Molecular Mechanisms of Glucocorticoid-Induced Insulin Resistance

 ,
, 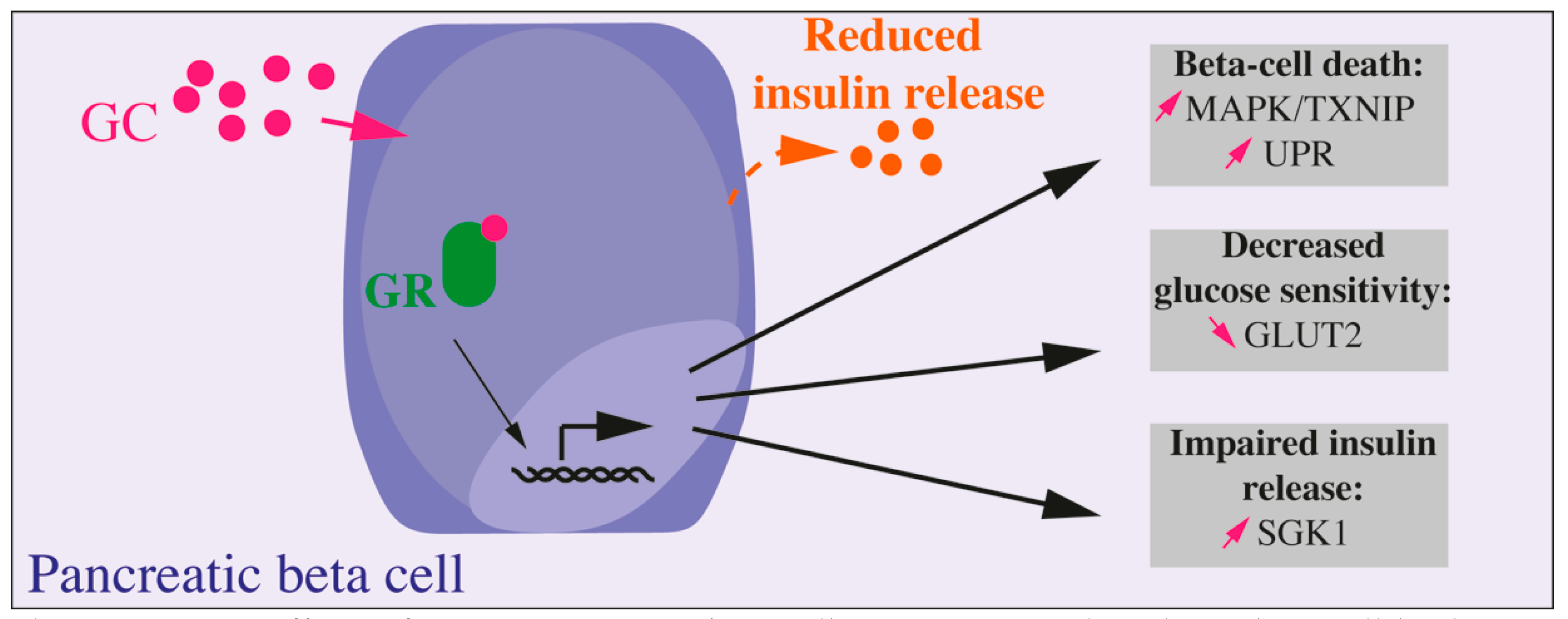{kind=link}
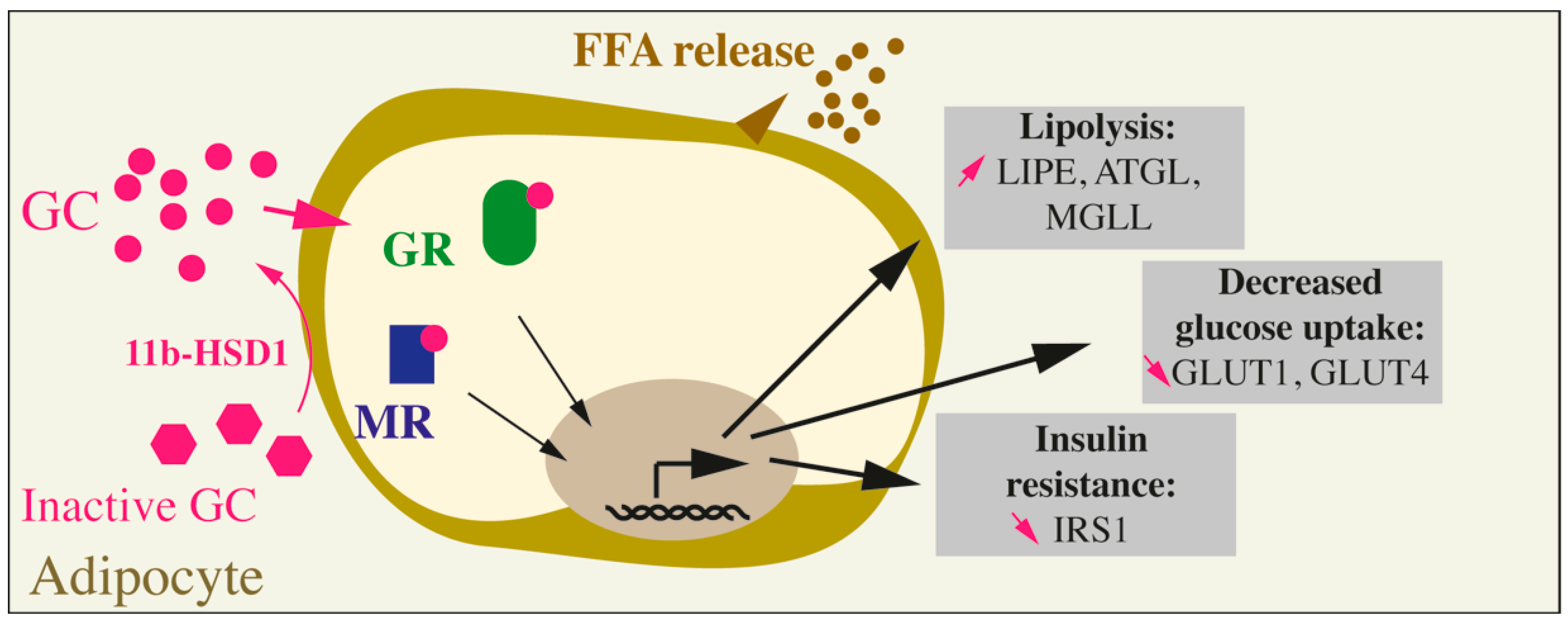{kind=link}
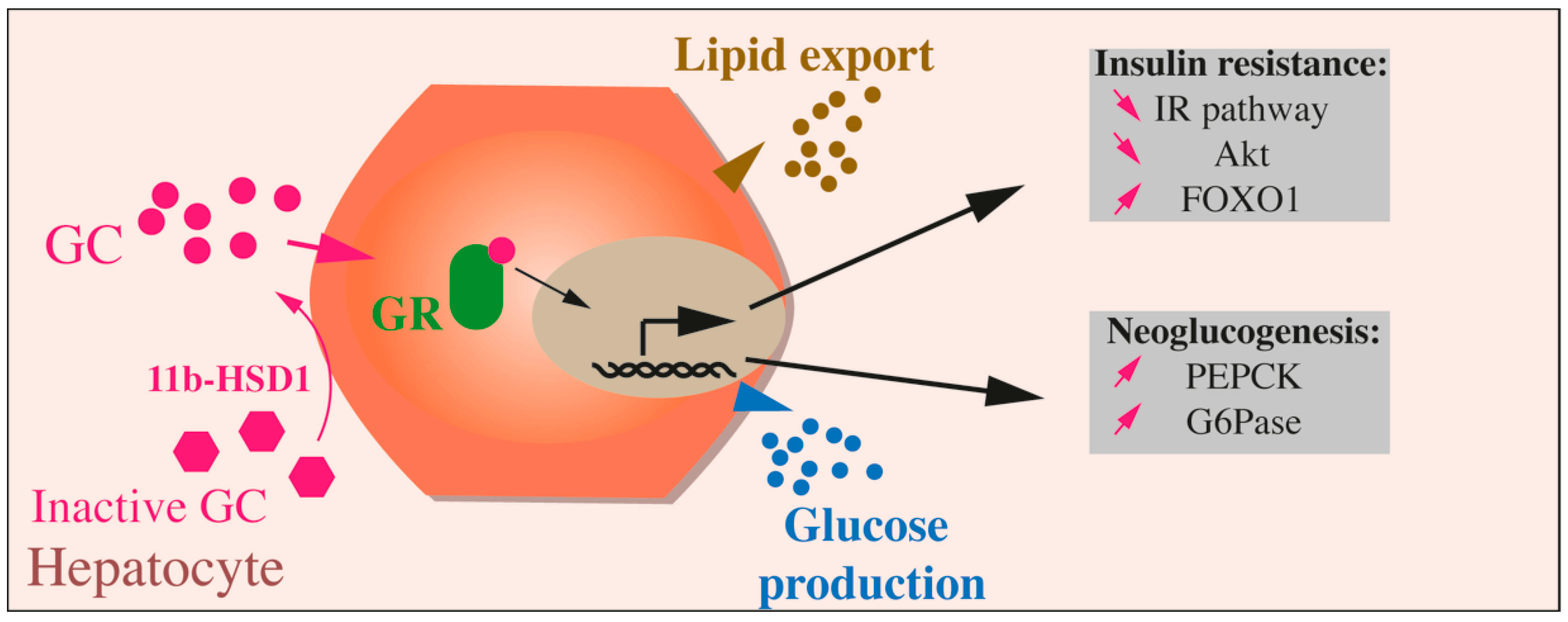{kind=link}
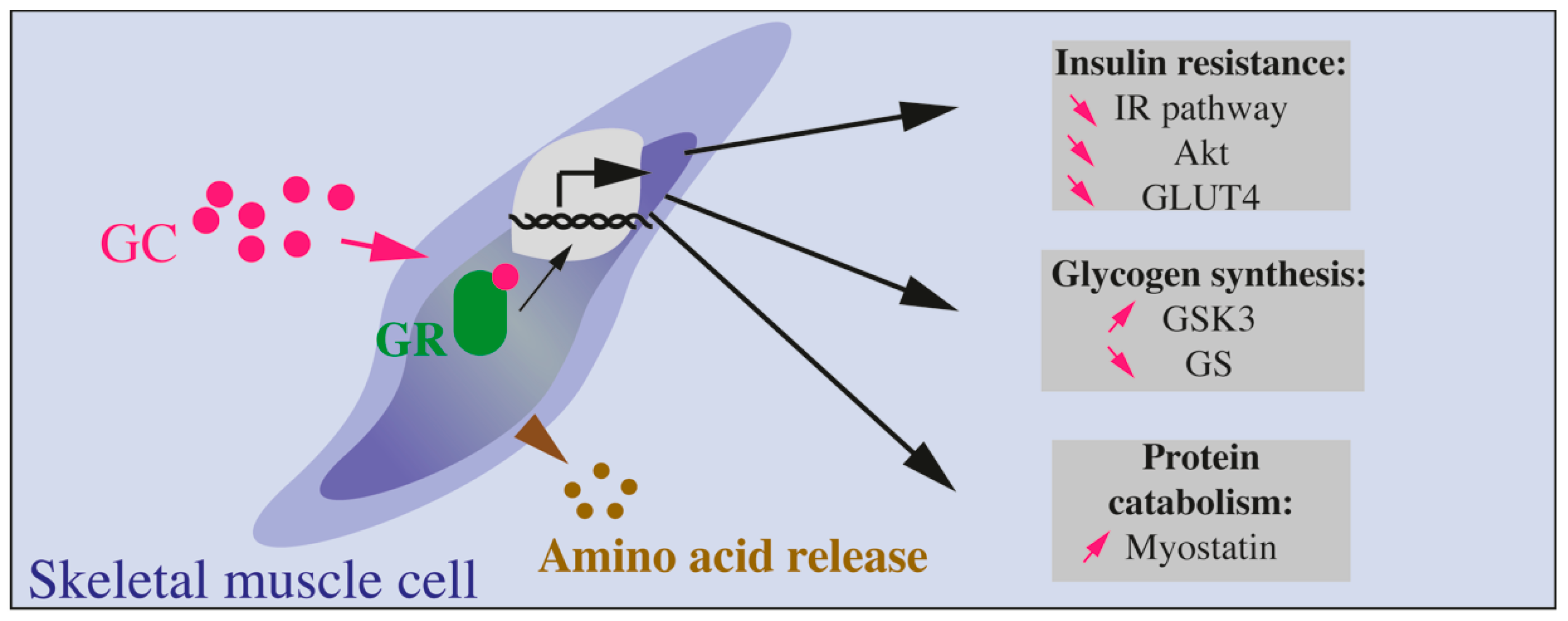{kind=link}
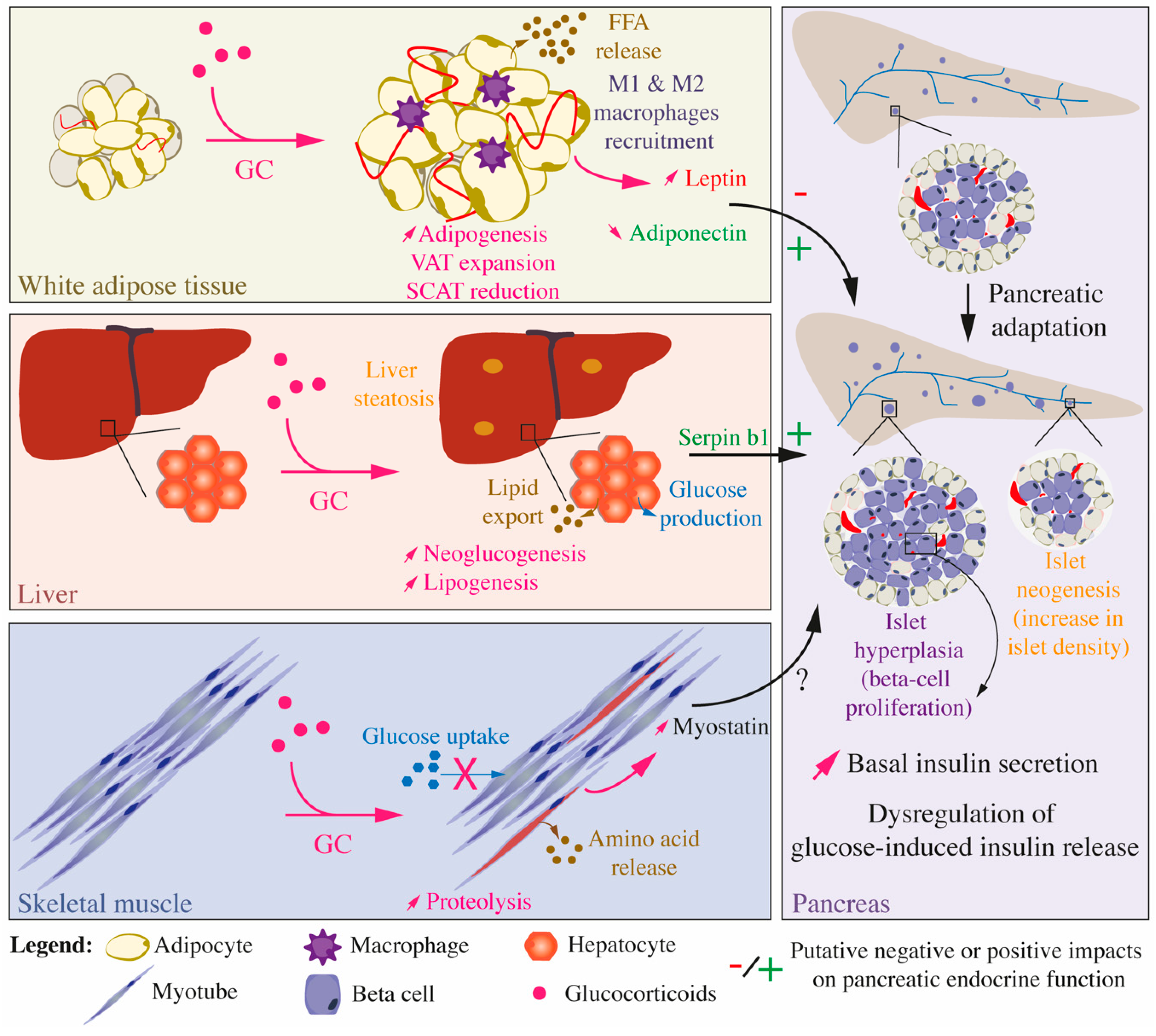{kind=link}
Abstract
1. Introduction
2. Glucocorticoid Production and Action
3. Pharmacological Use of Glucocorticoids and Main Side Effects
4. Direct and Indirect Effects of Glucocorticoids upon Pancreatic Endocrine Function
5. A Brief Molecular View on the Insulin Signaling Pathway
6. Long-Term Glucocorticoid Exposure and Insulin Resistance of The Adipose Tissue
6.1. Glucocorticoids and Insulin Resistance in Adipose Tissues In Vivo
6.2. Glucocorticoids and Insulin Resistance in Adipose Tissues at the Cellular Level
6.3. Glucocorticoid Impact on Non-White Adipose Tissues
6.4. Glucocorticoid Impact on the Endocrine Function Of Adipose Tissues
7. Effects of Glucocorticoids on Liver Function
8. Effect of Glucocorticoids on Skeletal Muscles
9. Glucocorticoid-Induced Insulin Resistance in Other Tissues
9.1. Bone
9.2. Gut
9.3. Brain
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 11b-HSD1 | Type 1 11β-hydroxysteroid dehydrogenase |
| 11b-HSD2 | Type 2 11β-hydroxysteroid dehydrogenase |
| ACC | Acetyl-CoA carboxylase |
| AMPK | AMP-activated protein kinase |
| Angptl4 | Angiopoietin-like 4 |
| ASC | Adipose stem cells |
| ATGL | Adipose triacylglycerol lipase |
| BAT | Brown adipose tissue |
| BMAT | Bone marrow adipose tissue |
| C/EBP | CCAAT-enhancer-binding protein |
| CBG | Corticosteroid-binding globulin |
| ChREBP | Carbohydrate response element-binding potein |
| DEX | Dexamethasone |
| DM | Diabetes mellitus |
| FAS | Fatty acid synthase |
| FFA | Free-fatty acid |
| FoxO | Forkhead box proteins |
| G6Pase | Glucose 6-phosphatase |
| GC | Glucocorticoid(s) |
| Gilz | Glucocorticoid-induced leucine zipper |
| GK | Glucokinase |
| GLP-1 | Glucagon-like peptide-1 |
| GLUT | Glucose transporter |
| GR | Glucocorticoid Receptor |
| GRE | Glucocorticoid response element |
| GSIS | Glucose-stimulated insulin secretion |
| GSK3 | Glycogen synthase kinase 3 |
| HFD | High fat diet |
| HIR | High-insulin responders |
| HPA | Hypothalamic-pituitary-adrenal axis |
| HSL | Hormone-sensitive lipase |
| IECs | Intestinal epithelial cells |
| IR | Insulin receptor |
| IRS | Insulin receptor substrate |
| LIPE | Gene encoding hormone sensitive lipase |
| LIR | Low-insulin responders |
| LPL | Lipoprotein lipase |
| LXR | Liver X receptors |
| LMO3 | LIM domain only protein 3 |
| MAPK | Mitogen-activated protein kinase |
| MED1 | Mediator 1 |
| MGLL | Monoglyceride Lipase |
| MR | Mineralocorticoid receptor |
| MSC | Muscle stem cells |
| Ngre | Negative glucocorticoid response element |
| OPG | Osteoprotegerin |
| PDX1 | Pancreatic and duodenal homeobox 1 |
| PEPCK | Phosphoenolpyruvate carboxykinase |
| PI3K | phosphatidylinositol 3-kinase |
| PIK3R1 | Phosphatidylinositol 3-kinase regulatory subunit alpha |
| PPAR | Peroxisome proliferator-activated receptor |
| RANK | Receptor activator of NF-kappaB |
| RANKL | Receptor activator of NF-kappaB ligand |
| ROS | Reactive oxygen species |
| SCAT | Subcutaneous adipose tissue |
| SREBP-1c | Sterol regulatory element-binding protein-1c |
| TAG | Triacylglycerol |
| T2DM | Type 2 diabetes mellitus |
| TXNIP | Thioredoxin Interacting Protein |
| UCP-1 | Uncoupling protein-1 |
| VAT | Visceral adipose tissue |
| WAT | White adipose tissue |
References
- Harrell, C.; Gillespie, C.F.; Neigh, G.N. Energetic stress: The reciprocal relationship between energy availability and the stress response. Physiol. Behav. 2016, 166, 43–55. [Google Scholar] [CrossRef]
- Lewis, J.G.; Bagley, C.J.; Elder, P.A.; Bachmann, A.W.; Torpy, D.J. Plasma free cortisol fraction reflects levels of functioning corticosteroid-binding globulin. Clin. Chim. Acta 2005, 359, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Meyer, E.J.; Nenke, M.A.; Lewis, J.G.; Torpy, D.J. Corticosteroid-binding globulin: Acute and chronic inflammation. Expert Rev. Endocrinol. Metab. 2017, 12, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Pemberton, P.A.; Stein, P.E.; Pepys, M.B.; Potter, J.M.; Carrell, R.W. Hormone binding globulins undergo serpin conformational change in inflammation. Nat. Cell Biol. 1988, 336, 257–258. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.M.; Krozowski, Z.S. 11 beta-Hydroxysteroid dehydrogenase. Vitam. Horm. 1999, 57, 249–324. [Google Scholar]
- de Kloet, E.R.; de Kloet, S.F.; de Kloet, C.S.; de Kloet, A.D. Top-down and bottom-up control of stress-coping. J. Neuroendocrinol. 2019, 31, e12675. [Google Scholar] [CrossRef]
- Presul, E.; Schmidt, S.; Kofler, R.; Helmberg, A. Identification, tissue expression, and glucocorticoid responsiveness of alternative first exons of the human glucocorticoid receptor. J. Mol. Endocrinol. 2007, 38, 79–90. [Google Scholar] [CrossRef][Green Version]
- Bockmühl, Y.; Murgatroyd, C.; Kuczynska, A.; Adcock, I.M.; Almeida, O.F.X.; Spengler, D. Differential Regulation and Function of 5′-Untranslated GR-Exon 1 Transcripts. Mol. Endocrinol. 2011, 25, 1100–1110. [Google Scholar] [CrossRef]
- Turner, J.D.; Muller, C.P. Structure of the glucocorticoid receptor (NR3C1) gene 5′ untranslated region: Identification, and tissue distribution of multiple new human exon 1. J. Mol. Endocrinol. 2005, 35, 283–292. [Google Scholar] [CrossRef]
- Oakley, R.H.; Cidlowski, J.A. Cellular Processing of the Glucocorticoid Receptor Gene and Protein: New Mechanisms for Generating Tissue-specific Actions of Glucocorticoids. J. Biol. Chem. 2011, 286, 3177–3184. [Google Scholar] [CrossRef]
- Oakley, R.H.; Webster, J.C.; Sar, M.; Parker, C.R., Jr.; Cidlowski, J.A. Expression and subcellular distribution of the beta-isoform of the human glucocorticoid receptor. Endocrinology 1997, 138, 5028–5038. [Google Scholar] [CrossRef] [PubMed]
- Webster, J.C.; Oakley, R.H.; Jewell, C.M.; Cidlowski, J.A. Proinflammatory cytokines regulate human glucocorticoid receptor gene expression and lead to the accumulation of the dominant negative beta isoform: A mechanism for the generation of glucocorticoid resistance. Proc. Natl. Acad. Sci. USA 2001, 98, 6865–6870. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.Z.; Cidlowski, J.A. Translational Regulatory Mechanisms Generate N-Terminal Glucocorticoid Receptor Isoforms with Unique Transcriptional Target Genes. Mol. Cell 2005, 18, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Duma, D.; Jewell, C.M.; Cidlowski, J.A. Multiple glucocorticoid receptor isoforms and mechanisms of post-translational modification. J. Steroid Biochem. Mol. Biol. 2006, 102, 11–21. [Google Scholar] [CrossRef]
- Vandevyver, S.; Dejager, L.; Libert, C. Comprehensive Overview of the Structure and Regulation of the Glucocorticoid Receptor. Endocr. Rev. 2014, 35, 671–693. [Google Scholar] [CrossRef]
- Scheschowitsch, K.; Leite, J.A.; Assreuy, J. New Insights in Glucocorticoid Receptor Signaling—More Than Just a Ligand-Binding Receptor. Front. Endocrinol. 2017, 8, 16. [Google Scholar] [CrossRef]
- Liu, D.; Ahmet, A.; Ward, L.M.; Krishnamoorthy, P.; Mandelcorn, E.D.; Leigh, R.; Brown, J.P.; Cohen, A.; Kim, H. A practical guide to the monitoring and management of the complications of systemic corticosteroid therapy. Allergy Asthma Clin. Immunol. 2013, 9, 30. [Google Scholar] [CrossRef]
- Gulliford, M.C.; Charlton, J.; Latinovic, R. Risk of Diabetes Associated With Prescribed Glucocorticoids in a Large Population. Diabetes Care 2006, 29, 2728–2729. [Google Scholar] [CrossRef]
- Donihi, A.C.; Raval, D.; Saul, M.; Korytkowski, M.T.; DeVita, M.A. Prevalence and predictors of corticosteroid-related hyperglycemia in hospitalized patients. Endocr. Pract 2006, 12, 358–362. [Google Scholar] [CrossRef]
- Perez, A.L.; Jansen-Chaparro, S.; Saigi, I.; Bernal-Lopez, M.R.; Miñambres, I.; Gomez-Huelgas, R. Glucocorticoid-induced hyperglycemia. J. Diabetes 2014, 6, 9–20. [Google Scholar] [CrossRef]
- Bruno, A.; Carucci, P.; Cassader, M.; Cavallo-Perin, P.; Gruden, G.; Olivetti, C.; Pagano, G. Serum glucose, insulin and C-peptide response to oral glucose after intravenous administration of hydrocortisone and methylprednisolone in man. Eur. J. Clin. Pharmacol. 1994, 46, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Amiche, M.A.; Albaum, J.M.; Tadrous, M.; Pechlivanoglou, P.; E Levesque, L.; Adachi, J.D.; Cadarette, S.M. Fracture risk in oral glucocorticoid users: A Bayesian meta-regression leveraging control arms of osteoporosis clinical trials. Osteoporos. Int. 2016, 27, 1709–1718. [Google Scholar] [CrossRef] [PubMed]
- Curtis, J.R.; Westfall, A.O.; Allison, J.; Bijlsma, J.W.; Freeman, A.; George, V.; Saag, K.G. Population-based assessment of adverse events associated with long-term glucocorticoid use. Arthritis Rheum. 2006, 55, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Löfberg, E.; Gutierrez, A.; Wernerman, J.; Anderstam, B.; Mitch, W.E.; Price, S.R.; Bergström, J.; Alvestrand, A. Effects of high doses of glucocorticoids on free amino acids, ribosomes and protein turnover in human muscle. Eur. J. Clin. Investig. 2002, 32, 345–353. [Google Scholar] [CrossRef]
- van Raalte, D.H.; Ouwens, D.M.; Diamant, M. Novel insights into glucocorticoid-mediated diabetogenic effects: Towards expansion of therapeutic options? Eur. J. Clin. Investig. 2009, 39, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nat. Cell Biol. 2014, 510, 84–91. [Google Scholar] [CrossRef]
- Fichna, M.; Fichna, P. Glucocorticoids and beta-cell function. Endokrynol. Pol. 2017, 68, 568–573. [Google Scholar] [CrossRef]
- Biering, H.; Knappe, G.; Gerl, H.; Lochs, H. Prevalence of diabetes in acromegaly and Cushing syndrome. Acta Med. Austriaca 2000, 27, 27–31. [Google Scholar] [CrossRef]
- Wajngot, A.; Giacca, A.; Grill, V.; Vranic, M.; Efendic, S. The diabetogenic effects of glucocorticoids are more pronounced in low- than in high-insulin responders. Proc. Natl. Acad. Sci. USA 1992, 89, 6035–6039. [Google Scholar] [CrossRef]
- Friedman, T.C.; Mastorakos, G.; Newman, T.D.; Mullen, N.M.; Horton, E.G.; Costello, R.; Papadopoulos, N.M.; Chrousos, G.P. Carbohydrate and Lipid Metabolism in Endogenous Hypercortisolism: Shared Features with Metabolic Syndrome X and NIDDM. Endocr. J. 1996, 43, 645–655. [Google Scholar] [CrossRef]
- Page, R.; Boolell, M.; Kalfas, A.; Sawyer, S.; Pestell, R.; Ward, G.; Alford, F. Insulin secretion, insulin sensitivity and glucose-mediated glucose disposal in Cushing’s disease: A minimal model analysis. Clin. Endocrinol. 1991, 35, 509–517. [Google Scholar] [CrossRef] [PubMed]
- van Raalte, D.H.; Nofrate, V.; Bunck, M.C.; van Iersel, T.; Schaap, J.E.; Nassander, U.K.; Diamant, M. Acute and 2-week exposure to prednisolone impair different aspects of beta-cell function in healthy men. Eur. J. Endocrinol. 2010, 162, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Wise, J.K.; Hendler, R.; Felig, P. Influence of Glucocorticoids on Glucagon Secretion and Plasma Amino Acid Concentrations in Man. J. Clin. Investig. 1973, 52, 2774–2782. [Google Scholar] [CrossRef]
- Akalestou, E.; Genser, L.; A Rutter, G. Glucocorticoid Metabolism in Obesity and Following Weight Loss. Front. Endocrinol. 2020, 11, 59. [Google Scholar] [CrossRef]
- Fine, N.H.F.; Doig, C.L.; Elhassan, Y.S.; Vierra, N.C.; Marchetti, P.; Bugliani, M.; Lavery, G.G. Glucocorticoids Reprogram beta-Cell Signaling to Preserve Insulin Secretion. Diabetes 2018, 67, 278–290. [Google Scholar] [CrossRef]
- Lambillotte, C.; Gilon, P.; Henquin, J.-C. Direct glucocorticoid inhibition of insulin secretion. An in vitro study of dexamethasone effects in mouse islets. J. Clin. Investig. 1997, 99, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Reich, E.; Tamary, A.; Sionov, R.V.; Melloul, D. Involvement of thioredoxin-interacting protein (TXNIP) in glucocorticoid-mediated beta cell death. Diabetologia 2012, 55, 1048–1057. [Google Scholar] [CrossRef]
- Avram, D.; Ranta, F.; Hennige, A.M.; Berchtold, S.; Hopp, S.; Häring, H.-U.; Lang, F.; Ullrich, S. IGF-1 Protects Against Dexamethasone-Induced Cell Death in Insulin Secreting INS-1 Cells Independent of AKT/PKB Phosphorylation. Cell. Physiol. Biochem. 2008, 21, 455–462. [Google Scholar] [CrossRef]
- Guo, B.; Zhang, W.; Xu, S.; Lou, J.; Wang, S.; Men, X. GSK-3beta mediates dexamethasone-induced pancreatic beta cell apoptosis. Life Sci. 2016, 144, 1–7. [Google Scholar] [CrossRef]
- Ranta, F.; Avram, D.; Berchtold, S.; Düfer, M.; Drews, G.; Lang, F.; Ullrich, S. Dexamethasone Induces Cell Death in Insulin-Secreting Cells, an Effect Reversed by Exendin-4. Diabetes 2006, 55, 1380–1390. [Google Scholar] [CrossRef]
- Zhang, X.; Yong, W.; Lv, J.; Zhu, Y.; Zhang, J.; Chen, F.; Han, X. Inhibition of forkhead box O1 protects pancreatic beta-cells against dexamethasone-induced dysfunction. Endocrinology 2009, 150, 4065–4073. [Google Scholar] [CrossRef]
- Gremlich, S.; Roduit, R.; Thorens, B. Dexamethasone induces posttranslational degradation of GLUT2 and inhibition of insulin secretion in isolated pancreatic beta cells. Comparison with the effects of fatty acids. J. Biol. Chem. 1997, 272, 3216–3222. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, S.; Berchtold, S.; Ranta, F.; Seebohm, G.; Henke, G.; Lupescu, A.; Mack, A.F.; Chao, C.-M.; Su, J.; Nitschke, R.; et al. Serum- and Glucocorticoid-Inducible Kinase 1 (SGK1) Mediates Glucocorticoid-Induced Inhibition of Insulin Secretion. Diabetes 2005, 54, 1090–1099. [Google Scholar] [CrossRef] [PubMed]
- Linssen, M.M.; Van Raalte, D.H.; Toonen, E.; Alkema, W.; Van Der Zon, G.C.; Dokter, W.H.; Diamant, M.; Guigas, B.; Ouwens, D.M. Prednisolone-induced beta cell dysfunction is associated with impaired endoplasmic reticulum homeostasis in INS-1E cells. Cell. Signal. 2011, 23, 1708–1715. [Google Scholar] [CrossRef] [PubMed]
- Delaunay, F.; Khan, A.; Cintra, A.; Davani, B.; Ling, Z.C.; Andersson, A.; Ostenson, C.G.; Gustafsson, J.; Efendic, S.; Okret, S. Pancreatic beta cells are important targets for the diabetogenic effects of glucocorticoids. J. Clin. Investig. 1997, 100, 2094–2098. [Google Scholar] [CrossRef]
- Rafacho, A.; Abrantes, J.L.F.; Ribeiro, D.L.; Paula, F.M.; Pinto-Fochi, M.E.; Boschero, A.C.; Bosqueiro, J.R. Morphofunctional Alterations in Endocrine Pancreas of Short- and Long-term Dexamethasone-treated Rats. Horm. Metab. Res. 2011, 43, 275–281. [Google Scholar] [CrossRef]
- Rafacho, A.; Cestari, T.M.; Taboga, S.R.; Boschero, A.C.; Bosqueiro, J.R. High doses of dexamethasone induce increased beta-cell proliferation in pancreatic rat islets. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E681–E689. [Google Scholar] [CrossRef]
- Beaudry, J.L.; D’Souza, A.M.; Teich, T.; Tsushima, R.; Riddell, M.C. Exogenous Glucocorticoids and a High-Fat Diet Cause Severe Hyperglycemia and Hyperinsulinemia and Limit Islet Glucose Responsiveness in Young Male Sprague-Dawley Rats. Endocrinology 2013, 154, 3197–3208. [Google Scholar] [CrossRef]
- Courty, E.; Besseiche, A.; Do, T.T.H.; Liboz, A.; Aguid, F.M.; Quilichini, E.; Haumaitre, C. Adaptive beta-Cell Neogenesis in the Adult Mouse in Response to Glucocorticoid-Induced Insulin Resistance. Diabetes 2019, 68, 95–108. [Google Scholar] [CrossRef]
- Davani, B.; Portwood, N.; Bryzgalova, G.; Reimer, M.K.; Heiden, T.; Östenson, C.-G.; Okret, S.; Ahrén, B.; Efendic, S.; Khan, A. Aged transgenic mice with increased glucocorticoid sensitivity in pancreatic beta-cells develop diabetes. Diabetes 2004, 53, S51–S59. [Google Scholar] [CrossRef]
- Blondeau, B.; Sahly, I.; Massouridès, E.; Singh-Estivalet, A.; Valtat, B.; Dorchène, D.; Jaisser, F.; Breant, B.; Tronche, F. Novel Transgenic Mice for Inducible Gene Overexpression in Pancreatic Cells Define Glucocorticoid Receptor-Mediated Regulations of Beta Cells. PLoS ONE 2012, 7, e30210. [Google Scholar] [CrossRef] [PubMed]
- Gesina, E.; Tronche, F.; Herrera, P.; Duchene, B.; Tales, W.; Czernichow, P.; Breant, B. Dissecting the Role of Glucocorticoids on Pancreas Development. Diabetes 2004, 53, 2322–2329. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Valtat, B.; Riveline, J.P.; Zhang, P.; Singh-Estivalet, A.; Armanet, M.; Venteclef, N.; Gautier, J.F. Fetal PGC-1alpha overexpression programs adult pancreatic beta-cell dysfunction. Diabetes 2013, 62, 1206–1216. [Google Scholar] [CrossRef] [PubMed]
- Besseiche, A.; Riveline, J.-P.; Delavallée, L.; Foufelle, F.; Gautier, J.-F.; Blondeau, B. Oxidative and energetic stresses mediate beta-cell dysfunction induced by PGC-1α. Diabetes Metab. 2018, 44, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Jornayvaz, F.R.; Vollenweider, P.; Bochud, M.; Mooser, V.; Waeber, G.; Marques-Vidal, P. Low birth weight leads to obesity, diabetes and increased leptin levels in adults: The CoLaus study. Cardiovasc. Diabetol. 2016, 15, 1–10. [Google Scholar] [CrossRef]
- Wang, X.; Garfinkel, S.N.; King, A.P.; Angstadt, M.; Dennis, M.J.; Xie, H.; Welsh, R.C.; Tamburrino, M.B.; Liberzon, I. A multiple-plane approach to measure the structural properties of functionally active regions in the human cortex. NeuroImage 2010, 49, 3075–3085. [Google Scholar] [CrossRef]
- Blondeau, B.; Lesage, J.; Czernichow, P.; Dupouy, J.P.; Breant, B. Glucocorticoids impair fetal beta-cell development in rats. Am. J. Physiol. Metab. 2001, 281, E592–E599. [Google Scholar]
- Valtat, B.; Dupuis, C.; Zenaty, D.; Singh-Estivalet, A.; Tronche, F.; Bréant, B.; Blondeau, B. Genetic evidence of the programming of beta cell mass and function by glucocorticoids in mice. Diabetologia 2010, 54, 350–359. [Google Scholar] [CrossRef]
- Riveline, J.-P.; Baz, B.; Nguewa, J.-L.; Vidal-Trecan, T.; Ibrahim, F.; Boudou, P.; Vicaut, E.; De La Perrière, A.B.; Fetita, S.; Bréant, B.; et al. Exposure to Glucocorticoids in the First Part of Fetal Life is Associated with Insulin Secretory Defect in Adult Humans. J. Clin. Endocrinol. Metab. 2019, 105, e191–e199. [Google Scholar] [CrossRef]
- Rosen, E.D.; Spiegelman, B.M. What We Talk About When We Talk About Fat. Cell 2014, 156, 20–44. [Google Scholar] [CrossRef]
- Kahn, C.R.; Wang, G.; Lee, K.Y. Altered adipose tissue and adipocyte function in the pathogenesis of metabolic syndrome. J. Clin. Investig. 2019, 129, 3990–4000. [Google Scholar] [CrossRef]
- Suchacki, K.J.; Tavares, A.A.S.; Mattiucci, D.; Scheller, E.L.; Papanastasiou, G.; Gray, C.; Sinton, M.C.; Ramage, L.E.; McDougald, W.A.; Lovdel, A.; et al. Bone marrow adipose tissue is a unique adipose subtype with distinct roles in glucose homeostasis. Nat. Commun. 2020, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Zwick, R.K.; Guerrero-Juarez, C.F.; Horsley, V.; Plikus, M.V. Anatomical, Physiological, and Functional Diversity of Adipose Tissue. Cell Metab. 2018, 27, 68–83. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.T.; Yamamoto, Y.; Gesta, S.; Kahn, C.R. Beneficial Effects of Subcutaneous Fat Transplantation on Metabolism. Cell Metab. 2008, 7, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Czech, M.P. Mechanisms of insulin resistance related to white, beige, and brown adipocytes. Mol. Metab. 2020, 34, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Softic, S.; Boucher, J.; Solheim, M.H.; Fujisaka, S.; Haering, M.-F.; Homan, E.P.; Winnay, J.; Perez-Atayde, A.R.; Kahn, C.R. Lipodystrophy Due to Adipose Tissue–Specific Insulin Receptor Knockout Results in Progressive NAFLD. Diabetes 2016, 65, 2187–2200. [Google Scholar] [CrossRef]
- Guerra, C.; Navarro, P.; Valverde, A.M.; Arribas, M.; Bruning, J.; Kozak, L.P.; Benito, M. Brown adipose tissue-specific insulin receptor knockout shows diabetic phenotype without insulin resistance. J. Clin. Investig. 2001, 108, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Hazlehurst, J.M.; Gathercole, L.L.; Nasiri, M.; Armstrong, M.J.; Borrows, S.; Yu, J.; Wagenmakers, A.J.; Stewart, P.M.; Tomlinson, J.W. Glucocorticoids Fail to Cause Insulin Resistance in Human Subcutaneous Adipose Tissue In Vivo. J. Clin. Endocrinol. Metab. 2013, 98, 1631–1640. [Google Scholar] [CrossRef]
- Rebuffé-Scrive, M.; Brönnegård, M.; Nilsson, A.; Eldh, J.; Gustafsson, J.-Å.; Björntorp, P. Steroid Hormone Receptors in Human Adipose Tissues. J. Clin. Endocrinol. Metab. 1990, 71, 1215–1219. [Google Scholar] [CrossRef]
- Campbell, J.E.; Peckett, A.J.; D’Souza, A.M.; Hawke, T.J.; Riddell, M.C. Adipogenic and lipolytic effects of chronic glucocorticoid exposure. Am. J. Physiol. Physiol. 2011, 300, C198–C209. [Google Scholar] [CrossRef]
- Chimin, P.; Farias, T.D.S.M.; Torres-Leal, F.L.; Bolsoni-Lopes, A.; Campaña, A.B.; Andreotti, S.; Lima, F.B. Chronic glucocorticoid treatment enhances lipogenic activity in visceral adipocytes of male Wistar rats. Acta Physiol. 2014, 211, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Dalle, H.; Garcia, M.; Antoine, B.; Boehm, V.; Do, T.T.H.; Buyse, M.; Ledent, T.; Lamazière, A.; Magnan, C.; Postic, C.; et al. Adipocyte Glucocorticoid Receptor Deficiency Promotes Adipose Tissue Expandability and Improves the Metabolic Profile Under Corticosterone Exposure. Diabetes 2019, 68, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-J.; Pramyothin, P.; Karastergiou, K.; Fried, S.K. Deconstructing the roles of glucocorticoids in adipose tissue biology and the development of central obesity. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Marcondes-De-Mello, M.L.-D.-F.; Serafim-Costa, M.C.; Alves-E-Silva, M.M.; Oliveira, N.R.; Bertolucci-Caldo, N.V.; Ferraz, R.K.; Chaves, V.E. Effect of glucocorticoids on glyceroneogenesis in adipose tissue: A systematic review. Biochimie 2020, 168, 210–219. [Google Scholar] [CrossRef]
- Balachandran, A.; Guan, H.; Sellan, M.; van Uum, S.; Yang, K. Insulin and dexamethasone dynamically regulate adipocyte 11beta-hydroxysteroid dehydrogenase type 1. Endocrinology 2008, 149, 4069–4079. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chapman, K.; Holmes, M.; Seckl, J. 11beta-hydroxysteroid dehydrogenases: Intracellular gate-keepers of tissue glucocorticoid action. Physiol. Rev. 2013, 93, 1139–1206. [Google Scholar] [CrossRef]
- Do, T.T.H.; Marie, G.; Héloïse, D.; Guillaume, D.; Marthe, M.; Fève, B.; Marion, B. Glucocorticoid-induced insulin resistance is related to macrophage visceral adipose tissue infiltration. J. Steroid Biochem. Mol. Biol. 2019, 185, 150–162. [Google Scholar] [CrossRef]
- Morgan, S.A.; McCabe, E.L.; Gathercole, L.L.; Hassan-Smith, Z.K.; Larner, D.P.; Bujalska, I.J.; Lavery, G.G. 11beta-HSD1 is the major regulator of the tissue-specific effects of circulating glucocorticoid excess. Proc. Natl. Acad. Sci. USA 2014, 111, E2482–E2491. [Google Scholar] [CrossRef]
- Peng, K.; Pan, Y.; Li, J.; Khan, Z.; Fan, M.; Yin, H.; Zheng, C. 11beta-Hydroxysteroid Dehydrogenase Type 1(11beta-HSD1) mediates insulin resistance through JNK activation in adipocytes. Sci. Rep. 2016, 6, 37160. [Google Scholar] [CrossRef]
- Urbanet, R.; Cat, A.N.D.; Feraco, A.; Venteclef, N.; El Mogrhabi, S.; Sierra-Ramos, C.; De La Rosa, D.A.; Adler, G.K.; Quilliot, D.; Rossignol, P.; et al. Adipocyte Mineralocorticoid Receptor Activation Leads to Metabolic Syndrome and Induction of Prostaglandin D2 Synthase. Hypertension 2015, 66, 149–157. [Google Scholar] [CrossRef]
- Guo, C.; Ricchiuti, V.; Lian, C.G.; Yao, T.M.; Coutinho, P.; Romero, J.R.; Li, J.; Williams, G.H.; Adler, G.K. Mineralocorticoid Receptor Blockade Reverses Obesity-Related Changes in Expression of Adiponectin, Peroxisome Proliferator-Activated Receptor-γ, and Proinflammatory Adipokines. Circulation 2008, 117, 2253–2261. [Google Scholar] [CrossRef] [PubMed]
- Lefranc, C.; Friederich-Persson, M.; Braud, L.; Palacios-Ramírez, R.; Karlsson, S.; Boujardine, N.; Motterlini, R.; Jaisser, F.; Cat, A.N.D. MR (Mineralocorticoid Receptor) Induces Adipose Tissue Senescence and Mitochondrial Dysfunction Leading to Vascular Dysfunction in Obesity. Hypertension 2019, 73, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.K.; Hutson, I.; Harris, C.A. Hepatic Glucocorticoid Receptor Plays a Greater Role Than Adipose GR in Metabolic Syndrome Despite Renal Compensation. Endocrinology 2016, 157, 4943–4960. [Google Scholar] [CrossRef] [PubMed]
- Desarzens, S.; Faresse, N. Adipocyte glucocorticoid receptor has a minor contribution in adipose tissue growth. J. Endocrinol. 2016, 230, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mueller, K.M.; Hartmann, K.; Kaltenecker, D.; Vettorazzi, S.; Bauer, M.; Mauser, L.; Amann, S.; Jall, S.; Fischer, K.; Esterbauer, H.; et al. Adipocyte Glucocorticoid Receptor Deficiency Attenuates Aging- and HFD-Induced Obesity and Impairs the Feeding-Fasting Transition. Diabetes 2016, 66, 272–286. [Google Scholar] [CrossRef]
- Shen, Y.; Roh, H.C.; Kumari, M.; Rosen, E.D. Adipocyte glucocorticoid receptor is important in lipolysis and insulin resistance due to exogenous steroids, but not insulin resistance caused by high fat feeding. Mol. Metab. 2017, 6, 1150–1160. [Google Scholar] [CrossRef]
- Takeshita, Y.; Watanabe, S.; Hattori, T.; Nagasawa, K.; Matsuura, N.; Takahashi, K.; Murohara, T.; Nagata, K. Blockade of glucocorticoid receptors with RU486 attenuates cardiac damage and adipose tissue inflammation in a rat model of metabolic syndrome. Hypertens. Res. 2015, 38, 741–750. [Google Scholar] [CrossRef]
- Lundgren, M.; Burén, J.; Ruge, T.; Myrnäs, T.; Eriksson, J.W. Glucocorticoids Down-Regulate Glucose Uptake Capacity and Insulin-Signaling Proteins in Omental But Not Subcutaneous Human Adipocytes. J. Clin. Endocrinol. Metab. 2004, 89, 2989–2997. [Google Scholar] [CrossRef]
- Ma, X.; Xu, L.; Mueller, E. Forkhead box A3 mediates glucocorticoid receptor function in adipose tissue. Proc. Natl. Acad. Sci. USA 2016, 113, 3377–3382. [Google Scholar] [CrossRef]
- Lindroos, J.; Husa, J.; Mitterer, G.; Haschemi, A.; Rauscher, S.; Haas, R.; Gröger, M.; Loewe, R.; Kohrgruber, N.; Schrögendorfer, K.F.; et al. Human but Not Mouse Adipogenesis Is Critically Dependent on LMO3. Cell Metab. 2013, 18, 62–74. [Google Scholar] [CrossRef]
- Sakoda, H.; Ogihara, T.; Anai, M.; Funaki, M.; Inukai, K.; Katagiri, H.; Fukushima, Y.; Onishi, Y.; Ono, H.; Fujishiro, M.; et al. Dexamethasone-induced insulin resistance in 3T3-L1 adipocytes is due to inhibition of glucose transport rather than insulin signal transduction. Diabetes 2000, 49, 1700–1708. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Sumuano, J.-T.; Velez-Delvalle, C.; Beltrán-Langarica, A.; Marsch-Moreno, M.; Hernandez-Mosqueira, C.; Kuri-Harcuch, W. Glucocorticoid Paradoxically Recruits Adipose Progenitors and Impairs Lipid Homeostasis and Glucose Transport in Mature Adipocytes. Sci. Rep. 2013, 3, srep02573. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.-K.; Ge, K. Glucocorticoid Receptor Accelerates, but Is Dispensable for, Adipogenesis. Mol. Cell. Biol. 2016, 37. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.K.; Wang, L.; Giampietro, A.; Lai, B.; Lee, J.E.; Ge, K. Distinct Roles of Transcription Factors KLF4, Krox20, and Peroxisome Proliferator-Activated Receptor gamma in Adipogenesis. Mol. Cell Biol. 2017, 37, e00554-16. [Google Scholar] [CrossRef] [PubMed]
- Perpetuini, C.A.; Giuliani, G.; Reggio, A.; Cerretani, M.; Santoriello, M.; Stefanelli, R.; Bresciani, A. Janus effect of glucocorticoids on differentiation of muscle fibro/adipogenic progenitors. Sci. Rep. 2020, 10, 5363. [Google Scholar] [CrossRef]
- Bauerle, K.T.; Hutson, I.; Scheller, E.L.; Harris, C.A. Glucocorticoid Receptor Signaling Is Not Required for In Vivo Adipogenesis. Endocrinology 2018, 159, 2050–2061. [Google Scholar] [CrossRef]
- Caprio, M.; Antelmi, A.; Chetrite, G.; Muscat, A.; Mammi, C.; Marzolla, V.; Fabbri, A.; Zennaro, M.-C.; Fève, B. Antiadipogenic Effects of the Mineralocorticoid Receptor Antagonist Drospirenone: Potential Implications for the Treatment of Metabolic Syndrome. Endocrinology 2011, 152, 113–125. [Google Scholar] [CrossRef]
- Caprio, M.; Fève, B.; Claës, A.; Viengchareun, S.; Lombès, M.; Zennaro, M.-C. Pivotal role of the mineralocorticoid receptor in corticosteroid-induced adipogenesis. FASEB J. 2007, 21, 2185–2194. [Google Scholar] [CrossRef]
- Jia, G.; Aroor, A.R.; Sowers, J.R. The role of mineralocorticoid receptor signaling in the cross-talk between adipose tissue and the vascular wall. Cardiovasc. Res. 2017, 113, 1055–1063. [Google Scholar] [CrossRef]
- Lee, R.A.; Harris, C.A.; Wang, A.J.-C. Glucocorticoid Receptor and Adipocyte Biology. Nucl. Recept. Res. 2018, 5. [Google Scholar] [CrossRef]
- Ngo, S.; Barry, J.B.; Nisbet, J.; Prins, J.; Whitehead, J.P. Reduced phosphorylation of AS160 contributes to glucocorticoid-mediated inhibition of glucose uptake in human and murine adipocytes. Mol. Cell. Endocrinol. 2009, 302, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Tsai, L.T.; Zhou, Y.; Evertts, A.G.; Xu, S.; Griffin, M.J.; Issner, R.; Whitton, H.J.; Garcia, B.A.; Epstein, C.B.; et al. Identification of nuclear hormone receptor pathways causing insulin resistance by transcriptional and epigenomic analysis. Nat. Cell Biol. 2015, 17, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Ramage, L.E.; Akyol, M.; Fletcher, A.M.; Forsythe, J.; Nixon, M.; Carter, R.N.; Van Beek, E.J.; Morton, N.M.; Walker, B.R.; Stimson, R.H. Glucocorticoids Acutely Increase Brown Adipose Tissue Activity in Humans, Revealing Species-Specific Differences in UCP-1 Regulation. Cell Metab. 2016, 24, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Glantschnig, C.; Mattijssen, F.; Vogl, E.S.; Khan, A.A.; Garcia, M.R.; Fischer, K.; Müller, T.; Uhlenhaut, H.; Nawroth, P.; Scheideler, M.; et al. The glucocorticoid receptor in brown adipocytes is dispensable for control of energy homeostasis. EMBO Rep. 2019, 20, e48552. [Google Scholar] [CrossRef] [PubMed]
- Justesen, J.; Mosekilde, L.; Holmes, M.; Stenderup, K.; Gasser, J.; Mullins, J.J.; Kassem, M. Mice deficient in 11beta-hydroxysteroid dehydrogenase type 1 lack bone marrow adipocytes, but maintain normal bone formation. Endocrinology 2004, 145, 1916–1925. [Google Scholar] [CrossRef] [PubMed]
- Bönisch, C.; Irmler, M.; Brachthäuser, L.; Neff, F.; Bamberger, M.T.; Marschall, S.; De Angelis, M.H.; Beckers, J. Dexamethasone treatment alters insulin, leptin, and adiponectin levels in male mice as observed in DIO but does not lead to alterations of metabolic phenotypes in the offspring. Mamm. Genome 2015, 27, 17–28. [Google Scholar] [CrossRef]
- de Kloet, A.D.; Herman, J.P. Fat-brain connections: Adipocyte glucocorticoid control of stress and metabolism. Front. Neuroendocrinol. 2018, 48, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-J.; Fried, S.K. The glucocorticoid receptor, not the mineralocorticoid receptor, plays the dominant role in adipogenesis and adipokine production in human adipocytes. Int. J. Obes. 2014, 38, 1228–1233. [Google Scholar] [CrossRef] [PubMed]
- Dunmore, S.J.; Browz, J.E. The role of adipokines in beta-cell failure of type 2 diabetes. J. Endocrinol. 2013, 216, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef]
- Gregori, C.; Guillet-Deniau, I.; Girard, J.; Decaux, J.-F.; Pichard, A.-L. Insulin regulation of glucokinase gene expression: Evidence against a role for sterol regulatory element binding protein 1 in primary hepatocytes. FEBS Lett. 2005, 580, 410–414. [Google Scholar] [CrossRef] [PubMed]
- InSug, O.; Zhang, W.; Wasserman, D.H.; Liew, C.W.; Liu, J.; Paik, J.; Unterman, T.G. FoxO1 integrates direct and indirect effects of insulin on hepatic glucose production and glucose utilization. Nat. Commun. 2015, 6, 7079. [Google Scholar]
- Vincent, E.E.; Elder, D.J.E.; Thomas, E.C.; Phillips, L.V.; Morgan, C.T.; Pawade, J.; Sohail, M.; May, M.T.; Hetzel, M.; Tavare, J.M. Akt phosphorylation on Thr308 but not on Ser473 correlates with Akt protein kinase activity in human non-small cell lung cancer. Br. J. Cancer 2011, 104, 1755–1761. [Google Scholar] [CrossRef] [PubMed]
- Yung, H.W.; Charnock-Jones, D.S.; Burton, G.J. Regulation of AKT Phosphorylation at Ser473 and Thr308 by Endoplasmic Reticulum Stress Modulates Substrate Specificity in a Severity Dependent Manner. PLoS ONE 2011, 6, e17894. [Google Scholar] [CrossRef] [PubMed]
- Woods, C.; Hazlehurst, J.M.; Tomlinson, J.W. Glucocorticoids and non-alcoholic fatty liver disease. J. Steroid Biochem. Mol. Biol. 2015, 154, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Fain, J.N.; Scow, R.O.; Chernick, S.S. Effects of Glucocorticoids on Metabolism of Adipose Tissue In vitro. J. Biol. Chem. 1963, 238, 54–58. [Google Scholar] [CrossRef]
- Carter-Su, C.; Okamoto, K. Effect of glucocorticoids on hexose transport in rat adipocytes. Evidence for decreased transporters in the plasma membrane. J. Biol. Chem. 1985, 260, 11091–11098. [Google Scholar] [CrossRef]
- Pagano, G.; Cavallo-Perin, P.; Cassader, M.; Bruno, A.; Ozzello, A.; Masciola, P.; Dall’Omo, A.M.; Imbimbo, B. An in vivo and in vitro study of the mechanism of prednisone-induced insulin resistance in healthy subjects. J. Clin. Investig. 1983, 72, 1814–1820. [Google Scholar] [CrossRef]
- Höppner, W.; Süssmuth, W.; O’Brien, C.; Seitz, H.J.; Luda, D.; Harneit, A. Cooperative effect of thyroid and glucocorticoid hormones on the induction of hepatic phosphoenolpyruvate carboxykinase in vivo and in cultured hepatocytes. JBIC J. Biol. Inorg. Chem. 1986, 159, 399–405. [Google Scholar] [CrossRef]
- Czegle, I.; Piccirella, S.; Senesi, S.; Csala, M.; Mandl, J.; Banhegyi, G.; Benedetti, A. Cooperativity between 11beta-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphate dehydrogenase is based on a common pyridine nucleotide pool in the lumen of the endoplasmic reticulum. Mol. Cell Endocrinol. 2006, 248, 24–25. [Google Scholar] [CrossRef]
- Liu, Y.; Nakagawa, Y.; Wang, Y.; Sakurai, R.; Tripathi, P.V.; Lutfy, K.; Friedman, T.C. Increased glucocorticoid receptor and 11{beta}-hydroxysteroid dehydrogenase type 1 expression in hepatocytes may contribute to the phenotype of type 2 diabetes in db/db mice. Diabetes 2005, 54, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Whorwood, C.B.; Donovan, S.J.; Flanagan, D.; Phillips, D.I.; Byrne, C.D. Increased glucocorticoid receptor expression in human skeletal muscle cells may contribute to the pathogenesis of the metabolic syndrome. Diabetes 2002, 51, 1066–1075. [Google Scholar] [CrossRef] [PubMed]
- Anil, T.M.; Dandu, A.; Harsha, K.; Singh, J.; Shree, N.; Kumar, V.S.; Lakshmi, M.N.; Sunil, V.; Harish, C.; Balamurali, G.V.; et al. A novel 11β-hydroxysteroid dehydrogenase type1 inhibitor CNX-010-49 improves hyperglycemia, lipid profile and reduces body weight in diet induced obese C57B6/J mice with a potential to provide cardio protective benefits. BMC Pharmacol. Toxicol. 2014, 15, 1–43. [Google Scholar] [CrossRef] [PubMed]
- Kotelevtsev, Y.; Holmes, M.C.; Burchell, A.; Houston, P.M.; Schmoll, D.; Jamieson, P.; Mullins, J.J. 11beta-hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc. Natl. Acad. Sci. USA. 1997, 94, 14924–14929. [Google Scholar] [CrossRef] [PubMed]
- Lavery, G.G.; Zielinska, A.E.; Gathercole, L.L.; Hughes, B.; Semjonous, N.; Guest, P.; Tomlinson, J.W. Lack of significant metabolic abnormalities in mice with liver-specific disruption of 11beta-hydroxysteroid dehydrogenase type 1. Endocrinology 2012, 153, 3236–3248. [Google Scholar] [CrossRef]
- Paterson, J.M.; Morton, N.M.; Fievet, C.; Kenyon, C.J.; Holmes, M.C.; Staels, B.; Mullins, J.J. Metabolic syndrome without obesity: Hepatic overexpression of 11beta-hydroxysteroid dehydrogenase type 1 in transgenic mice. Proc. Natl. Acad. Sci. USA 2004, 101, 7088–7093. [Google Scholar] [CrossRef]
- Saad, M.J.; Folli, F.; A Kahn, J.; Kahn, C.R. Modulation of insulin receptor, insulin receptor substrate-1, and phosphatidylinositol 3-kinase in liver and muscle of dexamethasone-treated rats. J. Clin. Investig. 1993, 92, 2065–2072. [Google Scholar] [CrossRef]
- Coutinho, A.E.; Chapman, K.E. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol. Cell. Endocrinol. 2011, 335, 2–13. [Google Scholar] [CrossRef]
- Liang, Y.; Osborne, M.C.; Monia, B.P.; Bhanot, S.; Watts, L.M.; She, P.; Decarlo, S.O.; Chen, X.; Demarest, K. Antisense oligonucleotides targeted against glucocorticoid receptor reduce hepatic glucose production and ameliorate hyperglycemia in diabetic mice. Metabolism 2005, 54, 848–855. [Google Scholar] [CrossRef]
- Opherk, C.; Tronche, F.; Kellendonk, C.; Kohlmüller, D.; Schulze, A.; Schmid, W.; Schütz, G. Inactivation of the Glucocorticoid Receptor in Hepatocytes Leads to Fasting Hypoglycemia and Ameliorates Hyperglycemia in Streptozotocin-Induced Diabetes Mellitus. Mol. Endocrinol. 2004, 18, 1346–1353. [Google Scholar] [CrossRef]
- Watts, L.M.; Manchem, V.P.; Leedom, T.A.; Rivard, A.L.; McKay, R.A.; Bao, D.; Neroladakis, T.; Monia, B.P.; Bodenmiller, D.M.; Cao, J.X.-C.; et al. Reduction of Hepatic and Adipose Tissue Glucocorticoid Receptor Expression With Antisense Oligonucleotides Improves Hyperglycemia and Hyperlipidemia in Diabetic Rodents without Causing Systemic Glucocorticoid Antagonism. Diabetes 2005, 54, 1846–1853. [Google Scholar] [CrossRef] [PubMed]
- Zinker, B.; Mika, A.; Nguyen, P.; Wilcox, D.; Öhman, L.; Von Geldern, T.W.; Opgenorth, T.; Jacobson, P. Liver-selective glucocorticoid receptor antagonism decreases glucose production and increases glucose disposal, ameliorating insulin resistance. Metabolism 2007, 56, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Petersen, D.D.; A Magnuson, M.; Granner, D.K. Location and characterization of two widely separated glucocorticoid response elements in the phosphoenolpyruvate carboxykinase gene. Mol. Cell. Biol. 1988, 8, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Magomedova, L.; Tsai, R.; Angers, S.; Orellana, A.; Cummins, C.L. Separating the Anti-Inflammatory and Diabetogenic Effects of Glucocorticoids Through LXRbeta Antagonism. Endocrinology 2017, 158, 1034–1047. [Google Scholar] [CrossRef][Green Version]
- Li, X.; Monks, B.; Ge, Q.; Birnbaum, M.J. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1α transcription coactivator. Nat. Cell Biol. 2007, 447, 1012–1016. [Google Scholar] [CrossRef]
- Southgate, R.J.; Bruce, C.R.; Carey, A.L.; Steinberg, G.R.; Walder, K.; Monks, R.; Febbraio, M.A. PGC-1alpha gene expression is down-regulated by Akt- mediated phosphorylation and nuclear exclusion of FoxO1 in insulin-stimulated skeletal muscle. FASEB J. 2005, 19, 2072–2074. [Google Scholar] [CrossRef]
- Yoon, J.C.; Puigserver, P.; Chen, G.; Donovan, J.; Wu, Z.; Rhee, J.; Adelmant, G.; Stafford, J.E.H.; Kahn, C.R.; Granner, D.K.; et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nat. Cell Biol. 2001, 413, 131–138. [Google Scholar] [CrossRef]
- Besse-Patin, A.; Jeromson, S.; Levesque-Damphousse, P.; Secco, B.; Laplante, M.; Estall, J.L. PGC1A regulates the IRS1:IRS2 ratio during fasting to influence hepatic metabolism downstream of insulin. Proc. Natl. Acad. Sci. USA 2019, 116, 4285–4290. [Google Scholar] [CrossRef]
- Jia, Y.; Viswakarma, N.; Fu, T.; Yu, S.; Rao, M.S.; Borensztajn, J.; Reddy, J.K. Conditional Ablation of Mediator Subunit MED1 (MED1/PPARBP) Gene in Mouse Liver Attenuates Glucocorticoid Receptor Agonist Dexamethasone-Induced Hepatic Steatosis. Gene Expr. 2009, 14, 291–306. [Google Scholar] [CrossRef]
- Hemmer, M.C.; Wierer, M.; Schachtrup, K.; Downes, M.; Hübner, N.; Evans, R.M.; Uhlenhaut, N.H. E47 modulates hepatic glucocorticoid action. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Christ-Crain, M.; Kola, B.; Lolli, F.; Fekete, C.; Seboek, D.; Wittmann, G.; Kunos, G. AMP-activated protein kinase mediates glucocorticoid-induced metabolic changes: A novel mechanism in Cushing’s syndrome. FASEB J. 2008, 22, 1672–1683. [Google Scholar] [CrossRef] [PubMed]
- Kahn, B.B.; Alquier, T.; Carling, D.; Hardie, D.G. AMP-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005, 1, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Wang, Y.; Sheikhahmadi, A.; Li, X.; Buyse, J.; Lin, H.; Song, Z. Effects of glucocorticoids on lipid metabolism and AMPK in broiler chickens’ liver. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2019, 232, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Patel, M.; Tsai, R.; Lin, V.; Bookout, A.L.; Zhang, Y.; Mangelsdorf, D.J. LXRbeta is required for glucocorticoid-induced hyperglycemia and hepatosteatosis in mice. J. Clin. Investig. 2011, 121, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Luan, G.; Li, G.; Ma, X.; Jin, Y.; Hu, N.; Li, J.; Wang, Z.; Wang, H. Dexamethasone-Induced Mitochondrial Dysfunction and Insulin Resistance-Study in 3T3-L1 Adipocytes and Mitochondria Isolated from Mouse Liver. Molecules 2019, 24, 1982. [Google Scholar] [CrossRef]
- El Ouaamari, A.; Dirice, E.; Gedeon, N.; Hu, J.; Zhou, J.Y.; Shirakawa, J.; Boucher, J. SerpinB1 Promotes Pancreatic beta Cell Proliferation. Cell metabolism. 2016, 23, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Takebayashi, K.; Hara, K.; Terasawa, T.; Naruse, R.; Suetsugu, M.; Tsuchiya, T.; Inukai, T. Circulating SerpinB1 levels and clinical features in patients with type 2 diabetes. BMJ Open Diabetes Res. Care 2016, 4, e000274. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Tripathy, D. Skeletal Muscle Insulin Resistance Is the Primary Defect in Type 2 Diabetes. Diabetes Care 2009, 32, S157–S163. [Google Scholar] [CrossRef]
- Braun, T.P.; Marks, D.L. The regulation of muscle mass by endogenous glucocorticoids. Front. Physiol. 2015, 6, 12. [Google Scholar] [CrossRef]
- Weinstein, S.P.; Paquin, T.; Pritsker, A.; Haber, R.S. Glucocorticoid-induced insulin resistance: Dexamethasone inhibits the activation of glucose transport in rat skeletal muscle by both insulin- and non-insulin-related stimuli. Diabetes 1995, 44, 441–445. [Google Scholar] [CrossRef]
- Burén, J.; Lai, Y.-C.; Lundgren, M.; Eriksson, J.; Jensen, J. Insulin action and signalling in fat and muscle from dexamethasone-treated rats. Arch. Biochem. Biophys. 2008, 474, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Vestergaard, H.; Bratholm, P.; Christensen, N.J. Increments in insulin sensitivity during intensive treatment are closely correlated with decrements in glucocorticoid receptor mRNA in skeletal muscle from patients with Type II diabetes. Clin. Sci. 2001, 101, 533. [Google Scholar] [CrossRef] [PubMed]
- Giorgino, F.; Almahfouz, A.; Goodyear, L.J.; Smith, R.J. Glucocorticoid regulation of insulin receptor and substrate IRS-1 tyrosine phosphorylation in rat skeletal muscle In vivo. J. Clin. Investig. 1993, 91, 2020–2030. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo Transcription Factors Induce the Atrophy-Related Ubiquitin Ligase Atrogin-1 and Cause Skeletal Muscle Atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef]
- Hu, Z.; Wang, H.; Lee, I.H.; Du, J.; Mitch, W.E. Endogenous glucocorticoids and impaired insulin signaling are both required to stimulate muscle wasting under pathophysiological conditions in mice. J. Clin. Investig. 2009, 119, 3059–3069. [Google Scholar] [CrossRef]
- Kuo, T.; Lew, M.J.; Mayba, O.; Harris, C.A.; Speed, T.P.; Wang, J.-C. Genome-wide analysis of glucocorticoid receptor-binding sites in myotubes identifies gene networks modulating insulin signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 11160–11165. [Google Scholar] [CrossRef]
- Coderre, L.; Srivastava, A.K.; Chiasson, J.L. Effect of hypercorticism on regulation of skeletal muscle glycogen metabolism by insulin. Am. J. Physiol. Metab. 1992, 262, E427–E433. [Google Scholar] [CrossRef]
- Ohshima, K.; Shargill, N.S.; Chan, T.M.; Bray, G.A. Adrenalectomy reverses insulin resistance in muscle from obese (ob/ob) mice. Am. J. Physiol. Metab. 1984, 246, E193–E197. [Google Scholar] [CrossRef]
- Kuo, T.; Harris, C.A.; Wang, J.-C. Metabolic functions of glucocorticoid receptor in skeletal muscle. Mol. Cell. Endocrinol. 2013, 380, 79–88. [Google Scholar] [CrossRef]
- Morgan, S.A.; Sherlock, M.; Gathercole, L.L.; Lavery, G.G.; Lenaghan, C.; Bujalska, I.J.; Hegyi, K. 11beta-hydroxysteroid dehydrogenase type 1 regulates glucocorticoid-induced insulin resistance in skeletal muscle. Diabetes 2009, 58, 2506–2515. [Google Scholar] [CrossRef]
- Walsh, F.S.; Celeste, A.J. Myostatin: A modulator of skeletal-muscle stem cells. Biochem. Soc. Trans. 2005, 33 Pt 6, 1513–1517. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.L.; Loh, A.S. Posttranscriptional mechanisms involving microRNA-27a and b contribute to fast-specific and glucocorticoid-mediated myostatin expression in skeletal muscle. Am. J. Physiol. Physiol. 2011, 300, C124–C137. [Google Scholar] [CrossRef] [PubMed]
- Amirouche, A.; Durieux, A.-C.; Banzet, S.; Koulmann, N.; Bonnefoy, R.; Mouret, C.; Bigard, X.A.; Peinnequin, A.; Freyssenet, D. Down-Regulation of Akt/Mammalian Target of Rapamycin Signaling Pathway in Response to Myostatin Overexpression in Skeletal Muscle. Endocrinology 2009, 150, 286–294. [Google Scholar] [CrossRef]
- Ma, K.; Mallidis, C.; Artaza, J.; Taylor, W.; Gonzalez-Cadavid, N.; Bhasin, S. Characterization of 5′-regulatory region of human myostatin gene: Regulation by dexamethasone In vitro. Am. J. Physiol. Metab. 2001, 281, E1128–E1136. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.L.; Cleary, A.S.; Hanson, A.M.; Lindsay, S.F.; Reed, J.M. CCAAT/enhancer binding protein-delta expression is increased in fast skeletal muscle by food deprivation and regulates myostatin transcription In vitro. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R1592–R1601. [Google Scholar] [CrossRef]
- McFarlane, C.; Plummer, E.; Thomas, M.; Hennebry, A.; Ashby, M.; Ling, N.; Kambadur, R. Myostatin induces cachexia by activating the ubiquitin proteolytic system through an NF-kappaB-independent, FoxO1-dependent mechanism. J. Cell Physiol. 2006, 209, 501–514. [Google Scholar] [CrossRef]
- Hardy, R.S.; Zhou, H.; Seibel, M.J.; Cooper, M.S. Glucocorticoids and Bone: Consequences of Endogenous and Exogenous Excess and Replacement Therapy. Endocr. Rev. 2018, 39, 519–548. [Google Scholar] [CrossRef]
- Wei, J.; Ferron, M.; Clarke, C.J.; Hannun, Y.A.; Jiang, H.; Blaner, W.S.; Karsenty, G. Bone-specific insulin resistance disrupts whole-body glucose homeostasis via decreased osteocalcin activation. J. Clin. Investig. 2014, 124, 1781–1793. [Google Scholar] [CrossRef]
- Parker, L.; Lin, X.; Garnham, A.; McConell, G.; Stepto, N.K.; Hare, D.L.; Byrnes, E.; Ebeling, P.R.; Seeman, E.; Brennan-Speranza, T.C.; et al. Glucocorticoid-Induced Insulin Resistance in Men Is Associated With Suppressed Undercarboxylated Osteocalcin. J. Bone Miner. Res. 2018, 34, 49–58. [Google Scholar] [CrossRef]
- Nguyen, N.Q.; Debreceni, T.L.; Bambrick, J.E.; Chia, B.; Wishart, J.; Deane, A.M.; Rayner, C.K.; Horowitz, M.; Young, R.L. Accelerated Intestinal Glucose Absorption in Morbidly Obese Humans: Relationship to Glucose Transporters, Incretin Hormones, and Glycemia. J. Clin. Endocrinol. Metab. 2015, 100, 968–976. [Google Scholar] [CrossRef]
- Tobin, V.; Le Gall, M.; Fioramonti, X.; Stolarczyk, E.; Blazquez, A.G.; Klein, C.; Prigent, M.; Serradas, P.; Cuif, M.-H.; Magnan, C.; et al. Insulin Internalizes GLUT2 in the Enterocytes of Healthy but Not Insulin-Resistant Mice. Diabetes 2007, 57, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Luck, H.; Tsai, S.; Chung, J.; Clemente-Casares, X.; Ghazarian, M.; Revelo, X.S.; Lei, H.; Luk, C.T.; Shi, S.Y.; Surendra, A.; et al. Regulation of Obesity-Related Insulin Resistance with Gut Anti-inflammatory Agents. Cell Metab. 2015, 21, 527–542. [Google Scholar] [CrossRef] [PubMed]
- Monteiro-Sepulveda, M.; Touch, S.; Mendes-Sá, C.; André, S.; Poitou, C.; Allatif, O.; Cotillard, A.; Fohrer-Ting, H.; Hubert, E.-L.; Remark, R.; et al. Jejunal T Cell Inflammation in Human Obesity Correlates with Decreased Enterocyte Insulin Signaling. Cell Metab. 2015, 22, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Winer, D.A.; Winer, S.; Dranse, H.J.; Lam, T.K. Immunologic impact of the intestine in metabolic disease. J. Clin. Investig. 2017, 127, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Filaretova, L.; Bagaeva, T. The Realization of the Brain-Gut Interactions with Corticotropin-Releasing Factor and Glucocorticoids. Curr. Neuropharmacol. 2016, 14, 876–881. [Google Scholar] [CrossRef]
- Slominski, R.M.; Tuckey, R.C.; Manna, P.R.; Jetten, A.M.; Postlethwaite, A.; Raman, C.; Slominski, A.T. Extra-adrenal glucocorticoid biosynthesis: Implications for autoimmune and inflammatory disorders. Genes Immun. 2020, 21, 150–168. [Google Scholar] [CrossRef]
- Mukherji, A.; Kobiita, A.; Ye, T.; Chambon, P. Homeostasis in Intestinal Epithelium Is Orchestrated by the Circadian Clock and Microbiota Cues Transduced by TLRs. Cell 2013, 153, 812–827. [Google Scholar] [CrossRef]
- Kappe, C.; Fransson, L.; Wolbert, P.; Ortsäter, H. Glucocorticoids suppress GLP-1 secretion: Possible contribution to their diabetogenic effects. Clin. Sci. 2015, 129, 405–414. [Google Scholar] [CrossRef]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the brain: The role of glucose in physiological and pathological brain function. Trends Neurosci. 2013, 36, 587–597. [Google Scholar] [CrossRef]
- Pearson-Leary, J.; McNay, E.C. Novel Roles for the Insulin-Regulated Glucose Transporter-4 in Hippocampally Dependent Memory. J. Neurosci. 2016, 36, 11851–11864. [Google Scholar] [CrossRef]
- Reno, C.M.; Puente, E.C.; Sheng, Z.; Daphna-Iken, D.; Bree, A.J.; Routh, V.H.; Kahn, B.B.; Fisher, S.J. Brain GLUT4 Knockout Mice Have Impaired Glucose Tolerance, Decreased Insulin Sensitivity, and Impaired Hypoglycemic Counterregulation. Diabetes 2017, 66, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.-Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Obici, S.; Zhang, B.B.; Karkanias, G.; Rossetti, L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat. Med. 2002, 8, 1376–1382. [Google Scholar] [CrossRef] [PubMed]
- Scherer, T.; O’Hare, J.; Diggs-Andrews, K.; Schweiger, M.; Cheng, B.; Lindtner, C.; Zielinski, E.; Vempati, P.; Su, K.; Dighe, S.; et al. Brain Insulin Controls Adipose Tissue Lipolysis and Lipogenesis. Cell Metab. 2011, 13, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Air, E.L.; Benoit, S.C.; Smith, K.A.B.; Clegg, D.J.; Woods, S.C. Acute third ventricular administration of insulin decreases food intake in two paradigms. Pharmacol. Biochem. Behav. 2002, 72, 423–429. [Google Scholar] [CrossRef]
- DeBons, A.F.; Krimsky, I.; From, A. A direct action of insulin on the hypothalamic satiety center. Am. J. Physiol. Content 1970, 219, 938–943. [Google Scholar] [CrossRef][Green Version]
- Obici, S.; Feng, Z.; Karkanias, G.; Baskin, D.G.; Rossetti, L. Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat Neurosci. 2002, 5, 566–572. [Google Scholar] [CrossRef]
- Coker, G.T., 3rd; Studelska, D.; Harmon, S.; Burke, W.; O’Malley, K.L. Analysis of tyrosine hydroxylase and insulin transcripts in human neuroendocrine tissues. Brain Res. Mol. Brain Res. 1990, 8, 93–98. [Google Scholar] [CrossRef]
- Devaskar, S.U.; Giddings, S.J.; A Rajakumar, P.; Carnaghi, L.R.; Menon, R.K.; Zahm, D.S. Insulin gene expression and insulin synthesis in mammalian neuronal cells. J. Biol. Chem. 1994, 269, 8445–8454. [Google Scholar] [CrossRef]
- Yazdani, S.; Jaldin-Fincati, J.R.; Pereira, R.V.S.; Klip, A. Endothelial cell barriers: Transport of molecules between blood and tissues. Traffic 2019, 20, 390–403. [Google Scholar] [CrossRef]
- Jaldin-Fincati, J.R.; Pereira, R.V.S.; Bilan, P.J.; Klip, A. Insulin uptake and action in microvascular endothelial cells of lymphatic and blood origin. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E204–E217. [Google Scholar] [CrossRef] [PubMed]
- Baura, G.D.; Foster, D.M.; Kaiyala, K.; Porte, D., Jr.; Kahn, S.E.; Schwartz, M.W. Insulin transport from plasma into the central nervous system is inhibited by dexamethasone in dogs. Diabetes 1996, 45, 86–90. [Google Scholar] [CrossRef]
- Oitzl, M.S.; Fluttert, M.; Sutanto, W.; De Kloet, E.R. Continuous blockade of brain glucocorticoid receptors facilitates spatial learning and memory in rats. Eur. J. Neurosci. 1998, 10, 3759–3766. [Google Scholar] [CrossRef]
- Pavlides, C.; Watanabe, Y.; McEwen, B.S. Effects of glucocorticoids on hippocampal long-term potentiation. Hippocampus 1993, 3, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Wright, R.L.; Lightner, E.N.; Harman, J.S.; Meijer, O.C.; Conrad, C.D. Attenuating corticosterone levels on the day of memory assessment prevents chronic stress-induced impairments in spatial memory. Eur. J. Neurosci. 2006, 24, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Piroli, G.G.; Grillo, C.A.; Reznikov, L.R.; Adams, S.; Bs, M.; Charron, M.J.; Reagan, L.P. Corticosterone Impairs Insulin-Stimulated Translocation of GLUT4 in the Rat Hippocampus. Neuroendocrinology 2007, 85, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Biessels, G.J.; Reagan, L.P. Hippocampal insulin resistance and cognitive dysfunction. Nat. Rev. Neurosci. 2015, 16, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Stranahan, A.M.; Arumugam, T.V.; Cutler, R.G.; Lee, K.; Egan, J.M.; Mattson, M.P. Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat. Neurosci. 2008, 11, 309–317. [Google Scholar] [CrossRef] [PubMed]
- de Quervain, D.J.; Roozendaal, B.; Nitsch, R.M.; McGaugh, J.L.; Hock, C. Acute cortisone administration impairs retrieval of long-term declarative memory in humans. Nat. Neurosci. 2000, 3, 313–314. [Google Scholar] [CrossRef] [PubMed]
- Newcomer, J.W.; Craft, S.; Hershey, T.; Askins, K.; E Bardgett, M. Glucocorticoid-induced impairment in declarative memory performance in adult humans. J. Neurosci. 1994, 14, 2047–2053. [Google Scholar] [CrossRef] [PubMed]
- Newcomer, J.W.; Selke, G.; Melson, A.K.; Hershey, T.; Craft, S.; Richards, K.; Alderson, A.L. Decreased memory performance in healthy humans induced by stress-level cortisol treatment. Arch. Gen. Psychiatry. 1999, 56, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Ragnarsson, O.; Berglund, P.; Eder, D.N.; Johannsson, G. Long-term cognitive impairments and attentional deficits in patients with Cushing’s disease and cortisol-producing adrenal adenoma in remission. J. Clin. Endocrinol. Metab. 2012, 97, E1640–E1648. [Google Scholar] [CrossRef] [PubMed]
- Chiodini, I.; Adda, G.; Scillitani, A.; Coletti, F.; Morelli, V.; Di Lembo, S.; Ambrosi, B. Cortisol secretion in patients with type 2 diabetes: Relationship with chronic complications. Diabetes Care 2007, 30, 83–88. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beaupere, C.; Liboz, A.; Fève, B.; Blondeau, B.; Guillemain, G. Molecular Mechanisms of Glucocorticoid-Induced Insulin Resistance. Int. J. Mol. Sci. 2021, 22, 623. https://doi.org/10.3390/ijms22020623
Beaupere C, Liboz A, Fève B, Blondeau B, Guillemain G. Molecular Mechanisms of Glucocorticoid-Induced Insulin Resistance. International Journal of Molecular Sciences. 2021; 22(2):623. https://doi.org/10.3390/ijms22020623
Chicago/Turabian StyleBeaupere, Carine, Alexandrine Liboz, Bruno Fève, Bertrand Blondeau, and Ghislaine Guillemain. 2021. "Molecular Mechanisms of Glucocorticoid-Induced Insulin Resistance" International Journal of Molecular Sciences 22, no. 2: 623. https://doi.org/10.3390/ijms22020623
APA StyleBeaupere, C., Liboz, A., Fève, B., Blondeau, B., & Guillemain, G. (2021). Molecular Mechanisms of Glucocorticoid-Induced Insulin Resistance. International Journal of Molecular Sciences, 22(2), 623. https://doi.org/10.3390/ijms22020623