMed]
- Le Grand, J.N.; Bon, K.; Fraichard, A.; Zhang, J.; Jouvenot, M.; Risold, P.Y.; Boyer-Guittaut, M.; Delage-Mourroux, R. Specific Distribution of the Autophagic Protein GABARAPL1/GEC1 in the Developing and Adult Mouse Brain and Identification of Neuronal Populations Expressing GABARAPL1/GEC1. PLoS ONE 2013, 8, e63133. [Google Scholar] [CrossRef] [PubMed]
- Villa, E.; Marchetti, S.; Ricci, J.E. No Parkin Zone: Mitophagy without Parkin. Trends Cell Biol. 2018, 28, 882–895. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T. Mechanisms of Selective Autophagy and Mitophagy: Implications for Neurodegenerative Diseases. Neurobiol. Dis. 2019, 122, 23–34. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Polymenidou, M.; Hutt, K.R.; Vu, A.Q.; Baughn, M.; Huelga, S.C.; Clutario, K.M.; Ling, S.-C.; Liang, T.Y.; Mazur, C. Divergent Roles of ALS-Linked Proteins FUS/TLS and TDP-43 Intersect in Processing Long Pre-MRNAs. Nat. Neurosci. 2012, 15, 1488–1497. [Google Scholar] [CrossRef]
- Palomo, G.M.; Granatiero, V.; Kawamata, H.; Konrad, C.; Kim, M.; Arreguin, A.J.; Zhao, D.; Milner, T.A.; Manfredi, G. Parkin Is a Disease Modifier in the Mutant SOD 1 Mouse Model of ALS. EMBO Mol. Med. 2018, 10, e8888. [Google Scholar] [CrossRef]
- Chen, Y.; Deng, J.; Wang, P.; Yang, M.; Chen, X.; Zhu, L.; Liu, J.; Lu, B.; Shen, Y.; Fushimi, K.; et al. PINK1 and Parkin Are Genetic Modifiers for FUS-Induced Neurodegeneration. Hum. Mol. Genet. 2016, 25, 5059–5068. [Google Scholar] [CrossRef]
- Cha, S.J.; Choi, H.J.; Kim, H.J.; Choi, E.J.; Song, K.H.; Im, D.S.; Kim, K. Parkin Expression Reverses Mitochondrial Dysfunction in Fused in Sarcoma-Induced Amyotrophic Lateral Sclerosis. Insect Mol. Biol. 2020, 29, 56–65. [Google Scholar] [CrossRef]
- Wong, Y.C.; Holzbaur, E.L.F. Optineurin Is an Autophagy Receptor for Damaged Mitochondria in Parkin-Mediated Mitophagy That Is Disrupted by an ALS-Linked Mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef] [PubMed]
- Sirohi, K.; Swarup, G. Defects in Autophagy Caused by Glaucoma-Associated Mutations in Optineurin. Exp. Eye Res. 2016, 144, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Tak, Y.J.; Park, J.H.; Rhim, H.; Kang, S. ALS-Related Mutant SOD1 Aggregates Interfere with Mitophagy by Sequestering the Autophagy Receptor Optineurin. Int. J. Mol. Sci. 2020, 21, 7525. [Google Scholar] [CrossRef] [PubMed]
- Hadano, S.; Mitsui, S.; Pan, L.; Otomo, A.; Kubo, M.; Sato, K.; Ono, S.; Onodera, W.; Abe, K.; Chen, X.P.; et al. Functional Links between SQSTM1 and ALS2 in the Pathogenesis of ALS: Cumulative Impact on the Protection against Mutant SOD1-Mediated Motor Dysfunction in Mice. Hum. Mol. Genet. 2016, 25, 3321–3340. [Google Scholar] [CrossRef]
- Moore, A.S.; Holzbaur, E.L.F. Dynamic Recruitment and Activation of ALS-Associated TBK1 with Its Target Optineurin Are Required for Efficient Mitophagy. Proc. Natl. Acad. Sci. USA 2016, 113, E3349–E3358. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.K.H.; Thombre, R.; Wang, J. Autophagy as a Common Pathway in Amyotrophic Lateral Sclerosis. Neurosci. Lett. 2019, 697, 34–48. [Google Scholar] [CrossRef]
- Balendra, R.; Isaacs, A.M. C9orf72-Mediated ALS and FTD: Multiple Pathways to Disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef]
- Huang, C.; Yan, S.; Zhang, Z. Maintaining the Balance of TDP-43, Mitochondria, and Autophagy: A Promising Therapeutic Strategy for Neurodegenerative Diseases. Transl. Neurodegener. 2020, 9, 40. [Google Scholar] [CrossRef]
- Wang, W.; Arakawa, H.; Wang, L.; Okolo, O.; Siedlak, S.L.; Jiang, Y.; Gao, J.; Xie, F.; Petersen, R.B.; Wang, X. Motor-Coordinative and Cognitive Dysfunction Caused by Mutant TDP-43 Could Be Reversed by Inhibiting Its Mitochondrial Localization. Mol. Ther. 2017, 25, 127–139. [Google Scholar] [CrossRef]
- Akizuki, M.; Yamashita, H.; Uemura, K.; Maruyama, H.; Kawakami, H.; Ito, H.; Takahashi, R. Optineurin Suppression Causes Neuronal Cell Death via NF-ΚB Pathway. J. Neurochem. 2013, 126, 699–704. [Google Scholar] [CrossRef]
- Nakazawa, S.; Oikawa, D.; Ishii, R.; Ayaki, T.; Takahashi, H.; Takeda, H.; Ishitani, R.; Kamei, K.; Takeyoshi, I.; Kawakami, H.; et al. Linear Ubiquitination Is Involved in the Pathogenesis of Optineurin-Associated Amyotrophic Lateral Sclerosis. Nat. Commun. 2016, 7, 12547. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, L.; Zhang, S.-Y.; Casanova, J.-L.; Sancho-Shimizu, V. Human TBK1: A Gatekeeper of Neuroinflammation. Trends Mol. Med. 2016, 22, 511–527. [Google Scholar] [CrossRef] [PubMed]
- Beghi, E.; Chiò, A.; Inghilleri, M.; Mazzini, L.; Micheli, A.; Mora, G.; Poloni, M. A Randomized Controlled Trial of Recombinant Interferon Beta-1a in ALS. Neurology 2000, 54, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Poutiainen, E.; Hokkanen, L.; Niemi, M.L.; Färkkilä, M. Reversible Cognitive Decline during High-Dose α-Interferon Treatment. Pharmacol. Biochem. Behav. 1994, 47, 901–905. [Google Scholar] [CrossRef]
- Yu, J.; Zhou, X.; Chang, M.; Nakaya, M.; Chang, J.H.; Xiao, Y.; William, L.J.; Dorta-Estremera, S.; Cao, W.; Zal, A.; et al. Regulation of T-Cell Activation and Migration by the Kinase TBK1 during Neuroinflammation. Nat. Commun. 2015, 6, 6074. [Google Scholar] [CrossRef]
- Xu, D.; Jin, T.; Zhu, H.; Chen, H.; Ofengeim, D.; Zou, C.; Mifflin, L.; Pan, L.; Amin, P.; Li, W.; et al. TBK1 Suppresses RIPK1-Driven Apoptosis and Inflammation during Development and in Aging. Cell 2018, 174, 1477–1491. [Google Scholar] [CrossRef]
- Sasaki, S. Autophagy in Spinal Cord Motor Neurons in Sporadic Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 2011, 70, 349–359. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of Basal Autophagy in Neural Cells Causes Neurodegenerative Disease in Mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.I.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of Autophagy in the Central Nervous System Causes Neurodegeneration in Mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Amin, A.; Perera, N.D.; Beart, P.M.; Turner, B.J.; Shabanpoor, F. Amyotrophic Lateral Sclerosis and Autophagy: Dysfunction and Therapeutic Targeting. Cells 2020, 9, 2413. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, S.; Lu, K.; Wang, F.; Deng, J.; Xu, Z.; Wang, X.; Zhou, Q.; Le, W.; Zhao, Y. Verapamil Ameliorates Motor Neuron Degeneration and Improves Lifespan in the SOD1G93A Mouse Model of Als by Enhancing Autophagic Flux. Aging Dis. 2019, 10, 1159–1173. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Lee, J.H. Calcium Channel Blockers as Potential Therapeutics for Obesity-Associated Autophagy Defects and Fatty Liver Pathologies. Autophagy 2014, 10, 2385–2386. [Google Scholar] [CrossRef] [PubMed]
- Perera, N.D.; Sheean, R.K.; Lau, C.L.; Shin, Y.S.; Beart, P.M.; Horne, M.K.; Turner, B.J. Rilmenidine Promotes MTOR-Independent Autophagy in the Mutant SOD1 Mouse Model of Amyotrophic Lateral Sclerosis without Slowing Disease Progression. Autophagy 2018, 14, 534–551. [Google Scholar] [CrossRef] [PubMed]
- Apolloni, S.; Fabbrizio, P.; Amadio, S.; Volonté, C. Actions of the Antihistaminergic Clemastine on Presymptomatic SOD1-G93A Mice Ameliorate ALS Disease Progression. J. Neuroinflamm. 2016, 13, 191. [Google Scholar] [CrossRef] [PubMed]
- Castillo, K.; Nassif, M.; Valenzuela, V.; Rojas, F.; Matus, S.; Mercado, G.; Court, F.A.; Van Zundert, B.; Hetz, C. Trehalose Delays the Progression of Amyotrophic Lateral Sclerosis by Enhancing Autophagy in Motoneurons. Autophagy 2013, 9, 1308–1320. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, S.; Song, L.; Tang, Y.; Shen, Y.; Jia, L.; Le, W. MTOR-Independent, Autophagic Enhancer Trehalose Prolongs Motor Neuron Survival and Ameliorates the Autophagic Flux Defect in a Mouse Model of Amyotrophic Lateral Sclerosis. Autophagy 2014, 10, 588–602. [Google Scholar] [CrossRef]
- Li, Y.; Guo, Y.; Wang, X.; Yu, X.; Duan, W.; Hong, K.; Wang, J.; Han, H.; Li, C. Trehalose Decreases Mutant SOD1 Expression and Alleviates Motor Deficiency in Early but Not End-Stage Amyotrophic Lateral Sclerosis in a SOD1-G93A Mouse Model. Neuroscience 2015, 298, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Thau, N.; Knippenberg, S.; Körner, S.; Rath, K.J.; Dengler, R.; Petri, S. Decreased MRNA Expression of PGC-1α and PGC-1αregulated Factors in the SOD1G93A ALS Mouse Model and in Human Sporadic ALS. J. Neuropathol. Exp. Neurol. 2012, 71, 1064–1074. [Google Scholar] [CrossRef]
- Xie, Y.; Zhou, B.; Lin, M.-Y.; Wang, S.; Foust, K.D.; Sheng, Z.-H. Endolysosomal Deficits Augment Mitochondria Pathology in Spinal Motor Neurons of Asymptomatic FALS Mice. Neuron 2015, 87, 355–370. [Google Scholar] [CrossRef]
- Evans, C.S.; Holzbaur, E.L.F. Lysosomal Degradation of Depolarized Mitochondria Is Rate-Limiting in OPTN-Dependent Neuronal Mitophagy. Autophagy 2020, 16, 962–964. [Google Scholar] [CrossRef]
- Fornai, F.; Longone, P.; Cafaro, L.; Kastsiuchenka, O.; Ferrucci, M.; Manca, M.L.; Lazzeri, G.; Spalloni, A.; Bellio, N.; Lenzi, P.; et al. Lithium Delays Progression of Amyotrophic Lateral Sclerosis. Proc. Natl. Acad. Sci. USA 2008, 105, 2052–2057. [Google Scholar] [CrossRef] [PubMed]
- Meira Martins, L.A.; Vieira, M.Q.; Ilha, M.; de Vasconcelos, M.; Biehl, H.B.; Lima, D.B.; Schein, V.; Barbé-Tuana, F.; Borojevic, R.; Guma, F.C.R. The Interplay Between Apoptosis, Mitophagy and Mitochondrial Biogenesis Induced by Resveratrol Can Determine Activated Hepatic Stellate Cells Death or Survival. Cell Biochem. Biophys. 2014, 71, 657–672. [Google Scholar] [CrossRef] [PubMed]
- Da Cruz, S.; Parone, P.A.; Lopes, V.S.; Lillo, C.; McAlonis-Downes, M.; Lee, S.K.; Vetto, A.P.; Petrosyan, S.; Marsala, M.; Murphy, A.N.; et al. Elevated PGC-1α Activity Sustains Mitochondrial Biogenesis and Muscle Function without Extending Survival in a Mouse Model of Inherited ALS. Cell Metab. 2012, 15, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.A.; Southam, K.A.; Blizzard, C.A.; King, A.E.; Dickson, T.C. Axonal Degeneration, Distal Collateral Branching and Neuromuscular Junction Architecture Alterations Occur Prior to Symptom Onset in the SOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. J. Chem. Neuroanat. 2016, 76, 35–47. [Google Scholar] [CrossRef]
- Martineau, É.; Di Polo, A.; Velde, C.V.; Robitaille, R. Dynamic Neuromuscular Remodeling Precedes Motor-Unit Loss in a Mouse Model of ALS. eLife 2018, 7, e41973. [Google Scholar] [CrossRef] [PubMed]
- Rudnick, N.D.; Griffey, C.J.; Guarnieri, P.; Gerbino, V.; Wang, X.; Piersaint, J.A.; Tapia, J.C.; Rich, M.M.; Maniatis, T. Distinct Roles for Motor Neuron Autophagy Early and Late in the SOD1G93A Mouse Model of ALS. Proc. Natl. Acad. Sci. USA 2017, 114, E8294–E8303. [Google Scholar] [CrossRef]
- Kanning, K.C.; Kaplan, A.; Henderson, C.E. Motor Neuron Diversity in Development and Disease. Annu. Rev. Neurosci. 2010, 33, 409–440. [Google Scholar] [CrossRef]
- Le Masson, G.; Przedborski, S.; Abbott, L.F. A Computational Model of Motor Neuron Degeneration. Neuron 2014, 83, 975–988. [Google Scholar] [CrossRef]
- Ravera, S.; Bonifacino, T.; Bartolucci, M.; Milanese, M.; Gallia, E.; Provenzano, F.; Cortese, K.; Panfoli, I.; Bonanno, G. Characterization of the Mitochondrial Aerobic Metabolism in the Pre- and Perisynaptic Districts of the SOD1 G93A Mouse Model of Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2018, 55, 9220–9233. [Google Scholar] [CrossRef]
- Ravera, S.; Torazza, C.; Bonifacino, T.; Provenzano, F.; Rebosio, C.; Milanese, M.; Usai, C.; Panfoli, I.; Bonanno, G. Altered Glucose Catabolism in the Presynaptic and Perisynaptic Compartments of SOD1G93A Mouse Spinal Cord and Motor Cortex Indicates That Mitochondria Are the Site of Bioenergetic Imbalance in ALS. J. Neurochem. 2019, 151, 336–350. [Google Scholar] [CrossRef]
- Purves, D.; Augustine, G.J.; Fitzpatrick, D.; Katz, L.C.; LaMantia, A.-S.; McNamara, J.O.; Williams, M. Neuroscience, 2nd ed.; Sinauer Associates: Sunderland, UK, 2001. [Google Scholar]
- Rogers, R.S.; Tungtur, S.; Tanaka, T.; Nadeau, L.L.; Badawi, Y.; Wang, H.; Ni, H.M.; Ding, W.X.; Nishimune, H. Impaired Mitophagy Plays a Role in Denervation of Neuromuscular Junctions in ALS Mice. Front. Neurosci. 2017, 11, 447. [Google Scholar] [CrossRef] [PubMed]
- Nijssen, J.; Comley, L.H.; Hedlund, E. Motor Neuron Vulnerability and Resistance in Amyotrophic Lateral Sclerosis. Acta Neuropathol. 2017, 133, 863–885. [Google Scholar] [CrossRef] [PubMed]
- Grosskreutz, J.; Van Den Bosch, L.; Keller, B.U. Calcium Dysregulation in Amyotrophic Lateral Sclerosis. Cell Calcium 2010, 47, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Barrett, E.F.; Barrett, J.N.; David, G. Dysfunctional Mitochondrial Ca2+ Handling in Mutant SOD1 Mouse Models of FALS: Integration of Findings from Motor Neuron Somata and Motor Terminals. Front. Cell. Neurosci. 2014, 8, 184. [Google Scholar] [CrossRef] [PubMed]
- Strohm, L.; Behrends, C. Glia-Specific Autophagy Dysfunction in ALS. Semin. Cell Dev. Biol. 2020, 99, 172–182. [Google Scholar] [CrossRef]
- Beers, D.R.; Henkel, J.S.; Xiao, Q.; Zhao, W.; Wang, J.; Yen, A.A.; Siklos, L.; McKercher, S.R.; Appel, S.H. Wild-Type Microglia Extend Survival in PU.1 Knockout Mice with Familial Amyotrophic Lateral Sclerosis. Proc. Natl. Acad. Sci. USA 2006, 103, 16021–16026. [Google Scholar] [CrossRef]
- Yamanaka, K.; Chun, S.J.; Boillee, S.; Fujimori-Tonou, N.; Yamashita, H.; Gutmann, D.H.; Takahashi, R.; Misawa, H.; Cleveland, D.W. Astrocytes as Determinants of Disease Progression in Inherited Amyotrophic Lateral Sclerosis. Nat. Neurosci. 2008, 11, 251–253. [Google Scholar] [CrossRef]
- Boillée, S.; Yamanaka, K.; Lobsiger, C.S.; Copeland, N.G.; Jenkins, N.A.; Kassiotis, G.; Kollias, G.; Cleveland, D.W. Onset and Progression in Inherited ALS Determined by Motor Neurons and Microglia. Science 2006, 312, 1389–1392. [Google Scholar] [CrossRef]
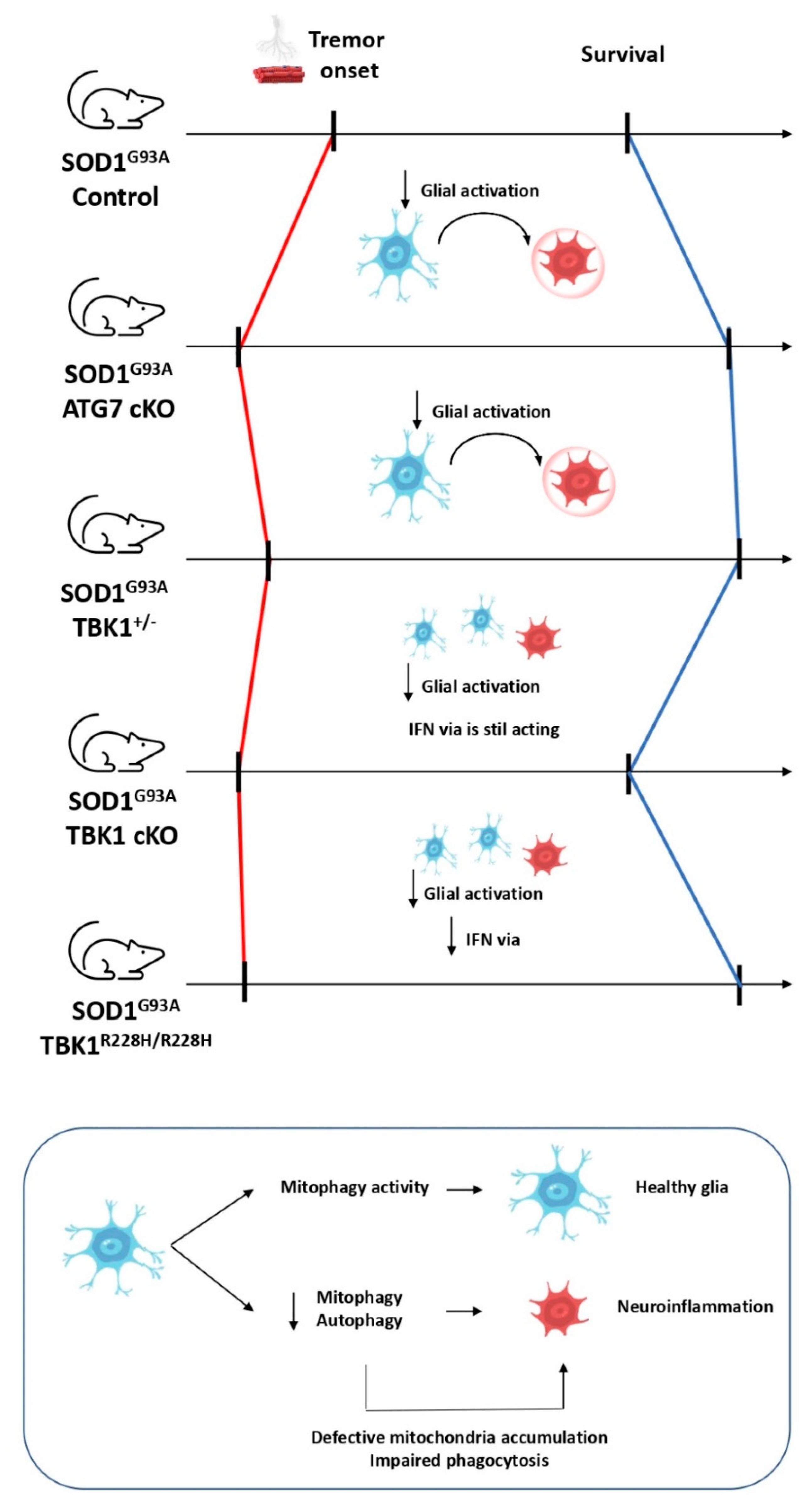
- Brenner, D.; Sieverding, K.; Bruno, C.; Lüningschrör, P.; Buck, E.; Mungwa, S.; Fischer, L.; Brockmann, S.J.; Ulmer, J.; Bliederhäuser, C.; et al. Heterozygous Tbk1 Loss Has Opposing Effects in Early and Late Stages of ALS in Mice. J. Exp. Med. 2019, 216, 267–278. [Google Scholar] [CrossRef]
- Gerbino, V.; Kaunga, E.; Ye, J.; Canzio, D.; O’Keeffe, S.; Rudnick, N.D.; Guarnieri, P.; Lutz, C.M.; Maniatis, T. The Loss of TBK1 Kinase Activity in Motor Neurons or in All Cell Types Differentially Impacts ALS Disease Progression in SOD1 Mice. Neuron 2020, 106, 789–805.e5. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, J.W.; Nam, H.; Yu, S.W.; Yu, S.W. Cx3cr1 CreERT2-Driven Atg7 Deletion in Adult Mice Induces Intestinal Adhesion. Mol. Brain 2020, 13, 88. [Google Scholar] [CrossRef] [PubMed]
- Lautrup, S.; Lou, G.; Aman, Y.; Nilsen, H.; Tao, J.; Fang, E.F. Microglial Mitophagy Mitigates Neuroinflammation in Alzheimer’s Disease. Neurochem. Int. 2019, 129, 104469. [Google Scholar] [CrossRef] [PubMed]
- Choong, C.-J.; Okuno, T.; Ikenaka, K.; Baba, K.; Hayakawa, H.; Koike, M.; Yokota, M.; Doi, J.; Kakuda, K.; Takeuchi, T.; et al. Alternative Mitochondrial Quality Control Mediated by Extracellular Release. Autophagy 2020, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bajwa, E.; Pointer, C.B.; Klegeris, A. The Role of Mitochondrial Damage-Associated Molecular Patterns in Chronic Neuroinflammation. Mediat. Inflamm. 2019, 2019, 4050796. [Google Scholar] [CrossRef] [PubMed]
- Morales, I.; Sanchez, A.; Puertas-Avendaño, R.; Rodriguez-Sabate, C.; Perez-Barreto, A.; Rodriguez, M. Neuroglial Transmitophagy and Parkinson’s Disease. Glia 2020, 68, 2277–2299. [Google Scholar] [PubMed]
- Jackson, J.G.; Robinson, M.B. Regulation of Mitochondrial Dynamics in Astrocytes: Mechanisms, Consequences, and Unknowns. Glia 2018, 66, 1213–1234. [Google Scholar] [CrossRef]
- Plaza-Zabala, A.; Sierra-Torre, V.; Sierra, A. Autophagy and Microglia: Novel Partners in Neurodegeneration and Aging. Int. J. Mol. Sci. 2017, 18, 598. [Google Scholar] [CrossRef]
- Berglund, R.; Guerreiro-Cacais, A.O.; Adzemovic, M.Z.; Zeitelhofer, M.; Lund, H.; Ewing, E.; Ruhrmann, S.; Nutma, E.; Parsa, R.; Thessen-Hedreul, M.; et al. Microglial Autophagy-Associated Phagocytosis Is Essential for Recovery from Neuroinflammation. Sci. Immunol. 2020, 5, eabb5077. [Google Scholar] [CrossRef]
- Lutz, C. Mouse Models of ALS: Past, Present and Future. Brain Res. 2018, 1693, 1–10. [Google Scholar] [CrossRef]
- Sieverding, K.; Ulmer, J.; Bruno, C.; Satoh, T.; Tsao, W.; Freischmidt, A.; Akira, S.; Wong, P.C.; Ludolph, A.C.; Danzer, K.M.; et al. Hemizygous Deletion of Tbk1 Worsens Neuromuscular Junction Pathology in TDP-43G298S Transgenic Mice. Exp. Neurol. 2020, 335, 113496. [Google Scholar] [CrossRef]
- Shellikeri, S.; Karthikeyan, V.; Martino, R.; Black, S.E.; Zinman, L.; Keith, J.; Yunusova, Y. The Neuropathological Signature of Bulbar-Onset ALS: A Systematic Review. Neurosci. Biobehav. Rev. 2017, 75, 378–392. [Google Scholar] [CrossRef] [PubMed]
- Smittkamp, S.E.; Spalding, H.N.; Brown, J.W.; Gupte, A.A.; Chen, J.; Nishimune, H.; Geiger, P.C.; Stanford, J.A. Measures of Bulbar and Spinal Motor Function, Muscle Innervation, and Mitochondrial Function in ALS Rats. Behav. Brain Res. 2010, 211, 48–57. [Google Scholar] [CrossRef]
- De Marchi, E.; Orioli, E.; Dal Ben, D.; Adinolfi, E. P2X7 Receptor as a Therapeutic Target. Adv. Protein Chem. Struct. Biol. 2016, 104, 39–79. [Google Scholar] [PubMed]
- Fabbrizio, P.; Amadio, S.; Apolloni, S.; Volonté, C. P2X7 Receptor Activation Modulates Autophagy in SOD1-G93a Mouse Microglia. Front. Cell. Neurosci. 2017, 11, 249. [Google Scholar] [CrossRef] [PubMed]
- Sekar, P.; Huang, D.Y.; Hsieh, S.L.; Chang, S.F.; Lin, W.W. AMPK-Dependent and Independent Actions of P2X7 in Regulation of Mitochondrial and Lysosomal Functions in Microglia. Cell Commun. Signal. 2018, 16, 83. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madruga, E.; Maestro, I.; Martínez, A. Mitophagy Modulation, a New Player in the Race against ALS. Int. J. Mol. Sci. 2021, 22, 740. https://doi.org/10.3390/ijms22020740
Madruga E, Maestro I, Martínez A. Mitophagy Modulation, a New Player in the Race against ALS. International Journal of Molecular Sciences. 2021; 22(2):740. https://doi.org/10.3390/ijms22020740
Chicago/Turabian StyleMadruga, Enrique, Inés Maestro, and Ana Martínez. 2021. "Mitophagy Modulation, a New Player in the Race against ALS" International Journal of Molecular Sciences 22, no. 2: 740. https://doi.org/10.3390/ijms22020740
APA StyleMadruga, E., Maestro, I., & Martínez, A. (2021). Mitophagy Modulation, a New Player in the Race against ALS. International Journal of Molecular Sciences, 22(2), 740. https://doi.org/10.3390/ijms22020740