Extracellular Vesicle-Mediated Purinergic Signaling Contributes to Host Microenvironment Plasticity and Metastasis in Triple Negative Breast Cancer



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
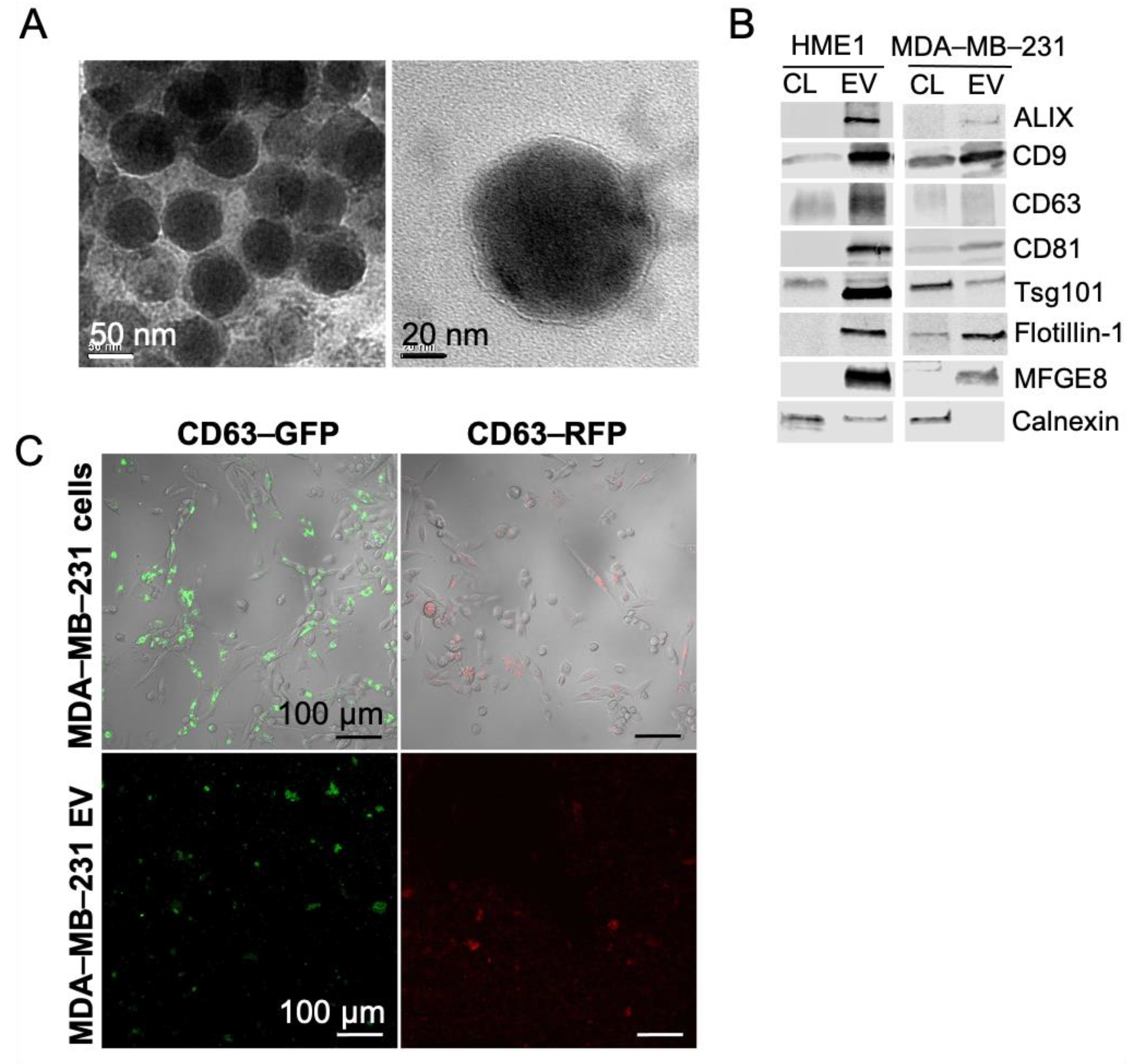
2.1. Extracellular Vesicles Secreted by Non-Tumorigenic Human Mammary Cells and Triple Negative Breast Cancer Cells Demonstrate Classical Morphological and Molecular Features
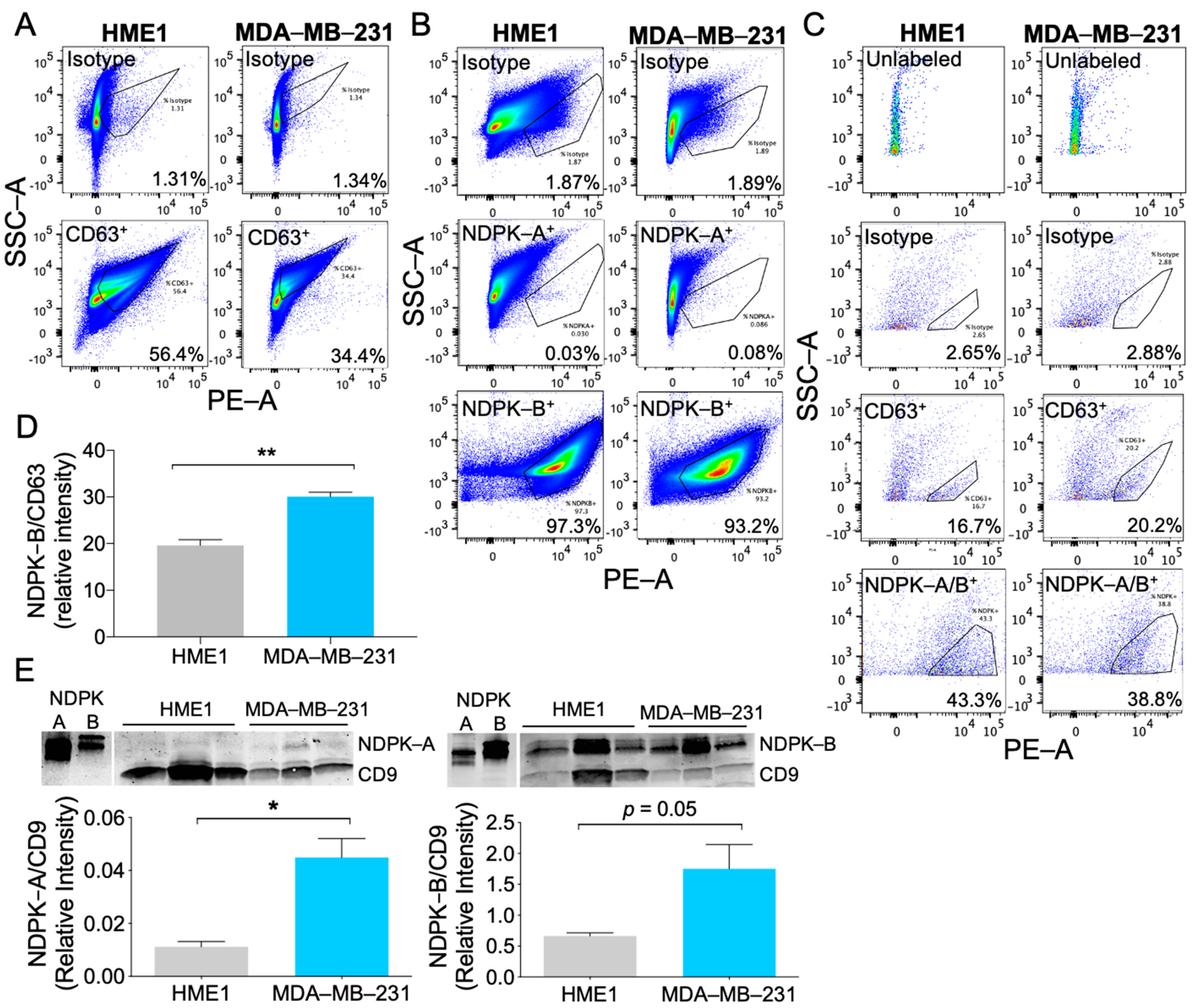
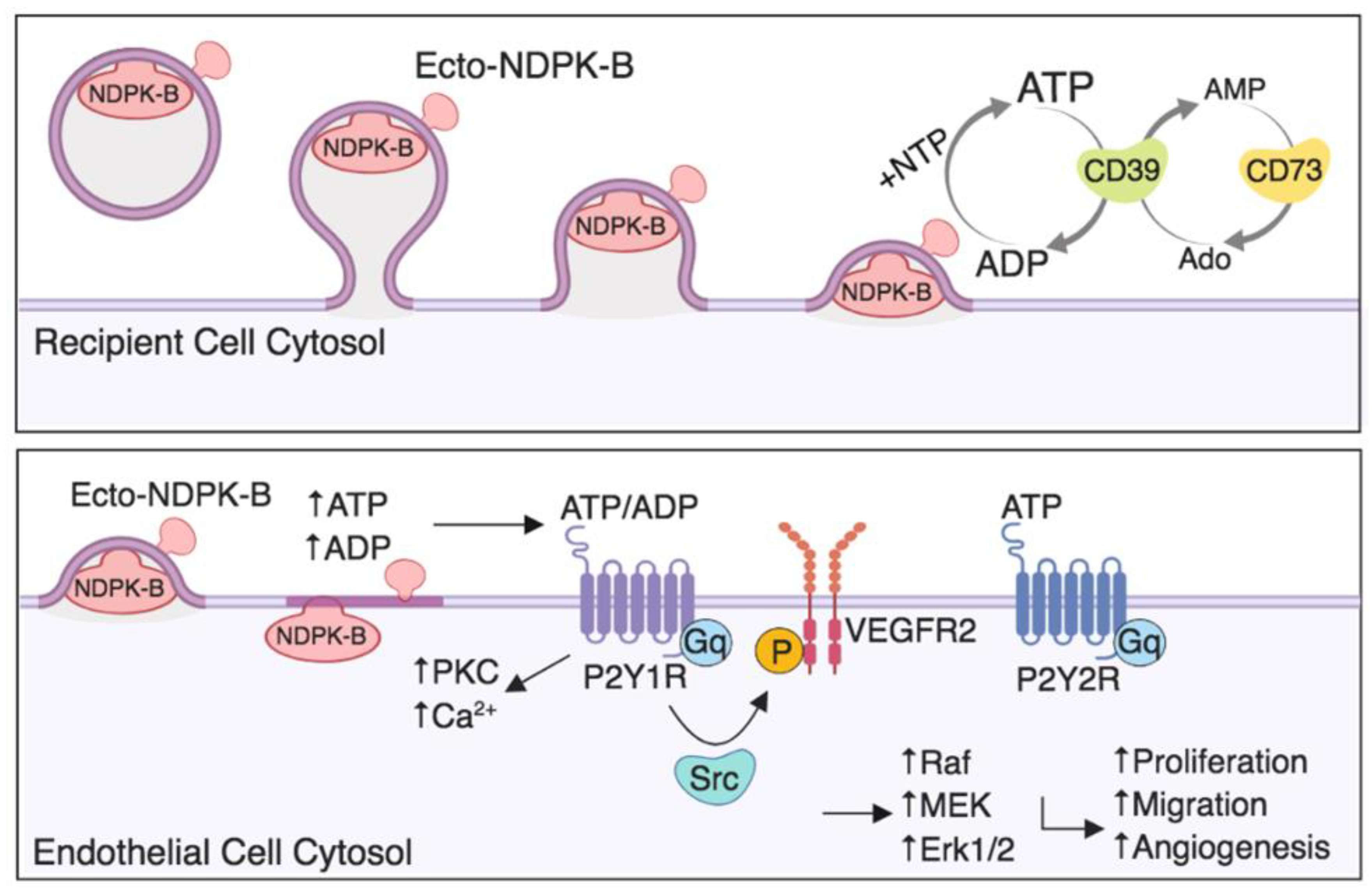
2.2. MDA-MB-231 EVs Are Enriched in NDPK-B Expression Compared to Non-Tumorigenic Mammary EVs
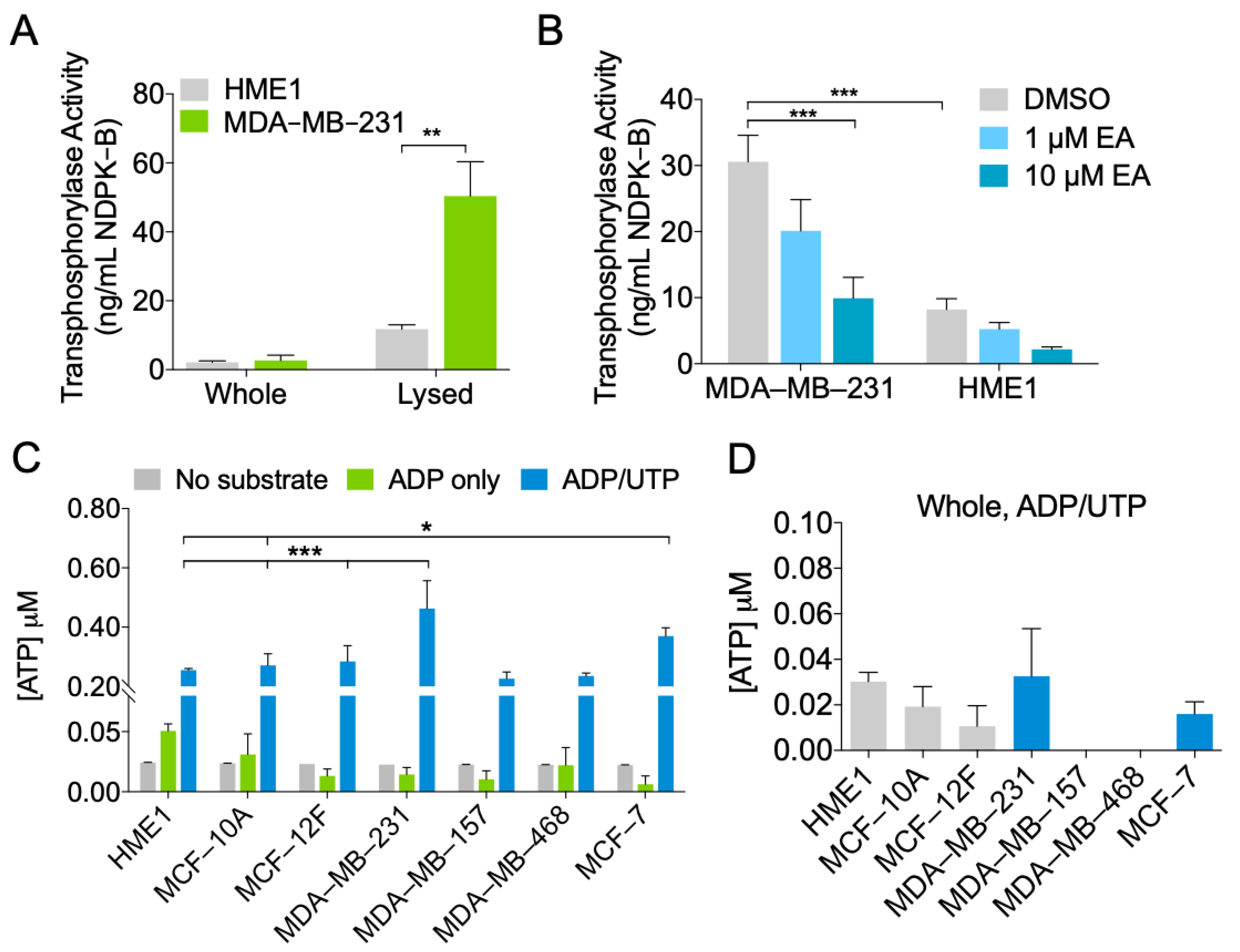
2.3. MDA-MB-231 EVs Are Enriched in NDPK Phosphotransferase Activity
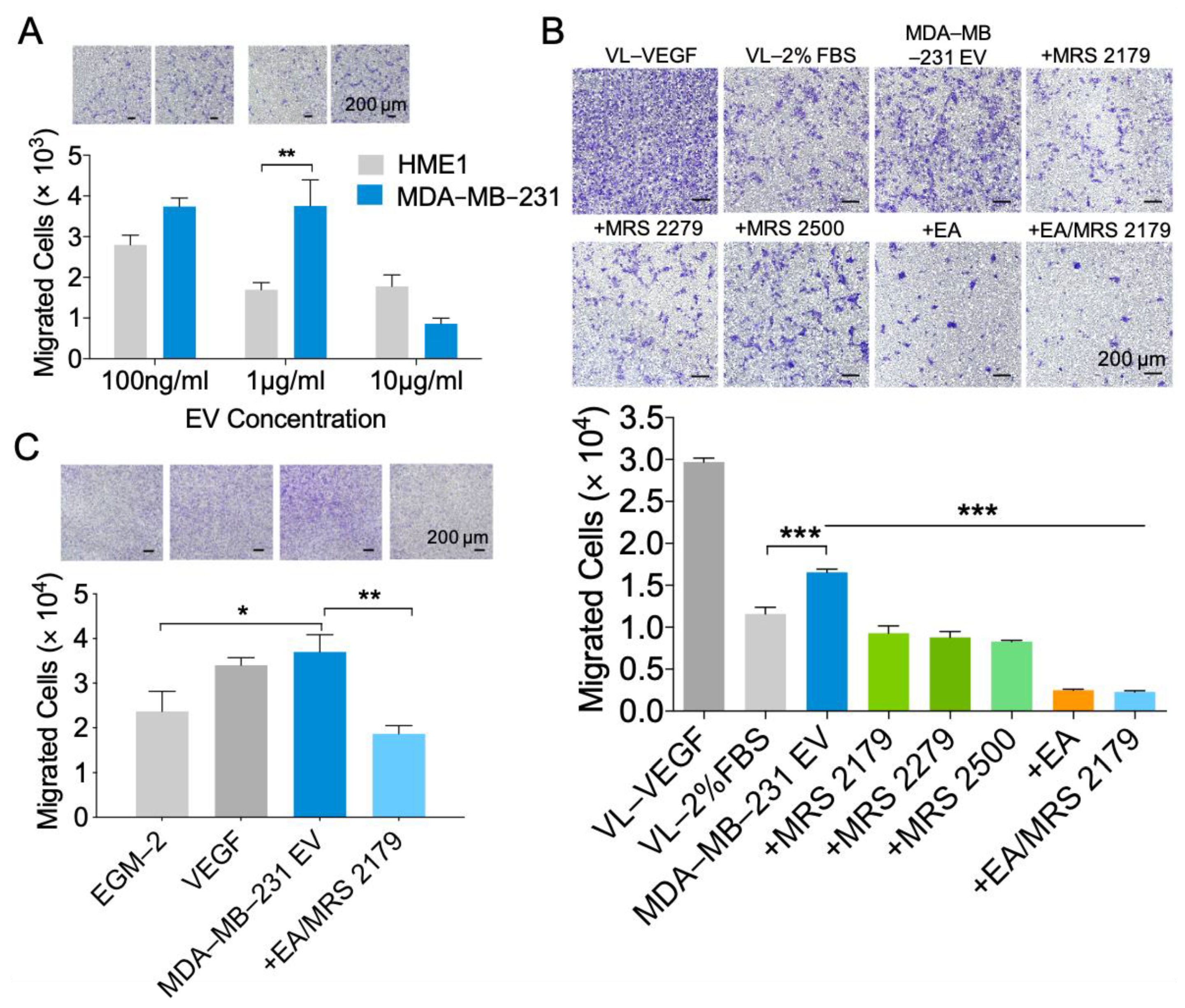
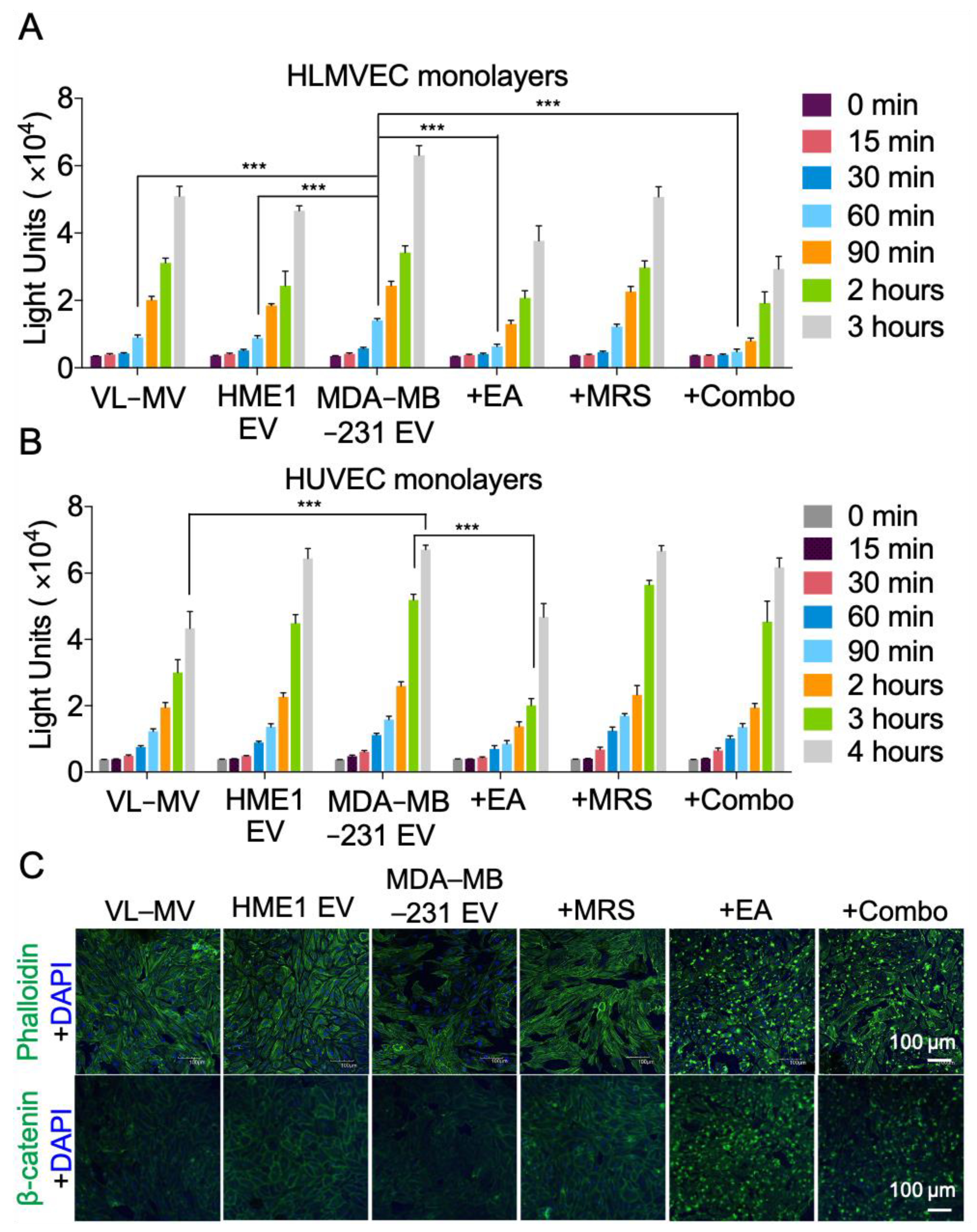
2.4. NDPK Inhibition and P2Y1 Receptor Antagonism Reverse the Pro-Migratory Effect of MDA-MB-231 EVs on Vascular Endothelial Cells
2.5. NDPK Inhibition Rescues MDA-MB-231 EV-Mediated Permeabilization of Vascular Endothelial Cell Monolayers
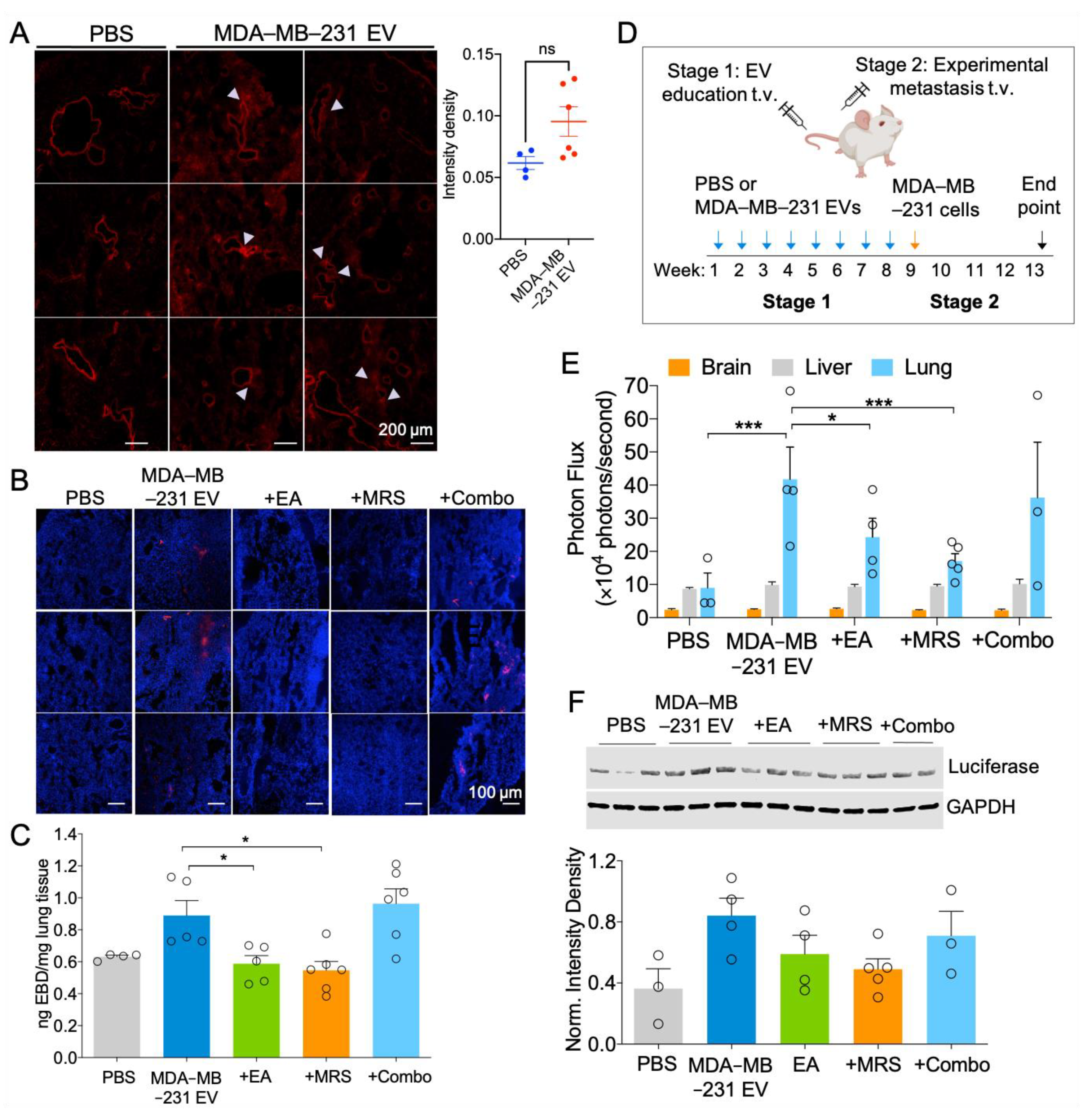
2.6. MDA-MB-231 EVs Enhance Vascular Leakage in the Lung and NDPK Inhibition or P2Y1 Receptor Antagonism Ameliorates This Effect
2.7. Attenuation of eNDPK Activity or P2Y1 Receptor Activation Blunts the Development of Lung Metastases in an Experimental Metastasis Model
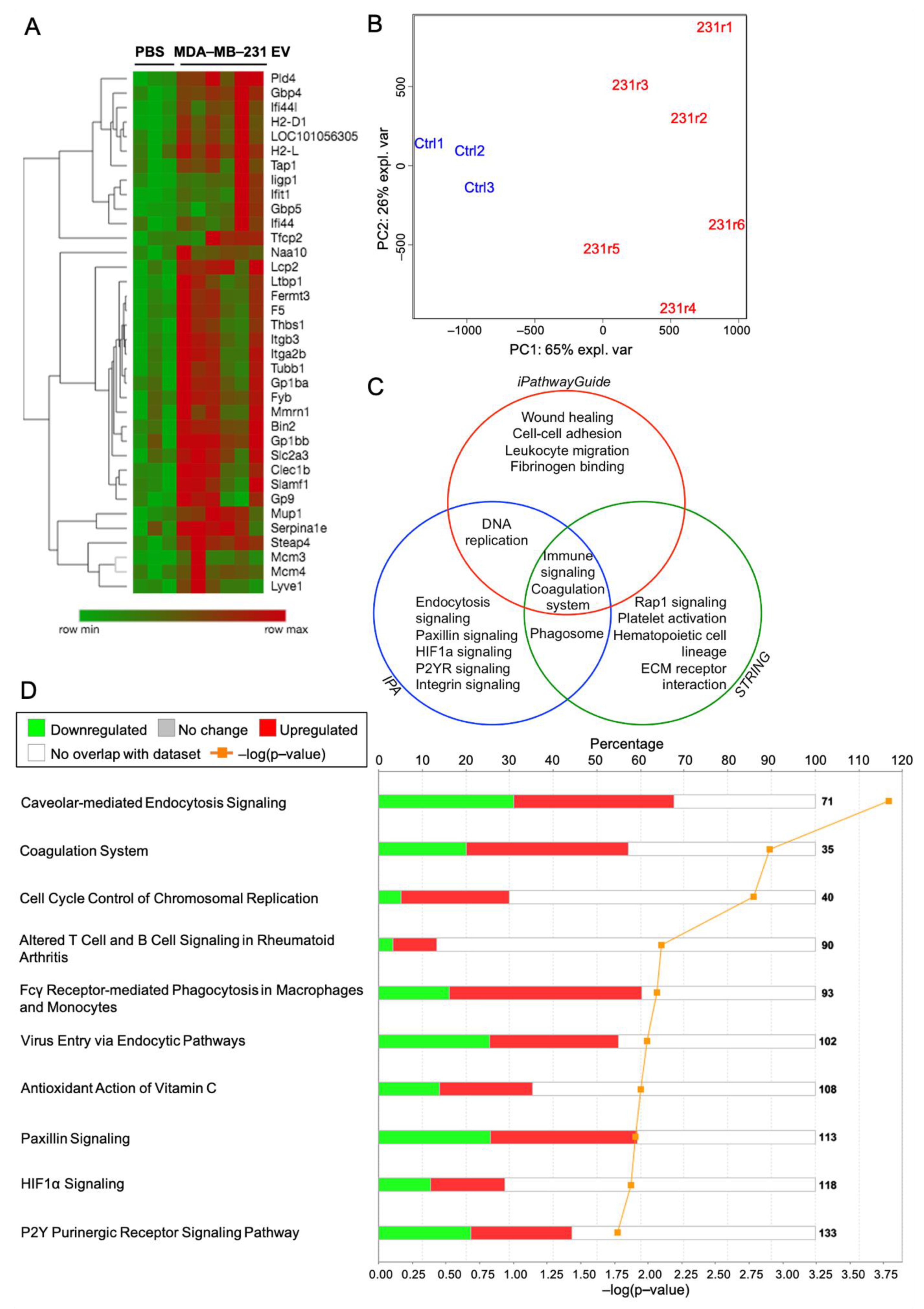
2.8. Proteomic Analysis of the Lung Following MDA-MB-231 EV Treatment Identifies Purinergic Events Known to Support Pre-Metastatic Niche Formation
3. Discussion
4. Materials and Methods
4.1. Drug Reagents
4.2. Cell Culture of Human Cell Lines
4.3. Neonatal Pulmonary Endothelial Cell Isolation
4.4. Extracellular Vesicle Isolation
4.5. Transmission Electron Microscopy (TEM)
4.6. EV Analysis with Flow Cytometry
4.7. Western Blot Analysis
4.8. Wes Protein Assay
4.9. NDPK Transphosphorylation Activity Assay
4.10. Transwell Migration Assay
4.11. Transwell Permeability Assay
4.12. Imaging of Endothelial Barrier
4.13. Confocal Laser Scanning Microscopy (CLSM)
4.14. Animal Studies
4.15. TMT-Labeled Mass Spectrometry
4.16. Evans Blue Dye (EBD) Extravasation Study
4.17. Experimental Metastasis Study
4.18. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef]
- Hüsemann, Y.; Geigl, J.B.; Schubert, F.; Musiani, P.; Meyer, M.; Burghart, E.; Forni, G.; Eils, R.; Fehm, T.; Riethmüller, G.; et al. Systemic spread is an early step in breast cancer. Cancer Cell 2008, 13, 58–68. [Google Scholar] [CrossRef] [Green Version]
- Hosseini, H.; Obradović, M.; Hoffmann, M.; Harper, K.L.; Sosa, M.S.; Werner-Klein, M.; Nanduri, L.K.; Werno, C.; Ehrl, C.; Maneck, M.; et al. Early dissemination seeds metastasis in breast cancer. Nature 2016. [Google Scholar] [CrossRef] [Green Version]
- Harper, K.L.; Sosa, M.S.; Entenberg, D.; Hosseini, H.; Cheung, J.F.; Nobre, R.; Avivar-Valderas, A.; Nagi, C.; Girnius, N.; Davis, R.J.; et al. Mechanism of early dissemination and metastasis in Her2+ mammary cancer. Nature 2016. [Google Scholar] [CrossRef]
- Xu, R.; Rai, A.; Chen, M.; Suwakulsiri, W.; Greening, D.W.; Simpson, R.J. Extracellular vesicles in cancer—Implications for future improvements in cancer care. Nat. Rev. Clin. Oncol. 2018, 15, 617–638. [Google Scholar] [CrossRef]
- TKach, M.; Thery, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [Green Version]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Steeg, P.S.; Bevilacqua, G.; Kopper, L.; Thorgeirsson, U.P.; Talmadge, J.E.; Liotta, L.A.; Sobel, M.E. Evidence for a novel gene associated with low tumor metastatic potential. J. Natl. Cancer Inst. 1988, 80, 200–204. [Google Scholar] [CrossRef]
- Stahl, J.A.; Leone, A.; Rosengard, A.M.; Porter, L.; King, C.R.; Steeg, P.S. Identification of a second human nm23 gene, nm23-H2. Cancer Res. 1991, 51, 445–449. [Google Scholar] [PubMed]
- Boissan, M.; Schlattner, U.; Lacombe, M.L. The NDPK/NME superfamily: State of the art. Lab Investig. 2018, 98, 164–174. [Google Scholar] [CrossRef]
- Sharma, S.; Sengupta, A.; Chowdhury, S. NM23/NDPK proteins in transcription regulatory functions and chromatin modulation: Emerging trends. Lab Invest. 2017, 98, 175–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruger, S.; Abd Elmageed, Z.Y.; Hawke, D.H.; Wörner, P.M.; Jansen, D.A.; Abdel-Mageed, A.B.; Alt, E.U.; Izadpanah, R. Molecular characterization of exosome-like vesicles from breast cancer cells. BMC Cancer 2014, 14, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palazzolo, G.; Albanese, N.N.; DI Cara, G.; Gygax, D.; Vittorelli, M.L.; Pucci-Minafra, I. Proteomic analysis of exosome-like vesicles derived from breast cancer cells. Anticancer Res. 2012, 32, 847–860. [Google Scholar] [PubMed]
- Hurwitz, S.N.; Rider, M.A.; Bundy, J.L.; Liu, X.; Singh, R.K.; Meckes, D.G., Jr. Proteomic profiling of NCI-60 extracellular vesicles uncovers common protein cargo and cancer type-specific biomarkers. Oncotarget 2016, 7, 86999–87015. [Google Scholar] [CrossRef]
- Demory Beckler, M.; Higginbotham, J.N.; Franklin, J.L.; Ham, A.J.; Halvey, P.J.; Imasuen, I.E.; Whitwell, C.; Li, M.; Liebler, D.C.; Coffey, R.J. Proteomic Analysis of Exosomes from Mutant KRAS Colon Cancer Cells Identifies Intercellular Transfer of Mutant KRAS. Mol. Cell Proteom. 2013, 12, 343–355. [Google Scholar] [CrossRef] [Green Version]
- Liang, B.; Peng, P.; Chen, S.; Li, L.; Zhang, M.; Cao, D.; Yang, J.; Li, H.; Gui, T.; Li, X.; et al. Characterization and proteomic analysis of ovarian cancer-derived exosomes. J. Proteom. 2013, 80, 171–182. [Google Scholar] [CrossRef]
- He, M.; Qin, H.; Poon, T.C.; Sze, S.C.; Ding, X.; Co, N.N.; Ngai, S.M.; Chan, T.F.; Wong, N. Hepatocellular carcinoma-derived exosomes promote motility of immortalized hepatocyte through transfer of oncogenic proteins and RNAs. Carcinogenesis 2015, 36, 1008–1018. [Google Scholar] [CrossRef] [Green Version]
- Lazar, I.; Clement, E.; Ducoux-Petit, M.; Denat, L.; Soldan, V.; Dauvillier, S.; Balor, S.; Burlet-Schiltz, O.; Larue, L.; Muller, C.; et al. Proteome characterization of melanoma exosomes reveals a specific signature for metastatic cell lines. Pigment Cell Melanoma Res. 2015, 28, 464–475. [Google Scholar] [CrossRef]
- Buxton, I.L.; Kaiser, R.A.; Oxhorn, B.C.; Cheek, D.J. Evidence supporting the Nucleotide Axis Hypothesis: ATP release and metabolism by coronary endothelium. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H1657–H1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Virgilio, F.; Sarti, A.C.; Falzoni, S.; De Marchi, E.; Adinolfi, E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat. Rev. Cancer 2018, 18, 601–618. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.L.; Liu, Y.; Yang, H.; Zhang, H.Q.; Tian, X.X.; Fang, W.G. ATP-P2Y2-β-catenin axis promotes cell invasion in breast cancer cells. Cancer Sci. 2017, 108, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Eun, S.Y.; Lee, J.S.; Park, S.W.; Lee, J.H.; Chang, K.C.; Kim, H.J. P2Y2 receptor activation by nucleotides released from highly metastatic breast cancer cells increases tumor growth and invasion via crosstalk with endothelial cells. Breast Cancer Res. 2014, 16, R77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adinolfi, E.; Raffaghello, L.; Giuliani, A.L.; Cavazzini, L.; Capece, M.; Chiozzi, P.; Bianchi, G.; Kroemer, G.; Pistoia, V.; Di Virgilio, F. Expression of P2X7 receptor increases in vivo tumor growth. Cancer Res. 2012, 72, 2957–2969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buvinic, S.; Bravo-Zehnder, M.; Boyer, J.L.; Huidobro-Toro, J.P. Nucleotide P2Y1 receptor regulates EGF receptor mitogenic signaling and expression in epithelial cells. J. Cell Sci. 2007, 120, 4289–4301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rumjahn, S.M.; Javed, M.A.; Wong, N.; Law, W.E.; Buxton, I.L. Purinergic regulation of angiogenesis by human breast carcinoma-secreted nucleoside diphosphate kinase. Br. J. Cancer 2007, 97, 1372–1380. [Google Scholar] [CrossRef] [PubMed]
- Rumjahn, S.M.; Yokdang, N.; Baldwin, K.A.; Thai, J.; Buxton, I.L. Purinergic regulation of vascular endothelial growth factor signaling in angiogenesis. Br. J. Cancer 2009, 100, 1465–1470. [Google Scholar] [CrossRef] [Green Version]
- Yokdang, N.; Tellez, J.D.; Tian, H.; Norvell, J.; Barsky, S.H.; Valencik, M.; Buxton, I.L. A role for nucleotides in support of breast cancer angiogenesis: Heterologous receptor signalling. Br. J. Cancer 2011, 104, 1628–1640. [Google Scholar] [CrossRef] [Green Version]
- Burnstock, G.; Ralevic, V. Purinergic signaling and blood vessels in health and disease. Pharmacol. Rev. 2014, 66, 102–192. [Google Scholar] [CrossRef]
- Yokdang, N.; Nordmeier, S.; Speirs, K.; Burkin, H.R.; Buxton, I.L. Blockade of extracellular NM23 or its endothelial target slows breast cancer growth and metastasis. Integr. Cancer Sci.Ther. 2015, 2, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Pellegatti, P.; Raffaghello, L.; Bianchi, G.; Piccardi, F.; Pistoia, V.; Di Virgilio, F. Increased Level of Extracellular ATP at Tumor Sites: In Vivo Imaging with Plasma Membrane Luciferase. PLoS ONE 2008, 3, e2599. [Google Scholar] [CrossRef] [PubMed]
- Joo, Y.N.; Jin, H.; Eun, S.Y.; Park, S.W.; Chang, K.C.; Kim, H.J. P2Y2R activation by nucleotides released from the highly metastatic breast cancer cell contributes to pre-metastatic niche formation by mediating lysyl oxidase secretion, collagen crosslinking, and monocyte recruitment. Oncotarget 2014, 5, 9322–9334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otero-Estévez, O.; De Chiara, L.; Barcia-Castro, L.; Páez de la Cadena, M.; Rodríguez-Berrocal, F.J.; Cubiella, J.; Hernández, V.; Martínez-Zorzano, V.S. Evaluation of serum nucleoside diphosphate kinase A for the detection of colorectal cancer. Sci. Rep. 2016, 6, 26703. [Google Scholar] [CrossRef]
- Alvarez-Chaver, P.; Rodríguez-Piñeiro, A.M.; Rodríguez-Berrocal, F.J.; García-Lorenzo, A.; Páez de la Cadena, M.; Martínez-Zorzano, V.S. Selection of putative colorectal cancer markers by applying PCA on the soluble proteome of tumors: NDK A as a promising candidate. J. Proteom. 2011, 74, 874–886. [Google Scholar] [CrossRef]
- Okabe-Kado, J.; Kasukabe, T.; Honma, Y.; Hanada, R.; Nakagawara, A.; Kaneko, Y. Clinical significance of serum NM23-H1 protein in neuroblastoma. Cancer Sci. 2005, 96, 653–660. [Google Scholar] [CrossRef]
- Su Kim, D.; Choi, Y.D.; Moon, M.; Kang, S.; Lim, J.B.; Kim, K.M.; Park, K.M.; Cho, N.H. Composite three-marker assay for early detection of kidney cancer. Cancer Epidemiol. Biomark. Prev. 2013, 22, 390–398. [Google Scholar] [CrossRef] [Green Version]
- Niitsu, N.; Nakamine, H.; Okamoto, M. Expression of nm23-H1 is associated with poor prognosis in peripheral T-cell lymphoma, not otherwise specified. Clin. Cancer Res. 2011, 17, 2893–2899. [Google Scholar] [CrossRef] [Green Version]
- Yokdang, N.; Buxton, N.D.; Buxton, I.L. Measurement of human breast tumor cell-secreted shNDPK-B in a murine breast cancer model suggests its role in metastatic progression. Proc. West Pharmacol. Soc. 2009, 52, 88–91. [Google Scholar]
- Anzinger, J.; Malmquist, N.A.; Gould, J.; Buxton, I.L. Secretion of a nucleoside diphosphate kinase (Nm23-H2) by cells from human breast, colon, pancreas and lung tumors. Proc. West Pharmacol. Soc. 2001, 44, 61–63. [Google Scholar]
- Ludwig, N.; Yerneni, S.S.; Razzo, B.M.; Whiteside, T.L. Exosomes from HNSCC promote angiogenesis through reprogramming of endothelial cells. Mol. Cancer Res. 2018, 16, 1798–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Fong, M.Y.; Min, Y.; Somlo, G.; Liu, L.; Palomares, M.R.; Yu, Y.; Chow, A.; O’Connor, S.T.; Chin, A.R.; et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 2014, 25, 501–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hippe, H.J.; Wolf, N.M.; Abu-Taha, I.; Mehringer, R.; Just, S.; Lutz, S.; Niroomand, F.; Postel, E.H.; Katus, H.A.; Rottbauer, W.; et al. The interaction of nucleoside diphosphate kinase B with Gβγ dimers controls heterotrimeric G protein function. Proc. Natl. Acad. Sci. USA 2009, 106, 16269–16274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Gross, S.; Wolf, N.M.; Butenschön, V.M.; Qiu, Y.; Devraj, K.; Liebner, S.; Kroll, J.; Skolnik, E.Y.; Hammes, H.P.; et al. Nucleoside diphosphate kinase B regulates angiogenesis through modulation of vascular endothelial growth factor receptor type 2 and endothelial adherens junction proteins. Arter. Thromb Vasc Biol. 2014, 34, 2292–2300. [Google Scholar] [CrossRef] [Green Version]
- Francois-Moutal, L.; Ouberai, M.M.; Maniti, O.; Welland, M.E.; Strzelecka-Kiliszek, A.; Wos, M.; Pikula, S.; Bandorowicz-Pikula, J.; Marcillat, O.; Granjon, T. Two-Step Membrane Binding of NDPK-B Induces Membrane Fluidity Decrease and Changes in Lipid Lateral Organization and Protein Cluster Formation. Langmuir ACS J. Surf. Colloids 2016, 32, 12923–12933. [Google Scholar] [CrossRef]
- Gross, S.; Devraj, K.; Feng, Y.; Macas, J.; Liebner, S.; Wieland, T. Nucleoside diphosphate kinase B regulates angiogenic responses in the endothelium via caveolae formation and c-Src-mediated caveolin-1 phosphorylation. J. Cereb Blood Flow Metab. 2016, 37, 2471–2484. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, K.A.; Deflorian, F.; Mishra, S.; Costanzi, S. Pharmacochemistry of the platelet purinergic receptors. Purinergic. Signal. 2011, 7, 305–324. [Google Scholar] [CrossRef] [Green Version]
- Labelle, M.; Begum, S.; Hynes, R.O. Platelets guide the formation of early metastatic niches. Proc. Natl. Acad. Sci. USA 2014, 111, E3053–E3061. [Google Scholar] [CrossRef] [Green Version]
- Ward, Y.; Lake, R.; Faraji, F.; Sperger, J.; Martin, P.; Gilliard, C.; Ku, K.P.; Rodems, T.; Niles, D.; Tillman, H.; et al. Platelets Promote Metastasis via Binding Tumor CD97 Leading to Bidirectional Signaling that Coordinates Transendothelial Migration. Cell Rep. 2018, 23, 808–822. [Google Scholar] [CrossRef]
- Gomes, F.G.; Sandim, V.; Almeida, V.H.; Rondon, A.; Succar, B.B.; Hottz, E.D.; Leal, A.C.; Verçoza, B.; Rodrigues, J.; Bozza, P.T.; et al. Breast-cancer extracellular vesicles induce platelet activation and aggregation by tissue factor-independent and -dependent mechanisms. Thromb Res. 2017, 159, 24–32. [Google Scholar] [CrossRef]
- Blay, J.; White, T.D.; Hoskin, D.W. The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res. 1997, 57, 2602–2605. [Google Scholar] [PubMed]
- De Mello, P.A.; Coutinho-Silva, R.; Savio, L.E.B. Multifaceted Effects of Extracellular Adenosine Triphosphate and Adenosine in the Tumor-Host Interaction and Therapeutic Perspectives. Front Immunol. 2017, 8, 1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobczak, M.; Dargatz, J.; Chrzanowska-Wodnicka, M. Isolation and Culture of Pulmonary Endothelial Cells from Neonatal Mice. J. Vis. Exp. 2010, 46, e2316. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 30, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Martins-Green, M.; Petreaca, M.; Yao, M. An assay system for in vitro detection of permeability in human “endothelium”. Methods Enzymol. 2008, 443, 137–153. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duan, S.; Nordmeier, S.; Byrnes, A.E.; Buxton, I.L.O. Extracellular Vesicle-Mediated Purinergic Signaling Contributes to Host Microenvironment Plasticity and Metastasis in Triple Negative Breast Cancer. Int. J. Mol. Sci. 2021, 22, 597. https://doi.org/10.3390/ijms22020597
Duan S, Nordmeier S, Byrnes AE, Buxton ILO. Extracellular Vesicle-Mediated Purinergic Signaling Contributes to Host Microenvironment Plasticity and Metastasis in Triple Negative Breast Cancer. International Journal of Molecular Sciences. 2021; 22(2):597. https://doi.org/10.3390/ijms22020597
Chicago/Turabian StyleDuan, Suzann, Senny Nordmeier, Aidan E. Byrnes, and Iain L. O. Buxton. 2021. "Extracellular Vesicle-Mediated Purinergic Signaling Contributes to Host Microenvironment Plasticity and Metastasis in Triple Negative Breast Cancer" International Journal of Molecular Sciences 22, no. 2: 597. https://doi.org/10.3390/ijms22020597
APA StyleDuan, S., Nordmeier, S., Byrnes, A. E., & Buxton, I. L. O. (2021). Extracellular Vesicle-Mediated Purinergic Signaling Contributes to Host Microenvironment Plasticity and Metastasis in Triple Negative Breast Cancer. International Journal of Molecular Sciences, 22(2), 597. https://doi.org/10.3390/ijms22020597