Pluripotent Stem Cells for Disease Modeling and Drug Discovery in Niemann-Pick Type C1



Abstract
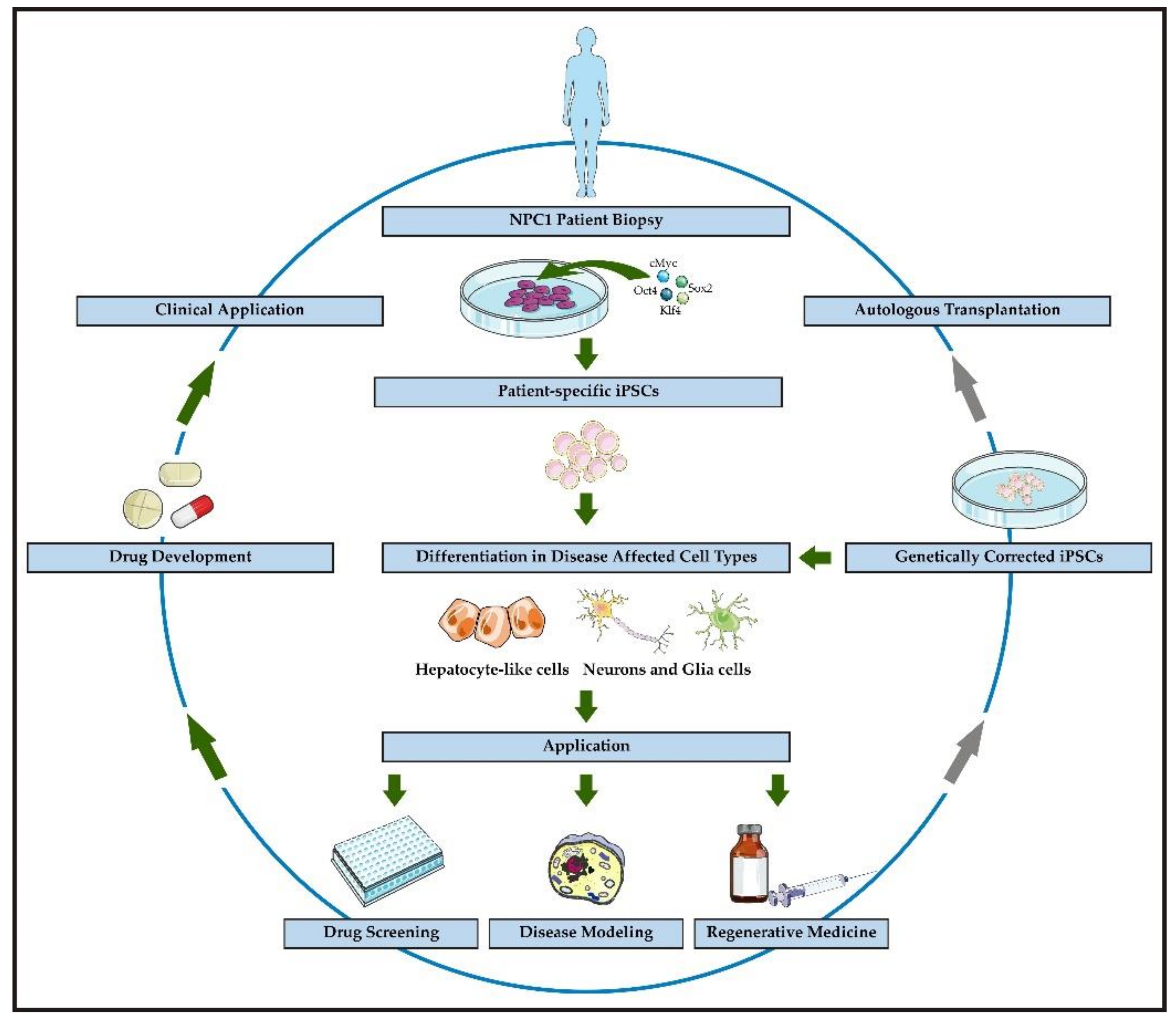
1. Introduction
2. Commonly Used NPC1 Model Systems
3. NPC1 Disease Modeling Using Pluripotent Stem Cells
3.1. Lipid Accumulation
3.2. Alterations of Autophagy
3.3. Defective Mitochondrial Homeostasis and Oxidative Stress
3.4. Gliosis
3.5. Functional Aspects of NPC1-Deficient Neurons
4. Treatment Strategies for NPC1 and the Use of iPSCs for Drug Discovery
4.1. Administration of Miglustat
4.2. Cyclodextrins
4.3. Histone Deacetylase Inhibitors
4.4. Pharmacological Chaperones
5. Limitations and Perspectives of iPSCs Used for Disease Modeling and Drug Discovery
5.1. Limitation of the Applicability of iPSCs and Derived Cells
5.2. Perspectives of the Applicability of iPSC-Derived Cells
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 24(S)-HC | 24(S)-hydroxycholesterol |
| 25-HC | 25-hydroxycholesterol |
| 3-MA | 3-Methyladenine |
| AD | Alzheimer’s disease |
| ALS | Amyotrophic lateral sclerosis |
| AMPAR | AMPA-receptor |
| AMPK | AMP-activated protein kinase |
| Apo E | Apolipoprotein E |
| APP | Amyloid precursor protein |
| Cas | CRISPR-associated gene |
| CBZ | Carbamazepine |
| CD | Cyclodextrin |
| CNS | Central nervous system |
| CoQ10 | Coenzyme Q10 |
| cPCs | Cerebellar Purkinje cells |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| ER | Endoplasmic reticulum |
| GABAAR | GABAA receptor |
| GD | Gaucher’s Disease |
| GFAP | Glial fibrillary acidic protein |
| GlyR | Glycine receptor |
| HAT | Histone acetyl transferase |
| HDACi | Histone deacetylase inhibitor |
| hESC | Human embryonic stem cell |
| Hex A | Hexosaminidase A |
| HLC | Hepatocyte-like cell |
| hSKIN-MASCS | Human skin multipotent adult stem cells |
| HP-α-CD | Hydroxypropyl-alpha-cyclodextrin |
| HP-β-CD | Hydroxypropyl-beta-cyclodextrin |
| HP-γ-CD | Hydroxypropyl-gamma-cyclodextrin |
| HSEM | Horizontal saccadic eye movement |
| iPSC | Induced pluripotent stem cell |
| KD | Knock down |
| LC3B | Microtubule-associated protein a light chain 3B |
| LE/LY | Late endosome/lysosome |
| LSD | Lysosomal storage disorder |
| M-β-CD | Methyl-beta-cyclodextrin |
| NDCs | Neuronal differentiated cells |
| NPC | Neural progenitor cell |
| NPC1 | Niemann-Pick type C1 |
| NPC2 | Niemann-Pick type C2 |
| NSS | Neurological Severity Scores |
| OS | Oxidative stress |
| PC | Pharmacological chaperone |
| PD | Parkinson’s disease |
| PMA | Phorbol 12-myristate 13-acetate |
| PSCs | Pluripotent stem cells |
| Rap | Rapamycin |
| RIP1/3 | Receptor interacting protein kinase 1/3 |
| ROS | Reactive oxygen species |
| S1P | Sphingosine-1-phosphate |
| SAHA | Suberanilohydroxamic acid |
| SOD | Superoxide-Dismutase |
| SphK | Sphingosine kinase |
| SRT | Substrate reduction therapy |
| TALEN | Transcription activator-like effector nuclease |
| Tre | Trehalose |
| VEGF | Vascular endothelial growth factor |
| Ver | Verapamil |
| VSGP | Vertical supranuclear gaze palsy |
References
- Vanier, M.T. Niemann-Pick disease type C. Orphanet J. Rare Dis. 2010, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- The Human Gene Mutation Database. Gene Database. Available online: http://www.hgmd.cf.ac.uk/ac/index.php (accessed on 23 November 2020).
- Wraith, J.E.; Baumgartner, M.R.; Bembi, B.; Covanis, A.; Levade, T.; Mengel, E.; Pineda, M.; Sedel, F.; Topçu, M.; Vanier, M.T.; et al. Recommendations on the diagnosis and management of Niemann-Pick disease type C. Mol. Genet. Metab. 2009, 98, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Carstea, E.D.; Morris, J.A.; Coleman, K.G.; Loftus, S.K.; Zhang, D.; Cummings, C.; Gu, J.; Rosenfeld, M.A.; Pavan, W.J.; Krizman, D.B.; et al. Niemann-Pick C1 disease gene: Homology to mediators of cholesterol homeostasis. Science 1997, 277, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Park, I.-H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q. Disease-specific induced pluripotent stem cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef]
- Kawagoe, S.; Higuchi, T.; Meng, X.-L.; Shimada, Y.; Shimizu, H.; Hirayama, R.; Fukuda, T.; Chang, H.; Nakahata, T.; Fukada, S.; et al. Generation of induced pluripotent stem (iPS) cells derived from a murine model of Pompe disease and differentiation of Pompe-iPS cells into skeletal muscle cells. Mol. Genet. Metab. 2011, 104, 123–128. [Google Scholar] [CrossRef]
- Doerr, J.; Böckenhoff, A.; Ewald, B.; Ladewig, J.; Eckhardt, M.; Gieselmann, V.; Matzner, U.; Brüstle, O.; Koch, P. Arylsulfatase A Overexpressing Human iPSC-derived Neural Cells Reduce CNS Sulfatide Storage in a Mouse Model of Metachromatic Leukodystrophy. Mol. Ther. 2015, 23, 1519–1531. [Google Scholar] [CrossRef]
- Lojewski, X.; Staropoli, J.F.; Biswas-Legrand, S.; Simas, A.M.; Haliw, L.; Selig, M.K.; Coppel, S.H.; Goss, K.A.; Petcherski, A.; Chandrachud, U.; et al. Human iPSC models of neuronal ceroid lipofuscinosis capture distinct effects of TPP1 and CLN3 mutations on the endocytic pathway. Hum. Mol. Genet. 2014, 23, 2005–2022. [Google Scholar] [CrossRef]
- Tolar, J.; Park, I.-H.; Xia, L.; Lees, C.J.; Peacock, B.; Webber, B.; McElmurry, R.T.; Eide, C.R.; Orchard, P.J.; Kyba, M.; et al. Hematopoietic differentiation of induced pluripotent stem cells from patients with mucopolysaccharidosis type I (Hurler syndrome). Blood 2011, 117, 839–847. [Google Scholar] [CrossRef]
- Long, Y.; Xu, M.; Li, R.; Dai, S.; Beers, J.; Chen, G.; Soheilian, F.; Baxa, U.; Wang, M.; Marugan, J.J.; et al. Induced Pluripotent Stem Cells for Disease Modeling and Evaluation of Therapeutics for Niemann-Pick Disease Type A. Stem Cells Transl. Med. 2016, 5, 1644–1655. [Google Scholar] [CrossRef]
- Trilck, M.; Hübner, R.; Seibler, P.; Klein, C.; Rolfs, A.; Frech, M.J. Niemann-Pick type C1 patient-specific induced pluripotent stem cells display disease specific hallmarks. Orphanet J. Rare Dis. 2013, 8, 144. [Google Scholar] [CrossRef]
- Fog, C.K.; Kirkegaard, T. Animal models for Niemann-Pick type C: Implications for drug discovery & development. Expert Opin. Drug Discov. 2019, 14, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Pallottini, V.; Pfrieger, F.W. Understanding and Treating Niemann-Pick Type C Disease: Models Matter. Int. J. Mol. Sci. 2020, 21, 8979. [Google Scholar] [CrossRef] [PubMed]
- Chiba, Y.; Komori, H.; Takei, S.; Hasegawa-Ishii, S.; Kawamura, N.; Adachi, K.; Nanba, E.; Hosokawa, M.; Enokido, Y.; Kouchi, Z.; et al. Niemann-Pick disease type C1 predominantly involving the frontotemporal region, with cortical and brainstem Lewy bodies: An autopsy case. Neuropathology 2014, 34, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Cologna, S.M.; Cluzeau, C.V.M.; Yanjanin, N.M.; Blank, P.S.; Dail, M.K.; Siebel, S.; Toth, C.L.; Wassif, C.A.; Lieberman, A.P.; Porter, F.D. Human and mouse neuroinflammation markers in Niemann-Pick disease, type C1. J. Inherit. Metab. Dis. 2014, 37, 83–92. [Google Scholar] [CrossRef]
- Pentchev, P.G.; Gal, A.E.; Booth, A.D.; Omodeo-Sale, F.; Fours, J.; Neumeyer, B.A.; Quirk, J.M.; Dawson, G.; Brady, R.O. A lysosomal storage disorder in mice characterized by a dual deficiency of sphingomyelinase and glucocerebrosidase. Biochim. Biophys. Acta (BBA)-Lipids Lipid Metab. 1980, 619, 669–679. [Google Scholar] [CrossRef]
- Võikar, V.; Rauvala, H.; Ikonen, E. Cognitive deficit and development of motor impairment in a mouse model of Niemann-Pick type C disease. Behav. Brain Res. 2002, 132, 1–10. [Google Scholar] [CrossRef]
- Morris, M.D.; Bhuvaneswaran, C.; Shio, H.; Fowler, S. Lysosome lipid storage disorder in NCTR-BALB/c mice. I. Description of the disease and genetics. Am. J. Pathol. 1982, 108, 140–149. [Google Scholar]
- Rabenstein, M.; Peter, F.; Rolfs, A.; Frech, M.J. Impact of Reduced Cerebellar EAAT Expression on Purkinje Cell Firing Pattern of NPC1-deficient Mice. Sci. Rep. 2018, 8, 4661. [Google Scholar] [CrossRef]
- Rabenstein, M.; Peter, F.; Joost, S.; Trilck, M.; Rolfs, A.; Frech, M.J. Decreased calcium flux in Niemann-Pick type C1 patient-specific iPSC-derived neurons due to higher amount of calcium-impermeable AMPA receptors. Mol. Cell. Neurosci. 2017. [Google Scholar] [CrossRef]
- German, D.C.; Liang, C.L.; Song, T.; Yazdani, U.; Xie, C.; Dietschy, J.M. Neurodegeneration in the Niemann-Pick C mouse: Glial involvement. Neuroscience 2002, 109. [Google Scholar] [CrossRef]
- Brown, D.E.; Thrall, M.A.; Walkley, S.U.; Wenger, D.A.; Mitchell, T.W.; Smith, M.O.; Royals, K.L.; March, P.A.; Allison, R.W. Feline Niemann-Pick disease type C. Am. J. Pathol. 1994, 144, 1412–1415. [Google Scholar] [PubMed]
- Walkley, S.U.; Suzuki, K. Consequences of NPC1 and NPC2 loss of function in mammalian neurons. Biochim. Biophys. Acta 2004, 1685, 48–62. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, S.; Mitsuoka, S.; Sakiyama, T.; Kitagawa, T. Sphingomyelinosis, a new mutation in the mouse: A model of Niemann-Pick disease in humans. J. Hered. 1982, 73, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Brown, M.S.; Shelton, J.M.; Richardson, J.A.; Goldstein, J.L.; Liang, G. Amino acid substitution in NPC1 that abolishes cholesterol binding reproduces phenotype of complete NPC1 deficiency in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 15330–15335. [Google Scholar] [CrossRef]
- Praggastis, M.; Tortelli, B.; Zhang, J.; Fujiwara, H.; Sidhu, R.; Chacko, A.; Chen, Z.; Chung, C.; Lieberman, A.P.; Sikora, J.; et al. A murine Niemann-Pick C1 I1061T knock-in model recapitulates the pathological features of the most prevalent human disease allele. J. Neurosci. 2015, 35, 8091–8106. [Google Scholar] [CrossRef]
- Maue, R.A.; Burgess, R.W.; Wang, B.; Wooley, C.M.; Seburn, K.L.; Vanier, M.T.; Rogers, M.A.; Chang, C.C.; Chang, T.-Y.; Harris, B.T.; et al. A novel mouse model of Niemann-Pick type C disease carrying a D1005G-Npc1 mutation comparable to commonly observed human mutations. Hum. Mol. Genet. 2012, 21, 730–750. [Google Scholar] [CrossRef]
- Saito, Y.; Suzuki, K.; Nanba, E.; Yamamoto, T.; Ohno, K.; Murayama, S. Niemann-Pick type C disease: Accelerated neurofibrillary tangle formation and amyloid beta deposition associated with apolipoprotein E epsilon 4 homozygosity. Ann. Neurol. 2002, 52, 351–355. [Google Scholar] [CrossRef]
- Lopez, M.E.; Scott, M.P. Genetic dissection of a cell-autonomous neurodegenerative disorder: Lessons learned from mouse models of Niemann-Pick disease type C. Dis. Model. Mech. 2013, 6, 1089–1100. [Google Scholar] [CrossRef]
- Singh, V.K.; Kalsan, M.; Kumar, N.; Saini, A.; Chandra, R. Induced pluripotent stem cells: Applications in regenerative medicine, disease modeling, and drug discovery. Front. Cell Dev. Biol. 2015, 3, 2. [Google Scholar] [CrossRef]
- Omole, A.E.; Fakoya, A.O.J. Ten years of progress and promise of induced pluripotent stem cells: Historical origins, characteristics, mechanisms, limitations, and potential applications. PeerJ 2018, 6, e4370. [Google Scholar] [CrossRef]
- Ordonez, M.P.; Roberts, E.A.; Kidwell, C.U.; Yuan, S.H.; Plaisted, W.C.; Goldstein, L.S.B. Disruption and therapeutic rescue of autophagy in a human neuronal model of Niemann Pick type C1. Hum. Mol. Genet. 2012, 21, 2651–2662. [Google Scholar] [CrossRef] [PubMed]
- Maetzel, D.; Sarkar, S.; Wang, H.; Abi-Mosleh, L.; Xu, P.; Cheng, A.W.; Gao, Q.; Mitalipova, M.; Jaenisch, R. Genetic and chemical correction of cholesterol accumulation and impaired autophagy in hepatic and neural cells derived from Niemann-Pick Type C patient-specific iPS cells. Stem Cell Rep. 2014, 2, 866–880. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Swaroop, M.; Wang, M.; Baxa, U.; Yang, R.; Yan, Y.; Coksaygan, T.; DeTolla, L.; Marugan, J.J.; Austin, C.P.; et al. Niemann-Pick Disease Type C: Induced Pluripotent Stem Cell-Derived Neuronal Cells for Modeling Neural Disease and Evaluating Drug Efficacy. J. Biomol. Screen. 2014, 19, 1164–1173. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, J.K.; Park, M.H.; Hong, Y.R.; Marti, H.H.; Kim, H.; Okada, Y.; Otsu, M.; Seo, E.-J.; Park, J.-H.; et al. Pathological roles of the VEGF/SphK pathway in Niemann-Pick type C neurons. Nat. Commun. 2014, 5, 5514. [Google Scholar] [CrossRef] [PubMed]
- Soga, M.; Ishitsuka, Y.; Hamasaki, M.; Yoneda, K.; Furuya, H.; Matsuo, M.; Ihn, H.; Fusaki, N.; Nakamura, K.; Nakagata, N.; et al. HPGCD outperforms HPBCD as a potential treatment for Niemann-Pick disease type C during disease modeling with iPS cells. Stem Cells 2015, 33, 1075–1088. [Google Scholar] [CrossRef] [PubMed]
- Efthymiou, A.G.; Steiner, J.; Pavan, W.J.; Wincovitch, S.; Larson, D.M.; Porter, F.D.; Rao, M.S.; Malik, N. Rescue of an in vitro neuron phenotype identified in Niemann-Pick disease, type C1 induced pluripotent stem cell-derived neurons by modulating the WNT pathway and calcium signaling. Stem Cells Transl. Med. 2015, 4, 230–238. [Google Scholar] [CrossRef]
- Cougnoux, A.; Cluzeau, C.; Mitra, S.; Li, R.; Williams, I.; Burkert, K.; Xu, X.; Wassif, C.A.; Zheng, W.; Porter, F.D. Necroptosis in Niemann-Pick disease, type C1: A potential therapeutic target. Cell Death Dis. 2016, 7, e2147. [Google Scholar] [CrossRef]
- Trilck, M.; Peter, F.; Zheng, C.; Frank, M.; Dobrenis, K.; Mascher, H.; Rolfs, A.; Frech, M.J. Diversity of glycosphingolipid GM2 and cholesterol accumulation in NPC1 patient-specific iPSC-derived neurons. Brain Res. 2017, 1657, 52–61. [Google Scholar] [CrossRef]
- Peter, F.; Trilck, M.; Rabenstein, M.; Rolfs, A.; Frech, M.J. Dataset in support of the generation of Niemann-Pick disease Type C1 patient-specific iPS cell lines carrying the novel NPC1 mutation c.1180TC or the prevalent c.3182TC mutation-Analysis of pluripotency and neuronal differentiation. Data Brief 2017, 12, 123–131. [Google Scholar] [CrossRef]
- Peter, F.; Rost, S.; Rolfs, A.; Frech, M.J. Activation of PKC triggers rescue of NPC1 patient specific iPSC derived glial cells from gliosis. Orphanet J. Rare Dis. 2017, 12, 145. [Google Scholar] [CrossRef]
- Dai, S.; Dulcey, A.E.; Hu, X.; Wassif, C.A.; Porter, F.D.; Austin, C.P.; Ory, D.S.; Marugan, J.; Zheng, W. Methyl-β-cyclodextrin restores impaired autophagy flux in Niemann-Pick C1-deficient cells through activation of AMPK. Autophagy 2017, 13, 1435–1451. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Pradhan, M.; Xu, M.; Roeder, A.; Beers, J.; Zou, J.; Liu, C.; Porter, F.D.; Zheng, W. An induced pluripotent stem cell line (TRNDi001-D) from a Niemann-Pick disease type C1 (NPC1) patient carrying a homozygous p. I1061T (c. 3182TC) mutation in the NPC1 gene. Stem Cell Res. 2020, 44, 101737. [Google Scholar] [CrossRef] [PubMed]
- Völkner, C.; Liedtke, M.; Petters, J.; Huth, K.; Knuebel, G.; Murua Escobar, H.; Bullerdiek, J.; Lukas, J.; Hermann, A.; Frech, M.J. Generation of an iPSC line (AKOSi006-A) from fibroblasts of a NPC1 patient, carrying the homozygous mutation p.I1061T (c.3182 T C) and a control iPSC line (AKOSi007-A) using a non-integrating Sendai virus system. Stem Cell Res. 2020, 49, 102056. [Google Scholar] [CrossRef] [PubMed]
- Jürs, A.V.; Völkner, C.; Liedtke, M.; Huth, K.; Lukas, J.; Hermann, A.; Frech, M.J. Oxidative Stress and Alterations in the Antioxidative Defense System in Neuronal Cells Derived from NPC1 Patient-Specific Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2020, 21, 7667. [Google Scholar] [CrossRef] [PubMed]
- Völkner, C.; Liedtke, M.; Petters, J.; Lukas, J.; Murua Escobar, H.; Knuebel, G.; Bullerdiek, J.; Holzmann, C.; Hermann, A.; Frech, M.J. Generation of an iPSC line (AKOSi004-A) from fibroblasts of a female adult NPC1 patient, carrying the compound heterozygous mutation p.Val1023Serfs*15/p.Gly992Arg and of an iPSC line (AKOSi005-A) from a female adult control individual. Stem Cell Res. 2021, 50, 102127. [Google Scholar] [CrossRef] [PubMed]
- Völkner, C.; Peter, F.; Liedtke, M.; Krohn, S.; Lindner, I.; Murua Escobar, H.; Cimmaruta, C.; Lukas, J.; Hermann, A.; Frech, M.J. Generation of the Niemann-Pick type C2 patient-derived iPSC line AKOSi001-A. Stem Cell Res. 2019, 41, 101606. [Google Scholar] [CrossRef]
- Bergamin, N.; Dardis, A.; Beltrami, A.; Cesselli, D.; Rigo, S.; Zampieri, S.; Domenis, R.; Bembi, B.; Beltrami, C.A. A human neuronal model of Niemann Pick C disease developed from stem cells isolated from patient’s skin. Orphanet J. Rare Dis. 2013, 8. [Google Scholar] [CrossRef]
- Sung, E.-A.; Yu, K.-R.; Shin, J.-H.; Seo, Y.; Kim, H.-S.; Guen Koog, M.; Kang, I.; Kim, J.-J.; Lee, B.-C.; Shin, T.-H.; et al. Generation of patient specific human neural stem cells from Niemann-Pick disease type C patient-derived fibroblasts. Oncotarget 2017. [Google Scholar] [CrossRef]
- Bräuer, A.U.; Kuhla, A.; Holzmann, C.; Wree, A.; Witt, M. Current Challenges in Understanding the Cellular and Molecular Mechanisms in Niemann-Pick Disease Type C1. Int. J. Mol. Sci. 2019, 20, 4392. [Google Scholar] [CrossRef]
- Maxfield, F.R.; Wüstner, D. Analysis of cholesterol trafficking with fluorescent probes. Methods Cell Biol. 2012, 108, 367–393. [Google Scholar] [CrossRef]
- Millat, G.; Marçais, C.; Tomasetto, C.; Chikh, K.; Fensom, A.H.; Harzer, K.; Wenger, D.A.; Ohno, K.; Vanier, M.T. Niemann-Pick C1 disease: Correlations between NPC1 mutations, levels of NPC1 protein, and phenotypes emphasize the functional significance of the putative sterol-sensing domain and of the cysteine-rich luminal loop. Am. J. Hum. Genet. 2001, 68, 1373–1385. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Marks, D.L.; Park, W.D.; Wheatley, C.L.; Puri, V.; O’Brien, J.F.; Kraft, D.L.; Lundquist, P.A.; Patterson, M.C.; Pagano, R.E.; et al. Niemann-Pick C variant detection by altered sphingolipid trafficking and correlation with mutations within a specific domain of NPC1. Am. J. Hum. Genet. 2001, 68, 1361–1372. [Google Scholar] [CrossRef] [PubMed]
- Barton, B.R. Niemann–Pick Type C. In Encyclopedia of Movement Disorders; Kompoliti, K., Verhagen, L., Eds.; Elsevier Science: Burlington, ON, Canada, 2010; pp. 301–304. ISBN 9780123741059. [Google Scholar]
- Genaro-Mattos, T.C.; Anderson, A.; Allen, L.B.; Korade, Z.; Mirnics, K. Cholesterol Biosynthesis and Uptake in Developing Neurons. ACS Chem. Neurosci. 2019, 10, 3671–3681. [Google Scholar] [CrossRef] [PubMed]
- Pfrieger, F.W.; Ungerer, N. Cholesterol metabolism in neurons and astrocytes. Prog. Lipid Res. 2011, 50, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Vanier, M.T. Lipid changes in Niemann-Pick disease type C brain: Personal experience and review of the literature. Neurochem. Res. 1999, 24, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Taniguchi, M.; Akaboshi, S.; Vanier, M.T.; Tai, T.; Sakuraba, H.; Ohno, K. Accumulation of GM2 Ganglioside Accumulation of GM2 in Niemann-Pick Type C Fibroblasts. Proc. Jpn. Acad. 1996, 20, 50–52. [Google Scholar]
- Taniguchi, M.; Shinoda, Y.; Ninomiya, H.; Vanier, M.T.; Ohno, K. Sites and temporal changes of gangliosides GM1/GM2 storage in the Niemann-Pick disease type C mouse brain. Brain Dev. 2001, 23. [Google Scholar] [CrossRef]
- Jiménez-Moreno, N.; Stathakos, P.; Caldwell, M.A.; Lane, J.D. Induced Pluripotent Stem Cell Neuronal Models for the Study of Autophagy Pathways in Human Neurodegenerative Disease. Cells 2017, 6, 24. [Google Scholar] [CrossRef]
- Füllgrabe, J.; Klionsky, D.J.; Joseph, B. Histone post-translational modifications regulate autophagy flux and outcome. Autophagy 2013, 9, 1621–1623. [Google Scholar] [CrossRef][Green Version]
- Loos, B.; Du Toit, A.; Hofmeyr, J.-H.S. Defining and measuring autophagosome flux—concept and reality. Autophagy 2014, 10, 2087–2096. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Anglade, P.; Vyas, S.; Javoy-Agid, F.; Herrero, M.T.; Michel, P.P.; Marquez, J.; Mouatt-Prigent, A.; Ruberg, M.; Hirsch, E.C.; Agid, Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histopathol. 1997, 12, 25–31. [Google Scholar] [PubMed]
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; de Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 2010, 13, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Guo, J.; Liu, Y.; Tang, B. Autophagy and Its Comprehensive Impact on ALS. Int. J. Neurosci. 2012, 122, 695–703. [Google Scholar] [CrossRef]
- Ko, D.C.; Milenkovic, L.; Beier, S.M.; Manuel, H.; Buchanan, J.; Scott, M.P. Cell-autonomous death of cerebellar purkinje neurons with autophagy in Niemann-Pick type C disease. PLoS Genet. 2005, 1, 81–95. [Google Scholar] [CrossRef]
- Liao, G.; Yao, Y.; Liu, J.; Yu, Z.; Cheung, S.; Xie, A.; Liang, X.; Bi, X. Cholesterol accumulation is associated with lysosomal dysfunction and autophagic stress in Npc1 -/- mouse brain. Am. J. Pathol. 2007, 171, 962–975. [Google Scholar] [CrossRef]
- Pacheco, C.D.; Lieberman, A.P. Lipid trafficking defects increase Beclin-1 and activate autophagy in Niemann-Pick type C disease. Autophagy 2007, 3, 487–489. [Google Scholar] [CrossRef]
- Pacheco, C.D.; Kunkel, R.; Lieberman, A.P. Autophagy in Niemann-Pick C disease is dependent upon Beclin-1 and responsive to lipid trafficking defects. Hum. Mol. Genet. 2007, 16, 1495–1503. [Google Scholar] [CrossRef]
- Evans, C.S.; Holzbaur, E.L.F. Autophagy and mitophagy in ALS. Neurobiol. Dis. 2019, 122, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Stachowiak, A.; Mamun, A.A.; Tzvetkov, N.T.; Takeda, S.; Atanasov, A.G.; Bergantin, L.B.; Abdel-Daim, M.M.; Stankiewicz, A.M. Autophagy and Alzheimer’s Disease: From Molecular Mechanisms to Therapeutic Implications. Front. Aging Neurosci. 2018, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Moors, T.E.; Hoozemans, J.J.M.; Ingrassia, A.; Beccari, T.; Parnetti, L.; Chartier-Harlin, M.-C.; van de Berg, W.D.J. Therapeutic potential of autophagy-enhancing agents in Parkinson’s disease. Mol. Neurodegener. 2017, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.H.; Wang, X.; Zhu, X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic. Biol. Med. 2013, 62, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Yanjanin, N.M.; Bianconi, S.; Pavan, W.J.; Porter, F.D. Oxidative stress in Niemann-Pick disease, type C. Mol. Genet. Metab. 2010, 101, 214–218. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 2019, 49, 35–45. [Google Scholar] [CrossRef]
- Elfawy, H.A.; Das, B. Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: Etiologies and therapeutic strategies. Life Sci. 2019, 218, 165–184. [Google Scholar] [CrossRef]
- Ordonez, M.P. Defective mitophagy in human Niemann-Pick Type C1 neurons is due to abnormal autophagy activation. Autophagy 2012, 8, 1157–1158. [Google Scholar] [CrossRef]
- Yu, W.; Gong, J.-S.; Ko, M.; Garver, W.S.; Yanagisawa, K.; Michikawa, M. Altered cholesterol metabolism in Niemann-Pick type C1 mouse brains affects mitochondrial function. J. Biol. Chem. 2005, 280, 11731–11739. [Google Scholar] [CrossRef]
- Marí, M.; Caballero, F.; Colell, A.; Morales, A.; Caballeria, J.; Fernandez, A.; Enrich, C.; Fernandez-Checa, J.C.; García-Ruiz, C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006, 4, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Vázquez, M.C.; Balboa, E.; Alvarez, A.R.; Zanlungo, S. Oxidative stress: A pathogenic mechanism for Niemann-Pick type C disease. Oxidative Med. Cell. Longev. 2012, 2012, 205713. [Google Scholar] [CrossRef] [PubMed]
- Fernández, A.; Llacuna, L.; Fernández-Checa, J.C.; Colell, A. Mitochondrial cholesterol loading exacerbates amyloid beta peptide-induced inflammation and neurotoxicity. J. Neurosci. 2009, 29. [Google Scholar] [CrossRef]
- Zampieri, S.; Mellon, S.H.; Butters, T.D.; Nevyjel, M.; Covey, D.F.; Bembi, B.; Dardis, A. Oxidative stress in NPC1 deficient cells: Protective effect of allopregnanolone. J. Cell. Mol. Med. 2009, 13, 3786–3796. [Google Scholar] [CrossRef] [PubMed]
- Cologna, S.M.; Jiang, X.-S.; Backlund, P.S.; Cluzeau, C.V.M.; Dail, M.K.; Yanjanin, N.M.; Siebel, S.; Toth, C.L.; Jun, H.; Wassif, C.A.; et al. Quantitative proteomic analysis of Niemann-Pick disease, type C1 cerebellum identifies protein biomarkers and provides pathological insight. PLoS ONE 2012, 7, e47845. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Sakiyama, T.; Harada, N.; Abe, M.; Tadokoro, M. Pathologic changes of glial cells in murine model of Niemann-Pick disease type C: Immunohistochemical, lectin-histochemical and ultrastructural observations. Pediatr. Int. 2003, 45, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Tamari, F.; Chen, F.W.; Li, C.; Chaudhari, J.; Ioannou, Y.A. PKC activation in Niemann pick C1 cells restores subcellular cholesterol transport. PLoS.ONE. 2013, 8, e74169. [Google Scholar] [CrossRef]
- Walter, M.; Chen, F.W.; Tamari, F.; Wang, R.; Ioannou, Y.A. Endosomal lipid accumulation in NPC1 leads to inhibition of PKC, hypophosphorylation of vimentin and Rab9 entrapment. Biol. Cell 2009, 101, 141–152. [Google Scholar] [CrossRef]
- Ioannou, Y.A.; Altstiel, L.; Crockford, D.R.; Kongsamut, M. Methods and Compositions for Treatment of Lipid Storage Disorders. U.S. Patent Application No. US000009724328B220170808, 8 August 2017. [Google Scholar]
- Pentchev, P.G.; Boothe, A.D.; Kruth, H.S.; Weintroub, H.; Stivers, J.; Brady, R.O. A genetic storage disorder in BALB/C mice with a metabolic block in esterification of exogenous cholesterol. J. Biol. Chem. 1984, 259, 5784–5791. [Google Scholar] [CrossRef]
- Davidson, C.D.; Ali, N.F.; Micsenyi, M.C.; Stephney, G.; Renault, S.; Dobrenis, K.; Ory, D.S.; Vanier, M.T.; Walkley, S.U. Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS ONE 2009, 4, e6951. [Google Scholar] [CrossRef]
- Sarna, J.R.; Larouche, M.; Marzban, H.; Sillitoe, R.V.; Rancourt, D.E.; Hawkes, R. Patterned Purkinje cell degeneration in mouse models of Niemann-Pick type C disease. J. Comp. Neurol. 2003, 456, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Perkins, E.; Suminaite, D.; Jackson, M. Cerebellar ataxias: Β-III spectrin’s interactions suggest common pathogenic pathways. J. Physiol. 2016, 594, 4661–4676. [Google Scholar] [CrossRef] [PubMed]
- Rabenstein, M.; Murr, N.; Hermann, A.; Rolfs, A.; Frech, M.J. Alteration of GABAergic Input Precedes Neurodegeneration of Cerebellar Purkinje Cells of NPC1-Deficient Mice. Int. J. Mol. Sci. 2019, 20, 6288. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Yang, F.; Rabenstein, M.; Wang, Z.; Frech, M.J.; Wree, A.; Bräuer, A.U.; Witt, M.; Gläser, A.; Hermann, A.; et al. Stimulation of mGluR1/5 Improves Defective Internalization of AMPA Receptors in NPC1 Mutant Mouse. Cereb. Cortex 2020, 30, 1465–1480. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Bader, B.M.; Yang, F.; Segura, M.; Schultz, L.; Schröder, O.H.-U.; Rolfs, A.; Luo, J. Improvement of impaired electrical activity in NPC1 mutant cortical neurons upon DHPG stimulation detected by micro-electrode array. Brain Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Elstein, D.; Hollak, C.; Aerts, J.M.F.G.; van Weely, S.; Maas, M.; Cox, T.M.; Lachmann, R.H.; Hrebicek, M.; Platt, F.M.; Butters, T.D.; et al. Sustained therapeutic effects of oral miglustat (Zavesca, N-butyldeoxynojirimycin, OGT 918) in type I Gaucher disease. J. Inherit. Metab. Dis. 2004, 27, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Wraith, J.E.; Imrie, J. New therapies in the management of Niemann-Pick type C disease: Clinical utility of miglustat. Ther. Clin. Risk Manag. 2009, 5, 877–887. [Google Scholar] [CrossRef]
- Lyseng-Williamson, K.A. Miglustat: A review of its use in Niemann-Pick disease type C. Drugs 2014, 74, 61–74. [Google Scholar] [CrossRef]
- Zervas, M.; Somers, K.L.; Thrall, M.A.; Walkley, S.U. Critical role for glycosphingolipids in Niemann-Pick disease type C. Curr. Biol. 2001, 11, 1283–1287. [Google Scholar] [CrossRef]
- Patterson, M.C.; Vecchio, D.; Prady, H.; Abel, L.; Wraith, J.E. Miglustat for treatment of Niemann-Pick C disease: A randomised controlled study. Lancet Neurol. 2007, 6, 765–772. [Google Scholar] [CrossRef]
- Wraith, J.E.; Vecchio, D.; Jacklin, E.; Abel, L.; Chadha-Boreham, H.; Luzy, C.; Giorgino, R.; Patterson, M.C. Miglustat in adult and juvenile patients with Niemann-Pick disease type C: Long-term data from a clinical trial. Mol. Genet. Metab. 2010, 99, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Patterson, M.C.; Vecchio, D.; Jacklin, E.; Abel, L.; Chadha-Boreham, H.; Luzy, C.; Giorgino, R.; Wraith, J.E. Long-term miglustat therapy in children with Niemann-Pick disease type C. J. Child Neurol. 2010, 25, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Pineda, M.; Wraith, J.E.; Mengel, E.; Sedel, F.; Hwu, W.-L.; Rohrbach, M.; Bembi, B.; Walterfang, M.; Korenke, G.C.; Marquardt, T.; et al. Miglustat in patients with Niemann-Pick disease Type C (NP-C): A multicenter observational retrospective cohort study. Mol. Genet. Metab. 2009, 98, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Jansook, P.; Ogawa, N.; Loftsson, T. Cyclodextrins: Structure, physicochemical properties and pharmaceutical applications. Int. J. Pharm. 2018, 535, 272–284. [Google Scholar] [CrossRef]
- Szente, L.; Szejtli, J. Cyclodextrins as food ingredients. Trends Food Sci. Technol. 2004, 15, 137–142. [Google Scholar] [CrossRef]
- Del Valle, E. Cyclodextrins and their uses: A review. Process Biochem. 2004, 39, 1033–1046. [Google Scholar] [CrossRef]
- Ottinger, E.A.; Kao, M.L.; Carrillo-Carrasco, N.; Yanjanin, N.; Shankar, R.K.; Janssen, M.; Brewster, M.; Scott, I.; Xu, X.; Cradock, J.; et al. Collaborative development of 2-hydroxypropyl-β-cyclodextrin for the treatment of Niemann-Pick type C1 disease. Curr. Top. Med. Chem. 2014, 14, 330–339. [Google Scholar] [CrossRef]
- Griffin, L.D.; Gong, W.; Verot, L.; Mellon, S.H. Niemann-Pick type C disease involves disrupted neurosteroidogenesis and responds to allopregnanolone. Nat. Med. 2004, 10, 704–711. [Google Scholar] [CrossRef]
- Liu, B.; Turley, S.D.; Burns, D.K.; Miller, A.M.; Repa, J.J.; Dietschy, J.M. Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the npc1-/- mouse. Proc. Natl. Acad. Sci. USA 2009, 106, 2377–2382. [Google Scholar] [CrossRef]
- Liu, B.; Ramirez, C.M.; Miller, A.M.; Repa, J.J.; Turley, S.D.; Dietschy, J.M. Cyclodextrin overcomes the transport defect in nearly every organ of NPC1 mice leading to excretion of sequestered cholesterol as bile acid. J. Lipid Res. 2010, 51, 933–944. [Google Scholar] [CrossRef]
- Pontikis, C.C.; Davidson, C.D.; Walkley, S.U.; Platt, F.M.; Begley, D.J. Cyclodextrin alleviates neuronal storage of cholesterol in Niemann-Pick C disease without evidence of detectable blood-brain barrier permeability. J. Inherit. Metab. Dis. 2013, 36, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, A.I.; Zhang, G.; Warren, J.D.; Maxfield, F.R. Endocytosis of beta-cyclodextrins is responsible for cholesterol reduction in Niemann-Pick type C mutant cells. Proc. Natl. Acad. Sci. USA 2010, 107, 5477–5482. [Google Scholar] [CrossRef] [PubMed]
- Ory, D.S.; Ottinger, E.A.; Farhat, N.Y.; King, K.A.; Jiang, X.; Weissfeld, L.; Berry-Kravis, E.; Davidson, C.D.; Bianconi, S.; Keener, L.A.; et al. Intrathecal 2-hydroxypropyl-β-cyclodextrin decreases neurological disease progression in Niemann-Pick disease, type C1: A non-randomised, open-label, phase 1–2 trial. Lancet 2017, 390, 1758–1768. [Google Scholar] [CrossRef]
- Farmer, C.A.; Thurm, A.; Farhat, N.; Bianconi, S.; Keener, L.A.; Porter, F.D. Long-term neuropsychological outcomes from an open-label phase 1/2a trial of 2-hydoxypropyl-β-cyclodextrins (VTS-270) in Niemann-Pick Disease, Type C1. CNS Drugs 2019, 33, 677–683. [Google Scholar] [CrossRef]
- Davidson, C.D.; Fishman, Y.I.; Puskás, I.; Szemán, J.; Sohajda, T.; McCauliff, L.A.; Sikora, J.; Storch, J.; Vanier, M.T.; Szente, L.; et al. Efficacy and ototoxicity of different cyclodextrins in Niemann-Pick C disease. Ann. Clin. Transl. Neurol. 2016, 3, 366–380. [Google Scholar] [CrossRef]
- Peterson, C.L. HDAC’s at Work. Mol. Cell 2002, 9, 921–922. [Google Scholar] [CrossRef]
- Kawai, H.; Li, H.; Avraham, S.; Jiang, S.; Avraham, H.K. Overexpression of histone deacetylase HDAC1 modulates breast cancer progression by negative regulation of estrogen receptor alpha. Int. J. Cancer 2003, 107, 353–358. [Google Scholar] [CrossRef]
- Sugars, K.L.; Rubinsztein, D.C. Transcriptional abnormalities in Huntington disease. Trends Genet. 2003, 19, 233–238. [Google Scholar] [CrossRef]
- Kim, S.-J.; Lee, B.-H.; Lee, Y.-S.; Kang, K.-S. Defective cholesterol traffic and neuronal differentiation in neural stem cells of Niemann-Pick type C disease improved by valproic acid, a histone deacetylase inhibitor. Biochem. Biophys. Res. Commun. 2007, 360, 593–599. [Google Scholar] [CrossRef]
- Munkacsi, A.B.; Chen, F.W.; Brinkman, M.A.; Higaki, K.; Gutiérrez, G.D.; Chaudhari, J.; Layer, J.V.; Tong, A.; Bard, M.; Boone, C.; et al. An “Exacerbate-reverse” Strategy in Yeast Identifies Histone Deacetylase Inhibition as a Correction for Cholesterol and Sphingolipid Transport Defects in Human Niemann-Pick Type C Disease*. J. Biol. Chem. 2011, 286, 23842–23851. [Google Scholar] [CrossRef]
- Pipalia, N.H.; Cosner, C.C.; Huang, A.; Chatterjee, A.; Bourbon, P.; Farley, N.; Helquist, P.; Wiest, O.; Maxfield, F.R. Histone deacetylase inhibitor treatment dramatically reduces cholesterol accumulation in Niemann-Pick type C1 mutant human fibroblasts. Proc. Natl. Acad. Sci. USA 2011, 108, 5620–5625. [Google Scholar] [CrossRef] [PubMed]
- Hait, N.C.; Allegood, J.; Maceyka, M.; Strub, G.M.; Harikumar, K.B.; Singh, S.K.; Luo, C.; Marmorstein, R.; Kordula, T.; Milstien, S.; et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 2009, 325, 1254–1257. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G. Treating lysosomal storage diseases with pharmacological chaperones: From concept to clinics. EMBO Mol. Med. 2009, 1, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Valenzano, K.J.; Khanna, R.; Powe, A.C.; Boyd, R.; Lee, G.; Flanagan, J.J.; Benjamin, E.R. Identification and characterization of pharmacological chaperones to correct enzyme deficiencies in lysosomal storage disorders. ASSAY Drug Dev. Technol. 2011, 9, 213–235. [Google Scholar] [CrossRef] [PubMed]
- Millat, G.; Marcais, C.; Rafi, M.A.; Yamamoto, T.; Morris, J.A.; Pentchev, P.G.; Ohno, K.; Wenger, D.A.; Vanier, M.T. Niemann-Pick C1 disease: The I1061T substitution is a frequent mutant allele in patients of Western European descent and correlates with a classic juvenile phenotype. Am. J. Hum. Genet. 1999, 65, 1321–1329. [Google Scholar] [CrossRef]
- Gelsthorpe, M.E.; Baumann, N.; Millard, E.; Gale, S.E.; Langmade, S.J.; Schaffer, J.E.; Ory, D.S. Niemann-Pick type C1 I1061T mutant encodes a functional protein that is selected for endoplasmic reticulum-associated degradation due to protein misfolding. J. Biol. Chem. 2008, 283, 8229–8236. [Google Scholar] [CrossRef]
- Ohgane, K.; Karaki, F.; Dodo, K.; Hashimoto, Y. Discovery of oxysterol-derived pharmacological chaperones for NPC1: Implication for the existence of second sterol-binding site. Chem. Biol. 2013, 20. [Google Scholar] [CrossRef]
- Fukuda, H.; Karaki, F.; Dodo, K.; Noguchi-Yachide, T.; Ishikawa, M.; Hashimoto, Y.; Ohgane, K. Phenanthridin-6-one derivatives as the first class of non-steroidal pharmacological chaperones for Niemann-Pick disease type C1 protein. Bioorganic Med. Chem. Lett. 2017, 27. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- González, F.; Boué, S.; Belmonte, J.C.I. Methods for making induced pluripotent stem cells: Reprogramming à la carte. Nat. Rev. Genet. 2011, 12, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Raya, A.; Barrero, M.J.; Garreta, E.; Consiglio, A.; Gonzalez, F.; Vassena, R.; Bilić, J.; Pekarik, V.; Tiscornia, G.; et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat. Biotechnol. 2008, 26, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Su, R.J.; Neises, A.; Zhang, X.-B. Generation of iPS Cells from Human Peripheral Blood Mononuclear Cells Using Episomal Vectors. In Induced Pluripotent Stem (iPS) Cells: Methods and Protocols; Turksen, K., Nagy, A., Eds.; Humana Press: New York, NY, USA, 2016; pp. 57–69. ISBN 978-1-4939-3055-5. [Google Scholar]
- Steinle, H.; Weber, M.; Behring, A.; Mau-Holzmann, U.; von Ohle, C.; Popov, A.-F.; Schlensak, C.; Wendel, H.P.; Avci-Adali, M. Reprogramming of Urine-Derived Renal Epithelial Cells into iPSCs Using srRNA and Consecutive Differentiation into Beating Cardiomyocytes. Mol. Ther.-Nucleic Acids 2019, 17, 907–921. [Google Scholar] [CrossRef]
- Ben-Ari, Y. The GABA excitatory/inhibitory developmental sequence: A personal journey. Neuroscience 2014, 279, 187–219. [Google Scholar] [CrossRef]
- Miller, J.D.; Ganat, Y.M.; Kishinevsky, S.; Bowman, R.L.; Liu, B.; Tu, E.Y.; Mandal, P.K.; Vera, E.; Shim, J.; Kriks, S.; et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 2013, 13, 691–705. [Google Scholar] [CrossRef]
- Lopez-Leon, M.; Reggiani, P.C.; Herenu, C.B.; Goya, R.G. Regenerative Medicine for the Aging Brain. Enliven. J. Stem Cell Res. Regen. Med. 2014, 1, 1–9. [Google Scholar] [CrossRef]
- Hou, S.; Lu, P. Direct reprogramming of somatic cells into neural stem cells or neurons for neurological disorders. Neural Regen. Res. 2016, 11, 28–31. [Google Scholar] [CrossRef]
- Papapetrou, E.P.; Tomishima, M.J.; Chambers, S.M.; Mica, Y.; Reed, E.; Menon, J.; Tabar, V.; Mo, Q.; Studer, L.; Sadelain, M. Stoichiometric and temporal requirements of Oct4, Sox2, Klf4, and c-Myc expression for efficient human iPSC induction and differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 12759–12764. [Google Scholar] [CrossRef]
- Zhou, W.; Freed, C.R. Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells 2009, 27, 2667–2674. [Google Scholar] [CrossRef]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B 2009, 85, 348–362. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Nakagawa, M.; Hyenjong, H.; Ichisaka, T.; Yamanaka, S. Generation of mouse induced pluripotent stem cells without viral vectors. Science 2008, 322, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Si-Tayeb, K.; Noto, F.K.; Sepac, A.; Sedlic, F.; Bosnjak, Z.J.; Lough, J.W.; Duncan, S.A. Generation of human induced pluripotent stem cells by simple transient transfection of plasmid DNA encoding reprogramming factors. BMC Dev. Biol. 2010, 10, 81. [Google Scholar] [CrossRef] [PubMed]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.-H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef]
- Kim, D.; Kim, C.-H.; Moon, J.-I.; Chung, Y.-G.; Chang, M.-Y.; Han, B.-S.; Ko, S.; Yang, E.; Cha, K.Y.; Lanza, R.; et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell 2009, 4, 472–476. [Google Scholar] [CrossRef]
- Huangfu, D.; Maehr, R.; Guo, W.; Eijkelenboom, A.; Snitow, M.; Chen, A.E.; Melton, D.A. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat. Biotechnol. 2008, 26, 795–797. [Google Scholar] [CrossRef]
- Muguruma, K.; Nishiyama, A.; Ono, Y.; Miyawaki, H.; Mizuhara, E.; Hori, S.; Kakizuka, A.; Obata, K.; Yanagawa, Y.; Hirano, T.; et al. Ontogeny-recapitulating generation and tissue integration of ES cell-derived Purkinje cells. Nat. Neurosci. 2010, 13, 1171–1180. [Google Scholar] [CrossRef]
- Silva, T.P.; Bekman, E.P.; Fernandes, T.G.; Vaz, S.H.; Rodrigues, C.A.V.; Diogo, M.M.; Cabral, J.M.S.; Carmo-Fonseca, M. Maturation of Human Pluripotent Stem Cell-Derived Cerebellar Neurons in the Absence of Co-culture. Front. Bioeng. Biotechnol. 2020, 8, 70. [Google Scholar] [CrossRef]
- Hay, D.C.; Fletcher, J.; Payne, C.; Terrace, J.D.; Gallagher, R.C.J.; Snoeys, J.; Black, J.R.; Wojtacha, D.; Samuel, K.; Hannoun, Z.; et al. Highly efficient differentiation of hESCs to functional hepatic endoderm requires ActivinA and Wnt3a signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 12301–12306. [Google Scholar] [CrossRef]
- Toivonen, S.; Lundin, K.; Balboa, D.; Ustinov, J.; Tamminen, K.; Palgi, J.; Trokovic, R.; Tuuri, T.; Otonkoski, T. Activin A and Wnt-dependent specification of human definitive endoderm cells. Exp. Cell Res. 2013, 319, 2535–2544. [Google Scholar] [CrossRef]
- Benussi, A.; Alberici, A.; Premi, E.; Bertasi, V.; Cotelli, M.S.; Turla, M.; Dardis, A.; Zampieri, S.; Marchina, E.; Paghera, B.; et al. Phenotypic heterogeneity of Niemann-Pick disease type C in monozygotic twins. J. Neurol. 2015, 262, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Malnar, M.; Hecimovic, S.; Mattsson, N.; Zetterberg, H. Bidirectional links between Alzheimer’s disease and Niemann-Pick type C disease. Neurobiol. Dis. 2014, 72 Pt A, 37–47. [Google Scholar] [CrossRef]


{kind=link}
| Reference | Donor Cells Used | Reprogramming Method | Stem Cells Used | Differentiation Target(s) | Results/ Observations | Compounds Used | Genotypes Studied |
|---|---|---|---|---|---|---|---|
| Models Based on Pluripotent Stem Cells | |||||||
| Ordonez et al. 2012 [32] | N/A | N/A | NPC1 KD hESCs | NSCs neurons | Accumulation of lysotracker positive organelles Mitochondrial fragments Increased filipin staining Defective autophagy | M-β-CD | N/A |
| Bergamin et al. 2013 [48] | N/A | N/A | hSKIN-MASCs | neurons | Cholesterol and GM2 accumulation Morphological changes | N/A | I1061T/I1061T I1061T/I1061T V1165M |
| Models Based on Transdifferentiation | |||||||
| Sung et al. 2017 [49] | Fibroblasts | Trans-differentiation | N/A | neurons | Cholesterol accumulation Deficiencies in self-renewal of NSCs | SB202190 L-NAME valproic acid | P237S/I1061T I1061T/I1061T |
| Models Based on Induced Pluripotent Stem Cells | |||||||
| Trilck et al. 2013 [11] | Fibroblasts | Retroviral | iPSCs | NPCs NDCs | Cholesterol accumulation in fibroblasts, iPSCs and NPCs | N/A | E612D/P543Rfs*20 |
| Maetzel et al. 2014 [33] | Fibroblasts | Cre-excisable lentivirus | iPSCs | HLCs NDCs | Decreased cell viability Defects in cholesterol metabolism and autophagy | H-β-CD rapamycin bafilomycin A1 carbamazepine verapamil trehalose SMER28 | I1061T/I1061T P237S/I1061T 1920delG/1009G>A |
| Yu et al. 2014 [34] | Fibroblasts | Sendai virus | iPSCs | NSCs neurons | Cholesterol accumulation | HP-β-CD M-β-CD δ-tocopherol | P237S/I1061T |
| Lee et al. 2014 [35] | Fibroblasts | Retroviral | iPSCs | neurons | Defects in VEGF signaling Defective autophagic flux | SphK inhibitor VEGF | P237S/I1061T |
| Soga et al. 2015 [36] | Fibroblasts | Sendai virus | iPSCs | HLCs NPCs | Cholesterol accumulation Impaired autophagy | HP-β-CD HP-γ-CD | S667L/C1161Y Y1088C/581_592delinsG |
| Efthymiou et al. 2015 [37] | Fibroblasts | Lentiviral | iPSCs | NSCs neurons | Impaired Ca2+ and Wnt3a signaling | Curcumin dantrolene BIO | I1061T/I1061T |
| Cougnoux et al. 2016 [38] | Fibroblasts | Sendai virus | iPSCs | NPCs | Decreased viability Increased necroptosis | Nec1 | P237S/I1061T |
| Trilck et al. 2017 [39] | Fibroblasts | Retroviral | iPSCs | NPCs NDCs | Cholesterol and GM2 accumulation in NDCs Reduced Hex A activity | N/A | I1061T/I1061T Y394H/Y394H E612D/P543Rfs*20 |
| Peter et al. 2017 [40] | Fibroblasts | Retroviral | iPSCs | NPCs NDCs | Methodological report of reprogramming | N/A | I1061T/I1061T Y394H/Y394H |
| Peter et al. 2017 [41] | Fibroblasts | Retroviral | iPSCs | NPCs NDCs | Increased number of reactive astrocytes Hypophosphorylation of GFAP and vimentin | PMA | I1061T/I1061T Y394H/Y394H E612D/P543Rfs*20 |
| Rabenstein et al. 2017 [20] | Fibroblasts | Retroviral | iPSCs | NPCs NDCs | Decreased Ca2+ flux through AMPAR Increased amount of GluA2 | N/A | I1061T/I1061T Y394H/Y394H E612D/P543Rfs*20 |
| Dai et al. 2017 [42] | Fibroblasts | Sendai virus | iPSCs | NSCs neurons | Impaired macroautophagy/autophagy | HP-β-CD M-β-CD | P237S/I1061T |
| Li et al. 2020 [43] | Fibroblasts | Sendai virus | iPSCs | N/A | Methodological report of reprogramming | N/A | I1061T/I1061T |
| Völkner et al. 2020 [44] | Fibroblasts | Sendai virus | iPSCs | N/A | Methodological report of reprogramming | N/A | I1061T/I1061T |
| Jürs et al. 2020 [45] | Fibroblasts | Retroviral | iPSCs | NPCs NDCs | Increased level of ROS Down regulation of catalase | N/A | I1061T/I1061T Y394H/Y394H E612D/P543Rfs*20 |
| Völkner et al. 2020 [46] | Fibroblasts | Retroviral | iPSCs | N/A | Methodological report of reprogramming | N/A | V1023Sfs*15/G992R |
| Pluripotent Stem Cell-Based Models in NPC2 Disease | |||||||
| Völkner et al. 2019 [47] | Fibroblasts | Retroviral | iPSCs | N/A | Methodological report of reprogramming | N/A | E20X/C47F |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Völkner, C.; Liedtke, M.; Hermann, A.; Frech, M.J. Pluripotent Stem Cells for Disease Modeling and Drug Discovery in Niemann-Pick Type C1. Int. J. Mol. Sci. 2021, 22, 710. https://doi.org/10.3390/ijms22020710
Völkner C, Liedtke M, Hermann A, Frech MJ. Pluripotent Stem Cells for Disease Modeling and Drug Discovery in Niemann-Pick Type C1. International Journal of Molecular Sciences. 2021; 22(2):710. https://doi.org/10.3390/ijms22020710
Chicago/Turabian StyleVölkner, Christin, Maik Liedtke, Andreas Hermann, and Moritz J. Frech. 2021. "Pluripotent Stem Cells for Disease Modeling and Drug Discovery in Niemann-Pick Type C1" International Journal of Molecular Sciences 22, no. 2: 710. https://doi.org/10.3390/ijms22020710
APA StyleVölkner, C., Liedtke, M., Hermann, A., & Frech, M. J. (2021). Pluripotent Stem Cells for Disease Modeling and Drug Discovery in Niemann-Pick Type C1. International Journal of Molecular Sciences, 22(2), 710. https://doi.org/10.3390/ijms22020710