Control of Mesenchymal Stromal Cell Senescence by Tryptophan Metabolites


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
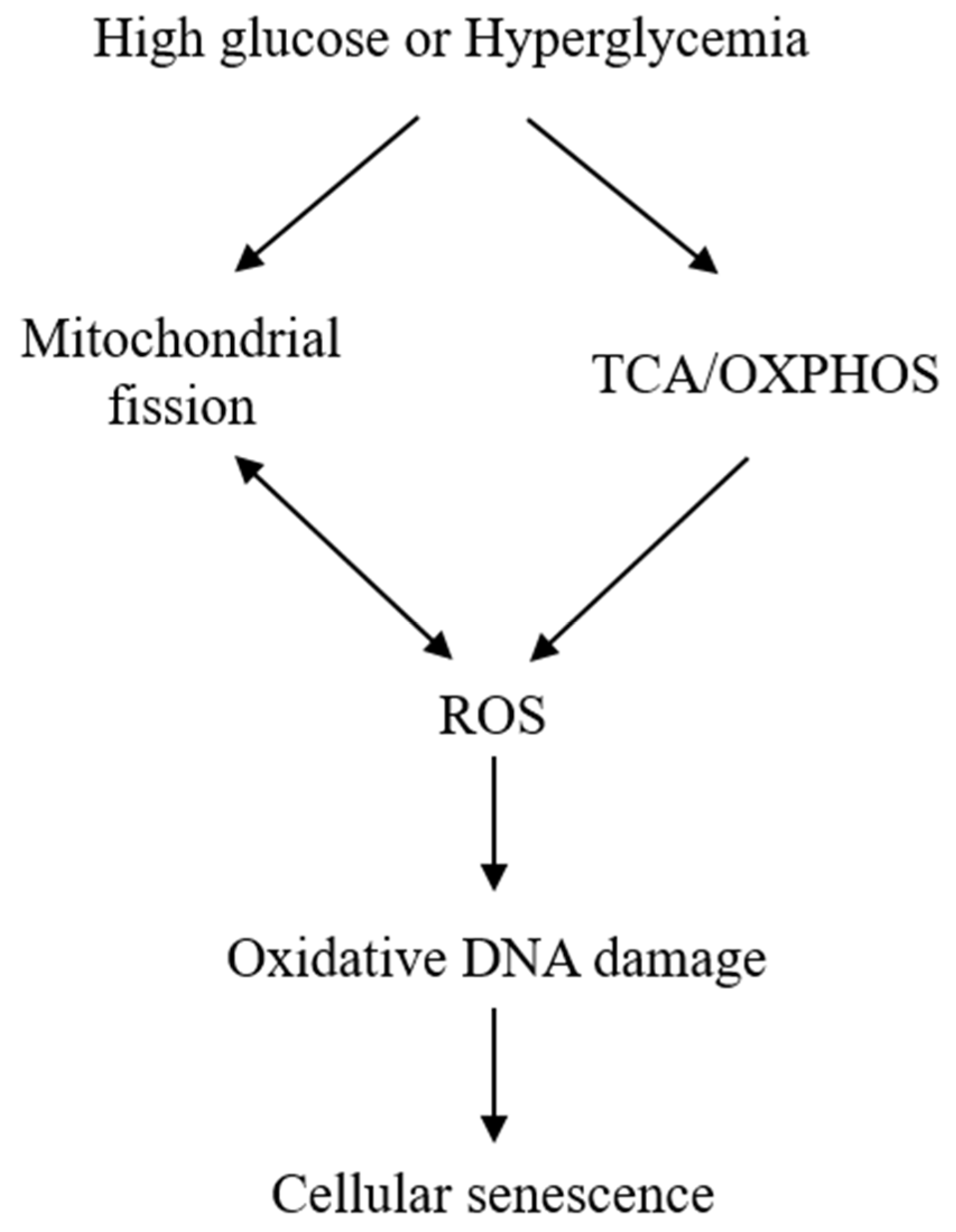
2. Hyperglycemia Induces Cellular Senescence
2.1. HG-Induced Cellular Senescence Is Attributed to Mitochondrial Dysfunction and ROS Generation
2.2. HG Induces MSC Senescence
3. 5-MTP Rescues MSCs from HG-Induced Senescence
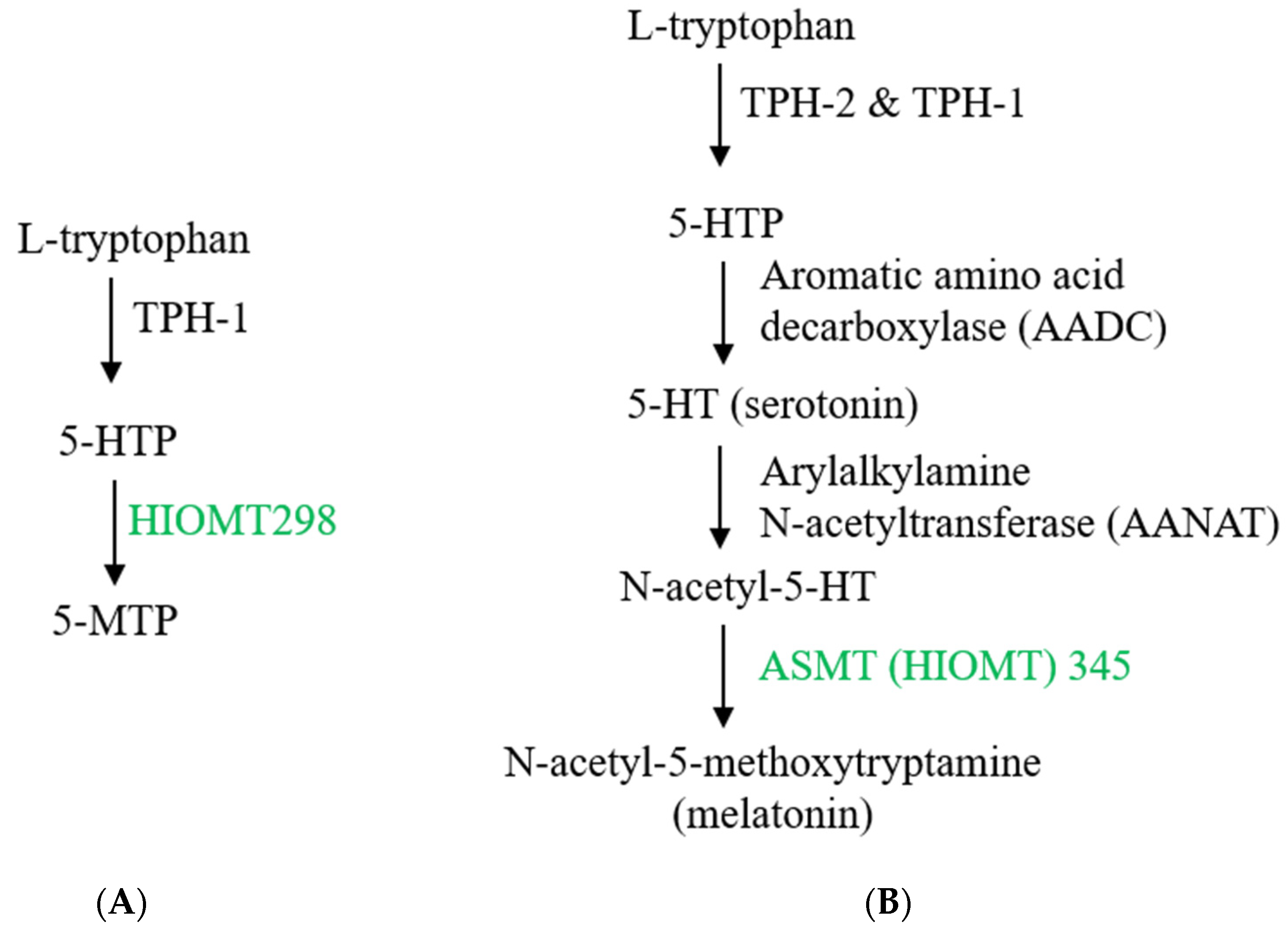
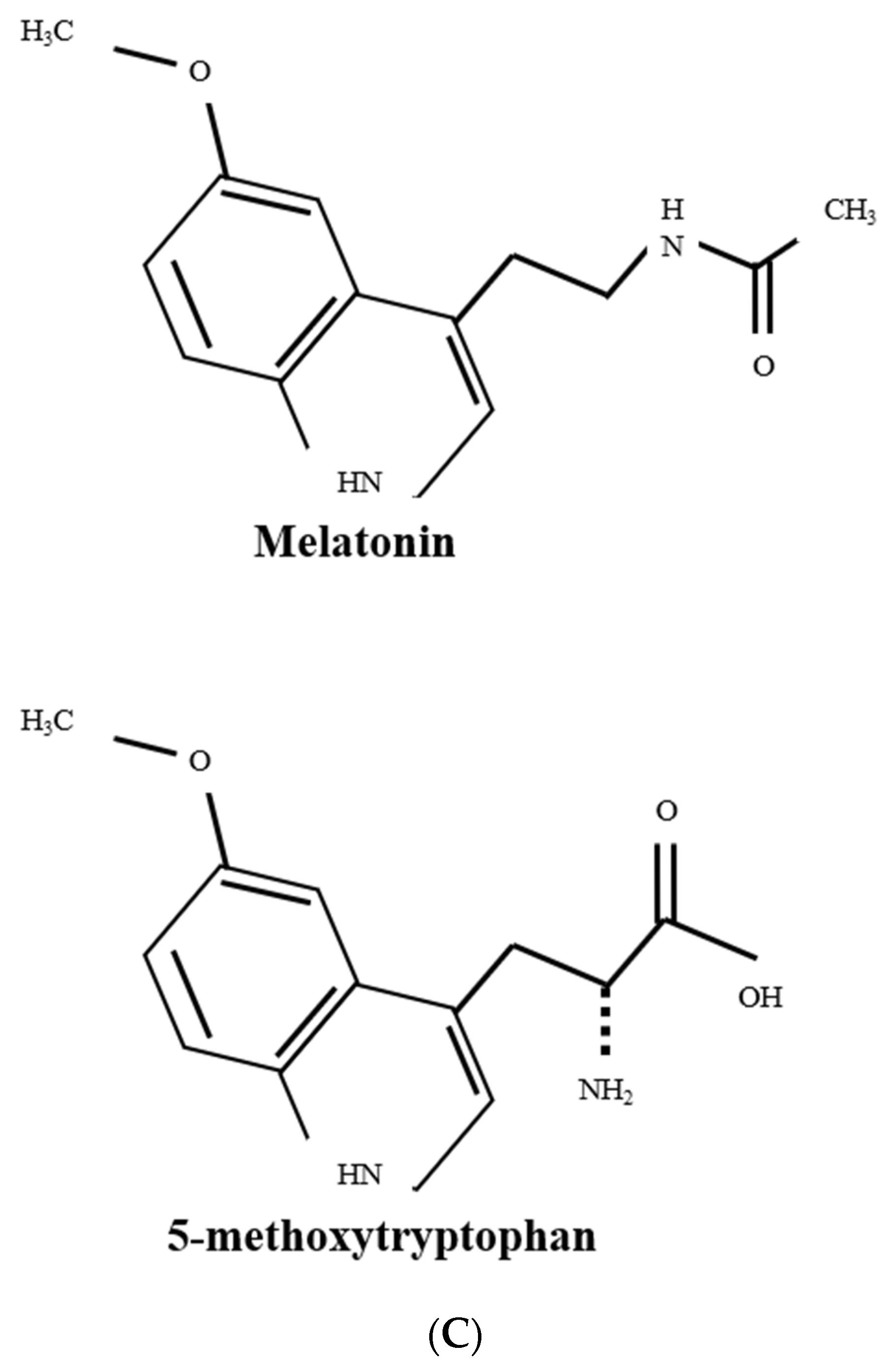
3.1. 5-MTP Biosynthesis and Its Perturbation by Environmental Stresses
3.2. 5-MTP Rescues BM-MSC from HG-Induced Cellular Senescence by Suppressing ROS Generation
3.3. 5-MTP Upregulates MnSOD and Catalase via FOXO3a
3.4. 5-MTP Restores BM-MSC Osteogenic Differentiation via Controlling ROS Levels
4. Melatonin Protects Against Replicative and Stress-Induced Cellular Senescence
4.1. Melatonin Attenuates MSC Replicative Senescence by Restoring Mitochondrial Function and Reducing ROS
4.2. Melatonin Controls HG- and Oxidant-Induced Cellular Senescence by Upregulating Antioxidant Enzymes
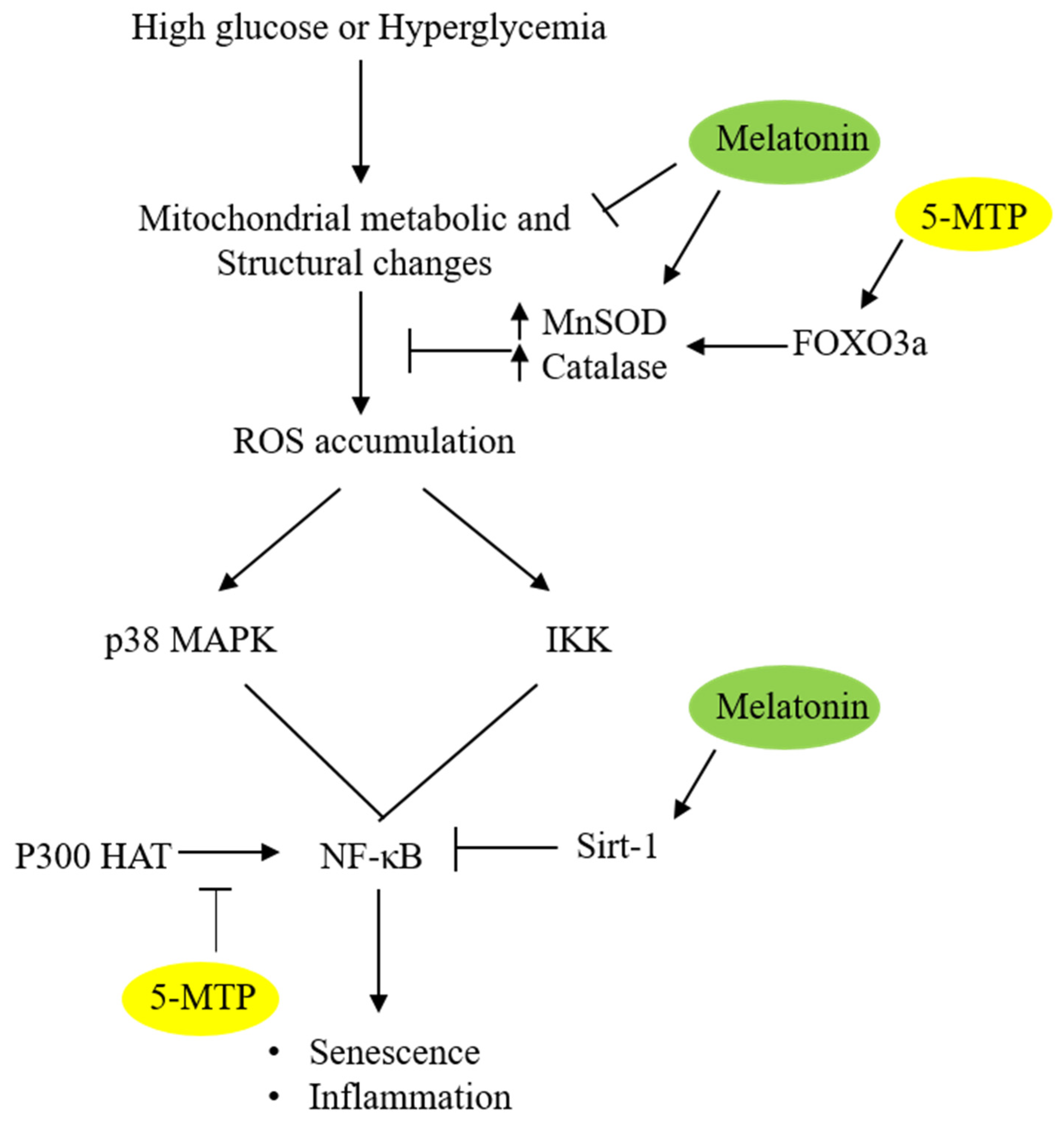
5. Melatonin and 5-MTP Target p38 MAPK
6. Melatonin and 5-MTP Control Cellular Senescence through Inhibition of NF-κB
Melatonin and 5-MTP Modify Histone and NF-κB Acetylation
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Behnia, F.; Taylor, B.D.; Woodson, M.; Kacerovsky, M.; Hawkins, H.; Fortunato, S.J.; Saade, G.R.; Menon, R. Chorioamniotic membrane senescence: A signal for parturition? Am. J. Obstet. Gynecol. 2015, 213, 359.e1–359.e16. [Google Scholar] [CrossRef] [PubMed]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Alessio, N.; Aprile, D.; Squillaro, T.; Di Bernardo, G.; Finicelli, M.; Melone, M.A.; Peluso, G.; Galderisi, U. The senescence-associated secretory phenotype (SASP) from mesenchymal stromal cells impairs growth of immortalized prostate cells but has no effect on metastatic prostatic cancer cells. Aging 2019, 11, 5817–5828. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Alfego, D.; Rodeck, U.; Kriete, A. Global mapping of transcription factor motifs in human aging. PLoS ONE 2018, 13, e0190457. [Google Scholar] [CrossRef] [PubMed]
- Lanigan, F.; Geraghty, J.G.; Bracken, A.P. Transcriptional regulation of cellular senescence. Oncogene 2011, 30, 2901–2911. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.K.; Tchkonia, T.; LeBrasseur, N.K.; Chini, E.N.; Xu, M.; Kirkland, J.L. Cellular Senescence in Type 2 Diabetes: A Therapeutic Opportunity. Diabetes 2015, 64, 2289–2298. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, E.M.; Le Blanc, K.; Dominici, M.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Deans, R.J.; Krause, D.S.; Keating, A.; International Society for Cellular Therapy. Clarification of the nomenclature for MSC: The International Society for Cellular Therapy position statement. Cytotherapy 2005, 7, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Squillaro, T.; Peluso, G.; Galderisi, U. Clinical Trials with Mesenchymal Stem Cells: An Update. Cell Transpl. 2016, 25, 829–848. [Google Scholar] [CrossRef]
- Uccelli, A.; Moretta, L.; Pistoia, V. Mesenchymal stem cells in health and disease. Nat. Rev. Immunol. 2008, 8, 726–736. [Google Scholar] [CrossRef]
- English, K. Mechanisms of mesenchymal stromal cell immunomodulation. Immunol. Cell Biol. 2013, 91, 19–26. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.M.; Marquess, D.; Dananberg, J.; van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef]
- Liu, J.; Ding, Y.; Liu, Z.; Liang, X. Senescence in Mesenchymal Stem Cells: Functional Alterations, Molecular Mechanisms, and Rejuvenation Strategies. Front. Cell Dev. Biol. 2020, 8, 258. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.C.; Hsu, M.F.; Shih, C.Y.; Wu, K.K. 5-methoxytryptophan protects MSCs from stress induced premature senescence by upregulating FoxO3a and mTOR. Sci. Rep. 2017, 7, 11133. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yoon, Y.M.; Song, K.H.; Noh, H.; Lee, S.H. Melatonin suppresses senescence-derived mitochondrial dysfunction in mesenchymal stem cells via the HSPA1L-mitophagy pathway. Aging Cell 2020, 19, e13111. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef]
- Kitada, K.; Nakano, D.; Ohsaki, H.; Hitomi, H.; Minamino, T.; Yatabe, J.; Felder, R.A.; Mori, H.; Masaki, T.; Kobori, H.; et al. Hyperglycemia causes cellular senescence via a SGLT2- and p21-dependent pathway in proximal tubules in the early stage of diabetic nephropathy. J. Diabetes Complicat. 2014, 28, 604–611. [Google Scholar] [CrossRef]
- Shosha, E.; Xu, Z.; Narayanan, S.P.; Lemtalsi, T.; Fouda, A.Y.; Rojas, M.; Xing, J.; Fulton, D.; Caldwell, R.W.; Caldwell, R.B. Mechanisms of Diabetes-Induced Endothelial Cell Senescence: Role of Arginase 1. Int. J. Mol. Sci. 2018, 19, 1215. [Google Scholar] [CrossRef]
- Blazer, S.; Khankin, E.; Segev, Y.; Ofir, R.; Yalon-Hacohen, M.; Kra-Oz, Z.; Gottfried, Y.; Larisch, S.; Skorecki, K.L. High glucose-induced replicative senescence: Point of no return and effect of telomerase. Biochem. Biophys. Res. Commun. 2002, 296, 93–101. [Google Scholar] [CrossRef]
- Zhong, W.; Zou, G.; Gu, J.; Zhang, J. L-arginine attenuates high glucose-accelerated senescence in human umbilical vein endothelial cells. Diabetes Res. Clin. Pract. 2010, 89, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Maeda, M.; Hayashi, T.; Mizuno, N.; Hattori, Y.; Kuzuya, M. Intermittent high glucose implements stress-induced senescence in human vascular endothelial cells: Role of superoxide production by NADPH oxidase. PLoS ONE 2015, 10, e0123169. [Google Scholar] [CrossRef]
- Palmer, A.K.; Gustafson, B.; Kirkland, J.L.; Smith, U. Cellular senescence: At the nexus between ageing and diabetes. Diabetologia 2019, 62, 1835–1841. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar] [CrossRef] [PubMed]
- Abuarab, N.; Munsey, T.S.; Jiang, L.H.; Li, J.; Sivaprasadarao, A. High glucose-induced ROS activates TRPM2 to trigger lysosomal membrane permeabilization and Zn 2+-mediated mitochondrial fission. Sci. Signal. 2017, 10, eaal4161. [Google Scholar] [CrossRef] [PubMed]
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000, 49, 1939–1945. [Google Scholar] [CrossRef] [PubMed]
- Balteau, M.; Tajeddine, N.; de Meester, C.; Ginion, A.; Des Rosiers, C.; Brady, N.R.; Sommereyns, C.; Horman, S.; Vanoverschelde, J.L.; Gailly, P.; et al. NADPH oxidase activation by hyperglycaemia in cardiomyocytes is independent of glucose metabolism but requires SGLT1. Cardiovasc. Res. 2011, 9, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.M.; Bartholomew, J.C.; Campisi, J.; Acosta, M.; Reagan, J.D.; Ames, B.N. Molecular analysis of H2O2-induced senescent-like growth arrest in normal human fibroblasts: p53 and Rb control G1 arrest but not cell replication. Biochem. J. 1998, 332, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.C.; Fenster, B.E.; Ito, H.; Takeda, K.; Bae, N.S.; Hirai, T.; Yu, Z.X.; Ferrans, V.J.; Howard, B.H.; Finkel, T. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J. Biol. Chem. 1999, 274, 7936–7940. [Google Scholar] [CrossRef] [PubMed]
- Macip, S.; Igarashi, M.; Berggren, P.; Yu, J.; Lee, S.W.; Aaronson, S.A. Influence of induced reactive oxygen species in p53-mediated cell fate decisions. Mol. Cell Biol. 2003, 23, 8576–8585. [Google Scholar] [CrossRef]
- Chen, Q.; Fischer, A.; Reagan, J.D.; Yan, L.J.; Ames, B.N. Oxidative DNA damage and senescence of human diploid fibroblast cells. Proc. Natl. Acad. Sci. USA 1995, 92, 4337–4341. [Google Scholar] [CrossRef]
- Ziegler, D.V.; Wiley, C.D.; Velarde, M.C. Mitochondrial effectors of cellular senescence: Beyond the free radical theory of aging. Aging Cell 2015, 14, 1–7. [Google Scholar] [CrossRef]
- Chen, H.; Chomyn, A.; Chan, D.C. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 2005, 280, 26185–26192. [Google Scholar] [CrossRef]
- Yoon, Y.S.; Yoon, D.S.; Lim, I.K.; Yoon, S.H.; Chung, H.Y.; Rojo, M.; Malka, F.; Jou, M.J.; Martinou, J.C.; Yoon, G. Formation of elongated giant mitochondria in DFO-induced cellular senescence: Involvement of enhanced fusion process through modulation of Fis1. J. Cell Physiol. 2006, 209, 468–480. [Google Scholar] [CrossRef]
- Stöckl, P.; Hütter, E.; Zwerschke, W.; Jansen-Dürr, P. Sustained inhibition of oxidative phosphorylation impairs cell proliferation and induces premature senescence in human fibroblasts. Exp. Gerontol. 2006, 41, 674–682. [Google Scholar] [CrossRef]
- Borradaile, N.M.; Pickering, J.G. Nicotinamide phosphoribosyltransferase imparts human endothelial cells with extended replicative lifespan and enhanced angiogenic capacity in a high glucose environment. Aging Cell 2009, 8, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.; van der Veer, E.; Akawi, O.; Pickering, J.G. SIRT1 markedly extends replicative lifespan if the NAD+ salvage pathway is enhanced. FEBS Lett. 2009, 583, 3081–3085. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.T.; Shih, Y.R.; Kuo, T.K.; Lee, O.K.; Wei, Y.H. Coordinated changes of mitochondrial biogenesis and antioxidant enzymes during osteogenic differentiation of human mesenchymal stem cells. Stem Cells 2008, 26, 960–968. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Suda, T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol. 2014, 15, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Pattappa, G.; Heywood, H.K.; de Bruijn, J.D.; Lee, D.A. The metabolism of human mesenchymal stem cells during proliferation and differentiation. J. Cell Physiol. 2011, 226, 2562–2570. [Google Scholar] [CrossRef]
- Stolzing, A.; Coleman, N.; Scutt, A. Glucose-induced replicative senescence in mesenchymal stem cells. Rejuvenation Res. 2006, 9, 31–35. [Google Scholar] [CrossRef]
- Zhang, D.; Lu, H.; Chen, Z.; Wang, Y.; Lin, J.; Xu, S.; Zhang, C.; Wang, B.; Yuan, Z.; Feng, X.; et al. High glucose induces the aging of mesenchymal stem cells via Akt/mTOR signaling. Mol. Med. Rep. 2017, 16, 1685–1690. [Google Scholar] [CrossRef]
- Ferraro, F.; Lymperi, S.; Méndez-Ferrer, S.; Saez, B.; Spencer, J.A.; Yeap, B.Y.; Masselli, E.; Graiani, G.; Prezioso, L.; Rizzini, E.L.; et al. Diabetes impairs hematopoietic stem cell mobilization by altering niche function. Sci. Transl. Med. 2011, 3, 104ra101. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.G.; Saunders, M.; Gilroy, D.; He, X.Z.; Yeh, H.; Zhu, Y.; Shtivelband, M.I.; Ruan, K.H.; Wu, K.K. Purification and characterization of a cyclooxygenase-2 and angiogenesis suppressing factor produced by human fibroblasts. FASEB J. 2002, 16, 1286–1288. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.H.; Kuo, C.C.; Yan, J.L.; Chen, H.L.; Lin, W.C.; Wang, K.H.; Tsai, K.K.; Guvén, H.; Flaberg, E.; Szekely, L.; et al. Control of cyclooxygenase-2 expression and tumorigenesis by endogenous 5-methoxytryptophan. Proc. Nat. Acad. Sci. USA 2012, 109, 13231–13236. [Google Scholar] [CrossRef] [PubMed]
- Lovenberg, W.; Jequier, E.; Sjoerdsma, A. Tryptophan hydroxylation: Measurement in pineal gland, brainstem, and carcinoid tumor. Science 1967, 155, 217–219. [Google Scholar] [CrossRef] [PubMed]
- Walther, D.J.; Peter, J.U.; Bashammakh, S.; Hörtnagl, H.; Voits, M.; Fink, H.; Bader, M. Synthesis of serotonin by a second tryptophan hydroxylase isoform. Science 2003, 299, 76. [Google Scholar] [CrossRef]
- Rodriguez, I.R.; Mazuruk, K.; Schoen, T.J.; Chader, G.J. Structural analysis of the human hydroxyindole-O-methyltransferase gene. Presence of two distinct promoters. J. Biol. Chem. 1994, 269, 31969–31977. [Google Scholar] [CrossRef] [PubMed]
- Donohue, S.J.; Roseboom, P.H.; Illnerova, H.; Weller, J.L.; Klein, D.C. Human hydroxyindole-O-methyltransferase: Presence of LINE-1 fragment in a cDNA clone and pineal mRNA. DNA Cell Biol. 1993, 12, 715–727. [Google Scholar] [CrossRef]
- Chen, H.L.; Yuan, C.Y.; Cheng, H.H.; Chang, T.C.; Huang, S.K.; Kuo, C.C.; Wu, K.K. Restoration of hydroxyindole O-methyltransferase levels in human cancer cells induces a tryptophan-metabolic switch and attenuates cancer progression. J. Biol. Chem. 2018, 293, 11131–11142. [Google Scholar] [CrossRef]
- Botros, H.G.; Legrand, P.; Pagan, C.; Bondet, V.; Weber, P.; Ben-Abdallah, M.; Lemière, N.; Huguet, G.; Bellalou, J.; Maronde, E.; et al. Crystal structure and functional mapping of human ASMT, the last enzyme of the melatonin synthesis pathway. J. Pineal Res. 2013, 54, 46–57. [Google Scholar] [CrossRef]
- Wang, Y.F.; Hsu, Y.J.; Wu, H.F.; Lee, G.L.; Yang, Y.S.; Wu, J.Y.; Yet, S.F.; Wu, K.K.; Kuo, C.C. Endothelium-derived 5-methoxytryptophan is a circulating anti-inflammatory molecule that blocks systemic inflammation. Circ. Res. 2016, 119, 222–236. [Google Scholar] [CrossRef]
- Chu, L.Y.; Wang, Y.F.; Cheng, H.H.; Kuo, C.C.; Wu, K.K. Endothelium-derived 5-methoxytryptophan protects endothelial barrier function by blocking p38 MAPK activation. PLoS ONE 2016, 11, e0152166. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.C.; Hsu, M.F.; Wu, K.K. High glucose induces bone marrow-derived mesenchymal stem cell senescence by upregulating autophagy. PLoS ONE 2015, 10, e0126537. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [PubMed]
- Sivitz, W.I.; Yorek, M.A. Mitochondrial dysfunction in diabetes: From molecular mechanisms to functional significance and therapeutic opportunities. Antioxid. Redox Signal. 2010, 12, 537–577. [Google Scholar] [CrossRef]
- Patel, H.; Chen, J.; Das, K.C.; Kavdia, M. Hyperglycemia induces differential change in oxidative stress at gene expression and functional levels in HUVEC and HMVEC. Cardiovasc. Diabetol. 2013, 12, 142. [Google Scholar] [CrossRef]
- Eijkelenboom, A.; Burgering, B.M. FOXOs: Signaling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 2013, 14, 83–97. [Google Scholar] [CrossRef]
- Marinkovic, D.; Zhang, X.; Yalcin, S.; Luciano, J.P.; Brugnara, C.; Huber, T.; Ghaffari, S. Foxo3 is required for the regulation of oxidative stress in erythropoiesis. J. Clin. Investig. 2007, 117, 2133–2144. [Google Scholar] [CrossRef]
- Zhang, Y.; Marsboom, G.; Toth, P.T.; Rehman, J. Mitochondrial respiration regulates adipogenic differentiation of human mesenchymal stem cells. PLoS ONE 2013, 8, e77077. [Google Scholar] [CrossRef]
- Li, Q.; Gao, Z.; Chen, Y.; Guan, M.X. The role of mitochondria in osteogenic, adipogenic and chondrogenic differentiation of mesenchymal stem cells. Protein Cell 2017, 8, 439–445. [Google Scholar] [CrossRef]
- Martin, J.A.; Buckwalter, J.A. Aging, articular cartilage chondrocyte senescence and osteoarthritis. Biogerontology 2002, 3, 257–264. [Google Scholar] [CrossRef]
- Luchetti, F.; Canonico, B.; Bartolini, D.; Arcangeletti, M.; Ciffolilli, S.; Murdolo, G.; Piroddi, M.; Papa, S.; Reiter, R.J.; Galli, F. Melatonin regulates mesenchymal stem cell differentiation: A review. J. Pineal Res. 2014, 56, 382–397. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R.; Pandi-Perumal, S.R.; Cardinali, D.P. Melatonin. Int. J. Biochem. Cell Biol. 2006, 38, 313–316. [Google Scholar] [CrossRef] [PubMed]
- García, J.J.; López-Pingarrón, L.; Almeida-Souza, P.; Tres, A.; Escudero, P.; García-Gil, F.A.; Tan, D.X.; Reiter, R.J.; Ramírez, J.M.; Bernal-Pérez, M. Protective effects of melatonin in reducing oxidative stress and in preserving the fluidity of biological membranes: A review. J. Pineal Res. 2014, 56, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Mayo, J.C.; Tan, D.X.; Sainz, R.M.; Alatorre-Jimenez, M.; Qin, L. Melatonin as an antioxidant: Under promises but over delivers. J. Pineal Res. 2016, 61, 253–278. [Google Scholar] [CrossRef]
- Blask, D.E.; Sauer, L.A.; Dauchy, R.T.; Holowachuk, E.W.; Ruhoff, M.S.; Kopff, H.S. Melatonin inhibition of cancer growth in vivo involves suppression of tumor fatty acid metabolism via melatonin receptor-mediated signal transduction events. Cancer Res. 1999, 59, 4693–4701. [Google Scholar]
- Reiter, R.J.; Rosales-Corral, S.A.; Tan, D.X.; Acuna-Castroviejo, D.; Qin, L.; Yang, S.F.; Xu, K. Melatonin, a Full Service Anti-Cancer Agent: Inhibition of Initiation, Progression and Metastasis. Int. J. Mol. Sci. 2017, 18, 843. [Google Scholar] [CrossRef]
- Mayer, M.P.; Bukau, B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell Mol. Life Sci. 2005, 62, 670–684. [Google Scholar] [CrossRef]
- Lee, Y.H.; Jung, H.S.; Kwon, M.J.; Jang, J.E.; Kim, T.N.; Lee, S.H.; Kim, M.K.; Park, J.H. Melatonin protects INS-1 pancreatic β-cells from apoptosis and senescence induced by glucotoxicity and glucolipotoxicity. Islets 2020, 12, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Yang, L.; Li, Y.; Yan, G.; Feng, C.; Liu, T.; Gong, R.; Yuan, Y.; Wang, N.; Idiiatullina, E.; et al. Melatonin protects bone marrow mesenchymal stem cells against iron overload-induced aberrant differentiation and senescence. J. Pineal Res. 2017, 63. [Google Scholar] [CrossRef]
- Yun, S.P.; Han, Y.S.; Lee, J.H.; Kim, S.M.; Lee, S.H. Melatonin Rescues Mesenchymal Stem Cells from Senescence Induced by the Uremic Toxin p-Cresol via Inhibiting mTOR-Dependent Autophagy. Biomol. Ther. 2018, 26, 389–398. [Google Scholar] [CrossRef]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signaling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Borodkina, A.; Shatrova, A.; Abushik, P.; Nikolsky, N.; Burova, E. Interaction between ROS dependent DNA damage, mitochondria and p38 MAPK underlies senescence of human adult stem cells. Aging 2014, 6, 481–495. [Google Scholar] [CrossRef]
- Xu, Y.; Li, N.; Xiang, R.; Sun, P. Emerging roles of the p38 MAPK and PI3K/AKT/mTOR pathways in oncogene-induced senescence. Trends Biochem. Sci. 2014, 39, 268–276. [Google Scholar] [CrossRef]
- Son, Y.; Cheong, Y.K.; Kim, N.H.; Chung, H.T.; Kang, D.G.; Pae, H.O. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J. Signal Transduct. 2011, 2011, 792639. [Google Scholar] [CrossRef] [PubMed]
- Hou, N.; Torii, S.; Saito, N.; Hosaka, M.; Takeuchi, T. Reactive oxygen species-mediated pancreatic beta-cell death is regulated by interactions between stress-activated protein kinases, p38 and c-Jun N-terminal kinase, and mitogen-activated protein kinase phosphatases. Endocrinology 2008, 149, 1654–1665. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.Y.; Liu, Y.; Wu, G.S. The role of mitogen-activated protein kinase phosphatase-1 in oxidative damage-induced cell death. Cancer Res. 2006, 66, 4888–4894. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Ho, Y.C.; Ho, H.H.; Liang, L.Y.; Jiang, W.C.; Lee, G.L.; Lee, J.K.; Hsu, Y.J.; Kuo, C.C.; Wu, K.K.; et al. Tryptophan metabolite 5-methoxytryptophan ameliorates arterial denudation-induced intimal hyperplasia via opposing effects on vascular endothelial and smooth muscle cells. Aging 2019, 11, 8604–8622. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.K.; Kuo, C.C.; Yet, S.F.; Lee, C.M.; Liou, J.Y. 5-methoxytryptophan: An arsenal against vascular injury and inflammation. J. Biomed. Sci. 2020, 27, 79. [Google Scholar] [CrossRef]
- Ho, Y.C.; Wu, M.L.; Su, C.H.; Chen, C.H.; Ho, H.H.; Lee, G.L.; Lin, W.S.; Lin, W.Y.; Hsu, Y.J.; Kuo, C.C.; et al. A Novel Protective Function of 5-Methoxytryptophan in Vascular Injury. Sci. Rep. 2016, 6, 25374. [Google Scholar] [CrossRef]
- Mao, L.; Yuan, L.; Slakey, L.M.; Jones, F.E.; Burow, M.E.; Hill, S.M. Inhibition of breast cancer cell invasion by melatonin is mediated through regulation of the p38 mitogen-activated protein kinase signaling pathway. Breast Cancer Res. 2010, 12, R107. [Google Scholar] [CrossRef]
- Vilar, A.; de Lemos, L.; Patraca, I.; Martínez, N.; Folch, J.; Junyent, F.; Verdaguer, E.; Pallàs, M.; Auladell, C.; Camins, A. Melatonin suppresses nitric oxide production in glial cultures by pro-inflammatory cytokines through p38 MAPK inhibition. Free Radic. Res. 2014, 48, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Chen, X.; Liu, T.; Gong, Y.; Chen, S.; Pan, G.; Cui, W.; Luo, Z.P.; Pei, M.; Yang, H.; et al. Melatonin reverses H2O2-induced premature senescence in mesenchymal stem cells via the SIRT1-dependent pathway. J. Pineal Res. 2015, 59, 190–205. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 2011, 25, 2125–2136. [Google Scholar] [CrossRef] [PubMed]
- Bernard, D.; Gosselin, K.; Monte, D.; Vercamer, C.; Bouali, F.; Pourtier, A.; Vandenbunder, B.; Abbadie, C. Involvement of Rel/nuclear factor-kappaB transcription factors in keratinocyte senescence. Cancer Res. 2004, 64, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Takada, Y.; Mukhopadhyay, A.; Kundu, G.C.; Mahabeleshwar, G.H.; Singh, S.; Aggarwal, B.B. Hydrogen peroxide activates NF-kappa B through tyrosine phosphorylation of I kappa B alpha and serine phosphorylation of p65: Evidence for the involvement of I kappa B alpha kinase and Syk protein-tyrosine kinase. J. Biol. Chem. 2003, 278, 24233–24241. [Google Scholar] [CrossRef]
- Reynaert, N.L.; van der Vliet, A.; Guala, A.S.; McGovern, T.; Hristova, M.; Pantano, C.; Heintz, N.H.; Heim, J.; Ho, Y.S.; Matthews, D.E.; et al. Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc. Natl. Acad. Sci. USA 2006, 103, 13086–13091. [Google Scholar] [CrossRef]
- Schoonbroodt, S.; Ferreira, V.; Best-Belpomme, M.; Boelaert, J.R.; Legrand-Poels, S.; Korner, M.; Piette, J. Crucial role of the amino-terminal tyrosine residue 42 and the carboxyl-terminal PEST domain of I kappa B alpha in NF-kappa B activation by an oxidative stress. J. Immunol. 2000, 164, 4292–4300. [Google Scholar] [CrossRef]
- Salotti, J.; Johnson, P.F. Regulation of senescence and the SASP by the transcription factor C/EBPβ. Exp. Gerontol. 2019, 128, 110752. [Google Scholar] [CrossRef]
- Goodman, R.H.; Smolik, S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000, 14, 1553–1577, Review. [Google Scholar]
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature 1997, 389, 349–352. [Google Scholar] [CrossRef]
- Deng, W.G.; Zhu, Y.; Wu, K.K. Up-regulation of p300 binding and p50 acetylation in tumor necrosis factor-alpha-induced cyclooxygenase-2 promoter activation. J. Biol. Chem. 2003, 278, 4770–4777. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.G.; Zhu, Y.; Wu, K.K. Role of p300 and PCAF in regulating cyclooxygenase-2 promoter activation by inflammatory mediators. Blood 2004, 103, 2135–2142. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Feng, B.; George, B.; Chakrabarti, R.; Chen, M.; Chakrabarti, S. Transcriptional coactivator p300 regulates glucose-induced gene expression in endothelial cells. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E127–E137. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.H.; Wang, K.H.; Chu, L.Y.; Chang, T.C.; Kuo, C.C.; Wu, K.K. Quiescent and proliferative fibroblasts exhibit differential p300 HAT activation through control of 5-methoxytryptophan production. PLoS ONE 2014, 9, e88507. [Google Scholar] [CrossRef]
- Favero, G.; Franceschetti, L.; Bonomini, F.; Rodella, L.F.; Rezzani, R. Melatonin as an Anti-Inflammatory Agent Modulating Inflammasome Activation. Int. J. Endocrinol. 2017, 2017, 1835195. [Google Scholar] [CrossRef]
- Deng, W.G.; Tang, S.T.; Tseng, H.P.; Wu, K.K. Melatonin suppresses macrophage cyclooxygenase-2 and inducible nitric oxide synthase expression by inhibiting p52 acetylation and binding. Blood 2006, 108, 518–524. [Google Scholar] [CrossRef]
- Wang, J.; Xiao, X.; Zhang, Y.; Shi, D.; Chen, W.; Fu, L.; Liu, L.; Xie, F.; Kang, T.; Huang, W.; et al. Simultaneous modulation of COX-2, p300, Akt, and Apaf-1 signaling by melatonin to inhibit proliferation and induce apoptosis in breast cancer cells. J. Pineal Res. 2012, 53, 77–90. [Google Scholar] [CrossRef]
- Nopparat, C.; Sinjanakhom, P.; Govitrapong, P. Melatonin reverses H2O2-induced senescence in SH-SY5Y cells by enhancing autophagy via sirtuin 1 deacetylation of the RelA/p65 subunit of NF-κB. J. Pineal Res. 2017, 63. [Google Scholar] [CrossRef]
- Rahman, S.; Islam, R. Mammalian Sirt1: Insights on its biological functions. Cell Commun. Signal 2011, 9, 11. [Google Scholar] [CrossRef]
- Xu, C.; Wang, L.; Fozouni, P.; Evjen, G.; Chandra, V.; Jiang, J.; Lu, C.; Nicastri, M.; Bretz, C.; Winkler, J.D.; et al. SIRT1 is downregulated by autophagy in senescence and ageing. Nat. Cell Biol. 2020, 22, 1170–1179. [Google Scholar] [CrossRef]
- Hayakawa, T.; Iwai, M.; Aoki, S.; Takimoto, K.; Maruyama, M.; Maruyama, W.; Motoyama, N. SIRT1 suppresses the senescence-associated secretory phenotype through epigenetic gene regulation. PLoS ONE 2015, 10, e0116480. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, K.K. Control of Mesenchymal Stromal Cell Senescence by Tryptophan Metabolites. Int. J. Mol. Sci. 2021, 22, 697. https://doi.org/10.3390/ijms22020697
Wu KK. Control of Mesenchymal Stromal Cell Senescence by Tryptophan Metabolites. International Journal of Molecular Sciences. 2021; 22(2):697. https://doi.org/10.3390/ijms22020697
Chicago/Turabian StyleWu, Kenneth K. 2021. "Control of Mesenchymal Stromal Cell Senescence by Tryptophan Metabolites" International Journal of Molecular Sciences 22, no. 2: 697. https://doi.org/10.3390/ijms22020697
APA StyleWu, K. K. (2021). Control of Mesenchymal Stromal Cell Senescence by Tryptophan Metabolites. International Journal of Molecular Sciences, 22(2), 697. https://doi.org/10.3390/ijms22020697