Abstract
Since the discovery of non-neuronal acetylcholine in the heart, this specific system has drawn scientific interest from many research fields, including cardiology, immunology, and pharmacology. This system, acquired by cardiomyocytes independent of the parasympathetic nervous system of the autonomic nervous system, helps us to understand unsolved issues in cardiac physiology and to realize that the system may be more pivotal for cardiac homeostasis than expected. However, it has been shown that the effects of this system may not be restricted to the heart, but rather extended to cover extra-cardiac organs. To this end, this system intriguingly influences brain function, specifically potentiating blood brain barrier function. Although the results reported appear to be unusual, this novel characteristic can provide us with another research interest and therapeutic application mode for central nervous system diseases. In this review, we discuss our recent studies and raise the possibility of application of this system as an adjunctive therapeutic modality.
1. What Is the Non-Neuronal ACh System in the Heart?
Acetylcholine (ACh) is one of neurotransmitters in the autonomic nervous system (ANS), which is composed of the sympathetic and parasympathetic nervous systems (SNS and PNS, respectively). In the SNS, the primary neurons release ACh to activate the secondary neurons possessing nicotinic receptors for ACh and transduce the signals to them. The activated secondary neurons then release noradrenaline or norepinephrine from their terminal ends that binds to a specific receptor on the effectors to execute their specific physiological actions. In contrast, the PNS releases ACh from the terminal ends. For instance, the heart is innervated by the ANS, and the SNS exerts positive chronotropic and inotropic actions, including upregulation of its contraction, heart rate, and conduction velocity. In contrast, PNS decreases heart rate and conduction velocity. Therefore, ACh in the heart plays a role as a counterpart in these functions.
The distribution mode of nerve ends of the PNS in the heart, that is, the vagus nerve (VN), is completely distinct from that of the SNS. Compared to the SNS nerve ends, which are distributed to the entire cardiac ventricles, the PNS nerve ends are predominantly located at the sinus node and atrioventricular node, but are very sparsely distributed in the cardiac ventricles [1,2,3,4,5]. This finding distinctively provides us a cue to consider a novel system in the heart in terms of ACh synthesis independent of the PNS; that is, a non-neuronal ACh (NNA) synthesis in the heart or non-neuronal cardiac cholinergic system (NNCCS). In other words, ACh is synthesized by cardiomyocytes [6]. According to previous studies, it is broadly confirmed that cardiomyocytes can synthesize ACh with the machineries equipped with cardiomyocytes, including choline transporter (CHT1), choline acetyltransferase (ChAT), a crucial enzyme for ACh synthesis, and vesicular ACh transporter (VAChT), a storage vesicle including ACh responsible for exocytosis, whereas it is rapidly degraded by acetylcholinesterase [7,8,9,10,11]. Furthermore, even rat primary cultured cardiomyocytes in vitro, which were cultured for one week, synthesized ACh with sufficiently detectable levels by High Performance Liquid Chromatography (HPLC) [6]. Based on these pioneering studies independently conducted by other researchers, the concept of the NNA system in the heart or NNCCS has been established [6,7,8,9,10,11].
Afterwards, significant functional evidence regarding this system has been further accumulated to indicate that the system is not an accessory but indispensable for homeostasis of cardiac functions using ChAT gene knockout or knockdown method in vitro. For example, the system decreases cellular energy metabolism by reducing oxygen consumption not only in contracting cardiomyocytes but also in non-contracting cells [6,12]. Second, as implicated with the first issue, this system preferentially uses glucose as an energy substrate through the upregulation of a glucose transporter [13,14,15]. Third, it sustains cell–cell interaction and maintains protein expression of connexin 43 and β-catenin up to a necessary level [12]. Fourth, it sustains cellular resiliency against serum starvation, energy starvation, hypoxic insults, and norepinephrine exposure [9,10,11,12,13,14,16]. Lastly, it modulates immune responses in the injured heart by suppressing cytokine expression [17]. This cardiac finding was partly shared with further findings about other cells, including microglia [18], endothelial cells [19], macrophages [20] and immune cells [21,22,23], suggesting that NNA generally possesses anti-inflammatory effects. Taken together with all these experimental results, it should be concluded that the NNA in the heart or NNCCS represents a basic cardiac machinery to play a self-defensive role against overshooting stress.
2. A Model Mouse Representing the Activated Non-Neuronal ACh System in the Heart
2.1. Cardiac Phenotypes of the Transgenic Mouse
To further investigate the physiological role of this system in the heart in vivo, we developed transgenic mice with the heart-specific overexpressing ChAT gene, a critical ACh synthesis enzyme gene, using the α-myosin heavy chain promoter, and labeled them as ChAT tg mice [14] (Figure 1). Another study modality was also conducted with deletions of the VAChT, a vesicle responsible for ACh storage and exocytosis, and ChAT genes, which clearly demonstrated completely opposite phenotypes to ours, leading us to the same conclusion regarding this system [16].
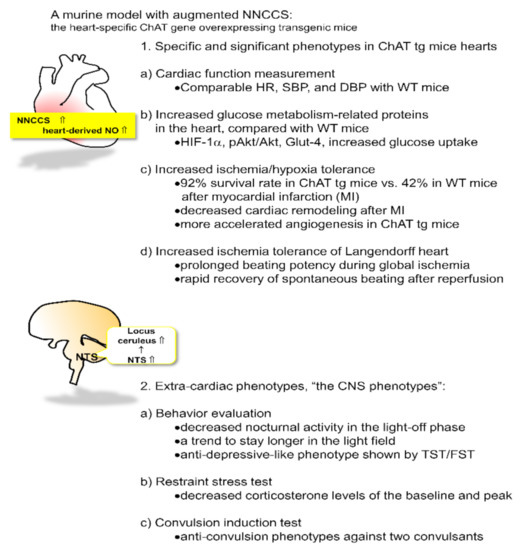
Figure 1.
A murine model of the activated non-neuronal cardiac cholinergic system (NNCCS) with characteristic phenotypes of the cardiac and non-cardiac organ, the central nervous system (CNS).
We consider that our ChAT tg mice represent a suitable model for the activation of this system because the transgene ChAT was confirmed to be expressed exclusively in the heart [14]. Although ChAT protein derived from the translated transgene was expected to be expressed both in atriums and ventricles [24,25], compared with the expression pattern of ChAT protein in wild-type (WT) mice, expression levels of the ChAT protein were increased predominantly in the ventricles. In contrast, atrial ChAT protein expression levels in ChAT tg were surprisingly not increased, as the ventricle ChAT expression did, to levels almost comparable with those in WT mice (unpublished data). ChAT tg mice showed comparable levels of blood pressure (both systolic and diastolic pressures, SBP and DBP, respectively) and heart rate (HR) with WT mice; therefore, systemic hemodynamic parameters in ChAT tg mice were not influenced by augmentation of this system [14,26].
Despite the comparable hemodynamic parameters between ChAT tg and WT mice, ChAT tg mice showed intriguing and striking cardiac phenotypes [14] that were also predicted by the in vitro phenotypes of the activated NNA system [12,13]. First, as the specific signal transduction following activated NNA synthesis in the heart, hearts of ChAT tg mice increased protein expression levels of HIF-1α, pAkt, and glut-4 as well as cardiac ventricular ACh levels, compared with those of WT mice, suggesting that even during normoxic conditions, ChAT tg mice upregulated the anti-hypoxic/anti-ischemic self-defense machinery in the heart. Therefore, ChAT tg mice with myocardial infarction (MI) survived more than WT mice in the chronic phase, with a survival rate of 92.3% (two weeks after the onset of infarction). In contrast, WT mice with MI usually died with cardiac rapture soon after MI, showing 41.7% survival rate [14].
Even after MI, the ChAT tg hearts possessed more intact myocardium but less fibrotic and necrotic changes. Furthermore, cardiac remodeling after MI was more suppressed in the heart of ChAT tg mice than in WT mice. Moreover, the hearts isolated from ChAT tg mice continued to beat for longer duration, even after perfusion-off in the Langendorff apparatus, than those from WT mice; alternatively, the former restarted to beat faster than the latter following reperfusion [14]. This observation clearly indicates that ChAT tg hearts are resilient to ischemia and efficiently sustain cardiac function even during insults.
2.2. Extra-Cardiac Phenotypes of ChAT tg Mice
As previously mentioned, ChAT tg mice were established to express the transgene restrictively in the heart. The heart-limited expression of ChAT was accurately verified by western blot analysis and immunohistochemical analysis of ChAT protein in each organ of ChAT tg mice. On the other hand, immunoreactive signals of ChAT in the ChAT tg mouse brain were completely comparable with those in WT mouse brains, and no neuronal cells with stronger signals of ChAT were detected in ChAT tg brains [26].
Despite the heart-limited transgenic mice overexpressing the ChAT gene, ChAT tg mice seemed more docile and quieter, and less aggressive than WT mice [26]. This characteristic phenotype was specifically observed when the mice were grasped. The presence of extra-cardiac phenotypes prompted us to further investigate whether ChAT tg mice possess other central phenotypes related to ChAT tg temperaments as follows [26].
(1) The total walking distance of ChAT tg mice was comparable with that of WT mice at daytime in a light-on phase; however, the nocturnal activity in the light-off phase was significantly decreased in ChAT tg mice compared with that in WT mice, specifically during the initial several hours following light-off. In general, this means that mice accelerate their activity in the light-off phase because they are nocturnal [27,28,29]. When they are transferred into a cage, they vigorously walk around to check whether the circumstance is safe or if they are under dangerous conditions. Therefore, decreasing nocturnal activity suggests that ChAT tg mice feel less anxious. Together with neither motor deficits nor muscular weakness in ChAT tg mice, these findings suggest that ChAT tg mice suffer from some influence on CNS function due to the augmented NNCCS [26].
(2) When subjected to a light and dark transition test, WT mice stayed longer in the dark field and they did not get out into a light field because they preferred dark conditions. However, ChAT tg mice tend to spend more time in the light field. These findings suggest that ChAT tg mice are less anxious. This may be compatible with decreased nocturnal activity [26].
(3) A representative test to evaluate a depressive-like phenotype includes the tail suspension test (TST) and forced swimming test (FST) [30]. In both tests, immobility time was measured during a part of the test because more prolonged immobility by giving up escaping or swimming indicates a more depressive-like phenotype in mice. When ChAT tg mice were subjected to both TST and FST, their immobility time was significantly decreased compared with that of WT mice. They continuously tried to escape from the suspension and to swim until the end of the tests. Furthermore, in elevated plus maze test, the total time spent by the ChAT tg mice in the open arms [31] was significantly longer than that of the WT mice; however, in the closed arms they stayed comparably with WT mice. Therefore, the decreased immobility time and increased time spent in the open space strongly suggest that ChAT tg mice were less under depressive-like conditions than WT mice [26].
(4) When WT mice were subjected to restraint stress with a silver metal net, they usually increased blood corticosterone concentration within 60 min because the stress activated the hypothalamus-pituitary-adrenal gland axis [32]. In contrast, when subjected to the same stress, ChAT tg mice increased up to only 50% of the peak blood corticosterone concentration in WT mice, suggesting that ChAT tg mice are resistant to stress and less stressed than WT mice [26].
(5) Finally, mice were stimulated with two convulsants, pilocarpine and pentylenetetrazole, which are well known to induce convulsions in animals [33,34]. We evaluated the susceptibility of convulsion by measuring duration, incidence, and survival rate from status epilepticus as well as by brain neuronal activity imaging using transcranial flavoprotein fluorescence imaging [35,36]. As predicted, when subjected to those convulsants, WT mice experienced convulsions with a higher incidence and longer duration, more often resulting in death due to status epilepticus. In contrast, ChAT tg mice less frequently experienced convulsions with a lower incidence and shorter duration. Significantly, more ChAT tg mice survived status epileptics than WT mice because neuronal activity of the ChAT tg mice brain evaluated by brain imaging was also significantly attenuated even in treatment with convulsants, compared with the WT mice brains [26].
3. The Mechanisms by Which Activation of the Non-Neuronal Cardiac Cholinergic System Influences the Brain Function
As mentioned above, ChAT tg mice are a representative useful model of activated NNCCS alone [14]; however, how does this heart-limited activated system influence the brain or CNS? The heart and CNS are connected via the VN, part of the PNS. As an effector organ, the VN is known to innervate the abdominal and intrathoracic organs in order to sense signals from other effector organs and execute orders from the CNS. The VN is composed of afferent and efferent fibers, the ratio of which is about 80% and 20% of the VN, respectively [37]. Consequently, the VN plays a role in the heart, mainly as an afferent fiber, in the transduction of various signals from the heart to the CNS. Based on these, we speculated that ChAT tg mice hearts transduce more signals from the heart to the CNS via the VN.
(1) To assess this, the solitary tract nucleus of the brain stem was examined by immunohistochemistry using an anti-c-Fos antibody. The VN from the peripheral organs in the whole body terminates at the nucleus of the solitary tract (NTS), which is known as the center of the PNS and located at the dorsal part of the brain stem [38]. Different from directly measuring neuronal activity of the NTS, immunohistochemical analysis of c-Fos in the NTS is another tool to evaluate neuronal activation, with increased c-Fos immunoreactivity signals indicating activation of these cells [39]. With this c-Fos analysis, ChAT tg brain stem showed that the number of c-Fos positive neuronal cells around the NTS was significantly increased in ChAT tg mice compared with that in WT mice, indicating that the ChAT tg mice brain stem received more afferent signals of the VN, probably due to the activated NNCCS. In other words, the VN in ChAT tg mice may be activated to transduce afferent fibers triggered by the heart with activated NNCCS [26].
(2) To directly evaluate VN neuronal activity, the left ChAT tg mice VN was dissected and fitted with silver bipolar electrodes under anesthetic conditions. The frequency of neuronal firing signals was significantly higher in ChAT tg mice than in WT mice. This result indicated that ChAT tg mice VN hyperfunctioned with its elevated frequency, although it was not possible to conclusively state that the increased VN activity was derived mainly from afferent or efferent fibers [26].
(3) To examine whether the VN of ChAT tg mice was critical for representing the specific central phenotypes, ChAT tg mice were subjected to lateral (left) vagotomy because bilateral vagotomy usually caused death. Surprisingly, lateral vagotomy almost reversed the specific CNS phenotypes of ChAT tg mice; vagotomy reversed the anti-stress, anti-depressive-like, and anti-convulsion phenotypes of ChAT tg mice. These results clearly indicate that ChAT tg mouse CNS phenotypes are influenced by the afferent VN [26]. Taken together with the results thus far, the ChAT tg VN plays a crucial role in transducing the heart-derived signals to the CNS to induce significantly specific CNS phenotypes, that is, an anti-stress phenotype (Figure 2).

Figure 2.
The speculated mechanisms involving the NNCCS which influences the CNS function.
VN stimulation (VNS), a modality for influencing the VN function, has been known first as a method to treat drug-refractory uncontrolled epilepsy. This is accepted by the FDA as an adjunctive therapeutic tool for these patients [40,41]. Thereafter, VNS has been accepted to be used for patients with drug-refractory depression [42]. Moreover, it has been also applied to patients with chronic heart failure with sometimes beneficial effects, although the conclusion regarding VNS outcomes on chronic heart failure is contentious depending on clinical studies [43,44]. Then, we checked whether VNS-treated WT mice showed the same beneficial phenotypes as those in ChAT tg mice, that is, anti-stress, anti-depressive-like, and anti-convulsion phenotypes [45,46,47,48,49,50]. Finally, we confirmed that the VNS phenotypes of WT mice are all included in ChAT tg mice phenotypes. Therefore, these results prompted us to consider that ChAT tg mice may be subjected to so-called self VNS; however, the afferent fibers of ChAT tg mice should be predominantly activated in this specific case.
Which factor should be involved in triggering the activation of the VN afferent fibers in ChAT tg mice? ChAT tg mice hearts synthesized much more ACh by cardiomyocytes. Secreted ACh should bind to muscarinic receptors on cardiomyocytes, activating signal transduction, one of which is nitric oxide (NO) in response to ACh [51]. As also reported in our previous study, cardiomyocytes released NO soon after ACh treatment, which was blocked by atropine, a muscarinic receptor antagonist [52]. ChAT tg mice hearts produced significantly more NO than WT hearts [26]. Furthermore, NO has been reported to play a role as a neurotransmitter [53,54], however, via a non-receptor mediated fashion because NO is a gas-like substance.
To further investigate whether NO is critical for influencing VN activity, ChAT tg mice were treated with L-NAME, a NO synthase (NOS) inhibitor. As predicted, the NOS inhibitor significantly decreased VN activity frequency rapidly, indicating that intact NOS activity and production of adequate NO are indispensable for sustaining the VN activity. Our recent study supported the finding that NO derived from n (neuronal) NOS or NOS1 in the heart is critical for cardiac function and VN activity [55]. Based on these results, ChAT tg mouse heart-derived NO can trigger VN activation.
To further consolidate our speculation that NO derived from ACh synthesized by the activated system would be a trigger for VN activation, ChAT tg mice were treated with L-NAME and the CNS phenotypes were assessed. Intriguingly, the beneficial phenotypes were completely cancelled and reversed to those of WT mice. L-NAME completely suppressed such phenotypes as anti-stress, anti-depressive-like, and anti-convulsion phenotypes to levels comparable to those in WT mice [26]. These results clearly demonstrate that at least NO is one of the stimulators of VN nerve endings in the heart, causing activation of the VN afferent fibers. Furthermore, our recent study using heart-specific ChAT knockdown mice, which possessed a depressed NO content in the heart and downregulated VN activity, were susceptible to depressive-like phenotypes and more subjected to stress load [55].
4. Another Evidence of Potentiating the Non-Neuronal Cardiac Cholinergic System Involving Consolidation of Blood Brain Barrier
Thus far, we know that ChAT tg mice possess specific CNS phenotypes that are influenced by increased cardiac NO production and VN afferent activity. However, other than the anti-inflammatory effects of the cholinergic system, that are often reported and represented partly by the representative effects of VNS [56], it remains to be elucidated the mechanisms by which the CNS phenotypes of ChAT tg are influenced. We speculate that blood brain barrier (BBB) function may be more consolidated in ChAT tg because consolidation of the BBB, which is composed of tight junction components [57,58], can interfere with the extravasation of pro-inflammatory substances into the blood, resulting in anti-inflammatory responses in the brain. We further investigated CNS phenotypes of ChAT tg mice especially focusing on their BBB function [59].
(1) Initially, we performed Western blot analysis of the brain to evaluate the protein expression of claudin-5, one of the components of BBB [60,61,62,63]. Intriguingly, the ChAT tg brain expressed more claudin-5 protein than WT mice brains. This striking result prompted us to further examine the BBB function. As mentioned in our previous study, the beneficial CNS phenotypes are mediated by the VN afferent fibers of ChAT tg mice [13,26]; therefore, we checked the effects of lateral vagotomy on claudin-5 protein expression in the brain. Surprisingly, vagotomy caused downregulation of claudin-5 protein levels within 5 d following the vagotomy [59]. This also suggests the novel fact that the expression of claudin-5 in the brain is regulated by the VN, specifically the afferent fiber activation. In other words, here there is evidence of a tight link between the BBB and the VN [59].
The in vitro experiments also followed the in vivo features of BBB [62,64,65]. Murine brain endothelial cells from ChAT tg mice sustained enhancement of claudin-5 protein expression compared with those from WT mice. Furthermore, an in vitro reconstruction study of BBB using brain endothelial cells, astrocytes, and pericytes demonstrated that ChAT tg mice-derived reconstructed BBB was more resistant to Evans blue dye leakage than WT mice-derived reconstructed BBB [66]. These results clearly indicate that BBB function is more consolidated in ChAT tg mice than in WT mice via increased claudin-5 protein expression [59], although the precise mechanisms are still unknown. However, a recent study demonstrated that α7 nicotinic receptor stimulation increased claudin-5 expression in rat brain endothelial cells, and therefore, this may be considered one of the mechanisms [60,61].
(2) ChAT tg mice were subjected to brain cold injury, which is a BBB disrupting model [67,68,69]. The brain cold injury was developed using a cold metal cylinder with a diameter of 3–4 mm, which was attached to the surface of the right parietal bone strictly for 5 s. The effect of cold injury on BBB transient disruption peaked 24 h after injury, which was shown by the systemically distributed Evans blue dye. The area subjected to cold injury was restricted to blue color, indicating that the dye was extravasated into the brain parenchyma. With brain cold injury, WT mice brains showed remarkable blue staining in the surface of the injured brain; however, ChAT tg mice brains significantly attenuated Evans blue leakage, suggesting that the BBB function of ChAT tg mice was more strengthened and resistant to the injury [59].
Furthermore, the Parkinson’s disease model induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), which is another model influencing BBB [70,71], was also applied to ChAT tg mice. Likewise, the brains of WT mice 3 d after injection of MPTP showed increased extravasation of sodium fluorescein into the brain parenchyma. In contrast, the ChAT tg mice brains significantly decreased the fluorescein levels in the brain. Although it was not fully studied regarding the mechanisms by which BBB consolidation leads to neuroprotective effects, ChAT tg brain was significantly resistant to neurodegeneration by MPTP, showing that more neuronal cells as well as neuron fibers were left intact in the substantia nigra, which is a main target of this model. These data clearly indicate that ChAT tg mice BBB was resistant to two different types of BBB disruption models [19].
Compatible with the more consolidated characteristics of ChAT tg BBB associated with increased immunoreactivity of claudin-5, the appearance of astrocytes in the ChAT tg brain was less hypertrophic and immunoreactive with glial fibrillary acidic protein (GFAP) staining [72], suggesting that less extravasating substances from the blood into the parenchyma attenuated reactive astrocyte transformation. This phenomenon was also evidenced by Western blot analysis of GFAP expression levels. The right brain of WT mice increased GFAP protein expression in response to the right injury compared with the left brain without injury; however, in the brains of ChAT tg mice, the laterality of the right to left brain GFAP expression was significantly decreased compared with that of the WT mice brains [59]. This is a novel and specific finding in ChAT tg mice brains caused by strengthened BBB function.
5. Anti-Inflammatory Reactions of ChAT tg Mice against Systemic Injection of LPS and Cold Brain Injury
As mentioned earlier regarding ChAT tg mice, their hearts produced more ACh. However, when LPS (10 mg/kg/dose i.p.) was systemically administered [73,74], ChAT tg mice survived more than WT mice within 48 h after injection, and their blood concentrations of TNF-α and IL-6 were significantly attenuated compared to those of WT mice. Moreover, Kupffer cells and the liver in ChAT tg mice also downregulated cytokines gene expression more than that in WT mice [59].
Likewise, in cold brain injury, the right injured parietal brain of ChAT tg mice expressed less cytokine gene expression than that of WT mice [59]. These results all support our concept that ChAT tg mice possess systemic anti-inflammatory response potency.
6. Speculated Underlying Mechanisms Responsible for Anti-Inflammatory Potency
Recent studies have reported that VNS, which was initially used as an anti-convulsion but later anti-heart failure or anti-depression treatment, plays a role in inhibiting inflammation. Since then, many research papers have come out to reveal that VNS attenuates inflammation, although the precise mechanisms remain to be fully investigated [43,75,76,77]. Initially, peripheral inflammation was the main target for VNS, including sepsis [56,78,79]; however, VNS later suppresses central brain inflammation [80,81], although the mechanisms may be complicated as the ascending root of the VN is not directly targeted to inflammatory lesions using the terminal ends. Rather, it is first terminated to the NTS followed by the second innervation to the whole brain, including the locus coeruleus (LC), thalamus, and hypothalamus [82]. The LC is an intermediate nucleus, which again projects third neurons into the whole brain as a noradrenergic neuron. In contrast, LC is also connected with the cholinergic pathway, which includes the prefrontal cortex [82].
ACh from the terminal ends of the VN exerts anti-inflammatory actions. Among receptors of ACh, including nicotinic and muscarinic receptors, the α7 nicotinic receptor has been well known to execute anti-inflammatory effects [83,84,85]. The α7 nicotinic receptor is unique with its homoheptamer structure, which is different from the other heterodimer nicotinic receptors [86]. The anti-inflammatory effects of the receptor may contribute to its signal transduction; however, compared with other nicotinic receptors, the α7 nicotinic receptor does not seem to utilize specific signal transduction modes that have already been reported [86]. However, the following characteristics of the receptor are speculated to be responsible for its anti-inflammatory function: the homomeric α7 nicotinic receptor desensitizes very rapidly, compared with other heteromeric receptors, with its agonists [79,86]. Furthermore, the α7 nicotinic receptor activates the JAK2/STAT3 pathway [87,88] as well as the Akt pathway [89,90], the former suppressing NF-κB translocation into the nucleus and the latter activating the Nrf2 pathway [90], which upregulates antioxidative factor HO-1 [91]. Based on these, its short opening time, rapid desensitization, activation of cellular protecting signals, or suppression of NF-κB nuclear translocation may be responsible for anti-inflammatory effects [86].
Likewise, the speculated mechanisms underlying upregulation of BBB claudin-5 remain to be studied in detail because there are few studies that have revealed a direct interaction between cholinergic nerve ends and brain endothelial cells composing the BBB. However, one in vitro study reported that the α7 nicotinic receptor agonist or glycogen synthase kinase inhibition upregulated tight junction proteins, including claudin-5 and occludin [61,92]. However, another study reported that the α7 nicotinic receptor was involved in attenuating the expression of BBB components [93,94]. The reason these results were contradictory remains to be elucidated. In contrast to the involvement of cholinergic agonists in regulating tight junctions, adrenergic receptors, instead of cholinergic receptors, were also reported to be responsible for sustaining BBB functions [27,95].
However, our transgenic mice overexpressing the ChAT gene restrictively in the heart provide a novel significant clue; that is, the link between the activation of NNA in the heart and BBB consolidation as well as anti-inflammation, as comprehensively expressed in a schema (Figure 3). However, it remains to be determined whether these distinctive findings of upregulation of BBB function and anti-inflammatory response may contribute to only one or several factors. Therefore, further studies are needed to investigate and address these issues.
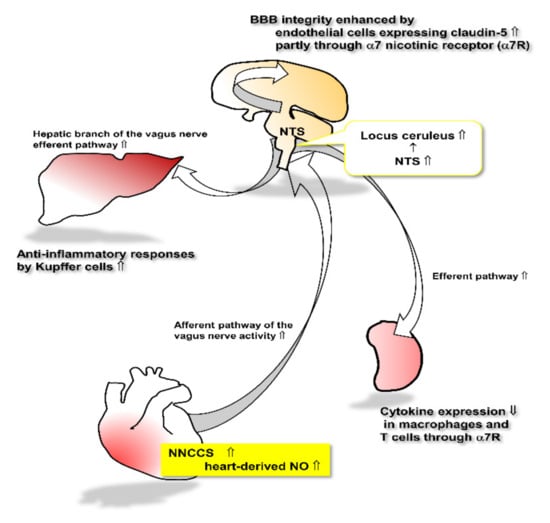
Figure 3.
This schema represents the speculated link between augmented NNCCS and other organs.
Despite the limitations of transgenic mice, the finding that the heart equipped with NNCCS influences higher levels of brain functions (e.g., mood and BBB) through the VN afferent pathway, may provide a novel concept to modulate the brain by the heart, not the brain–heart connection but heart–brain connection.
When the heart-derived non-neuronal ACh synthesis is activated, increased release of NO from cardiomyocytes stimulates the afferent pathway of the vagus nerve, leading to stimulation of the cholinergic and noradrenergic pathway in the brain through the NTS. The activated cholinergic system plays a role in increasing claudin-5 expression in brain endothelial cells, leading to the consolidation of BBB integrity. The efferent pathway of the vagus nerve, in turn, is activated to influence target organs including Kupffer cells in the liver as well as macrophages and T cells in the spleen. Those cells are regulated by the vagus nerve efferent pathway to attenuate inflammatory responses.
7. Concluding Remarks
Even though evidence of NNCCS or NNA in the heart has accumulated, there is no direct evidence of this system involving human diseases. Additionally, no answers have been found as to whether this system can be used as a therapeutic modality [10,96,97]. However, there are several studies to remind us of the possibility of addressing this system in human diseases. Donepezil, an Alzheimer’s disease drug known as an acetylcholinesterase inhibitor, can decrease the incidence and mortality of cardiovascular diseases in patients with Alzheimer’s disease [98,99,100]. Our study has already reported that donepezil upregulates NNCCS as an inducer of this system [4]. These results strengthen the speculation that the brain and heart are profoundly cross-talked through influencing each other, and the possibility of intervention in this system for both targets. Further studies focusing on this issue are anticipated.
Funding
This study was supported by the Japan Society for the Promotion of Science Grants-in-Aid for Scientific Research, Japan (JSPS KAKENHI) (C) Grant Number 16K08560, the Smoking Research Foundation 2019–2020, and partly by The Vehicle Racing Commemorative Foundation 2020.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Ethics Committee of Nippon Medical school (permission number: 27-003).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data available on request due to restrictions e.g., privacy or ethical.
Acknowledgments
We thank an English Editor from Tokyo, Japan, for editing our manuscript. This manuscript was critically checked by a native English speaker.
Conflicts of Interest
The author declares no conflict of interest.
Abbreviations
| ACh | Acetylcholine |
| ANS | Autonomic nervous system |
| SNS | Sympathetic nervous system |
| PNS | parasympathetic nervous system |
| NNA | a non-neuronal ACh |
| NNCCS | non-neuronal cardiac cholinergic system |
| CHT1 | choline transporter |
| ChAT | choline acetyltransferase |
| VAChT | vesicular ACh transporter |
| TST | tail suspension test |
| FST | Forced swimming test |
| NTS | The nucleus of the solitary tract |
| NO | Nitric oxide |
| NOS | NO synthase |
| VN | vagus nerve |
| WT | wild type |
| VNS | Vagus nerve stimulation |
| BBB | blood brain barrier |
| LC | locus coeruleus |
References
- Hoover, D.B.; Ganote, C.E.; Ferguson, S.M.; Blakely, R.D.; Parsons, R.L. Localization of cholinergic innervation in guinea pig heart by immunohistochemistry for high-affinity choline transporters. Cardiovasc. Res. 2004, 62, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Crick, S.J.; Wharton, J.; Sheppard, M.N.; Royston, D.; Yacoub, M.H.; Anderson, R.H.; Polak, J.M. Innervation of the human cardiac conduction system. A quantitative immunohistochemical and histochemical study. Circulation 1994, 89, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Kawano, H.; Okada, R.; Yano, K. Histological study on the distribution of autonomic nerves in the human heart. Heart Vessels 2003, 18, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Powley, T.L.; Schwaber, J.S.; Doyle, F.J., 3rd. Projections of the dorsal motor nucleus of the vagus to cardiac ganglia of rat atria: An anterograde tracing study. J. Comp. Neurol. 1999, 410, 320–341. [Google Scholar] [CrossRef]
- Hancock, J.C.; Hoover, D.B.; Hougland, M.W. Distribution of muscarinic receptors and acetylcholinesterase in the rat heart. J. Auton. Nerv. Syst. 1987, 19, 59–66. [Google Scholar] [CrossRef]
- Kakinuma, Y.; Akiyama, T.; Sato, T. Cholinoceptive and cholinergic properties of cardiomyocytes involving an amplification mechanism for vagal efferent effects in sparsely innervated ventricular myocardium. FEBS J. 2009, 276, 5111–5125. [Google Scholar] [CrossRef]
- Rana, O.R.; Schauerte, P.; Kluttig, R.; Schröder, J.W.; Koenen, R.R.; Weber, C.; Nolte, K.W.; Weis, J.; Hoffmann, R.; Marx, N.; et al. Acetylcholine as an age-dependent non-neuronal source in the heart. Auton. Neurosci. 2010, 156, 82–89. [Google Scholar] [CrossRef]
- Roy, A.; Fields, W.C.; Rocha-Resende, C.; Resende, R.R.; Guatimosim, S.; Prado, V.F.; Gros, R.; Prado, M.A. Cardiomyocyte-secreted acetylcholine is required for maintenance of homeostasis in the heart. FASEB J. 2013, 27, 5072–5082. [Google Scholar] [CrossRef]
- Rocha-Resende, C.; Roy, A.; Resende, R.; Ladeira, M.S.; Lara, A.; de Morais Gomes, E.R.; Prado, V.F.; Gros, R.; Guatimosim, C.; Prado, M.A.; et al. Non-neuronal cholinergic machinery present in cardiomyocytes offsets hypertrophic signals. J. Mol. Cell. Cardiol. 2012, 53, 206–216. [Google Scholar] [CrossRef]
- Saw, E.L.; Kakinuma, Y.; Fronius, M.; Katare, R. The non-neuronal cholinergic system in the heart: A comprehensive review. J. Mol. Cell Cardiol. 2018, 125, 129–139. [Google Scholar] [CrossRef]
- Gavioli, M.; Lara, A.; Almeida, P.W.; Lima, A.M.; Damasceno, D.D.; Rocha-Resende, C.; Ladeira, M.; Resende, R.R.; Martinelli, P.M.; Melo, M.B.; et al. Cholinergic signaling exerts protective effects in models of sympathetic hyperactivity-induced cardiac dysfunction. PLoS ONE 2014, 9, e100179. [Google Scholar] [CrossRef] [PubMed]
- Kakinuma, Y.; Akiyama, T.; Okazaki, K.; Arikawa, M.; Noguchi, T.; Sato, T. A non-neuronal cardiac cholinergic system plays a protective role in myocardium salvage during ischemic insults. PLoS ONE 2012, 7, e50761. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, S.; Iketani, M.; Kakinuma, Y. A non-neuronal cholinergic system regulates cellular ATP levels to maintain cell viability. Cell. Physiol. Biochem. 2014, 34, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Kakinuma, Y.; Tsuda, M.; Okazaki, K.; Akiyama, T.; Arikawa, M.; Noguchi, T.; Sato, T. Heart-specific, overexpression of choline acetyltransferase gene protects murine heart against ischemia through hypoxia-inducible factor-1α-related defense mechanisms. J. Am. Heart Assoc. 2013, 2, e004887. [Google Scholar] [CrossRef] [PubMed]
- Suchiang, K.; Sharma, R. Age-dependent modulation of fasting and long-term dietary restriction on acetylcholinesterase in non-neuronal tissues of mice. Mol. Cell Biochem. 2016, 419, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Dakroub, M.; Tezini, G.C.; Liu, Y.; Guatimosim, S.; Feng, Q.; Salgado, H.C.; Prado, V.F.; Prado, M.A.; Gros, R. Cardiac acetylcholine inhibits ventricular remodeling and dysfunction under pathologic conditions. FASEB J. 2016, 30, 688–701. [Google Scholar] [CrossRef]
- Rocha-Resende, C.; Weinheimer, C.; Bajpai, G.; Adamo, L.; Matkovich, S.J.; Schilling, J.; Barger, P.M.; Lavine, K.J.; Mann, D.L. Immunomodulatory role of non-neuronal cholinergic signaling in myocardial injury. JCI Insight 2019, 5, e128961. [Google Scholar] [CrossRef]
- De Simone, R.; Ajmone-Cat, M.A.; Carnevale, D.; Minghetti, L. Activation of alpha7 nicotinic acetylcholine receptor by nicotine selectively up-regulates cyclooxygenase-2 and prostaglandin E2 in rat microglial cultures. J. Neuroinflamm. 2005, 2, 4. [Google Scholar] [CrossRef]
- Wilson, C.; Lee, M.D.; McCarron, J.G. Acetylcholine released by endothelial cells facilitates flow-mediated dilatation. J. Physiol. 2016, 594, 7267–7307. [Google Scholar] [CrossRef]
- Arikawa, M.; Kakinuma, Y.; Noguchi, T.; Todaka, H.; Sato, T. Donepezil, an acetylcholinesterase inhibitor, attenuates LPS-induced inflammatory response in murine macrophage cell line RAW 264.7 through inhibition of nuclear factor kappa B translocation. Eur. J. Pharmacol. 2016, 789, 17–26. [Google Scholar] [CrossRef]
- Hecker, A.; Mikulski, Z.; Lips, K.S.; Pfeil, U.; Zakrzewicz, A.; Wilker, S.; Hartmann, P.; Padberg, W.; Wessler, I.; Kummer, W.; et al. Pivotal advance: Up-regulation of acetylcholine synthesis and paracrine cholinergic signaling in intravascular transplant leukocytes during rejection of rat renal allografts. J. Leukoc. Biol. 2009, 86, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Mashimo, M.; Moriwaki, Y.; Misawa, H.; Ono, S.; Horiguchi, K.; Kawashima, K. Physiological functions of the cholinergic system in immune cells. J. Pharmacol. Sci. 2017, 134, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Saracino, L.; Zorzetto, M.; Inghilleri, S.; Pozzi, E.; Stella, G.M. Non-neuronal cholinergic system in airways and lung cancer susceptibility. Transl. Lung Cancer Res. 2013, 2, 284–294. [Google Scholar] [PubMed]
- Nakajima, H.; Nakajima, H.O.; Salcher, O.; Dittiè, A.S.; Dembowsky, K.; Jing, S.; Field, L.J. Atrial but not ventricular fibrosis in mice expressing a mutant transforming growth factor-β1 transgene in the heart. Circ. Res. 2000, 86, 571–579. [Google Scholar] [CrossRef]
- Sah, V.P.; Minamisawa, S.; Tam, S.P.; Wu, T.H.; Dorn, G.W., 2nd; Ross, J., Jr.; Chien, K.R.; Brown, J.H. Cardiac-specific overexpression of RhoA results in sinus and atrioventricular nodal dysfunction and contractile failure. J. Clin. Investig. 1999, 103, 1627–1634. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, S.; Kai, Y.; Tsuda, M.; Ohata, H.; Mano, A.; Mizoguchi, N.; Sugama, S.; Nemoto, T.; Suzuki, K.; Kurabayashi, A.; et al. Non-neuronal cardiac cholinergic system influences CNS via the vagus nerve to acquire a stress-refractory propensity. Clin. Sci. 2016, 130, 1913–1928. [Google Scholar] [CrossRef]
- Hut, R.A.; Van der Zee, E.A. The cholinergic system, circadian rhythmicity, and time memory. Behav. Brain Res. 2011, 221, 466–480. [Google Scholar] [CrossRef]
- Murakami, N.; Takahashi, K.; Kawashima, K. Effect of light on the acetylcholine concentrations of the suprachiasmatic nucleus in the rat. Brain Res. 1984, 311, 358–360. [Google Scholar] [CrossRef][Green Version]
- Oikawa, S.; Kai, Y.; Mano, A.; Ohata, H.; Nemoto, T.; Kakinuma, Y. Various Regulatory Modes for Circadian Rhythmicity and Sexual Dimorphism in the Non-Neuronal Cardiac Cholinergic System. J. Cardiovasc. Transl. Res. 2017, 10, 411–422. [Google Scholar] [CrossRef]
- Sunal, R.; Gümüşel, B.; Kayaalp, S.O. Effect of changes in swimming area on results of “behavioral despair test”. Pharmacol. Biochem. Behav. 1994, 49, 891–896. [Google Scholar] [CrossRef]
- Lister, R.G. The use of a plus-maze to measure anxiety in the mouse. Psychopharmacology 1987, 92, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Sugama, S.; Sekiyama, K.; Kodama, T.; Takamatsu, Y.; Takenouchi, T.; Hashimoto, M.; Bruno, C.; Kakinuma, Y. Chronic restraint stress triggers dopaminergic and noradrenergic neurodegeneration: Possible role of chronic stress in the onset of Parkinson’s disease. Brain Behav. Immun. 2016, 51, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.J.; Gröticke, I.; Bankstahl, M.; Löscher, W. Behavioral and cognitive alterations, spontaneous seizures, and neuropathology developing after a pilocarpine-induced status epilepticus in C57BL/6 mice. Exp. Neurol. 2009, 219, 284–297. [Google Scholar] [CrossRef] [PubMed]
- Shafaroodi, H.; Moezi, L.; Bahremand, A.; Dehpour, A.R. The role of α2-adrenoceptors in the anti-convulsant effects of cannabinoids on pentylenetetrazole-induced seizure threshold in mice. Eur. J. Pharmacol. 2013, 714, 1–6. [Google Scholar] [CrossRef]
- Takahashi, K.; Hishida, R.; Kubota, Y.; Kudoh, M.; Takahashi, S.; Shibuki, K. Transcranial fluorescence imaging of auditory cortical plasticity regulated by acoustic environments in mice. Eur. J. Neurosci. 2006, 23, 1365–1376. [Google Scholar] [CrossRef]
- Zhao, M.; Nguyen, J.; Ma, H.; Nishimura, N.; Schaffer, C.B.; Schwartz, T.H. Preictal and ictal neurovascular and metabolic coupling surrounding a seizure focus. J. Neurosci. 2011, 31, 13292–13300. [Google Scholar] [CrossRef]
- Prechtl, J.C.; Powley, T.L. The fiber composition of the abdominal vagus of the rat. Anat. Embryol. 1990, 181, 101–115. [Google Scholar] [CrossRef]
- Sawchenko, P.E. Central connections of the sensory and motor nuclei of the vagus nerve. J. Auton. Nerv. Syst. 1983, 9, 13–26. [Google Scholar] [CrossRef]
- Molinari, C.; Sabbatini, M.; Grossini, E.; Mary, D.A.; Cannas, M.; Vacca, G. Cardiovascular effects and c-Fos expression in the rat hindbrain in response to innocuous stomach distension. Brain Res. Bull. 2006, 69, 140–146. [Google Scholar] [CrossRef]
- Wasade, V.S.; Schultz, L.; Mohanarangan, K.; Gaddam, A.; Schwalb, J.M.; Spanaki-Varelas, M. Long-term seizure and psychosocial outcomes of vagus nerve stimulation for intractable epilepsy. Epilepsy Behav. 2015, 53, 31–36. [Google Scholar] [CrossRef]
- Zanchetti, A.; Wang, S.C.; Moruzzi, G. The effect of vagal afferent stimulation on the EEG pattern of the cat. Electroencephalogr. Clin. Neurophysiol. 1952, 4, 357–361. [Google Scholar] [CrossRef]
- Berry, S.M.; Broglio, K.; Bunker, M.; Jayewardene, A.; Olin, B.; Rush, A.J. A patient-level meta-analysis of studies evaluating vagus nerve stimulation therapy for treatment-resistant depression. Med. Devices 2013, 6, 17–35. [Google Scholar]
- De Ferrari, G.M.; Crijns, H.J.; Borggrefe, M.; Milasinovic, G.; Smid, J.; Zabel, M.; Gavazzi, A.; Sanzo, A.; Dennert, R.; Kuschyk, J.; et al. Cardio Fit Multicenter Trial Investigators. Chronic vagus nerve stimulation: A new and promising therapeutic approach for chronic heart failure. Eur. Heart J. 2011, 32, 847–855. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zhao, M.; Bi, X.; Sun, L.; Yu, X.; Zhao, M.; Zang, W. Novel strategies and underlying protective mechanisms of modulation of vagal activity in cardiovascular diseases. Br. J. Pharmacol. 2015, 172, 5489–5500. [Google Scholar] [CrossRef] [PubMed]
- Peña, D.F.; Engineer, N.D.; McIntyre, C.K. Rapid remission of conditioned fear expression with extinction training paired with vagus nerve stimulation. Biol. Psychiatry 2013, 73, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Babygirija, R.; Sood, M.; Kannampalli, P.; Sengupta, J.N.; Miranda, A. Percutaneous electrical nerve field stimulation modulates central pain pathways and attenuates post-inflammatory visceral and somatic hyperalgesia in rats. Neuroscience 2017, 356, 11–21. [Google Scholar] [CrossRef]
- Albert, U.; Maina, G.; Aguglia, A.; Vitalucci, A.; Bogetto, F.; Fronda, C.; Ducati, A.; Lanotte, M. Vagus nerve stimulation for treatment-resistant mood disorders: A long-term naturalistic study. BMC Psychiatry 2015, 15, 64. [Google Scholar] [CrossRef]
- Musselman, E.D.; Pelot, N.A.; Grill, W.M. Empirically based guidelines for selecting vagus nerve stimulation parameters in epilepsy and heart failure. Cold Spring Harb. Perspect. Med. 2019, 9, a034264. [Google Scholar] [CrossRef]
- Johnson, R.L.; Wilson, C.G. A review of vagus nerve stimulation as a therapeutic intervention. J. Inflamm. Res. 2018, 11, 203–213. [Google Scholar] [CrossRef]
- Neren, D.; Johnson, M.D.; Legon, W.; Bachour, S.P.; Ling, G.; Divani, A.A. Vagus nerve stimulation and other neuromodulation methods for treatment of traumatic brain injury. Neurocrit. Care 2016, 24, 308–319. [Google Scholar] [CrossRef]
- Dedkova, E.N.; Ji, X.; Wang, Y.G.; Blatter, L.A.; Lipsius, S.L. Signaling mechanisms that mediate nitric oxide production induced by acetylcholine exposure and withdrawal in cat atrial myocytes. Circ. Res. 2003, 93, 1233–1240. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, M.; Kakinuma, Y.; Ando, M.; Katare, R.G.; Yamasaki, F.; Doi, Y.; Sato, T. Nitric oxide stimulates vascular endothelial growth factor production in cardiomyocytes involved in angiogenesis. J. Physiol. Sci. 2006, 56, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Petho, G.; Reeh, P.W. Sensory and signaling mechanisms of bradykinin, eicosanoids, platelet-activating factor, and nitric oxide in peripheral nociceptors. Physiol. Rev. 2012, 92, 1699–1775. [Google Scholar] [CrossRef] [PubMed]
- Dias, A.C.; Colombari, E.; Mifflin, S.W. Effect of nitric oxide on excitatory amino acid-evoked discharge of neurons in NTS. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H234–H240. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, S.; Kai, Y.; Mano, A.; Ohata, H.; Kurabayashi, A.; Tsuda, M.; Kakinuma, Y. Non-neuronal cardiac acetylcholine system playing indispensable roles in cardiac homeostasis confers resiliency to the heart. J. Physiol. Sci. 2020, in press. [Google Scholar]
- Bonaz, B.; Sinniger, V.; Pellissier, S. Anti-inflammatory properties of the vagus nerve: Potential therapeutic implications of vagus nerve stimulation. J. Physiol. 2016, 594, 5781–5790. [Google Scholar] [CrossRef]
- Aird, W.C. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ. Res. 2007, 100, 158–173. [Google Scholar] [CrossRef]
- Jia, W.G.; Lu, R.; Martin, T.A.; Jiang, W.G. The role of claudin-5 in blood brain barrier (BBB) and brain metastases. Mol. Med. Rep. 2014, 9, 779–785. [Google Scholar] [CrossRef]
- Oikawa, S.; Kai, Y.; Mano, A.; Sugama, S.; Mizoguchi, N.; Tsuda, M.; Muramoto, K.; Kakinuma, Y. Potentiating a non-neuronal cardiac cholinergic system reinforces the functional integrity of the blood brain barrier associated with systemic anti-inflammatory responses. Brain Behav. Immun. 2019, 81, 122–137. [Google Scholar] [CrossRef]
- Dash, P.K.; Zhao, J.; Kobori, N.; Redell, J.B.; Hylin, M.J.; Hood, K.N.; Moore, A.N. Activation of alpha 7 cholinergic nicotinic receptors reduce blood-brain barrier permeability following experimental traumatic brain injury. J. Neurosci. 2016, 36, 2809–2818. [Google Scholar] [CrossRef]
- Kimura, I.; Dohgu, S.; Takata, F.; Matsumoto, J.; Kawahara, Y.; Nishihira, M.; Sakada, S.; Saisho, T.; Yamauchi, A.; Kataoka, Y. Activation of the α7 nicotinic acetylcholine receptor upregulates blood-brain barrier function through increased claudin-5 and occludin expression in rat brain endothelial cells. Neurosci. Lett. 2019, 694, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Deli, M.A.; Nakao, S.; Honda, M.; Hayashi, K.; Nakaoke, R.; Kataoka, Y.; Niwa, M. Pericytes from brain microvessels strengthen the barrier integrity in primary cultures of rat brain endothelial cells. Cell. Mol. Neurobiol. 2007, 27, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.C.; Morris, A.P.; O’Neil, R.G. Tight junction protein expression and barrier properties of immortalized mouse brain microvessel endothelial cells. Brain Res. 2007, 1130, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Perrière, N.; Demeuse, P.; Garcia, E.; Regina, A.; Debray, M.; Andreux, J.P.; Couvreur, P.; Scherrmann, J.M.; Temsamani, J.; Couraud, P.O.; et al. Puromycin-based purification of rat brain capillary endothelial cell cultures. Effect on the expression of blood-brain barrier-specific properties. J. Neurochem. 2005, 93, 279–289. [Google Scholar] [CrossRef]
- Bernard-Patrzynski, F.; Lécuyer, M.A.; Puscas, I.; Boukhatem, I.; Charabati, M.; Bourbonnière, L.; Ramassamy, C.; Leclair, G.; Prat, A.; Roullin, V.G. Isolation of endothelial cells, pericytes and astrocytes from mouse brain. PLoS ONE 2019, 14, e0226302. [Google Scholar] [CrossRef]
- Czupalla, C.J.; Liebner, S.; Devraj, K. In vitro models of the blood-brain barrier. Methods Mol. Biol. 2014, 1135, 415–437. [Google Scholar]
- Kakinuma, Y.; Hama, H.; Sugiyama, F.; Yagami, K.; Goto, K.; Murakami, K.; Fukamizu, A. Impaired blood-brain barrier function in angiotensinogen-deficient mice. Nat. Med. 1998, 4, 1078–1080. [Google Scholar] [CrossRef]
- Murakami, K.; Kondo, T.; Yang, G.; Chen, S.F.; Morita-Fujimura, Y.; Chan, P.H. Cold injury in mice: A model to study mechanisms of brain edema and neuronal apoptosis. Prog. Neurobiol. 1999, 57, 289–299. [Google Scholar] [CrossRef]
- Liu, W.Y.; Wang, Z.B.; Wang, Y.; Tong, L.C.; Li, Y.; Wei, X.; Luan, P.; Li, L. Increasing the permeability of the blood-brain barrier in three different models in vivo. CNS Neurosci. Ther. 2015, 21, 568–574. [Google Scholar] [CrossRef]
- Sugama, S.; Yang, L.; Cho, B.P.; DeGiorgio, L.A.; Lorenzl, S.; Albers, D.S.; Beal, M.F.; Volpe, B.T.; Joh, T.H. Age-related microglial activation in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic neurodegeneration in C57BL/6 mice. Brain Res. 2003, 964, 288–294. [Google Scholar] [CrossRef]
- Liu, Z.; Qiu, A.W.; Huang, Y.; Yang, Y.; Chen, J.N.; Gu, T.T.; Cao, B.B.; Qiu, Y.H.; Peng, Y.P. IL-17A exacerbates neuroinflammation and neurodegeneration by activating microglia in rodent models of Parkinson’s disease. Brain Behav. Immun. 2019, 81, 630–645. [Google Scholar] [CrossRef] [PubMed]
- Krämer, T.J.; Hack, N.; Brühl, T.J.; Menzel, L.; Hummel, R.; Griemert, E.V.; Klein, M.; Thal, S.C.; Bopp, T.; Schäfer, M.K.E. Depletion of regulatory T cells increases T cell brain infiltration, reactive astrogliosis, and interferon-gamma gene expression in acute experimental traumatic brain injury. J. Neuroinflamm. 2019, 16, 163. [Google Scholar] [CrossRef] [PubMed]
- Adachi, Y.U.; Obata, Y.; Suzuki, K.; Katoh, H.; Itagaki, T.; Doi, M.; Sato, S. Nafamostat prevents hypothermia and improves survival time after administration of lipopolysaccharide in a mouse surgical model. J. Anesth. 2009, 23, 624–627. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, A.K.; Bettini, M.L.; Menon, R.T.; Gopal, V.Y.N.; Huang, S.; Edwards, D.P.; Pammi, M.; Barrios, R.; Shivanna, B. Consequences of early postnatal lipopolysaccharide exposure on developing lungs in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 316, L229–L244. [Google Scholar] [CrossRef] [PubMed]
- Meneses, G.; Bautista, M.; Florentino, A.; Díaz, G.; Acero, G.; Besedovsky, H.; Meneses, D.; Fleury, A.; Del Rey, A.; Gevorkian, G.; et al. Electric stimulation of the vagus nerve reduced mouse neuroinflammation induced by lipopolysaccharide. J. Inflamm. 2016, 13, 33. [Google Scholar] [CrossRef]
- Fonseca, R.C.; Bassi, G.S.; Brito, C.C.; Rosa, L.B.; David, B.A.; Araújo, A.M.; Nóbrega, N.; Diniz, A.B.; Jesus, I.C.G.; Barcelos, L.S.; et al. Vagus nerve regulates the phagocytic and secretory activity of resident macrophages in the liver. Brain Behav. Immun. 2019, 81, 444–454. [Google Scholar] [CrossRef]
- Frasch, M.G.; Szynkaruk, M.; Prout, A.P.; Nygard, K.; Cao, M.; Veldhuizen, R.; Hammond, R.; Richardson, B.S. Decreased neuroinflammation correlates to higher vagus nerve activity fluctuations in near-term ovine fetuses: A case for the afferent cholinergic anti-inflammatory pathway? J. Neuroinflamm. 2016, 13, 103. [Google Scholar] [CrossRef]
- Borovikova, L.V.; Ivanova, S.; Zhang, M.; Yang, H.; Botchkina, G.I.; Watkins, L.R.; Wang, H.; Abumrad, N.; Eaton, J.W.; Tracey, K.J. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000, 405, 458–462. [Google Scholar] [CrossRef]
- The, F.; Cailotto, C.; van der Vliet, J.; de Jonge, W.J.; Bennink, R.J.; Buijs, R.M.; Boeckxstaens, G.E. Central activation of the cholinergic anti-inflammatory pathway reduces surgical inflammation in experimental post-operative ileus. Br. J. Pharmacol. 2011, 163, 1007–1016. [Google Scholar] [CrossRef]
- Pavlov, V.A.; Tracey, K.J. The cholinergic anti-inflammatory pathway. Brain Behav. Immun. 2005, 19, 493–499. [Google Scholar] [CrossRef]
- Han, B.; Li, X.; Hao, J. The cholinergic anti-inflammatory pathway: An innovative treatment strategy for neurological diseases. Neurosci. Biobehav. Rev. 2017, 77, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Ruffoli, R.; Giorgi, F.S.; Pizzanelli, C.; Murri, L.; Paparelli, A.; Fornai, F. The chemical neuroanatomy of vagus nerve stimulation. J. Chem. Neuroanat. 2011, 42, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Giebelen, I.A.; van Westerloo, D.J.; LaRosa, G.J.; de Vos, A.F.; van der Poll, T. Local stimulation of alpha7 cholinergic receptors inhibits LPS-induced TNF-alpha release in the mouse lung. Shock 2007, 28, 700–703. [Google Scholar] [PubMed]
- Echeverria, V.; Yarkov, A.; Aliev, G. Positive modulators of the alpha7 nicotinic receptor against neuroinflammation and cognitive impairment in Alzheimer’s disease. Prog. Neurobiol. 2016, 144, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Egea, J.; Buendia, I.; Parada, E.; Navarro, E.; León, R.; Lopez, M.G. Anti-inflammatory role of microglial alpha7 nAChRs and its role in neuroprotection. Biochem. Pharmacol. 2015, 97, 463–472. [Google Scholar] [CrossRef]
- Arredondo, J.; Chernyavsky, A.I.; Jolkovsky, D.L.; Pinkerton, K.E.; Grando, S.A. Receptor-mediated tobacco toxicity: Cooperation of the Ras/Raf-1/MEK1/ERK and JAK-2/STAT-3 pathways downstream of alpha7 nicotinic receptor in oral keratinocytes. FASEB J. 2006, 20, 2093–2101. [Google Scholar] [CrossRef]
- Arredondo, J.; Chernyavsky, A.I.; Jolkovsky, D.L.; Pinkerton, K.E.; Grando, S.A. Receptor-mediated tobacco toxicity: Alterations of the NF-κB expression and activity downstream of alpha7 nicotinic receptor in oral keratinocytes. Life Sci. 2007, 80, 2191–2194. [Google Scholar] [CrossRef]
- Ren, Z.; Yang, M.; Guan, Z.; Yu, W. Astrocytic alpha7 Nicotinic Receptor Activation Inhibits Amyloid-beta Aggregation by Upregulating Endogenous αB-crystallin through the PI3K/Akt Signaling Pathway. Curr. Alzheimer Res. 2019, 16, 39–48. [Google Scholar] [CrossRef]
- Guo, J.; Yang, G.; He, Y.; Xu, H.; Fan, H.; An, J.; Zhang, L.; Zhang, R.; Cao, G.; Hao, D.; et al. Involvement of alpha7nAChR in the protective effects of genistein against beta-amyloid-induced oxidative stress in neurons via a PI3K/Akt/Nrf2 pathway-related mechanism. Cell Mol. Neurobiol. 2020, 1–17. [Google Scholar] [CrossRef]
- Wedn, A.M.; El-Gowilly, S.M.; El-Mas, M.M. Nicotine improves survivability, hypotension, and impaired adenosinergic renal vasodilations in endotoxic rats: Role of alpha7-nAChRs/HO-1 pathway. Shock 2020, 53, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, S.H.; Fan, S.; Dykstra, H.; Rom, S.; Mercer, A.; Reichenbach, N.L.; Gofman, L.; Persidsky, Y. Inhibition of glycogen synthase kinase 3β promotes tight junction stability in brain endothelial cells by half-life extension of occludin and claudin-5. PLoS ONE 2013, 8, e55972. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.H.; Jeong, H.S.; Kim, K.J.; Han, M.H.; Lee, E.H.; Lee, K.; Cho, C.H. β-Adrenergic receptor agonists attenuate pericyte loss in diabetic retinas through Akt activation. FASEB J. 2018, 32, 2324–2338. [Google Scholar] [CrossRef]
- Abbruscato, T.J.; Lopez, S.P.; Mark, K.S.; Hawkins, B.T.; Davis, T.P. Nicotine and cotinine modulate cerebral microvascular permeability and protein expression of ZO-1 through nicotinic acetylcholine receptors expressed on brain endothelial cells. J. Pharm. Sci. 2002, 91, 2525–2538. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Chen, X.; Zhang, X.; Shen, X.; Wang, M.; Wang, X.; Liu, W.C.; Liu, C.F.; Liu, J.; Liu, W.; et al. β2-adrenergic receptor-mediated HIF-1alpha upregulation mediates blood brain barrier damage in acute cerebral ischemia. Front. Mol. Neurosci. 2017, 10, 257. [Google Scholar] [CrossRef]
- Lewartowski, B.; Mackiewicz, U. The non-neuronal heart’s acetylcholine in health and disease. J. Physiol. Pharmacol. 2015, 66, 773–778. [Google Scholar]
- Kakinuma, Y. Future perspectives of a cardiac non-neuronal acetylcholine system targeting cardiovascular diseases as an adjunctive tool for metabolic intervention. Int. Immunopharmacol. 2015, 29, 185–188. [Google Scholar] [CrossRef]
- Nordström, P.; Religa, D.; Wimo, A.; Winblad, B.; Eriksdotter, M. The use of cholinesterase inhibitors and the risk of myocardial infarction and death: A nationwide cohort study in subjects with Alzheimer’s disease. Eur. Heart J. 2013, 34, 2585–2591. [Google Scholar] [CrossRef]
- Wu, P.H.; Lin, Y.T.; Hsu, P.C.; Yang, Y.H.; Lin, T.H.; Huang, C.T. Impact of acetylcholinesterase inhibitors on the occurrence of acute coronary syndrome in patients with dementia. Sci. Rep. 2015, 5, 15451. [Google Scholar] [CrossRef]
- Sato, K.; Urbano, R.; Yu, C.; Yamasaki, F.; Sato, T.; Jordan, J.; Robertson, D.; Diedrich, A. The effect of donepezil treatment on cardiovascular mortality. Clin. Pharmacol. Ther. 2010, 88, 335–338. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).