
Diffuse Axonal Injury: Clinical Prognostic Factors, Molecular Experimental Models and the Impact of the Trauma Related Oxidative Stress. An Extensive Review Concerning Milestones and Advances

 , , and
, , and 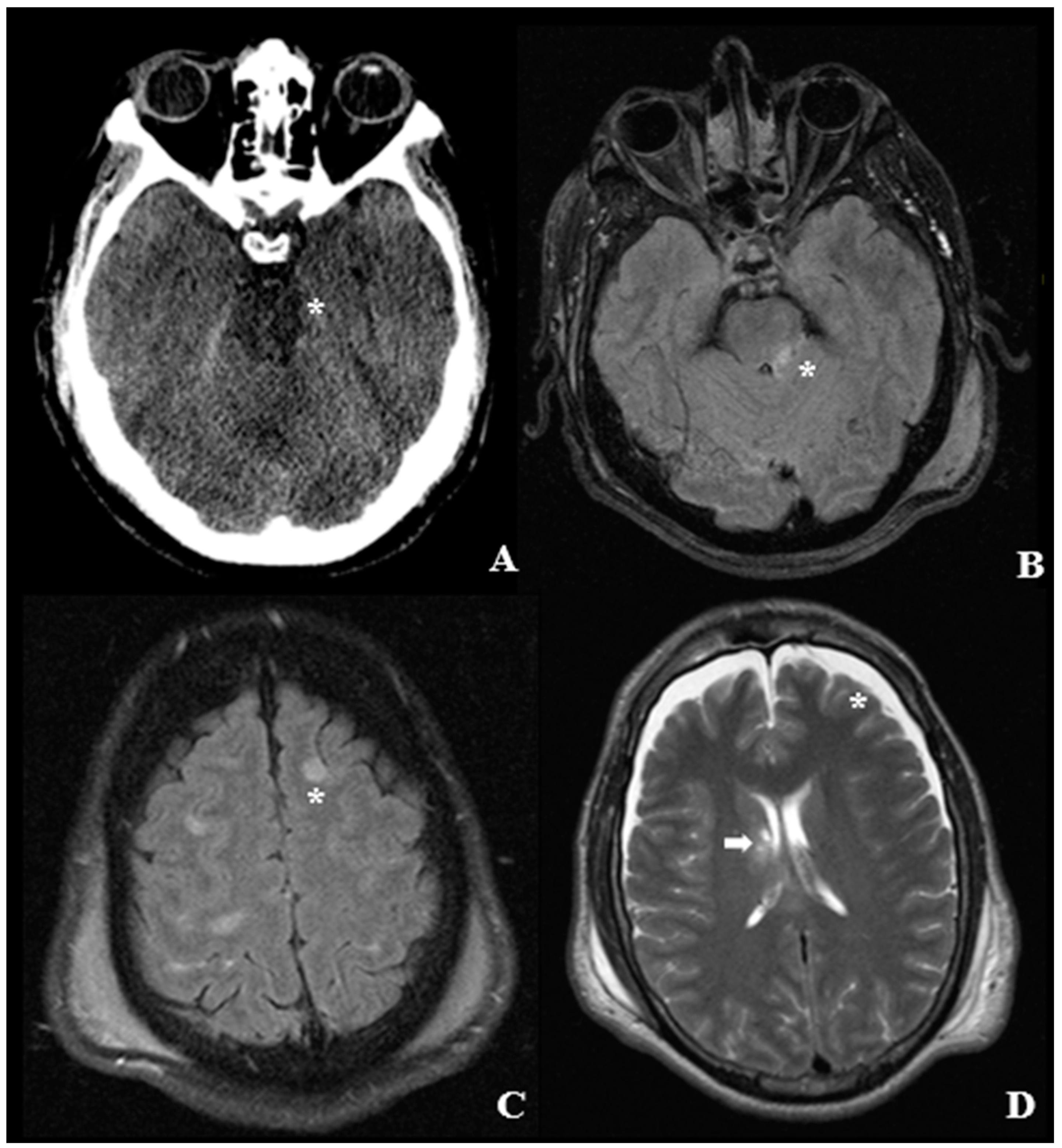{kind=link}
Abstract
:1. Introduction
2. Hypoxia and Blood Pressure
3. Glycemia
4. Pathological Anatomy and Morphologic Findings
- Diffused supratentorial damage to axons (grade I).
- A focal lesion in the corpus callosum (grade II).
- A focal lesion or multiple lesions in the rostral brain stem (grade III).
5. Time to Recover Consciousness
6. Severity of Trauma
7. GCS
8. Biomarkers and the Role of Oxidative Stress
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| TBI | Traumatic Brain Injury |
| DAI | Diffuse Axonal Injury |
| ROS | Reactive Oxygen Species |
| ISS | Injury Severity Score |
| NISS | New Injury Severity Score |
| MAIS | Maximum Abbreviated Injury Scale |
| GOSE | Glasgow Outcome Scale-Extended |
| GCS | Glasgow Coma Scale |
| mPTP | Mitochondrial Permeability Transition Pore |
| NF | Neurofilament Protein |
| pNF-H | Neurofilament Heavy Subunit |
| NF-L | Neurofilament Light Chain |
| GFAP | Glial Fibrillary Acid Protein |
| BBB | Blood–Brain Barrier |
| NSE | Neuron Specific Enolase |
| CSF | Cerebrospinal Fluid |
| Aβ42 | Amyloid β Peptide |
| MRI | Magnetic Resonance Imaging |
| CT | Computed Tomography |
| NCX | Reverse-mode Sodium Calcium Exchanger |
| Drp-1 | Dynamin Related Protein-1 |
| Nrf 2 | Nuclear factor erythroid 2-related factor 2 |
| HO-1 | Hemeoxygenase-1 |
| ARE | Antioxidant Response Element |
| SNF | Sulphoraphane |
| Drp-1 | Dynamin related protein-1 |
| DTI | Diffuse Tensor Imaging |
| oxLDLs | oxidized low-density lipoproteins |
| HNE | 4-Hydroxy-2-nonenal |
| NQO1 | NAD(P)H:quinone oxidoreductase 1 |
| Keap1 | Kelch sample related protein |
| PBS | Phosphate-Buffered Saline |
| AQP4 | Aquaporin-4 |
| TFP | Trifluoperazine |
| CNS | Central Nervous System |
References
- Gerber, L.M.; Chiu, Y.L.; Carney, N.; Härtl, R.; Ghajar, J. Marked reduction in mortality in patients with severe traumatic brain injury. J. Neurosurg. 2013, 119, 1583–1590. [Google Scholar] [CrossRef]
- Finfer, S.R.; Cohen, J. Severe traumatic brain injury. Resuscitation 2001, 48, 77–90. [Google Scholar] [CrossRef]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Axonal pathology in traumatic brain injury. Exp. Neurol. 2013, 246, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Finkelstein, E.A.; Corso, P.S.; Miller, T.R. The Incidence and Economic Burden of Injuries in the United States; Oxford University Press: New York, NY, USA, 2006. [Google Scholar]
- Park, S.J.; Hur, J.W.; Kwon, K.Y.; Rhee, J.J.; Lee, J.W.; Lee, H.K. Time to recover consciousness in patients with diffuse axonal injury: Assessment with reference to magnetic resonance grading. J. Korean Neurosurg. Soc. 2009, 46, 205–209. [Google Scholar] [CrossRef]
- Gennarelli, T.A.; Spielman, G.M.; Langfitt, T.W.; Gildenberg, P.L.; Harrington, T.; Jane, J.A.; Marshall, L.F.; Miller, J.D.; Pitts, L.H. Influence of the type of intracranial lesion on outcome from severe head injury. J. Neurosurg. 1982, 56, 26–32. [Google Scholar] [CrossRef]
- Levi, L.; Guilburd, J.N.; Lemberger, A.; Soustiel, J.F.; Feinsod, M. Diffuse axonal injury: Analysis of 100 patients with radiological signs. Neurosurgery 1990, 27, 429–432. [Google Scholar] [CrossRef]
- Park, S.W.; Park, K.; Kim, Y.B.; Min, B.K.; Hwang, S.N.; Suk, J.S.; Choi, D.Y. Prognostic factors in diffuse axonal injuries of brain. J. Korean Neurosurg. Soc. 1991, 20, 983–990. [Google Scholar]
- Williams, J.M.; Gomes, F.; Drudge, O.W.; Kessler, M. Predicting outcome from closed head injury by early assessment of trauma severity. J. Neurosurg. 1984, 61, 581–585. [Google Scholar] [CrossRef]
- Vieira, R.C.; Paiva, W.S.; de Oliveira, D.V.; Teixeira, M.J.; de Andrade, A.F.; de Sousa, R.M. Diffuse axonal injury: Epidemiology, outcome and associated risk factors. Front. Neurol. 2016, 20, 178. [Google Scholar] [CrossRef] [Green Version]
- Sobuwa, S.; Hartzenberg, H.B.; Geduld, H.; Uys, C. Predicting outcome in severe traumatic brain injury using a simple prognostic model. S. Afr. Med. J. 2014, 104, 492–494. [Google Scholar] [CrossRef]
- Manley, G.; Knudson, M.M.; Morabito, D.; Damron, S.; Erickson, V.; Pitts, L. Hypotension, hypoxia, and head injury: Frequency, duration, and consequences. Arch. Surg. 2001, 136, 1118–1123. [Google Scholar] [CrossRef] [Green Version]
- Newfield, P.; Pitts, L.; Kaktis, J.; Hoff, J. The influence of shock on mortality after head trauma. Crit. Care Med. 1980, 8, 254. [Google Scholar] [CrossRef]
- Jeffreys, R.V.; Jones, J.J. Avoidable factors contributing to the death of head injury patients in general hospitals in Mersey Region. Lancet 1981, 29, 459–461. [Google Scholar] [CrossRef]
- Hill, D.A.; Abraham, K.H.; West, R.H. Factors affecting outcome in the resuscitation of severely injured patients. Aust. N. Z. J. Surg. 1993, 63, 604–609. [Google Scholar] [CrossRef]
- Mayer, T.; Walke, M.L.; Shasha, I.; Matlak, M.; Johnson, D.G. Effect of multiple trauma on outcome of pediatric patients with neurologic injuries. Childs Brain 1981, 8, 189–197. [Google Scholar] [CrossRef]
- Haddad, S.H.; Arabi, Y.M. Critical care management of severe traumatic brain injury in adults. Scand. J. Trauma Resusc. Emerg. Med. 2012, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Carney, N.; Totten, A.M.; O’Reilly, C.; Ullman, J.S.; Hawryluk, G.W.; Bell, M.J.; Bratton, S.L.; Chesnut, R.; Harris, O.A.; Kissoon, N.; et al. Guidelines for the management of severe traumatic brain injury, fourth edition. Neurosurgery 2017, 1, 6–15. [Google Scholar] [CrossRef]
- Clifton, G.L.; Ziegler, M.G.; Grossman, R.G. Circulating catecholamines and sympathetic activity after head injury. Neurosurgery 1981, 8, 10–13. [Google Scholar] [CrossRef]
- Rosner, M.J.; Newsome, H.H.; Becker, D.P. Mechanical brain injury: The sympathoadrenal response. J. Neurosurg. 1984, 61, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Bilotta, F.; Giovannini, F.; Caramia, R.; Rosa, G. Glycemia management in neurocritical care patients: A review. J. Neurosurg. Anesthesiol. 2009, 21, 2–9. [Google Scholar] [CrossRef]
- Liu-DeRyke, X.; Collingridge, D.S.; Orme, J.; Roller, D.; Zurasky, J.; Rhoney, D.H. Clinical impact of early hyperglycemia during acute phase of traumatic brain injury. Neurocrit. Care 2009, 11, 151–157. [Google Scholar] [CrossRef]
- Young, B.; Ott, L.; Dempsey, R.; Haack, D.; Tibbs, P. Relationship between admission hyperglycemia and neurologic outcome of severely brain-injured patients. Ann. Surg. 1989, 210, 466–472. [Google Scholar] [CrossRef]
- Lam, A.M.; Winn, H.R.; Cullen, B.F.; Sundling, N. Hyperglycemia and neurological outcome in patients with head injury. J. Neurosurg. 1991, 75, 545–551. [Google Scholar] [CrossRef]
- Chelly, H.; Chaari, A.; Daoud, E.; Dammak, H.; Medhioub, F.; Mnif, J.; Hamida, C.B.; Bahloul, M.; Bouaziz, M. Diffuse axonal injury in patients with head injuries: An epidemiologic and prognosis study of 124 cases. J. Trauma 2011, 71, 838–846. [Google Scholar] [CrossRef]
- Adams, J.H.; Graham, D.I.; Murray, L.S.; Scott, G. Diffuse axonal injury due to nonmissile head injury in humans: An analysis of 45 cases. Ann. Neurol. 1982, 12, 557–563. [Google Scholar] [CrossRef]
- Adams, J.H.; Doyle, D.; Ford, I.; Gennarelli, T.A.; Graham, D.I.; McLellan, D.R. Diffuse axonal injury in head injury: Definition, diagnosis and grading. Histopathology 1989, 15, 49–59. [Google Scholar] [CrossRef]
- Adams, J.H.; Doyle, D.; Graham, D.I.; Lawrence, A.E.; McLellan, D.R. Microscopic diffuse axonal injury in cases of head injury. Med. Sci. Law 1985, 25, 265–269. [Google Scholar] [CrossRef]
- Hill, C.S.; Coleman, M.P.; Menon, D.K. Traumatic axonal injury: Mechanisms and translational opportunities. Trends Neurosci. 2016, 39, 311–324. [Google Scholar] [CrossRef] [Green Version]
- Frati, A.; Cerretani, D.; Fiaschi, A.I.; Frati, P.; Gatto, V.; La Russa, R.; Pesce, A.; Pinchi, E.; Santurro, A.; Fraschetti, F.; et al. Diffuse axonal injury and oxidative stress: A comprehensive review. Int. J. Mol. Sci. 2017, 18, 2600. [Google Scholar] [CrossRef] [Green Version]
- Davceva, N.; Basheska, N.; Balazic, J. Diffuse axonal injury—A distinct clinicopathological entity in closed head injuries. Am. J Forensic Med. Pathol. 2015, 36, 127–133. [Google Scholar] [CrossRef]
- Gennarelli, T.A.; Thibault, L.E.; Adams, J.H.; Graham, D.I.; Thompson, C.J.; Marcincin, R.P. Diffuse axonal injury and traumatic coma in the primate. Ann. Neurol. 1982, 12, 564–574. [Google Scholar] [CrossRef]
- Kim, C.H.; Lee, H.K.; Koh, Y.C.; Hwang, D.Y. Clinical analysis of diffuse axonal injury (DAI) diagnosed with magnetic resonance image (MRI). J. Korean Neurosurg. Soc. 1997, 26, 241–248. [Google Scholar]
- Kim, H.J.; Park, I.S.; Kim, J.H.; Kim, K.J.; Hwang, S.H.; Kim, E.S.; Jung, J.M.; Han, J.W. Clinical analysis of the prognosis of the patients with cerebral diffuse axonal injuries, based on gradient-echo MR imaging. J. Korean Neurosurg. Soc. 2001, 30, 168–172. [Google Scholar]
- Baker, S.P.; O’Neill, B.; Haddon, W.; Long, W.B. The injury severity score: A method for describing patients with multiple injuries and evaluating emergency care. J. Trauma 1974, 14, 187–196. [Google Scholar] [CrossRef]
- Osler, T.; Baker, S.P.; Long, W. A modification of the injury severity score that both improves accuracy and simplifies scoring. J. Trauma 1997, 43, 922–925. [Google Scholar] [CrossRef]
- Association for the Advancement of Automotive Medice—AAAM. The Abbreviated Injury Scale (AIS): 2005, Update 2008. Available online: https://www.aaam.org/abbreviated-injury-scale-ais/ (accessed on 5 June 2020).
- Nogueira, L.S.; Domingues, C.A.; Campos, M.A.; Sousa, R.M.C. Ten years of new injury severity score (NISS): Is it a possible change? Rev. Lat. Am. Enfermagem. 2008, 16, 314–319. [Google Scholar] [CrossRef]
- Wilson, J.T.; Pettigrew, L.E.; Teasdale, G.M. Structured interviews for the Glasgow Outcome Scale and the extended Glasgow Outcome Scale: Guidelines for their use. J. Neurotrauma 1998, 15, 573–585. [Google Scholar] [CrossRef]
- Wierzbicki, V.; Pesce, A.; Marrocco, L.; Piccione, E.; Colonnese, C.; Caruso, R. How old is your cervical spine? Cervical spine biological age: A new evaluation scale. Eur. Spine J. 2015, 24, 2763–2770. [Google Scholar] [CrossRef]
- Armocida, D.; Pesce, A.; Frati, A.; Miscusi, M.; Paglia, F.; Raco, A. Pneumoventricle of unknown origin: A personal experience and literature review of a clinical enigma. World Neurosurg. 2019, 122, 661–664. [Google Scholar] [CrossRef] [Green Version]
- Teasdale, G.; Jennett, B. Assessment of coma and impaired consciousness. A practical scale. Lancet 1974, 304, 81–84. [Google Scholar]
- Matsukawa, H.; Shinoda, M.; Fujii, M.; Takahashi, O.; Murakata, A.; Yamamoto, D. Acute alcohol intoxication, diffuse axonal injury and intraventricular bleeding in patients with isolated blunt traumatic brain injury. Brain Inj. 2013, 27, 1409–1414. [Google Scholar] [CrossRef]
- Calvi, M.R.; Beretta, L.; Dell’Acqua, A.; Anzalone, N.; Licini, G.; Gemma, M. Early prognosis after severe traumatic brain injury with minor or absent computed tomography scan lesions. J. Trauma 2011, 70, 447–451. [Google Scholar]
- Skandsen, T.; Kvistad, K.A.; Solheim, O.; Strand, I.H.; Folvik, M.; Vik, A. Prevalence and impact of diffuse axonal injury in patients with moderate and severe head injury: A cohort study of early magnetic resonance imaging findings and 1-year outcome. J. Neurosurg. 2010, 113, 556–563. [Google Scholar] [CrossRef]
- Pinchi, E.; Frati, A.; Cipolloni, L.; Aromatario, M.; Gatto, V.; La Russa, R.; Pesce, A.; Santurro, A.; Fraschetti, F.; Frati, P.; et al. Clinical-pathological study on β-APP, IL-1β, GFAP, NFL, Spectrin II, 8OHdG, TUNEL, miR-21, miR-16, miR-92 expressions to verify DAI-diagnosis, grade and prognosis. Sci. Rep. 2018, 5, 2387. [Google Scholar] [CrossRef] [Green Version]
- Cheng, G. Mitochondria in traumatic brain injury and mitochondrial-targeted multipotential therapeutic strategies. Br. J. Pharmacol. 2012, 167, 699–719. [Google Scholar] [CrossRef] [Green Version]
- Halestrap, A.P. What is the mitochondrial permeability transition pore? J. Mol. Cell Cardiol. 2009, 46, 821–831. [Google Scholar] [CrossRef]
- Mazzeo, A.T. The role of mitochondrial transition pore, and its modulation, in traumatic brain injury and delayed neurodegeneration after TBI. Exp. Neurol. 2009, 218, 363–370. [Google Scholar] [CrossRef]
- Barsukova, A.G. Focal increases of axoplasmic Ca2+, aggregation of sodium–calcium exchanger, N-type Ca2+ channel, and actin define the sites of spheroids in axons undergoing oxidative stress. J. Neurosci. 2012, 32, 12028–12037. [Google Scholar] [CrossRef] [Green Version]
- Büki, A.; Okonkwo, D.O.; Povlishock, J.T. Postinjury cyclosporin a administration limits axonal damage and disconnection in traumatic brain injury. J. Neurotrauma 1999, 16, 511–521. [Google Scholar] [CrossRef]
- Okonkwo, D.O.; Povlishock, J.T. An intrathecal bolus of cyclosporin A before injury preserves mitochondrial integrity and attenuates axonal disruption in traumatic brain injury. J. Cereb. Blood Flow Metab. 1999, 19, 443–451. [Google Scholar] [CrossRef] [Green Version]
- Okonkwo, D.O.; Buki, A.; Siman, R.; Povlishock, J.T. Cyclosporin A limits calcium-induced axonal damage following traumatic brain injury. Neuroreport 1999, 10, 353–358. [Google Scholar] [CrossRef]
- Barrientos, S.A. Axonal degeneration is mediated by the mitochondrial permeability transition pore. J. Neurosci. 2011, 31, 966–978. [Google Scholar] [CrossRef]
- Staal, J.A. Initial calcium release from intracellular stores followed by calcium dysregulation is linked to secondary axotomy following transient axonal stretch injury. J. Neurochem. 2010, 112, 1147–1155. [Google Scholar] [CrossRef]
- Forte, M.; Gold, B.G.; Marracci, G.; Chaudhary, P.; Basso, E.; Johnsen, D.; Yu, X.; Fowlkes, J.; Rahder, M.; Stem, K.; et al. Cyclophilin D inactivation protects axons in experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2007, 104, 7558–7563. [Google Scholar] [CrossRef] [Green Version]
- Aminmansour, B.; Fard, S.A.; Habibabadi, M.R.; Moein, P.; Norouzi, R.; Naderan, M. The efficacy of Cyclosporine-A on diffuse axonal injury after traumatic brain injury. Adv. Biomed. Res. 2014, 3, 35. [Google Scholar]
- Empey, P.E.; McNamara, P.J.; Young, B.; Rosbolt, M.B.; Hatton, J. Cyclosporin A disposition following acute traumatic brain injury. J. Neurotrauma 2006, 23, 109–116. [Google Scholar] [CrossRef]
- Hatton, J.; Rosbolt, B.; Empey, P.; Kryscio, R.; Young, B. Dosing and safety of cyclosporine in patients with severe brain injury. J. Neurosurg. 2008, 109, 699–707. [Google Scholar] [CrossRef]
- Mazzeo, A.T.; Brophy, G.M.; Gilman, C.B.; Alves, O.L.; Robles, J.R.; Hayes, R.L.; Povlishock, J.T.; Bullock, M.R. Safety and tolerability of cyclosporin a in severe traumatic brain injury patients: Results from a prospective randomized trial. J. Neurotrauma 2009, 26, 2195–2206. [Google Scholar] [CrossRef] [Green Version]
- Büki, A.; Okonkwo, D.; Wang, K.K.; Povlishock, J.T. Cytochrome c release and caspase activation in traumatic axonal injury. J. Neurosci. 2000, 20, 2825–2834. [Google Scholar] [CrossRef] [Green Version]
- McAllister, T.W. Neurobiological consequences of traumatic brain injury. Dialogues Clin. Neurosci. 2011, 13, 287–300. [Google Scholar]
- Gatson, J.W.; Barillas, J.; Hynan, L.S.; Diaz-Arrastia, R.; Wolf, S.E.; Minei, J.P. Detection of neurofilament-H in serum as a diagnostic tool to predict injury severity in patients who have suffered mild traumatic brain injury. J. Neurosurg. 2014, 121, 1232–1238. [Google Scholar] [CrossRef] [Green Version]
- Shibahashi, K.; Doi, T.; Tanaka, S.; Hoda, H.; Chikuda, H.; Sawada, Y.; Takasu, Y.; Chiba, K.; Nozaki, T.; Hamabe, Y.; et al. The serum phosphorylated neurofilament heavy subunit as a predictive marker for outcome in adult patients after traumatic brain injury. J. Neurotrauma 2016, 33, 1826–1833. [Google Scholar] [CrossRef]
- Ljungqvist, J.; Zetterberg, H.; Mitsis, M.; Blennow, K.; Skoglund, T. Serum neurofilament light protein as a marker for diffuse axonal injury: Results from a case series study. J. Neurotrauma 2017, 1, 1124–1127. [Google Scholar] [CrossRef]
- Nylén, K.; Ost, M.; Csajbok, L.Z.; Nilsson, I.; Blennow, K.; Nellgård, B.; Rosengren, L. Increased serum-GFAP in patients with severe traumatic brain injury is related to outcome. J. Neurol. Sci. 2006, 240, 85–91. [Google Scholar] [CrossRef]
- Neri, M.; Frati, A.; Turillazzi, E.; Cantatore, S.; Cipolloni, L.; Di Paolo, M.; Frati, P.; La Russa, R.; Maiese, A.; Scopetti, M.; et al. Immunohistochemical Evaluation of Aquaporin-4 and its Correlation with CD68, IBA-1, HIF-1α, GFAP, and CD15 Expressions in Fatal Traumatic Brain Injury. Int. J. Mol. Sci. 2018, 19, 3544. [Google Scholar] [CrossRef] [Green Version]
- Zemlan, F.P.; Jauch, E.C.; Mulchahey, J.J.; Gabbita, S.P.; Rosenberg, W.S.; Speciale, S.G.; Zuccarello, M. C-tau biomarker of neuronal damage in severe brain injured patients: Association with elevated intracranial pressure and clinical outcome. Brain Res. 2002, 947, 131–139. [Google Scholar] [CrossRef]
- Roberts, G.W.; Gentleman, S.M.; Lynch, A.; Murray, L.; Landon, M.; Graham, D.I. Beta amyloid protein deposition in the brain after severe head injury: Implications for the pathogenesis of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 1994, 57, 419–425. [Google Scholar] [CrossRef] [Green Version]
- Bogoslovsky, T.; Wilson, D.; Chen, Y.; Hanlon, D.; Gill, J.; Jeromin, A.; Song, L.; Moore, C.; Gong, Y.; Kenney, K.; et al. Increases of plasma levels of glial fibrillary acidic protein, tau, and amyloid ß up to 90 days after traumatic brain injury. J. Neurotrauma 2017, 34, 66–73. [Google Scholar] [CrossRef] [Green Version]
- Raabe, A.; Grolms, C.; Sorge, O.; Zimmermann, M.; Seifert, V. Serum S-100B protein in severe head injury. Neurosurgery 1999, 45, 477–483. [Google Scholar] [CrossRef]
- Romner, B.; Ingebrigtsen, T.; Kongstad, P.; Børgesen, S.E. Traumatic brain damage: Serum S-100 protein measurements related to neuroradiological findings. J. Neurotrauma 2000, 17, 641–647. [Google Scholar] [CrossRef]
- Ingebrigtsen, T.; Romner, B. Biochemical serum markers for brain damage: A short review with emphasis on clinical utility in mild head injury. Restor. Neurol. Neurosci. 2003, 21, 171–176. [Google Scholar]
- Kleindienst, A.; Schmidt, C.; Parsch, H.; Emtmann, I.; Xu, Y.; Buchfelder, M. The passage of S100B from brain to blood is not specifically related to the blood-brain barrier integrity. Cardiovasc. Psych. Neurol. 2010, 2010, 801295. [Google Scholar] [CrossRef]
- Anderson, R.E.; Hansson, L.O.; Nilsson, O.; Dijlai-Merzoug, R.; Settergren, G. High serum S100B levels for trauma patients without head injuries. Neurosurgery 2001, 48, 1255–1258. [Google Scholar]
- Thelin, E.P.; Jeppsson, E.; Frostell, A.; Svensson, M.; Mondello, S.; Bellander, B.M.; Nelson, D.W. Utility of neuron-specific enolase in traumatic brain injury; relations to S100B levels, outcome, and extracranial injury severity. Crit. Care 2016, 8, 285. [Google Scholar] [CrossRef] [Green Version]
- Pelinka, L.E.; Kroepfl, A.; Leixnering, M.; Buchinger, W.; Raabe, A.; Redl, H. GFAP versus S100B in serum after traumatic brain injury: Relationship to brain damage and outcome. J. Neurotrauma 2004, 21, 1553–1561. [Google Scholar] [CrossRef]
- Kövesdi, E.; Lückl, J.; Bukovics, P.; Farkas, O.; Pál, J.; Czeiter, E.; Szellár, D.; Dóczi, T.; Komoly, S.; Büki, A. Update on protein biomarkers in traumatic brain injury with emphasis on clinical use in adults and paediatrics. Acta Neurochir. 2010, 152, 1–17. [Google Scholar] [CrossRef]
- Berger, R.P.; Beers, S.R.; Richichi, R.; Wiesman, D.; Adelson, P.D. Serum biomarker concentrations and outcome after pediatric traumatic brain injury. J. Neurotrauma 2007, 24, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.M.; Zheng, Z.H.; Fan, Z.H.; Zhang, Z.F.; Chen, G.Q.; Lun, J. Sulforaphane administration alleviates diffuse axonal injury (DAI) via regulation signaling pathway of NRF2 and HO-1. J. Cell Biochem. 2020, 121, 430–442. [Google Scholar] [CrossRef]
- Ishii, T.; Itoh, K.; Ruiz, E.; Leake, D.S.; Unoky, H.; Yamamoto, M.; Mann, G.E. Role of Nrf2 in the regulation ofCD36 and stress protein expression in murine macrophages: Activation by oxidatively modified LDL and 4-hydroxynonenal. Circ. Res. 2004, 94, 609–616. [Google Scholar] [CrossRef] [Green Version]
- Wakabayashi, N.; Slocum, S.L.; Skoko, J.J.; Shin, S.; Kensler, T.W. When NRF2 talks, who’s listening? Antioxid. Redox Signal. 2010, 13, 1649–1663. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Zhou, M.; Ouyang, Y.; Wang, H. Activation of the nuclear factor erythroid 2-related factor 2-antioxidant response element signal in rats with diffuse axonal injury. Neuroreport 2019, 30, 389–396. [Google Scholar] [CrossRef]
- Kang, K.W.; Lee, S.J.; Kim, S.G. Molecular mechanism of nrf2 activation by oxidative stress. Antioxid. Redox Signal. 2005, 7, 1664–1673. [Google Scholar] [CrossRef]
- Dinkova Kostova, A.T.; Kostov, R.V. Glucosinolates and isothiocyanates in health and disease. Trends Mol. Med. 2012, 18, 337–347. [Google Scholar] [CrossRef]
- Omelchenko, A.; Shrirao, A.B.; Bhattiprolu, A.K.; Zhan, J.D.; Schloss, R.S.; Dickson, S.; Meaney, D.F.; Boustany, N.N.; Yarmush, L.L.; Firestein, B.L. Dynamin and reverse-mode sodium calcium exchanger blockade confers neuroprotection from diffuse axonal injury. Cell Death Dis. 2019, 10, 727. [Google Scholar] [CrossRef] [Green Version]
- Chou, A.C.; Ju, Y.T.; Pan, C.Y. Calmodulin interacts with the sodium/calcium exchanger NCX1 to regulate activity. PLoS ONE 2015, 10, e0138856. [Google Scholar]
- Morrison, B., III; Elkin, B.S.; Dolle, J.P.; Yarmush, M.L. In vitro models of traumatic brain injury. Annu. Rev. Biomed. Eng. 2011, 13, 91–126. [Google Scholar] [CrossRef]
- Jeffs, G.J.; Meloni, B.P.; Bakker, A.J.; Knuckey, N.W. The role of the Na+/Ca2+ exchanger (NCX) in neurons following ischaemia. J. Clin. Neurosci. 2007, 14, 507–514. [Google Scholar] [CrossRef]
- Weber, J.T. Altered calcium signaling following traumatic brain injury. Front. Pharmacol. 2012, 3, 60. [Google Scholar] [CrossRef] [Green Version]
- Kitchen, P.; Salman, M.M.; Halsey, A.M.; Clarke-Bland, C.; MacDonald, J.A.; Ishida, H.; Vogel, H.J.; Almutiri, S.; Logan, A.; Kreida, S.; et al. Targeting aquaporin-4 subcellular localization to treat central nervous system edema. Cell 2020, 181, 784–799.e19. [Google Scholar]
- Salman, M.M.; Kitchen, P.; Halsey, A.; Wang, M.X.; Tornroth-Horsefield, S.; Conner, A.C.; Badaut, J.; Iliff, J.J.; Bill, R.M. Emerging roles for dynamic aquaporin-4 subcellular relocalization in CNS water homeostasis. Brain 2021. online ahead of print. [Google Scholar] [CrossRef]
- Sylvain, N.J.; Salman, M.M.; Pushie, M.J.; Hou, H.; Meher, V.; Herlo, R.; Peeling, L.; Kelly, M.E. The effects of trifluoperazine on brain edema, aquaporin-4 expression and metabolic markers during the acute phase of stroke using photothrombotic mouse model. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183573. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palmieri, M.; Frati, A.; Santoro, A.; Frati, P.; Fineschi, V.; Pesce, A. Diffuse Axonal Injury: Clinical Prognostic Factors, Molecular Experimental Models and the Impact of the Trauma Related Oxidative Stress. An Extensive Review Concerning Milestones and Advances. Int. J. Mol. Sci. 2021, 22, 10865. https://doi.org/10.3390/ijms221910865
Palmieri M, Frati A, Santoro A, Frati P, Fineschi V, Pesce A. Diffuse Axonal Injury: Clinical Prognostic Factors, Molecular Experimental Models and the Impact of the Trauma Related Oxidative Stress. An Extensive Review Concerning Milestones and Advances. International Journal of Molecular Sciences. 2021; 22(19):10865. https://doi.org/10.3390/ijms221910865
Chicago/Turabian StylePalmieri, Mauro, Alessandro Frati, Antonio Santoro, Paola Frati, Vittorio Fineschi, and Alessandro Pesce. 2021. "Diffuse Axonal Injury: Clinical Prognostic Factors, Molecular Experimental Models and the Impact of the Trauma Related Oxidative Stress. An Extensive Review Concerning Milestones and Advances" International Journal of Molecular Sciences 22, no. 19: 10865. https://doi.org/10.3390/ijms221910865
APA StylePalmieri, M., Frati, A., Santoro, A., Frati, P., Fineschi, V., & Pesce, A. (2021). Diffuse Axonal Injury: Clinical Prognostic Factors, Molecular Experimental Models and the Impact of the Trauma Related Oxidative Stress. An Extensive Review Concerning Milestones and Advances. International Journal of Molecular Sciences, 22(19), 10865. https://doi.org/10.3390/ijms221910865