Neutrophil Extracellular Traps and Their Implications in Cardiovascular and Inflammatory Disease


Abstract
1. Introduction
2. Cellular Defense Functions of Neutrophils
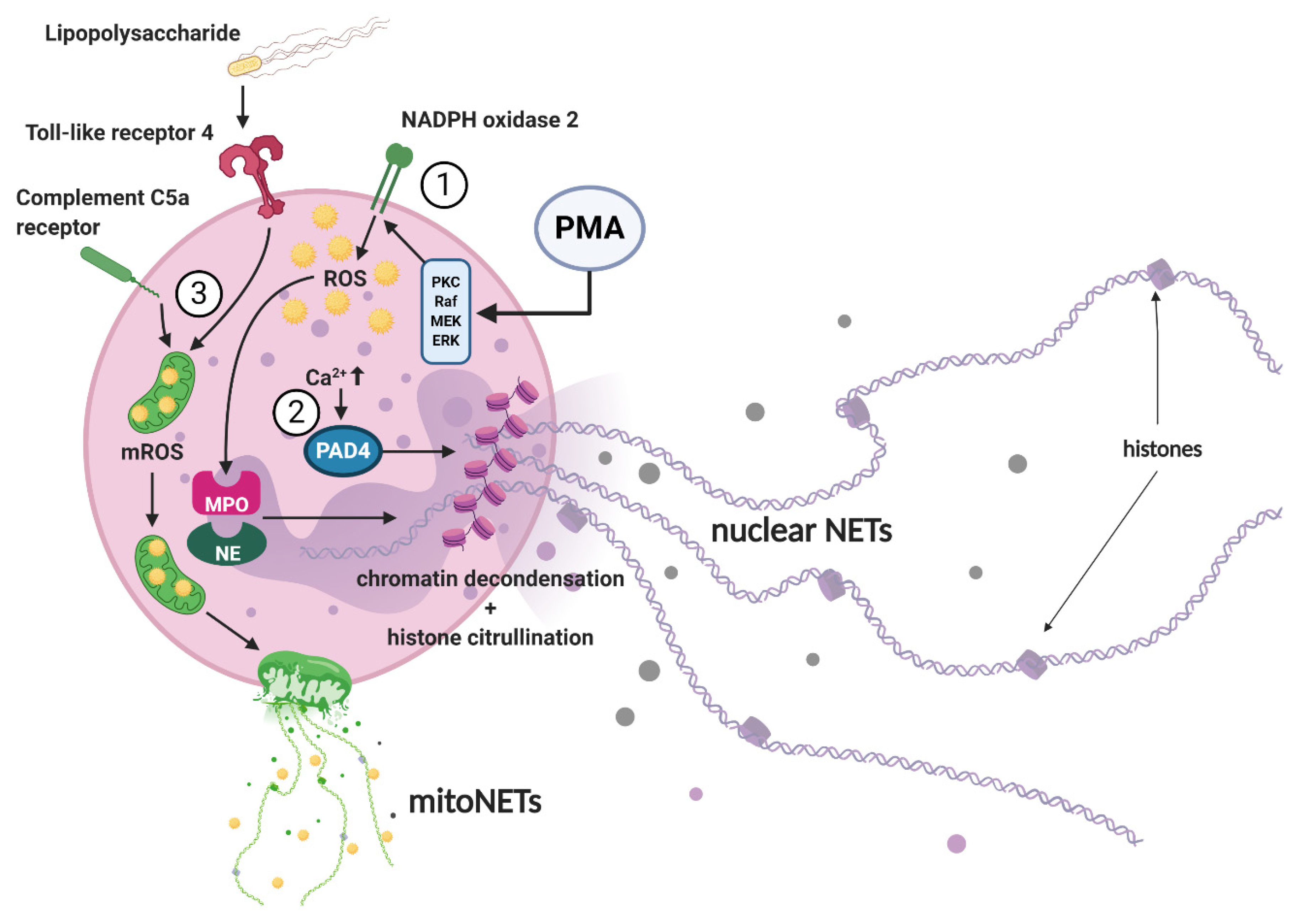
3. NET Morphology and Mechanisms of NET Formation
4. NET-Associated Diseases
4.1. NETs in Atherosclerosis
4.2. NETs Promote Vascular Disease
4.3. Autoimmunity and Autoinflammatory Diseases
4.4. Metabolic Diseases
4.5. Malignant Neoplasia
4.6. Sepsis
4.7. COVID-19
5. Current Perspective and Future Direction
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAA | abdominal aortic aneurysm |
| ACPAs | anti-citrullinated protein antibodies |
| AKIN | Acute Kidney Injury Network |
| ANCA | anti-neutrophil cytoplasmic antibody |
| APC | activated protein C |
| ApoE | apolipoprotein E |
| ARDS | acute respiratory distress syndrome |
| eCIRP | extracellular cold-inducible RNA-binding protein |
| G-CSF | granulocyte colony-stimulating factor |
| HMGB-1 | high-mobility group box 1 protein |
| IFN-α | interferon α |
| IL | interleukin |
| MPO | myeloperoxidase |
| mtDNA | mitochondrial DNA |
| NE | neutrophil elastase |
| NET | neutrophil extracellular trap |
| Nox2 | NADPH-oxidase 2 |
| oxLDL | oxidized low-density lipoprotein |
| PAD4 | protein arginine deiminase 4 |
| PMA | phorbol-12-myristate-13-acetate |
| RNP | anti-ribonucleoprotein |
| ROS | reactive oxygen species |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| SLE | systemic lupus erythematosus |
| SOFA | Sequential Organ Failure Assessment |
| TNF-α | tumor necrosis factor-α |
References
- Hidalgo, A.; Chilvers, E.R.; Summers, C.; Koenderman, L. The Neutrophil Life Cycle. Trends Immunol. 2019, 40, 584–597. [Google Scholar] [CrossRef] [PubMed]
- Rosales, C. Neutrophil: A Cell with Many Roles in Inflammation or Several Cell Types? Front. Physiol. 2018, 9, 113. [Google Scholar] [CrossRef] [PubMed]
- Mortaz, E.; Alipoor, S.D.; Adcock, I.M.; Mumby, S.; Koenderman, L. Update on Neutrophil Function in Severe Inflammation. Front. Immunol. 2018, 9, 2171. [Google Scholar] [CrossRef] [PubMed]
- Malech, H.L.; DeLeo, F.R.; Quinn, M.T. The Role of Neutrophils in the Immune System: An Overview. Methods Mol. Biol. 2020, 2087, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Mayadas, T.N.; Cullere, X.; Lowell, C.A. The multifaceted functions of neutrophils. Annu. Rev. Pathol. 2014, 9, 181–218. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Takei, H.; Araki, A.; Watanabe, H.; Ichinose, A.; Sendo, F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J. Leukoc. Biol. 1996, 59, 229–240. [Google Scholar] [CrossRef]
- Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps: Is immunity the second function of chromatin? J. Cell Biol. 2012, 198, 773–783. [Google Scholar] [CrossRef]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Hasler, P.; Giaglis, S.; Hahn, S. Neutrophil extracellular traps in health and disease. Swiss. Med. Wkly. 2016, 146, w14352. [Google Scholar] [CrossRef]
- Zeidler, C.; Germeshausen, M.; Klein, C.; Welte, K. Clinical implications of ELA2-, HAX1-, and G-CSF-receptor (CSF3R) mutations in severe congenital neutropenia. Br. J. Haematol. 2009, 144, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Borregaard, N. Neutrophils, from marrow to microbes. Immunity 2010, 33, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Hager, M.; Cowland, J.B.; Borregaard, N. Neutrophil granules in health and disease. J. Intern. Med. 2010, 268, 25–34. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.C.; Cheng, W.J.; Korinek, M.; Lin, C.Y.; Hwang, T.L. Neutrophils in Psoriasis. Front. Immunol. 2019, 10, 2376. [Google Scholar] [CrossRef] [PubMed]
- Borregaard, N.; Christensen, L.; Bejerrum, O.W.; Birgens, H.S.; Clemmensen, I. Identification of a highly mobilizable subset of human neutrophil intracellular vesicles that contains tetranectin and latent alkaline phosphatase. J. Clin. Investig. 1990, 85, 408–416. [Google Scholar] [CrossRef]
- Hoppenbrouwers, T.; Autar, A.S.A.; Sultan, A.R.; Abraham, T.E.; van Cappellen, W.A.; Houtsmuller, A.B.; van Wamel, W.J.B.; van Beusekom, H.M.M.; van Neck, J.W.; de Maat, M.P.M. In vitro induction of NETosis: Comprehensive live imaging comparison and systematic review. PLoS ONE 2017, 12, e0176472. [Google Scholar] [CrossRef]
- Kaplan, M.J.; Radic, M. Neutrophil extracellular traps: Double-edged swords of innate immunity. J. Immunol. 2012, 189, 2689–2695. [Google Scholar] [CrossRef]
- Pieterse, E.; Jeremic, I.; Czegley, C.; Weidner, D.; Biermann, M.H.; Veissi, S.; Maueröder, C.; Schauer, C.; Bilyy, R.; Dumych, T.; et al. Blood-borne phagocytes internalize urate microaggregates and prevent intravascular NETosis by urate crystals. Sci. Rep. 2016, 6, 38229. [Google Scholar] [CrossRef]
- An, Z.; Li, J.; Yu, J.; Wang, X.; Gao, H.; Zhang, W.; Wei, Z.; Zhang, J.; Zhang, Y.; Zhao, J.; et al. Neutrophil extracellular traps induced by IL-8 aggravate atherosclerosis via activation NF-κB signaling in macrophages. Cell Cycle 2019, 18, 2928–2938. [Google Scholar] [CrossRef]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Obama, T.; Ohinata, H.; Takaki, T.; Iwamoto, S.; Sawada, N.; Aiuchi, T.; Kato, R.; Itabe, H. Cooperative Action of Oxidized Low-Density Lipoproteins and Neutrophils on Endothelial Inflammatory Responses Through Neutrophil Extracellular Trap Formation. Front. Immunol. 2019, 10, 1899. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Lee, J.K. Role of HMGB1 in the Interplay between NETosis and Thrombosis in Ischemic Stroke: A Review. Cells 2020, 9, 1794. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Tan, J.; Diamond, S.L. Hemodynamic force triggers rapid NETosis within sterile thrombotic occlusions. J. Thromb. Haemost. 2018, 16, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Meher, A.K.; Spinosa, M.; Davis, J.P.; Pope, N.; Laubach, V.E.; Su, G.; Serbulea, V.; Leitinger, N.; Ailawadi, G.; Upchurch, G.R., Jr. Novel Role of IL (Interleukin)-1beta in Neutrophil Extracellular Trap Formation and Abdominal Aortic Aneurysms. Arter. Thromb. Vasc. Biol. 2018, 38, 843–853. [Google Scholar] [CrossRef]
- Delbosc, S.; Alsac, J.M.; Journe, C.; Louedec, L.; Castier, Y.; Bonnaure-Mallet, M.; Ruimy, R.; Rossignol, P.; Bouchard, P.; Michel, J.B.; et al. Porphyromonas gingivalis participates in pathogenesis of human abdominal aortic aneurysm by neutrophil activation. Proof of concept in rats. PLoS ONE 2011, 6, e18679. [Google Scholar] [CrossRef]
- Yu, Y.; Su, K. Neutrophil Extracellular Traps and Systemic Lupus Erythematosus. J. Clin. Cell. Immunol. 2013, 4, 139. [Google Scholar] [CrossRef]
- Rother, N.; Pieterse, E.; Lubbers, J.; Hilbrands, L.; van der Vlag, J. Acetylated Histones in Apoptotic Microparticles Drive the Formation of Neutrophil Extracellular Traps in Active Lupus Nephritis. Front. Immunol. 2017, 8, 1136. [Google Scholar] [CrossRef]
- Schön, M.P.; Erpenbeck, L. The Interleukin-23/Interleukin-17 Axis Links Adaptive and Innate Immunity in Psoriasis. Front. Immunol. 2018, 9, 1323. [Google Scholar] [CrossRef]
- Khandpur, R.; Carmona-Rivera, C.; Vivekanandan-Giri, A.; Gizinski, A.; Yalavarthi, S.; Knight, J.S.; Friday, S.; Li, S.; Patel, R.M.; Subramanian, V.; et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl. Med. 2013, 5, 178ra140. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Krumbholz, M.; Schonermarck, U.; Back, W.; Gross, W.L.; Werb, Z.; Grone, H.J.; Brinkmann, V.; Jenne, D.E. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 2009, 15, 623–625. [Google Scholar] [CrossRef] [PubMed]
- Menegazzo, L.; Ciciliot, S.; Poncina, N.; Mazzucato, M.; Persano, M.; Bonora, B.; Albiero, M.; Vigili de Kreutzenberg, S.; Avogaro, A.; Fadini, G.P. NETosis is induced by high glucose and associated with type 2 diabetes. Acta Diabetol. 2015, 52, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Njeim, R.; Azar, W.S.; Fares, A.H.; Azar, S.T.; Kassouf, H.K.; Eid, A.A. NETosis contributes to the pathogenesis of diabetes and its complications. J. Mol. Endocrinol. 2020, 65, R65. [Google Scholar] [CrossRef] [PubMed]
- Mutua, V.; Gershwin, L.J. A Review of Neutrophil Extracellular Traps (NETs) in Disease: Potential Anti-NETs Therapeutics. Clin. Rev. Allergy Immunol. 2020. [Google Scholar] [CrossRef]
- Dinallo, V.; Marafini, I.; Di Fusco, D.; Laudisi, F.; Franze, E.; Di Grazia, A.; Figliuzzi, M.M.; Caprioli, F.; Stolfi, C.; Monteleone, I.; et al. Neutrophil Extracellular Traps Sustain Inflammatory Signals in Ulcerative Colitis. J. Crohns Colitis 2019, 13, 772–784. [Google Scholar] [CrossRef]
- Joshi, M.B.; Baipadithaya, G.; Balakrishnan, A.; Hegde, M.; Vohra, M.; Ahamed, R.; Nagri, S.K.; Ramachandra, L.; Satyamoorthy, K. Elevated homocysteine levels in type 2 diabetes induce constitutive neutrophil extracellular traps. Sci. Rep. 2016, 6, 36362. [Google Scholar] [CrossRef]
- Park, J.; Wysocki, R.W.; Amoozgar, Z.; Maiorino, L.; Fein, M.R.; Jorns, J.; Schott, A.F.; Kinugasa-Katayama, Y.; Lee, Y.; Won, N.H.; et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci. Transl. Med. 2016, 8, 361ra138. [Google Scholar] [CrossRef]
- Gupta, A.K.; Joshi, M.B.; Philippova, M.; Erne, P.; Hasler, P.; Hahn, S.; Resink, T.J. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett. 2010, 584, 3193–3197. [Google Scholar] [CrossRef]
- Tohme, S.; Yazdani, H.O.; Al-Khafaji, A.B.; Chidi, A.P.; Loughran, P.; Mowen, K.; Wang, Y.; Simmons, R.L.; Huang, H.; Tsung, A. Neutrophil Extracellular Traps Promote the Development and Progression of Liver Metastases after Surgical Stress. Cancer Res. 2016, 76, 1367–1380. [Google Scholar] [CrossRef]
- Tadie, J.-M.; Bae, H.-B.; Jiang, S.; Park, D.W.; Bell, C.P.; Yang, H.; Pittet, J.-F.; Tracey, K.; Thannickal, V.J.; Abraham, E.; et al. HMGB1 promotes neutrophil extracellular trap formation through interactions with Toll-like receptor 4. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L342–L349. [Google Scholar] [CrossRef]
- Aziz, M.; Brenner, M.; Wang, P. Extracellular CIRP (eCIRP) and inflammation. J. Leukoc. Biol. 2019, 106, 133–146. [Google Scholar] [CrossRef]
- Al-Banna, N.; Lehmann, C. Oxidized LDL and LOX-1 in Experimental Sepsis. Mediat. Inflamm. 2013, 2013, 761789. [Google Scholar] [CrossRef]
- Cicco, S.; Cicco, G.; Racanelli, V.; Vacca, A. Neutrophil Extracellular Traps (NETs) and Damage-Associated Molecular Patterns (DAMPs): Two Potential Targets for COVID-19 Treatment. Mediat. Inflamm. 2020, 2020, 7527953. [Google Scholar] [CrossRef]
- Chicca, I.J.; Milward, M.R.; Chapple, I.L.C.; Griffiths, G.; Benson, R.; Dietrich, T.; Cooper, P.R. Development and Application of High-Content Biological Screening for Modulators of NET Production. Front. Immunol. 2018, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Castanheira, F.V.S.; Kubes, P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood 2019, 133, 2178–2185. [Google Scholar] [CrossRef] [PubMed]
- Pieterse, E.; Rother, N.; Yanginlar, C.; Gerretsen, J.; Boeltz, S.; Munoz, L.E.; Herrmann, M.; Pickkers, P.; Hilbrands, L.B.; van der Vlag, J. Cleaved N-terminal histone tails distinguish between NADPH oxidase (NOX)-dependent and NOX-independent pathways of neutrophil extracellular trap formation. Ann. Rheum. Dis. 2018, 77, 1790–1798. [Google Scholar] [CrossRef] [PubMed]
- Parker, H.; Dragunow, M.; Hampton, M.B.; Kettle, A.J.; Winterbourn, C.C. Requirements for NADPH oxidase and myeloperoxidase in neutrophil extracellular trap formation differ depending on the stimulus. J. Leukoc. Biol. 2012, 92, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Lewis, H.D.; Liddle, J.; Coote, J.E.; Atkinson, S.J.; Barker, M.D.; Bax, B.D.; Bicker, K.L.; Bingham, R.P.; Campbell, M.; Chen, Y.H.; et al. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat. Chem. Biol. 2015, 11, 189–191. [Google Scholar] [CrossRef]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef]
- Hamam, H.J.; Khan, M.A.; Palaniyar, N. Histone Acetylation Promotes Neutrophil Extracellular Trap Formation. Biomolecules 2019, 9, 32. [Google Scholar] [CrossRef]
- Yousefi, S.; Mihalache, C.; Kozlowski, E.; Schmid, I.; Simon, H.U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009, 16, 1438–1444. [Google Scholar] [CrossRef] [PubMed]
- Keshari, R.S.; Jyoti, A.; Kumar, S.; Dubey, M.; Verma, A.; Srinag, B.S.; Krishnamurthy, H.; Barthwal, M.K.; Dikshit, M. Neutrophil extracellular traps contain mitochondrial as well as nuclear DNA and exhibit inflammatory potential. Cytom. A 2012, 81, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Rosazza, T.; Warner, J.; Sollberger, G. NET formation—Mechanisms and how they relate to other cell death pathways. FEBS J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Van Avondt, K.; Hartl, D. Mechanisms and disease relevance of neutrophil extracellular trap formation. Eur. J. Clin. Investig. 2018, 48, e12919. [Google Scholar] [CrossRef]
- Thålin, C.; Lundström, S.; Seignez, C.; Daleskog, M.; Lundström, A.; Henriksson, P.; Helleday, T.; Phillipson, M.; Wallén, H.; Demers, M. Citrullinated histone H3 as a novel prognostic blood marker in patients with advanced cancer. PLoS ONE 2018, 13, e0191231. [Google Scholar] [CrossRef]
- Borissoff, J.I.; Joosen, I.A.; Versteylen, M.O.; Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Gallant, M.; Martinod, K.; Cate, H.t.; Hofstra, L.; et al. Elevated Levels of Circulating DNA and Chromatin Are Independently Associated With Severe Coronary Atherosclerosis and a Prothrombotic State. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2032–2040. [Google Scholar] [CrossRef]
- Lood, C.; Blanco, L.P.; Purmalek, M.M.; Carmona-Rivera, C.; De Ravin, S.S.; Smith, C.K.; Malech, H.L.; Ledbetter, J.A.; Elkon, K.B.; Kaplan, M.J. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 2016, 22, 146–153. [Google Scholar] [CrossRef]
- Roth, G.A.; Johnson, C.; Abajobir, A.; Abd-Allah, F.; Abera, S.F.; Abyu, G.; Ahmed, M.; Aksut, B.; Alam, T.; Alam, K.; et al. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J. Am. Coll. Cardiol. 2017, 70, 1–25. [Google Scholar] [CrossRef]
- Mortality, G.B.D.; Causes of Death, C. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1459–1544. [Google Scholar] [CrossRef]
- Rafieian-Kopaei, M.; Setorki, M.; Doudi, M.; Baradaran, A.; Nasri, H. Atherosclerosis: Process, indicators, risk factors and new hopes. Int. J. Prev. Med. 2014, 5, 927–946. [Google Scholar]
- Friedman, G.D.; Klatsky, A.L.; Siegelaub, A.B. The leukocyte count as a predictor of myocardial infarction. N. Engl. J. Med. 1974, 290, 1275–1278. [Google Scholar] [CrossRef] [PubMed]
- Giugliano, G.; Brevetti, G.; Lanero, S.; Schiano, V.; Laurenzano, E.; Chiariello, M. Leukocyte count in peripheral arterial disease: A simple, reliable, inexpensive approach to cardiovascular risk prediction. Atherosclerosis 2010, 210, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Drechsler, M.; Megens, R.T.A.; Zandvoort, M.V.; Weber, C.; Soehnlein, O. Hyperlipidemia-Triggered Neutrophilia Promotes Early Atherosclerosis. Circulation 2010, 122, 1837–1845. [Google Scholar] [CrossRef] [PubMed]
- Megens, R.T.A.; Vijayan, S.; Lievens, D.; Döring, Y.; van Zandvoort, M.A.M.J.; Grommes, J.; Weber, C.; Soehnlein, O. Presence of luminal neutrophil extracellular traps in atherosclerosis. Thromb. Haemost. 2012, 107, 597–598. [Google Scholar] [CrossRef] [PubMed]
- Quillard, T.; Araújo, H.A.; Franck, G.; Shvartz, E.; Sukhova, G.; Libby, P. TLR2 and neutrophils potentiate endothelial stress, apoptosis and detachment: Implications for superficial erosion. Eur. Heart J. 2015, 36, 1394–1404. [Google Scholar] [CrossRef]
- Franck, G.; Mawson, T.L.; Folco, E.J.; Molinaro, R.; Ruvkun, V.; Engelbertsen, D.; Liu, X.; Tesmenitsky, Y.; Shvartz, E.; Sukhova, G.K.; et al. Roles of PAD4 and NETosis in Experimental Atherosclerosis and Arterial Injury. Circ. Res. 2018, 123, 33–42. [Google Scholar] [CrossRef]
- Knight, J.S.; Luo, W.; O’Dell, A.A.; Yalavarthi, S.; Zhao, W.; Subramanian, V.; Guo, C.; Grenn, R.C.; Thompson, P.R.; Eitzman, D.T.; et al. Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ. Res. 2014, 114, 947–956. [Google Scholar] [CrossRef]
- Liu, Y.; Carmona-Rivera, C.; Moore, E.; Seto, N.L.; Knight, J.S.; Pryor, M.; Yang, Z.H.; Hemmers, S.; Remaley, A.T.; Mowen, K.A.; et al. Myeloid-Specific Deletion of Peptidylarginine Deiminase 4 Mitigates Atherosclerosis. Front. Immunol. 2018, 9, 1680. [Google Scholar] [CrossRef]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [Google Scholar] [CrossRef]
- Kimball, A.S.; Obi, A.T.; Diaz, J.A.; Henke, P.K. The Emerging Role of NETs in Venous Thrombosis and Immunothrombosis. Front. Immunol. 2016, 7, 236. [Google Scholar] [CrossRef]
- Brill, A.; Fuchs, T.A.; Chauhan, A.K.; Yang, J.J.; De Meyer, S.F.; Kollnberger, M.; Wakefield, T.W.; Lammle, B.; Massberg, S.; Wagner, D.D. von Willebrand factor-mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood 2011, 117, 1400–1407. [Google Scholar] [CrossRef] [PubMed]
- Etulain, J.; Martinod, K.; Wong, S.L.; Cifuni, S.M.; Schattner, M.; Wagner, D.D. P-selectin promotes neutrophil extracellular trap formation in mice. Blood 2015, 126, 242–246. [Google Scholar] [CrossRef] [PubMed]
- de Bont, C.M.; Boelens, W.C.; Pruijn, G.J.M. NETosis, complement, and coagulation: A triangular relationship. Cell. Mol. Immunol. 2019, 16, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Rossaint, J.; Kuhne, K.; Skupski, J.; Van Aken, H.; Looney, M.R.; Hidalgo, A.; Zarbock, A. Directed transport of neutrophil-derived extracellular vesicles enables platelet-mediated innate immune response. Nat. Commun. 2016, 7, 13464. [Google Scholar] [CrossRef] [PubMed]
- von Bruhl, M.L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Kollnberger, M.; et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012, 209, 819–835. [Google Scholar] [CrossRef]
- Massberg, S.; Grahl, L.; von Bruehl, M.L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef]
- Sambrano, G.R.; Huang, W.; Faruqi, T.; Mahrus, S.; Craik, C.; Coughlin, S.R. Cathepsin G activates protease-activated receptor-4 in human platelets. J. Biol. Chem. 2000, 275, 6819–6823. [Google Scholar] [CrossRef]
- Griffin, J.H.; Fernandez, J.A.; Gale, A.J.; Mosnier, L.O. Activated protein C. J. Thromb. Haemost. 2007, 5 (Suppl. 1), 73–80. [Google Scholar] [CrossRef]
- Griffin, J.H.; Zlokovic, B.V.; Mosnier, L.O. Activated protein C: Biased for translation. Blood 2015, 125, 2898–2907. [Google Scholar] [CrossRef]
- Healy, L.D.; Puy, C.; Fernandez, J.A.; Mitrugno, A.; Keshari, R.S.; Taku, N.A.; Chu, T.T.; Xu, X.; Gruber, A.; Lupu, F.; et al. Activated protein C inhibits neutrophil extracellular trap formation in vitro and activation in vivo. J. Biol. Chem. 2017, 292, 8616–8629. [Google Scholar] [CrossRef]
- Martinod, K.; Witsch, T.; Farley, K.; Gallant, M.; Remold-O’Donnell, E.; Wagner, D.D. Neutrophil elastase-deficient mice form neutrophil extracellular traps in an experimental model of deep vein thrombosis. J. Thromb. Haemost. 2016, 14, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Eliason, J.L.; Hannawa, K.K.; Ailawadi, G.; Sinha, I.; Ford, J.W.; Deogracias, M.P.; Roelofs, K.J.; Woodrum, D.T.; Ennis, T.L.; Henke, P.K.; et al. Neutrophil depletion inhibits experimental abdominal aortic aneurysm formation. Circulation 2005, 112, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhou, H.F.; Akk, A.; Hu, Y.; Springer, L.E.; Ennis, T.L.; Pham, C.T.N. Neutrophil Proteases Promote Experimental Abdominal Aortic Aneurysm via Extracellular Trap Release and Plasmacytoid Dendritic Cell Activation. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1660–1669. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.B.; Bartoli, M.A.; Ennis, T.L.; Mao, D.; Simmons, P.M.; Thompson, R.W.; Pham, C.T. Critical role of dipeptidyl peptidase I in neutrophil recruitment during the development of experimental abdominal aortic aneurysms. Proc. Natl. Acad. Sci. USA 2007, 104, 2855–2860. [Google Scholar] [CrossRef]
- Granger, V.; Peyneau, M.; Chollet-Martin, S.; de Chaisemartin, L. Neutrophil Extracellular Traps in Autoimmunity and Allergy: Immune Complexes at Work. Front. Immunol. 2019, 10, 2524. [Google Scholar] [CrossRef] [PubMed]
- Barnado, A.; Crofford, L.J.; Oates, J.C. At the Bedside: Neutrophil extracellular traps (NETs) as targets for biomarkers and therapies in autoimmune diseases. J. Leukoc. Biol. 2016, 99, 265–278. [Google Scholar] [CrossRef]
- Hakkim, A.; Furnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818. [Google Scholar] [CrossRef]
- Shao, S.; Fang, H.; Dang, E.; Xue, K.; Zhang, J.; Li, B.; Qiao, H.; Cao, T.; Zhuang, Y.; Shen, S.; et al. Neutrophil Extracellular Traps Promote Inflammatory Responses in Psoriasis via Activating Epidermal TLR4/IL-36R Crosstalk. Front. Immunol. 2019, 10, 746. [Google Scholar] [CrossRef]
- Li, T.; Wang, C.; Liu, Y.; Li, B.; Zhang, W.; Wang, L.; Yu, M.; Zhao, X.; Du, J.; Zhang, J.; et al. Neutrophil Extracellular Traps Induce Intestinal Damage and Thrombotic Tendency in Inflammatory Bowel Disease. J. Crohns Colitis 2020, 14, 240–253. [Google Scholar] [CrossRef]
- Lee, K.H.; Kronbichler, A.; Park, D.D.-Y.; Park, Y.; Moon, H.; Kim, H.; Choi, J.H.; Choi, Y.; Shim, S.; Lyu, I.S.; et al. Neutrophil extracellular traps (NETs) in autoimmune diseases: A comprehensive review. Autoimmun. Rev. 2017, 16, 1160–1173. [Google Scholar] [CrossRef]
- Wang, H.; Li, T.; Chen, S.; Gu, Y.; Ye, S. Neutrophil Extracellular Trap Mitochondrial DNA and Its Autoantibody in Systemic Lupus Erythematosus and a Proof-of-Concept Trial of Metformin. Arthritis Rheumatol. 2015, 67, 3190–3200. [Google Scholar] [CrossRef] [PubMed]
- Klopf, J.; Scheuba, A.; Brostjan, C.; Neumayer, C.; Eilenberg, W. Strategies so far and future Prospects for Reducing Growth Rates in abdominal Aortic Aneurysms A selective Literature Review and Discussion of the current Vienna MetAAA Trial. Gefasschirurgie 2020, 25, 446–449. [Google Scholar] [CrossRef]
- Glennon-Alty, L.; Hackett, A.P.; Chapman, E.A.; Wright, H.L. Neutrophils and redox stress in the pathogenesis of autoimmune disease. Free. Radic. Biol. Med. 2018, 125, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Apel, F.; Zychlinsky, A.; Kenny, E.F. The role of neutrophil extracellular traps in rheumatic diseases. Nat. Rev. Rheumatol. 2018, 14, 467–475. [Google Scholar] [CrossRef]
- Schauer, C.; Janko, C.; Munoz, L.E.; Zhao, Y.; Kienhofer, D.; Frey, B.; Lell, M.; Manger, B.; Rech, J.; Naschberger, E.; et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat. Med. 2014, 20, 511–517. [Google Scholar] [CrossRef]
- Garcia-Romo, G.S.; Caielli, S.; Vega, B.; Connolly, J.; Allantaz, F.; Xu, Z.; Punaro, M.; Baisch, J.; Guiducci, C.; Coffman, R.L.; et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 2011, 3, 73ra20. [Google Scholar] [CrossRef]
- Jennette, J.C.; Falk, R.J. Small-vessel vasculitis. N. Engl. J. Med. 1997, 337, 1512–1523. [Google Scholar] [CrossRef]
- Ralli, M.; Campo, F.; Angeletti, D.; Minni, A.; Artico, M.; Greco, A.; Polimeni, A.; de Vincentiis, M. Pathophysiology and therapy of systemic vasculitides. Excli J. 2020, 19, 817–854. [Google Scholar] [CrossRef]
- Nakazawa, D.; Tomaru, U.; Yamamoto, C.; Jodo, S.; Ishizu, A. Abundant neutrophil extracellular traps in thrombus of patient with microscopic polyangiitis. Front. Immunol. 2012, 3, 333. [Google Scholar] [CrossRef]
- Nakazawa, D.; Shida, H.; Tomaru, U.; Yoshida, M.; Nishio, S.; Atsumi, T.; Ishizu, A. Enhanced formation and disordered regulation of NETs in myeloperoxidase-ANCA-associated microscopic polyangiitis. J. Am. Soc. Nephrol. 2014, 25, 990–997. [Google Scholar] [CrossRef]
- Katsarou, A.; Gudbjornsdottir, S.; Rawshani, A.; Dabelea, D.; Bonifacio, E.; Anderson, B.J.; Jacobsen, L.M.; Schatz, D.A.; Lernmark, A. Type 1 diabetes mellitus. Nat. Rev. Dis. Primers 2017, 3, 17016. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Rizo, V.; Martinez-Guzman, M.A.; Iniguez-Gutierrez, L.; Garcia-Orozco, A.; Alvarado-Navarro, A.; Fafutis-Morris, M. Neutrophil Extracellular Traps and Its Implications in Inflammation: An Overview. Front. Immunol. 2017, 8, 81. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef]
- Berezin, A. Neutrophil extracellular traps: The core player in vascular complications of diabetes mellitus. Diabetes Metab. Syndr. 2019, 13, 3017–3023. [Google Scholar] [CrossRef]
- Gupta, S.; Kaplan, M.J. The role of neutrophils and NETosis in autoimmune and renal diseases. Nat. Rev. Nephrol. 2016, 12, 402–413. [Google Scholar] [CrossRef]
- Huang, J.; Xiao, Y.; Xu, A.; Zhou, Z. Neutrophils in type 1 diabetes. J. Diabetes Investig. 2016, 7, 652–663. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N. Environmental triggers for inflammatory bowel disease. Curr. Gastroenterol. Rep. 2013, 15, 302. [Google Scholar] [CrossRef]
- Baumgart, D.C.; Carding, S.R. Inflammatory bowel disease: Cause and immunobiology. Lancet 2007, 369, 1627–1640. [Google Scholar] [CrossRef]
- Nemati, S.; Teimourian, S. An Overview of Inflammatory Bowel Disease: General Consideration and Genetic Screening Approach in Diagnosis of Early Onset Subsets. Middle East J. Dig. Dis. 2017, 9, 69–80. [Google Scholar] [CrossRef][Green Version]
- Warnberg, J.; Marcos, A. Low-grade inflammation and the metabolic syndrome in children and adolescents. Curr. Opin. Lipidol. 2008, 19, 11–15. [Google Scholar] [CrossRef]
- Castro, A.M.; Macedo-de la Concha, L.E.; Pantoja-Meléndez, C.A. Low-grade inflammation and its relation to obesity and chronic degenerative diseases. Rev. Médica Hosp. Gen. México 2017, 80, 101–105. [Google Scholar] [CrossRef]
- Bonaventura, A.; Vecchie, A.; Abbate, A.; Montecucco, F. Neutrophil Extracellular Traps and Cardiovascular Diseases: An Update. Cells 2020, 9, 231. [Google Scholar] [CrossRef] [PubMed]
- D’Abbondanza, M.; Martorelli, E.E.; Ricci, M.A.; De Vuono, S.; Migliola, E.N.; Godino, C.; Corradetti, S.; Siepi, D.; Paganelli, M.T.; Maugeri, N.; et al. Increased plasmatic NETs by-products in patients in severe obesity. Sci. Rep. 2019, 9, 14678. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, Q.; Venugopal, J.; Wang, J.; Kleiman, K.; Guo, C.; Eitzman, D.T. Obesity-induced Endothelial Dysfunction is Prevented by Neutrophil Extracellular Trap Inhibition. Sci. Rep. 2018, 8, 4881. [Google Scholar] [CrossRef] [PubMed]
- Moorthy, A.N.; Tan, K.B.; Wang, S.; Narasaraju, T.; Chow, V.T. Effect of High-Fat Diet on the Formation of Pulmonary Neutrophil Extracellular Traps during Influenza Pneumonia in BALB/c Mice. Front. Immunol. 2016, 7, 289. [Google Scholar] [CrossRef]
- Olokoba, A.B.; Obateru, O.A.; Olokoba, L.B. Type 2 diabetes mellitus: A review of current trends. Oman. Med. J. 2012, 27, 269–273. [Google Scholar] [CrossRef]
- Carestia, A.; Frechtel, G.; Cerrone, G.; Linari, M.A.; Gonzalez, C.D.; Casais, P.; Schattner, M. NETosis before and after Hyperglycemic Control in Type 2 Diabetes Mellitus Patients. PLoS ONE 2016, 11, e0168647. [Google Scholar] [CrossRef]
- Karima, M.; Kantarci, A.; Ohira, T.; Hasturk, H.; Jones, V.L.; Nam, B.H.; Malabanan, A.; Trackman, P.C.; Badwey, J.A.; Van Dyke, T.E. Enhanced superoxide release and elevated protein kinase C activity in neutrophils from diabetic patients: Association with periodontitis. J. Leukoc. Biol. 2005, 78, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar] [CrossRef]
- Joshi, M.B.; Lad, A.; Bharath Prasad, A.S.; Balakrishnan, A.; Ramachandra, L.; Satyamoorthy, K. High glucose modulates IL-6 mediated immune homeostasis through impeding neutrophil extracellular trap formation. FEBS Lett. 2013, 587, 2241–2246. [Google Scholar] [CrossRef]
- Arpinati, L.; Shaul, M.E.; Kaisar-Iluz, N.; Mali, S.; Mahroum, S.; Fridlender, Z.G. NETosis in cancer: A critical analysis of the impact of cancer on neutrophil extracellular trap (NET) release in lung cancer patients vs. mice. Cancer Immunol. Immunother. 2020, 69, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Demers, M.; Krause, D.S.; Schatzberg, D.; Martinod, K.; Voorhees, J.R.; Fuchs, T.A.; Scadden, D.T.; Wagner, D.D. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc. Natl. Acad. Sci. USA 2012, 109, 13076–13081. [Google Scholar] [CrossRef] [PubMed]
- Mauracher, L.M.; Posch, F.; Martinod, K.; Grilz, E.; Daullary, T.; Hell, L.; Brostjan, C.; Zielinski, C.; Ay, C.; Wagner, D.D.; et al. Citrullinated histone H3, a biomarker of neutrophil extracellular trap formation, predicts the risk of venous thromboembolism in cancer patients. J. Thromb. Haemost. 2018, 16, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Guglietta, S.; Chiavelli, A.; Zagato, E.; Krieg, C.; Gandini, S.; Ravenda, P.S.; Bazolli, B.; Lu, B.; Penna, G.; Rescigno, M. Coagulation induced by C3aR-dependent NETosis drives protumorigenic neutrophils during small intestinal tumorigenesis. Nat. Commun. 2016, 7, 11037. [Google Scholar] [CrossRef] [PubMed]
- Cools-Lartigue, J.; Spicer, J.; McDonald, B.; Gowing, S.; Chow, S.; Giannias, B.; Bourdeau, F.; Kubes, P.; Ferri, L. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J. Clin. Investig. 2013, 123, 3446–3458. [Google Scholar] [CrossRef] [PubMed]
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Küttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361, eaao4227. [Google Scholar] [CrossRef]
- Wculek, S.K.; Malanchi, I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 2015, 528, 413–417. [Google Scholar] [CrossRef]
- Coffelt, S.B.; Kersten, K.; Doornebal, C.W.; Weiden, J.; Vrijland, K.; Hau, C.S.; Verstegen, N.J.M.; Ciampricotti, M.; Hawinkels, L.; Jonkers, J.; et al. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature 2015, 522, 345–348. [Google Scholar] [CrossRef]
- Wen, F.; Shen, A.; Choi, A.; Gerner, E.W.; Shi, J. Extracellular DNA in pancreatic cancer promotes cell invasion and metastasis. Cancer Res. 2013, 73, 4256–4266. [Google Scholar] [CrossRef]
- Yang, L.; Liu, Q.; Zhang, X.; Liu, X.; Zhou, B.; Chen, J.; Huang, D.; Li, J.; Li, H.; Chen, F.; et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature 2020, 583, 133–138. [Google Scholar] [CrossRef]
- Masucci, M.T.; Minopoli, M.; Del Vecchio, S.; Carriero, M.V. The Emerging Role of Neutrophil Extracellular Traps (NETs) in Tumor Progression and Metastasis. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Garley, M.; Jabłońska, E.; Dąbrowska, D. NETs in cancer. Tumor Biol. 2016, 37, 14355–14361. [Google Scholar] [CrossRef] [PubMed]
- Cedervall, J.; Olsson, A.K. Immunity Gone Astray—NETs in Cancer. Trends Cancer 2016, 2, 633–634. [Google Scholar] [CrossRef] [PubMed]
- New hope for sepsis. Lancet 2012, 379, 1462. [CrossRef]
- Cohen, J.; Vincent, J.L.; Adhikari, N.K.; Machado, F.R.; Angus, D.C.; Calandra, T.; Jaton, K.; Giulieri, S.; Delaloye, J.; Opal, S.; et al. Sepsis: A roadmap for future research. Lancet Infect. Dis. 2015, 15, 581–614. [Google Scholar] [CrossRef]
- Camicia, G.; Pozner, R.; de Larranaga, G. Neutrophil extracellular traps in sepsis. Shock 2014, 42, 286–294. [Google Scholar] [CrossRef]
- Czaikoski, P.G.; Mota, J.M.; Nascimento, D.C.; Sonego, F.; Castanheira, F.V.; Melo, P.H.; Scortegagna, G.T.; Silva, R.L.; Barroso-Sousa, R.; Souto, F.O.; et al. Neutrophil Extracellular Traps Induce Organ Damage during Experimental and Clinical Sepsis. PLoS ONE 2016, 11, e0148142. [Google Scholar] [CrossRef]
- Gollomp, K.; Sarkar, A.; Harikumar, S.; Seeholzer, S.H.; Arepally, G.M.; Hudock, K.; Rauova, L.; Kowalska, M.A.; Poncz, M. Fc-modified HIT-like monoclonal antibody as a novel treatment for sepsis. Blood 2020, 135, 743–754. [Google Scholar] [CrossRef]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Dassler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Pedersen, S.F.; Ho, Y.C. SARS-CoV-2: A storm is raging. J. Clin. Investig. 2020, 130, 2202–2205. [Google Scholar] [CrossRef]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar] [CrossRef] [PubMed]
- Leppkes, M.; Knopf, J.; Naschberger, E.; Lindemann, A.; Singh, J.; Herrmann, I.; Sturzl, M.; Staats, L.; Mahajan, A.; Schauer, C.; et al. Vascular occlusion by neutrophil extracellular traps in COVID-19. EBioMedicine 2020, 58, 102925. [Google Scholar] [CrossRef] [PubMed]
- Tomar, B.; Anders, H.J.; Desai, J.; Mulay, S.R. Neutrophils and Neutrophil Extracellular Traps Drive Necroinflammation in COVID-19. Cells 2020, 9, 1383. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, T.; Sperandio, M.; Mocsai, A. Neutrophils as emerging therapeutic targets. Nat. Rev. Drug Discov. 2020, 19, 253–275. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Category | Disease | Identified Triggers |
|---|---|---|
| Cardiovascular | Atherosclerosis | IL-8 [20], IL-1β, crystalized cholesterol [21], oxLDL [22] |
| Thrombosis | HMGB-1 [23], pathophysiologic hemodynamic forces [24] | |
| Abdominal aortic aneurysm | IL-1β [25], Porphyromonas gingivalis [26] | |
| Autoimmune & Autoinflammatory | Systemic lupus erythematosus & lupus nephritis | IFN-α, ANCAs, RNPs [27], acetylated histones & apoptotic microparticles [28] |
| Psoriasis | IL-8, IL-17, IL-23, TNF-α, autoantigens (keratin 17, LL37) [29] | |
| Rheumatoid arthritis & arthritis urica | ACPAs, ANCAs, RNPs, IL-17A, TNF-α [30], hyperuricemia and monosodium urate crystals [19] | |
| Small-vessel vasculitis | ANCAs [31] | |
| Diabetes mellitus type I | Hyperglycemia [32], β-cell death & autoantibodies [33] | |
| Crohn’s disease & ulcerative colitis | enhanced ROS production [34,35] | |
| Metabolic | Obesity & diabetes mellitus type II | Hyperglycemia [32], IL-6, IL-8, TNF-α, homocysteine [36] |
| Malignant neoplasia | metastatic breast cancer cells, metastatic colorectal cancer | G-CSF [37], IL-8 [38], intratumoral hypoxia [39] |
| Infectious | Sepsis | HMGB-1 [40], eCIRP [41], oxLDL [42] |
| COVID-19 | HMGB-1 [43], hypoxia [44] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klopf, J.; Brostjan, C.; Eilenberg, W.; Neumayer, C. Neutrophil Extracellular Traps and Their Implications in Cardiovascular and Inflammatory Disease. Int. J. Mol. Sci. 2021, 22, 559. https://doi.org/10.3390/ijms22020559
Klopf J, Brostjan C, Eilenberg W, Neumayer C. Neutrophil Extracellular Traps and Their Implications in Cardiovascular and Inflammatory Disease. International Journal of Molecular Sciences. 2021; 22(2):559. https://doi.org/10.3390/ijms22020559
Chicago/Turabian StyleKlopf, Johannes, Christine Brostjan, Wolf Eilenberg, and Christoph Neumayer. 2021. "Neutrophil Extracellular Traps and Their Implications in Cardiovascular and Inflammatory Disease" International Journal of Molecular Sciences 22, no. 2: 559. https://doi.org/10.3390/ijms22020559
APA StyleKlopf, J., Brostjan, C., Eilenberg, W., & Neumayer, C. (2021). Neutrophil Extracellular Traps and Their Implications in Cardiovascular and Inflammatory Disease. International Journal of Molecular Sciences, 22(2), 559. https://doi.org/10.3390/ijms22020559