
Adipokines as Immune Cell Modulators in Multiple Sclerosis


Abstract
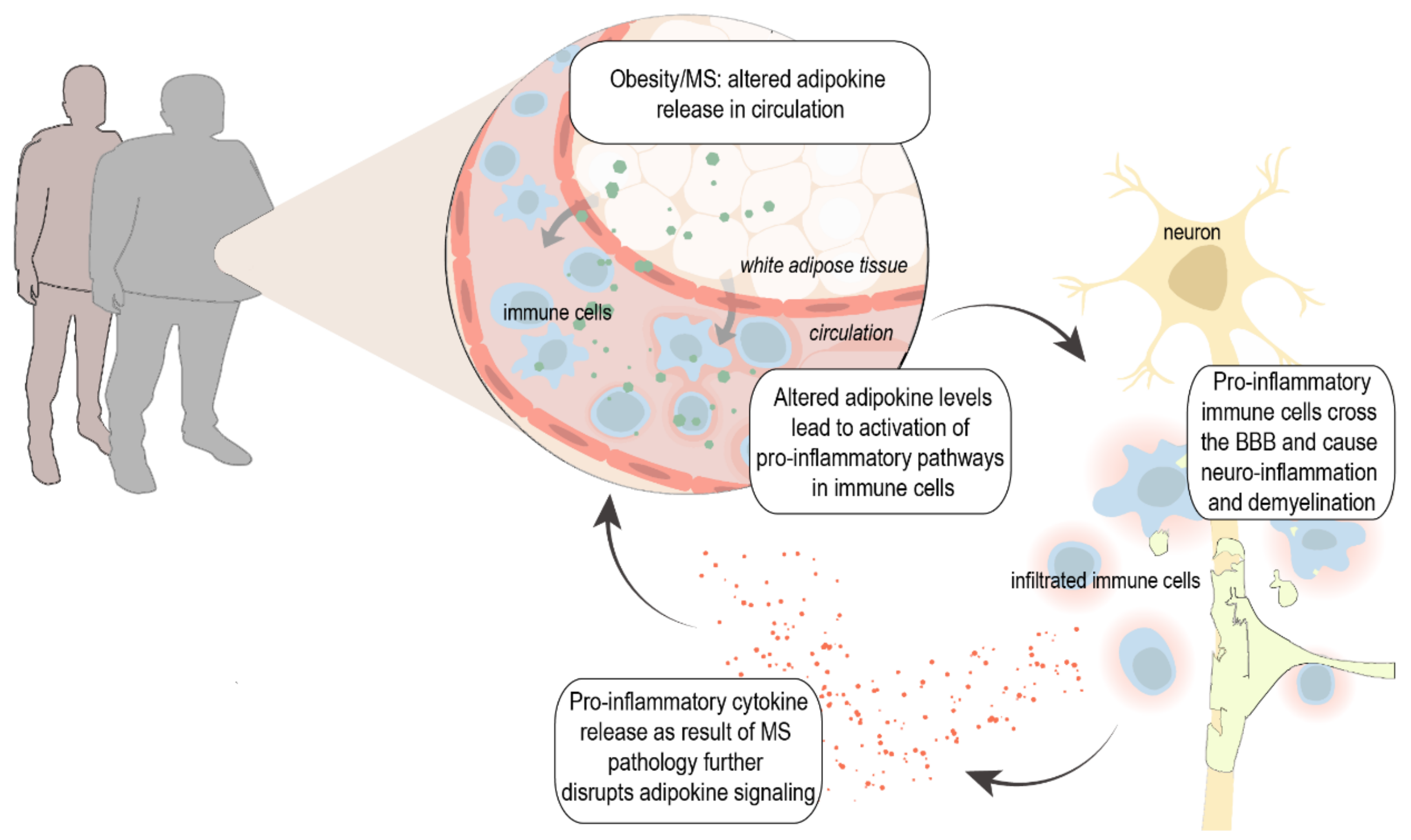
1. Introduction
2. MS Pathophysiology, Therapies, and Risk Factors
2.1. Pathophysiology
2.2. Disease-Modifying Treatments
2.3. Obesity as Risk Factor for MS
3. Adipokines, Inflammation, and MS
3.1. Adiponectin
3.2. Leptin
3.3. Resistin
3.4. Chemerin
3.5. Visfatin
3.6. Apelin
3.7. Confounding Factors
4. Adipokines as Therapeutic Target for MS—Evidence from Pre-Clinical Studies
4.1. Adiponectin
4.2. Leptin
4.3. Resistin
4.4. Chemerin
4.5. Visfatin
4.6. Apelin
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walton, C.; King, R.; Rechtman, L.; Kaye, W.; Leray, E.; Marrie, R.A.; Robertson, N.; La Rocca, N.; Uitdehaag, B.; Van Der Mei, I.; et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Mult. Scler. J. 2020, 26, 1816–1821. [Google Scholar] [CrossRef] [PubMed]
- Gianfrancesco, M.; Acuna, B.; Shen, L.; Briggs, F.; Quach, H.; Bellesis, K.H.; Bernstein, A.; Hedstrom, A.K.; Kockum, I.; Alfredsson, L.; et al. Obesity during childhood and adolescence increases susceptibility to multiple sclerosis after accounting for established genetic and environmental risk factors. Obes. Res. Clin. Pr. 2014, 8, e435–e447. [Google Scholar] [CrossRef] [PubMed]
- Munger, K.L.; Chitnis, T.; Ascherio, A. Body size and risk of MS in two cohorts of US women. Neurology 2009, 73, 1543–1550. [Google Scholar] [CrossRef] [PubMed]
- Olsson, T.; Barcellos, L.F.; Alfredsson, L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat. Rev. Neurol. 2017, 13, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Hedström, A.K.; Olsson, T.; Alfredsson, L. Body mass index during adolescence, rather than childhood, is critical in determining MS risk. Mult. Scler. J. 2015, 22, 878–883. [Google Scholar] [CrossRef]
- Munger, K.L. Childhood obesity is a risk factor for multiple sclerosis. Mult. Scler. J. 2013, 19, 1800. [Google Scholar] [CrossRef]
- Bassi, M.A.U.S.; Iezzi, E.; Buttari, F.; Gilio, L.; Simonelli, I.; Carbone, F.; Micillo, T.; De Rosa, V.; Sica, F.; Furlan, R.; et al. Obesity worsens central inflammation and disability in multiple sclerosis. Mult. Scler. J. 2020, 26, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- Mowry, E.M.; Azevedo, C.J.; McCulloch, C.E.; Okuda, D.T.; Lincoln, R.R.; Waubant, E.; Hauser, S.L.; Pelletier, D. Body mass index, but not vitamin D status, is associated with brain volume change in MS. Neurology 2018, 91, e2256–e2264. [Google Scholar] [CrossRef] [PubMed]
- Lago, F.; Diéguez, C.; Gómez-Reino, J.; Gualillo, O. Adipokines as emerging mediators of immune response and inflammation. Nat. Clin. Pr. Rheumatol. 2007, 3, 716–724. [Google Scholar] [CrossRef]
- Huang, S.; Wang, Y.; Gan, X.; Fang, D.; Zhong, C.; Wu, L.; Hu, G.; Sosunov, A.A.; McKhann, G.M.; Yu, H.; et al. Drp1-Mediated Mitochondrial Abnormalities Link to Synaptic Injury in Diabetes Model. Diabetes 2014, 64, 1728–1742. [Google Scholar] [CrossRef]
- Dayakar, A.; Chandrasekaran, S.; Veronica, J.; Maurya, R. Leptin induces the phagocytosis and protective immune response in Leishmania donovani infected THP-1 cell line and human PBMCs. Exp. Parasitol. 2016, 160, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Mastronardi, C.; Yu, W.H.; Rettori, V.; McCann, S. Lipopolysaccharide-Induced Leptin Release Is Not Mediated by Nitric Oxide, but Is Blocked by Dexamethasone. Neuroimmunomodulation 2000, 8, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef]
- Magliozzi, R.; Howell, O.; Durrenberger, P.; Aricò, E.; James, R.; Cruciani, C.; Reeves, C.; Roncaroli, F.; Nicholas, R.; Reynolds, R. Meningeal inflammation changes the balance of TNF signalling in cortical grey matter in multiple sclerosis. J. Neuroinflam. 2019, 16, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Bevan, R.; Evans, R.; Griffiths, L.; Watkins, L.M.; Rees, M.I.; Magliozzi, R.; Allen, I.; McDonnell, G.; Kee, R.; Naughton, M.; et al. Meningeal inflammation and cortical demyelination in acute multiple sclerosis. Ann. Neurol. 2018, 84, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Howell, O.W.; Reeves, C.A.; Nicholas, R.; Carassiti, D.; Radotra, B.; Gentleman, S.M.; Serafini, B.; Aloisi, F.; Roncaroli, F.; Magliozzi, R.; et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain 2011, 134, 2755–2771. [Google Scholar] [CrossRef]
- Silva, B.A.; Miglietta, E.; Ferrari, C.C. Insights into the role of B cells in the cortical pathology of Multiple sclerosis: Evidence from animal models and patients. Mult. Scler. Relat. Disord. 2021, 50, 102845. [Google Scholar] [CrossRef]
- Van Olst, L.; Rodriguez-Mogeda, C.; Picon, C.; Kiljan, S.; James, R.E.; Kamermans, A.; van der Pol, S.M.A.; Knoop, L.; Michailidou, I.; Drost, E.; et al. Meningeal inflammation in multiple sclerosis induces phenotypic changes in cortical microglia that differentially associate with neurodegeneration. Acta Neuropathol. 2021, 141, 881–899. [Google Scholar] [CrossRef]
- McGinley, M.P.; Goldschmidt, C.H.; Rae-Grant, A.D. Diagnosis and Treatment of Multiple Sclerosis: A Review. JAMA 2021, 325, 765–779. [Google Scholar] [CrossRef]
- Cohan, S.L.; Hendin, B.A.; Reder, A.T.; Smoot, K.; Avila, R.; Mendoza, J.P.; Weinstock-Guttman, B. Interferons and Multiple Sclerosis: Lessons from 25 Years of Clinical and Real-World Experience with Intramuscular Interferon Beta-1a (Avonex). CNS Drugs 2021, 35, 743–767. [Google Scholar] [CrossRef]
- Prod’Homme, T.; Zamvil, S.S. The Evolving Mechanisms of Action of Glatiramer Acetate. Cold Spring Harb. Perspect. Med. 2018, 9, a029249. [Google Scholar] [CrossRef]
- Mills, E.A.; Ogrodnik, M.A.; Plave, A.; Mao-Draayer, Y. Emerging Understanding of the Mechanism of Action for Dimethyl Fumarate in the Treatment of Multiple Sclerosis. Front. Neurol. 2018, 9, 5. [Google Scholar] [CrossRef]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; De Seze, J.; Giovannoni, G.; Hartung, H.-P.; Hemmer, B.; et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 209–220. [Google Scholar] [CrossRef]
- Hawker, K.; O’Connor, P.; Freedman, M.S.; Calabresi, P.A.; Antel, J.; Simon, J.; Hauser, S.; Waubant, E.; Vollmer, T.; Panitch, H.; et al. Rituximab in patients with primary progressive multiple sclerosis: Results of a randomized double-blind placebo-controlled multicenter trial. Ann. Neurol. 2009, 66, 460–471. [Google Scholar] [CrossRef]
- Hauser, S.L.; Waubant, E.; Arnold, D.L.; Vollmer, T.; Antel, J.; Fox, R.J.; Bar-Or, A.; Panzara, M.; Sarkar, N.; Agarwal, S.; et al. B-Cell Depletion with Rituximab in Relapsing–Remitting Multiple Sclerosis. N. Engl. J. Med. 2008, 358, 676–688. [Google Scholar] [CrossRef] [PubMed]
- Krumbholz, M.; Derfuss, T.; Hohlfeld, R.; Meinl, E. B cells and antibodies in multiple sclerosis pathogenesis and therapy. Nat. Rev. Neurol. 2012, 8, 613–623. [Google Scholar] [CrossRef]
- Myhr, K.-M.; Torkildsen, Ø.; Lossius, A.; Bø, L.; Holmøy, T. B cell depletion in the treatment of multiple sclerosis. Expert Opin. Biol. Ther. 2019, 19, 261–271. [Google Scholar] [CrossRef]
- Mokry, L.E.; Ross, S.; Timpson, N.J.; Sawcer, S.; Smith, G.D.; Richards, J.B. Obesity and Multiple Sclerosis: A Mendelian Randomization Study. PLoS Med. 2016, 13, e1002053. [Google Scholar] [CrossRef] [PubMed]
- Baranowska-Bik, A.; Uchman, D.; Litwiniuk, A.; Kalisz, M.; Martyńska, L.; Baranowska, B.; Bik, W.; Kochanowski, J. Peripheral levels of selected adipokines in patients with newly diagnosed multiple sclerosis. Endokrynol. Polska 2020, 71, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Kvistad, S.S.; Myhr, K.-M.; Holmøy, T.; Benth, J.Š.; Wergeland, S.; Beiske, A.G.; Bjerve, K.S.; Hovdal, H.; Lilleås, F.; Midgard, R.; et al. Body mass index influence interferon-beta treatment response in multiple sclerosis. J. Neuroimmunol. 2015, 288, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Asghar, A.; Sheikh, N. Role of immune cells in obesity induced low grade inflammation and insulin resistance. Cell. Immunol. 2017, 315, 18–26. [Google Scholar] [CrossRef]
- Bai, Z.; Chen, D.; Wang, L.; Zhao, Y.; Liu, T.; Yu, Y.; Yan, T.; Cheng, Y. Cerebrospinal Fluid and Blood Cytokines as Biomarkers for Multiple Sclerosis: A Systematic Review and Meta-Analysis of 226 Studies With 13,526 Multiple Sclerosis Patients. Front. Neurosci. 2019, 13, 1026. [Google Scholar] [CrossRef]
- Cryan, J.F.; O’Riordan, K.J.; Sandhu, K.; Peterson, V.; Dinan, T.G. The gut microbiome in neurological disorders. Lancet Neurol. 2020, 19, 179–194. [Google Scholar] [CrossRef]
- Fasshauer, M.; Blüher, M. Adipokines in health and disease. Trends Pharmacol. Sci. 2015, 36, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Kamermans, A.; Verhoeven, T.; Hof, B.V.H.; Koning, J.J.; Borghuis, L.; Witte, M.; Van Horssen, J.; De Vries, H.E.; Rijnsburger, M. Setmelanotide, a Novel, Selective Melanocortin Receptor-4 Agonist Exerts Anti-inflammatory Actions in Astrocytes and Promotes an Anti-inflammatory Macrophage Phenotype. Front. Immunol. 2019, 10, 2312. [Google Scholar] [CrossRef] [PubMed]
- Pineda-Torra, I.; Siddique, S.; Waddington, K.E.; Farrell, R.; Jury, E.C. Disrupted Lipid Metabolism in Multiple Sclerosis: A Role for Liver X Receptors? Front. Endocrinol. 2021, 12, 639757. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lam, K.S.L.; Yau, M.-H.; Xu, A. Post-translational modifications of adiponectin: Mechanisms and functional implications. Biochem. J. 2008, 409, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Heiker, J.T.; Kosel, D.; Beck-Sickinger, A.G. Molecular mechanisms of signal transduction via adiponectin and adiponectin receptors. Biol. Chem. 2010, 391, 1005–1018. [Google Scholar] [CrossRef] [PubMed]
- Simons, P.J.; Pangaart, P.S.V.D.; Aerts, J.M.F.G.; Boon, L. Pro-inflammatory delipidizing cytokines reduce adiponectin secretion from human adipocytes without affecting adiponectin oligomerization. J. Endocrinol. 2007, 192, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Surendar, J.; Frohberger, S.J.; Karunakaran, I.; Schmitt, V.; Stamminger, W.; Neumann, A.L.; Wilhelm, C.; Hoerauf, A.; Hubner, M.P. Adiponectin Limits IFN-gamma and IL-17 Producing CD4 T Cells in Obesity by Restraining Cell Intrinsic Glycolysis. Front. Immunol. 2019, 10, 2555. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Folco, E.J.; Shimizu, K.; Libby, P. Adiponectin Induces Pro-inflammatory Programs in Human Macrophages and CD4+ T Cells. J. Biol. Chem. 2012, 287, 36896–36904. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, X.; Lau, W.B.; Yuan, Y.; Booth, D.; Li, J.-J.; Scalia, R.; Preston, K.; Gao, E.; Koch, W.; et al. Adiponectin inhibits tumor necrosis factor-α-induced vascular inflammatory response via caveolin-mediated ceramidase recruitment and activation. Circ. Res. 2014, 114, 792–805. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, A.; Yamauchi, T.; Takekawa, S.; Hada, Y.; Ito, Y.; Maki, T.; Kadowaki, T. Peroxisome Proliferator-Activated Receptor (PPAR) Activation Increases Adiponectin Receptors and Reduces Obesity-Related Inflammation in Adipose Tissue: Comparison of Activation of PPAR, PPAR, and Their Combination. Diabetes 2005, 54, 3358–3370. [Google Scholar] [CrossRef]
- Yousefian, M.; Nemati, R.; Daryabor, G.; Gholijani, N.; Nikseresht, A.; Haghighi, A.B.; Kamali-Sarvestani, E. Gender-Specific Association of Leptin and Adiponectin Genes with Multiple Sclerosis. Am. J. Med. Sci. 2018, 356, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Çoban, A.; Düzel, B.; Tüzün, E.; Tamam, Y. Investigation of the prognostic value of adipokines in multiple sclerosis. Mult. Scler. Relat. Disord. 2017, 15, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Tehrani, A.R.; Gholipour, S.; Sharifi, R.; Yadegari, S.; Abbasi-Kolli, M.; Masoudian, N. Plasma levels of CTRP-3, CTRP-9 and apelin in women with multiple sclerosis. J. Neuroimmunol. 2019, 333, 576968. [Google Scholar] [CrossRef] [PubMed]
- Kraszula, Ł.; Jasińska, A.; Eusebio, M.-O.; Kuna, P.; Głąbiński, A.; Pietruczuk, M. Evaluation of the relationship between leptin, resistin, adiponectin and natural regulatory T cells in relapsing-remitting multiple sclerosis. Neurol. Neurochir. Polska 2012, 46, 22–28. [Google Scholar] [CrossRef]
- Musabak, U.H.; Demirkaya, S.; Genç, G.; Ilikci, R.S.; Odabasi, Z. Serum Adiponectin, TNF-α, IL-12p70, and IL-13 Levels in Multiple Sclerosis and the Effects of Different Therapy Regimens. Neuroimmunomodulation 2011, 18, 57–66. [Google Scholar] [CrossRef]
- Kvistad, S.S.; Myhr, K.-M.; Holmøy, T.; Benth, J.Š.; Wergeland, S.; Beiske, A.G.; Bjerve, K.S.; Hovdal, H.; Midgard, R.; Sagen, J.V.; et al. Serum levels of leptin and adiponectin are not associated with disease activity or treatment response in multiple sclerosis. J. Neuroimmunol. 2018, 323, 73–77. [Google Scholar] [CrossRef]
- Signoriello, E.; Lus, G.; Polito, R.; Casertano, S.; Scudiero, O.; Coletta, M.; Monaco, M.L.; Rossi, F.; Nigro, E.; Daniele, A. Adiponectin profile at baseline is correlated to progression and severity of multiple sclerosis. Eur. J. Neurol. 2019, 26, 348–355. [Google Scholar] [CrossRef]
- Düzel, B.; Tamam, Y.; Çoban, A.; Tüzün, E. Adipokines in Multiple Sclerosis Patients with and without Optic Neuritis as the First Clinical Presentation. Immunol. Investig. 2018, 48, 190–197. [Google Scholar] [CrossRef]
- Signoriello, E.; Mallardo, M.; Nigro, E.; Polito, R.; Casertano, S.; di Pietro, A.; Coletta, M.; Monaco, M.L.; Rossi, F.; Lus, G.; et al. Adiponectin in Cerebrospinal Fluid from Patients Affected by Multiple Sclerosis Is Correlated with the Progression and Severity of Disease. Mol. Neurobiol. 2021, 58, 2663–2670. [Google Scholar] [CrossRef]
- Hietaharju, A.; Kuusisto, H.; Nieminen, R.; Vuolteenaho, K.; Elovaara, I.; Moilanen, E. Elevated cerebrospinal fluid adiponectin and adipsin levels in patients with multiple sclerosis: A Finnish co-twin study. Eur. J. Neurol. 2009, 17, 332–334. [Google Scholar] [CrossRef] [PubMed]
- Francisco, V.; Pino, J.; Campos-Cabaleiro, V.; Ruiz-Fernández, C.; Mera, A.; Gonzalez-Gay, M.A.; Gómez, R.; Gualillo, O. Obesity, Fat Mass and Immune System: Role for Leptin. Front. Physiol. 2018, 9, 640. [Google Scholar] [CrossRef]
- Tartaglia, L.A.; Dembski, M.; Weng, X.; Deng, N.; Culpepper, J.; Devos, R.; Richards, G.J.; Campfield, L.; Clark, F.T.; Deeds, J.; et al. Identification and expression cloning of a leptin receptor, OB-R. Cell 1995, 83, 1263–1271. [Google Scholar] [CrossRef]
- Villanueva, E.C.; Myers, M.G. Leptin receptor signaling and the regulation of mammalian physiology. Int. J. Obes. 2008, 32, S8–S12. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Agrawal, S.; Gollapudi, S. Increased activation and cytokine secretion in B cells stimulated with leptin in aged humans. Immun. Ageing 2013, 10, 3. [Google Scholar] [CrossRef]
- Marrodan, M.; Farez, M.F.; Aguirre, M.E.B.; Correale, J. Obesity and the risk of Multiple Sclerosis. The role of Leptin. Ann. Clin. Transl. Neurol. 2021, 8, 406–424. [Google Scholar] [CrossRef] [PubMed]
- Shirshev, S.V.; Orlova, E.G. Molecular Mechanisms of Regulation of Functional Activity of Mononuclear Phagocytes by Leptin. Biochemistry (Moscow) 2005, 70, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Lafrance, V.; Inoue, W.; Kan, B.; Luheshi, G.N. Leptin modulates cell morphology and cytokine release in microglia. Brain Behav. Immun. 2010, 24, 358–365. [Google Scholar] [CrossRef]
- Tang, C.-H.; Lu, D.-Y.; Yang, R.-S.; Tsai, H.-Y.; Kao, M.-C.; Fu, W.-M.; Chen, Y.-F. Leptin-Induced IL-6 Production Is Mediated by Leptin Receptor, Insulin Receptor Substrate-1, Phosphatidylinositol 3-Kinase, Akt, NF-κB, and p300 Pathway in Microglia. J. Immunol. 2007, 179, 1292–1302. [Google Scholar] [CrossRef]
- Bahrami, E.; Zarkesh-Esfahani, S.H.; Kardi, M.T.; Mostajeran, M.; Triot, A.; Bouzari, M.; Maghzi, A.H.; Etemadifar, M. Leptin hormone level in serum of opticospinal, neuromyelitisoptica and multiple sclerosis patients. Clin. Exp. Neuroimmunol. 2014, 5, 77–83. [Google Scholar] [CrossRef]
- Chatzantoni, K.; Papathanassopoulos, P.; Gourzoulidou, E.; Mouzaki, A. Leptin and its soluble receptor in plasma of patients suffering from remitting–relapsing multiple sclerosis (MS): In vitro effects of leptin on type-1 and type-2 cytokine secretion by peripheral blood mononuclear cells, T-cells and monocytes of MS patients. J. Autoimmun. 2004, 23, 169–177. [Google Scholar] [CrossRef]
- Emamgholipour, S.; Eshaghi, S.M.; Hossein-nezhad, A.; Mirzaei, K.; Maghbooli, Z.; Sahraian, M.A. Adipocytokine profile, cytokine levels and foxp3 expression in multiple sclerosis: A possible link to susceptibility and clinical course of disease. PLoS ONE 2013, 8, e76555. [Google Scholar] [CrossRef]
- Joseph, D.; Kumar, S. Identifying clues to molecular etiology of multiple sclerosis in South Indian patients. Mult. Scler. Relat. Disord. 2016, 5, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Matarese, G.; Carrieri, P.B.; La Cava, A.; Perna, F.; Sanna, V.; De Rosa, V.; Aufiero, D.; Fontana, S.; Zappacosta, S. Leptin increase in multiple sclerosis associates with reduced number of CD4+CD25+ regulatory T cells. Proc. Natl. Acad. Sci. USA 2005, 102, 5150–5155. [Google Scholar] [CrossRef] [PubMed]
- Messina, S.; Vargas-Lowy, D.; Musallam, A.; Healy, B.C.; Kivisakk, P.; Gandhi, R.; Bove, R.; Gholipour, T.; Khoury, S.; Weiner, H.L.; et al. Increased leptin and A-FABP levels in relapsing and progressive forms of MS. BMC Neurol. 2013, 13, 172. [Google Scholar] [CrossRef]
- Penesova, A.; Vlcek, M.; Imrich, R.; Vernerova, L.; Marko, A.; Meskova, M.; Grunnerova, L.; Turčáni, P.; Jezova, D.; Kollár, B. Hyperinsulinemia in newly diagnosed patients with multiple sclerosis. Metab. Brain Dis. 2015, 30, 895–901. [Google Scholar] [CrossRef]
- Rotondi, M.; Batocchi, A.P.; Coperchini, F.; Caggiula, M.; Zerbini, F.; Sideri, R.; Leporati, P.; Nociti, V.; Frisullo, G.; Mirabella, M.; et al. Severe Disability in Patients with Relapsing-Remitting Multiple Sclerosis Is Associated with Profound Changes in the Regulation of Leptin Secretion. Neuroimmunomodulation 2013, 20, 341–347. [Google Scholar] [CrossRef]
- Dashti, M.; Alroughani, R.; Jacob, S.; Al-Temaimi, R. Leptin rs7799039 polymorphism is associated with multiple sclerosis risk in Kuwait. Mult. Scler. Relat. Disord. 2019, 36, 101409. [Google Scholar] [CrossRef]
- Evangelopoulos, M.E.; Koutsis, G.; Markianos, M. Serum Leptin Levels in Treatment-Naive Patients with Clinically Isolated Syndrome or Relapsing-Remitting Multiple Sclerosis. Autoimmune Dis. 2014, 2014, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Frisullo, G.; Mirabella, M.; Angelucci, F.; Caggiula, M.; Morosetti, R.; Sancricca, C.; Patanella, A.K.; Nociti, V.; Iorio, R.; Bianco, A.; et al. The effect of disease activity on leptin, leptin receptor and suppressor of cytokine signalling-3 expression in relapsing–remitting multiple sclerosis. J. Neuroimmunol. 2007, 192, 174–183. [Google Scholar] [CrossRef]
- Batocchi, A.P.; Rotondi, M.; Caggiula, M.; Frisullo, G.; Odoardi, F.; Nociti, V.; Carella, C.; Tonali, P.A.; Mirabella, M. Leptin as a marker of multiple sclerosis activity in patients treated with interferon-beta. J. Neuroimmunol. 2003, 139, 150–154. [Google Scholar] [CrossRef]
- Bahrami, E.; Mirmoghtadaee, P.; Ardalan, G.; Zarkesh-Esfahani, H.; Tajaddini, M.H.; Haghjooy-Javanmard, S.; Najafi, H.; Kelishadi, R. Insulin and leptin levels in overweight and normal-weight Iranian adolescents: The CASPIAN-III study. J. Res. Med. Sci. 2014, 19, 387–390. [Google Scholar] [PubMed]
- Lanzillo, R.; Carbone, F.; Quarantelli, M.; Bruzzese, D.; Carotenuto, A.; De Rosa, V.; Colamatteo, A.; Micillo, T.; Picione, C.D.L.; Saccà, F.; et al. Immunometabolic profiling of patients with multiple sclerosis identifies new biomarkers to predict disease activity during treatment with interferon beta-1a. Clin. Immunol. 2017, 183, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, F.; Mirabella, M.; Caggiula, M.; Frisullo, G.; Patanella, K.; Sancricca, C.; Nociti, V.; Tonali, P.A.; Batocchi, A.P. Evidence of involvement of leptin and IL-6 peptides in the action of interferon-beta in secondary progressive multiple sclerosis. Peptides 2005, 26, 2289–2293. [Google Scholar] [CrossRef]
- Steppan, C.M.; Brown, E.J.; Wright, C.M.; Bhat, S.; Banerjee, R.R.; Dai, C.Y.; Enders, G.H.; Silberg, D.G.; Wen, X.; Wu, G.D.; et al. A family of tissue-specific resistin-like molecules. Proc. Natl. Acad. Sci. USA 2001, 98, 502–506. [Google Scholar] [CrossRef]
- Park, H.K.; Ahima, R.S. Resistin in Rodents and Humans. Diabetes Metab. J. 2013, 37, 404–414. [Google Scholar] [CrossRef]
- Jamaluddin, S.; Weakley, S.M.; Yao, Q.; Chen, C. Resistin: Functional roles and therapeutic considerations for cardiovascular disease. Br. J. Pharmacol. 2012, 165, 622–632. [Google Scholar] [CrossRef]
- Lee, S.; Lee, H.-C.; Kwon, Y.-W.; Lee, S.E.; Cho, Y.; Kim, J.; Lee, S.; Kim, J.-Y.; Lee, J.; Yang, H.-M.; et al. Adenylyl Cyclase-Associated Protein 1 Is a Receptor for Human Resistin and Mediates Inflammatory Actions of Human Monocytes. Cell Metab. 2014, 19, 484–497. [Google Scholar] [CrossRef]
- Tarkowski, A.; Bjersing, J.; Shestakov, A.; Bokarewa, M.I. Resistin competes with lipopolysaccharide for binding to toll-like receptor 4. J. Cell. Mol. Med. 2009, 14, 1419–1431. [Google Scholar] [CrossRef]
- Bokarewa, M.; Nagaev, I.; Dahlberg, L.; Smith, U.; Tarkowski, A. Resistin, an Adipokine with Potent Proinflammatory Properties. J. Immunol. 2005, 174, 5789–5795. [Google Scholar] [CrossRef]
- Silswal, N.; Singh, A.K.; Aruna, B.; Mukhopadhyay, S.; Ghosh, S.; Ehtesham, N.Z. Human resistin stimulates the pro-inflammatory cytokines TNF-alpha and IL-12 in macrophages by NF-kappaB-dependent pathway. Biochem. Biophys. Res. Commun. 2005, 334, 1092–1101. [Google Scholar] [CrossRef]
- Kawanami, D.; Maemura, K.; Takeda, N.; Harada, T.; Nojiri, T.; Imai, Y.; Manabe, I.; Utsunomiya, K.; Nagai, R. Direct reciprocal effects of resistin and adiponectin on vascular endothelial cells: A new insight into adipocytokine–endothelial cell interactions. Biochem. Biophys. Res. Commun. 2004, 314, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, R.; Hagman, S.; Hämäläinen, M.; Leppänen, T.; Dastidar, P.; Moilanen, E.; Elovaara, I. Adipsin Is Associated with Multiple Sclerosis: A Follow-Up Study of Adipokines. Mult. Scler. Int. 2015, 2015, 1–9. [Google Scholar] [CrossRef]
- Vazquez-Villegas, M.L.; Gamez-Nava, J.I.; Saldaña-Cruz, A.M.; Celis, A.; Sanchez-Rodriguez, E.N.; Perez-Guerrero, E.E.; Ramirez-Villafaña, M.; Nava-Valdivia, C.A.; Contreras-Haro, B.; Vasquez-Jimenez, J.C.; et al. Functional disability is related to serum chemerin levels in rheumatoid arthritis. Sci. Rep. 2021, 11, 1–8. [Google Scholar] [CrossRef]
- Ha, Y.-J.; Kang, E.-J.; Song, J.-S.; Park, Y.-B.; Lee, S.-K.; Choi, S.T. Plasma chemerin levels in rheumatoid arthritis are correlated with disease activity rather than obesity. Jt. Bone Spine 2014, 81, 189–190. [Google Scholar] [CrossRef] [PubMed]
- Gu, P.; Jiang, W.; Lu, B.; Shi, Z. Chemerin is associated with inflammatory markers and metabolic syndrome phenotypes in hypertension patients. Clin. Exp. Hypertens. 2014, 36, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Zylla, S.; Pietzner, M.; Kühn, J.-P.; Völzke, H.; Dörr, M.; Nauck, M.; Friedrich, N. Serum chemerin is associated with inflammatory and metabolic parameters-results of a population-based study. Obesity 2017, 25, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Panigrahy, D.; Gilligan, M.M.; Serhan, C.N.; Kashfi, K. Resolution of inflammation: An organizing principle in biology and medicine. Pharmacol. Ther. 2021, 227, 107879. [Google Scholar] [CrossRef] [PubMed]
- Mariani, F.; Roncucci, L. Chemerin/chemR23 axis in inflammation onset and resolution. Inflamm. Res. 2015, 64, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Herova, M.; Schmid, M.; Gemperle, C.; Hersberger, M. ChemR23, the receptor for chemerin and resolvin E1, is expressed and functional on M1 but not on M2 macrophages. J. Immunol. 2015, 194, 2330–2337. [Google Scholar] [CrossRef]
- Rama, D.; Esendagli, G.; Guc, D. Expression of chemokine-like receptor 1 (CMKLR1) on J744A.1 macrophages co-cultured with fibroblast and/or tumor cells: Modeling the influence of microenvironment. Cell. Immunol. 2011, 271, 134–140. [Google Scholar] [CrossRef]
- Bondue, B.; De Henau, O.; Luangsay, S.; Devosse, T.; de Nadai, P.; Springael, J.-Y.; Parmentier, M.; Vosters, O. The Chemerin/ChemR23 System Does Not Affect the Pro-Inflammatory Response of Mouse and Human Macrophages Ex Vivo. PLoS ONE 2012, 7, e40043. [Google Scholar] [CrossRef] [PubMed]
- Rourke, J.L.; Dranse, H.J.; Sinal, C.J. CMKLR1 and GPR1 mediate chemerin signaling through the RhoA/ROCK pathway. Mol. Cell. Endocrinol. 2015, 417, 36–51. [Google Scholar] [CrossRef]
- Zabel, B.A.; Nakae, S.; Zuniga, L.; Kim, J.Y.; Ohyama, T.; Alt, C.; Pan, J.; Suto, H.; Soler, D.; Allen, S.J.; et al. Mast cell-expressed orphan receptor CCRL2 binds chemerin and is required for optimal induction of IgE-mediated passive cutaneous anaphylaxis. J. Exp. Med. 2008, 205, 2207–2220. [Google Scholar] [CrossRef] [PubMed]
- Tomalka-Kochanowska, J.; Baranowska, B.; Wolinska-Witort, E.; Uchman, D.; Litwiniuk, A.; Martyńska, L.; Kalisz, M.; Bik, W.; Kochanowski, J. Plasma chemerin levels in patients with multiple sclerosis. Neuro Endocrinol. Lett. 2014, 35, 218–223. [Google Scholar] [PubMed]
- Koskderelioglu, A.; Gedizlioglu, M.; Eskut, N.; Tamer, P.; Yalcin, G.; Bozkaya, G. Impact of chemerin, lipid profile, and insulin resistance on disease parameters in patients with multiple sclerosis. Neurol. Sci. 2020, 42, 1–9. [Google Scholar] [CrossRef]
- Weigert, J.; Neumeier, M.; Wanninger, J.; Filarsky, M.; Bauer, S.; Wiest, R.; Farkas, S.; Scherer, M.N.; Schäffler, A.; Aslanidis, C.; et al. Systemic chemerin is related to inflammation rather than obesity in type 2 diabetes. Clin. Endocrinol. 2010, 72, 342–348. [Google Scholar] [CrossRef]
- Fukuhara, A.; Matsuda, M.; Nishizawa, M.; Segawa, K.; Tanaka, M.; Kishimoto, K.; Matsuki, Y.; Murakami, M.; Ichisaka, T.; Murakami, H.; et al. Visfatin: A Protein Secreted by Visceral Fat That Mimics the Effects of Insulin. Science 2005, 307, 426–430. [Google Scholar] [CrossRef]
- McGee, K.C.; Harte, A.L.; da Silva, N.F.; Al-Daghri, N.; Creely, S.J.; Kusminski, C.M.; Tripathi, G.; Levick, P.L.; Khanolkar, M.; Evans, M.; et al. Visfatin is regulated by rosiglitazone in type 2 diabetes mellitus and influenced by NFkappaB and JNK in human abdominal subcutaneous adipocytes. PLoS ONE 2011, 6, e20287. [Google Scholar] [CrossRef]
- Nogueira, A.; Nokhbehsaim, M.; Damanaki, A.; Eick, S.; Kirschneck, C.; Schröder, A.; Jantsch, J.; Deschner, J. Filifactor alocis and Tumor Necrosis Factor-Alpha Stimulate Synthesis of Visfatin by Human Macrophages. Int. J. Mol. Sci. 2021, 22, 1235. [Google Scholar] [CrossRef]
- Rongvaux, A.; Galli, M.; Denanglaire, S.; Van Gool, F.; Drèze, P.L.; Szpirer, C.; Bureau, F.; Andris, F.; Leo, O. Nicotinamide phosphoribosyl transferase/pre-B cell colony-enhancing factor/visfatin is required for lymphocyte development and cellular resistance to genotoxic stress. J. Immunol. 2008, 181, 4685–4695. [Google Scholar] [CrossRef] [PubMed]
- Moschen, A.; Kaser, A.; Enrich, B.; Mosheimer, B.; Theurl, M.; Niederegger, H.; Tilg, H. Visfatin, an Adipocytokine with Proinflammatory and Immunomodulating Properties. J. Immunol. 2007, 178, 1748–1758. [Google Scholar] [CrossRef]
- Wu, X.-T.; Yang, Z.; Ansari, A.R.; Xiao, K.; Pang, X.-X.; Luo, Y.; Song, H. Visfatin regulates the production of lipopolysaccharide-induced inflammatory cytokines through p38 signaling in murine macrophages. Microb. Pathog. 2018, 117, 55–59. [Google Scholar] [CrossRef]
- Xu, Y.; Yu, L.; Liu, Y.; Tang, X.; Wang, X. Lipopolysaccharide-Induced Microglial Neuroinflammation: Attenuation by FK866. Neurochem. Res. 2021, 46, 1291–1304. [Google Scholar] [CrossRef]
- Kim, S.R.; Bae, Y.H.; Bae, S.K.; Choi, K.S.; Yoon, K.H.; Koo, T.H.; Jang, H.O.; Yun, I.; Kim, K.W.; Kwon, Y.G.; et al. Visfatin enhances ICAM-1 and VCAM-1 expression through ROS-dependent NF-kappaB activation in endothelial cells. Biochim. Biophys. Acta 2008, 1783, 886–895. [Google Scholar] [CrossRef]
- Chen, Y.; Pitzer, A.L.; Li, X.; Li, P.; Wang, L.; Zhang, Y. Instigation of endothelial Nlrp3 inflammasome by adipokine visfatin promotes inter-endothelial junction disruption: Role of HMGB 1. J. Cell. Mol. Med. 2015, 19, 2715–2727. [Google Scholar] [CrossRef] [PubMed]
- Arababadi, M.K.; Asadikaram, P.; Asadikaram, G. APLN/APJ pathway: The key regulator of macrophage functions. Life Sci. 2019, 232, 116645. [Google Scholar] [CrossRef] [PubMed]
- Izgüt-Uysal, V.N.; Gemici, B.; Birsen, I.; Acar, N.; Üstünel, I. The Effect of Apelin on the Functions of Peritoneal Macrophages. Physiol. Res. 2017, 66, 489–496. [Google Scholar] [CrossRef]
- Leeper, N.J.; Tedesco, M.M.; Kojima, Y.; Schultz, G.M.; Kundu, R.K.; Ashley, E.A.; Tsao, P.S.; Dalman, R.L.; Quertermous, T. Apelin prevents aortic aneurysm formation by inhibiting macrophage inflammation. Am. J. Physiol. Circ. Physiol. 2009, 296, H1329–H1335. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ye, Q.; Gong, D.; Lv, Y.; Cheng, H.; Huang, C.; Chen, L.; Zhao, Z.; Li, L.; Wei, X.; et al. Apelin-13 inhibits lipoprotein lipase expression via the APJ/PKCalpha/miR-361-5p signaling pathway in THP-1 macrophage-derived foam cells. Acta Biochim. Biophys. Sin (Shanghai) 2014, 9, 530–540. [Google Scholar]
- Obara, S.; Akifusa, S.; Ariyoshi, W.; Okinaga, T.; Usui, M.; Nakashima, K.; Nishihara, T. Pyroglutamated Apelin-13 Inhibits Lipopolysaccharide-Induced Production of Pro-Inflammatory Cytokines in Murine Macrophage J774.1 Cells. Mod. Res. Inflamm. 2014, 03, 59–66. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, S.; Zeng, M.; Lin, D.; Wang, Y.; Wen, X.; Xu, C.; Yang, L.; Fan, X.; Gong, Y.; et al. Apelin-13 Administration Protects Against LPS-Induced Acute Lung Injury by Inhibiting NF-kappaB Pathway and NLRP3 Inflammasome Activation. Cell Physiol. Biochem. 2018, 49, 1918–1932. [Google Scholar] [CrossRef]
- Chen, L.; Tao, Y.; Jiang, Y. Apelin activates the expression of inflammatory cytokines in microglial BV2 cells via PI-3K/Akt and MEK/Erk pathways. Sci. China Life Sci. 2015, 58, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, Y.; Fujii, T.; Kamimura, Y.; Kawashima, K. The endogenous, immunologically active peptide apelin inhibits lymphocytic cholinergic activity during immunological responses. J. Neuroimmunol. 2003, 144, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Guo, X.; Chen, S.; Xu, Z.; Duan, W.; Zeng, B. Apelin-13 regulates LPS-induced N9 microglia polarization involving STAT3 signaling pathway. Neuropeptides 2019, 76, 101938. [Google Scholar] [CrossRef]
- Alpua, M.; Turkel, Y.; Dag, E.; Kisa, U. Apelin-13: A promising biomarker for multiple sclerosis? Ann. Indian Acad. Neurol. 2018, 21, 126–129. [Google Scholar] [CrossRef]
- Valencak, T.G.; Osterrieder, A.; Schulz, T.J. Sex matters: The effects of biological sex on adipose tissue biology and energy metabolism. Redox Biol. 2017, 12, 806–813. [Google Scholar] [CrossRef]
- Chen, K.-H.E.; Lainez, N.M.; Coss, D. Sex Differences in Macrophage Responses to Obesity-Mediated Changes Determine Migratory and Inflammatory Traits. J. Immunol. 2021, 206, 141–153. [Google Scholar] [CrossRef]
- Han, J.; Fan, Y.; Zhou, K.; Blomgren, K.; Harris, R.A. Uncovering sex differences of rodent microglia. J. Neuroinflammation 2021, 18, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Guillot-Sestier, M.-V.; Araiz, A.R.; Mela, V.; Gaban, A.S.; O’Neill, E.; Joshi, L.; Chouchani, E.T.; Mills, E.L.; Lynch, M.A. Microglial metabolism is a pivotal factor in sexual dimorphism in Alzheimer’s disease. Commun. Biol. 2021, 4, 1–13. [Google Scholar] [CrossRef]
- Mancuso, P.; Bouchard, B. The Impact of Aging on Adipose Function and Adipokine Synthesis. Front. Endocrinol. 2019, 10, 137. [Google Scholar] [CrossRef] [PubMed]
- Rajappa, M.; Rathika, S.; Munisamy, M.; Chandrashekar, L.; Thappa, D. Effect of treatment with methotrexate and coal tar on adipokine levels and indices of insulin resistance and sensitivity in patients with psoriasis vulgaris. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Frühbeck, G.; Catalán, V.; Rodríguez, A.; Ramírez, B.; Becerril, S.; Salvador, J.; Colina, I.; Gómez-Ambrosi, J. Adiponectin-leptin Ratio is a Functional Biomarker of Adipose Tissue Inflammation. Nutrients 2019, 11, 454. [Google Scholar] [CrossRef]
- Frühbeck, G.; Catalán, V.; Rodríguez, A.; Gómez-Ambrosi, J. Adiponectin-leptin ratio: A promising index to estimate adipose tissue dysfunction. Relation with obesity-associated cardiometabolic risk. Adipocyte 2018, 7, 57–62. [Google Scholar] [CrossRef]
- Kuo, S.-M.; Halpern, M.M. Lack of association between body mass index and plasma adiponectin levels in healthy adults. Int. J. Obes. 2011, 35, 1487–1494. [Google Scholar] [CrossRef]
- Fehmann, H.-C.; Heyn, J. Plasma Resistin Levels in Patients with Type 1 and Type 2 Diabetes Mellitus and in Healthy Controls. Horm. Metab. Res. 2002, 34, 671–673. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Yang, G.; Dong, J.; Liu, Y.; Zong, H.; Liu, H.; Boden, G.; Li, L. Elevated plasma levels of chemerin in newly diagnosed type 2 diabetes mellitus with hypertension. J. Investig. Med. 2010, 58, 883–886. [Google Scholar] [CrossRef] [PubMed]
- Liakos, C.I.; Sanidas, E.A.; Perrea, D.N.; Grassos, C.A.; Chantziara, V.; Viniou, N.-A.; Barbetseas, J.D.; Papadopoulos, D.P. Apelin and Visfatin Plasma Levels in Healthy Individuals with High Normal Blood Pressure: Table 1. Am. J. Hypertens. 2015, 29, 549–552. [Google Scholar] [CrossRef]
- Kipp, M.; Nyamoya, S.; Hochstrasser, T.; Amor, S. Multiple sclerosis animal models: A clinical and histopathological perspective. Brain Pathol. 2017, 27, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Piccio, L.; Cantoni, C.; Henderson, J.G.; Hawiger, D.; Ramsbottom, M.; Mikesell, R.; Ryu, J.; Hsieh, C.-S.; Cremasco, V.; Haynes, W.; et al. Lack of adiponectin leads to increased lymphocyte activation and increased disease severity in a mouse model of multiple sclerosis. Eur. J. Immunol. 2013, 43, 2089–2100. [Google Scholar] [CrossRef]
- Zhang, K.; Guo, Y.; Ge, Z.; Zhang, Z.; Da, Y.; Li, W.; Zhang, Z.; Xue, Z.; Li, Y.; Ren, Y.; et al. Adiponectin Suppresses T Helper 17 Cell Differentiation and Limits Autoimmune CNS Inflammation via the SIRT1/PPARgamma/RORgammat Pathway. Mol. Neurobiol. 2017, 54, 4908–4920. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Luo, J.; Liu, H.; Cui, W.; Guo, W.; Zhao, L.; Guo, H.; Bai, H.; Guo, K.; Feng, D.; et al. Recombinant adiponectin peptide promotes neuronal survival after intracerebral haemorrhage by suppressing mitochondrial and ATF4-CHOP apoptosis pathways in diabetic mice via Smad3 signalling inhibition. Cell Prolif. 2020, 53, e12759. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Luo, J.; Liu, H.; Cui, W.; Guo, K.; Zhao, L.; Bai, H.; Guo, W.; Guo, H.; Feng, D.; et al. Recombinant Adiponectin Peptide Ameliorates Brain Injury Following Intracerebral Hemorrhage by Suppressing Astrocyte-Derived Inflammation via the Inhibition of Drp1-Mediated Mitochondrial Fission. Transl. Stroke Res. 2020, 11, 924–939. [Google Scholar] [CrossRef] [PubMed]
- Ng, R.C.; Jian, M.; Ma, O.K.; Bunting, M.; Kwan, J.S.; Zhou, G.J.; Senthilkumar, K.; Iyaswamy, A.; Chan, P.K.; Li, M.; et al. Chronic oral administration of adipoRon reverses cognitive impairments and ameliorates neuropathology in an Alzheimer’s disease mouse model. Mol. Psychiatry 2020, 1–21. [Google Scholar] [CrossRef]
- Matarese, G.; Di Giacomo, A.; Sanna, V.; Lord, G.M.; Howard, J.K.; Di Tuoro, A.; Bloom, S.R.; Lechler, R.I.; Zappacosta, S.; Fontana, S. Requirement for Leptin in the Induction and Progression of Autoimmune Encephalomyelitis. J. Immunol. 2001, 166, 5909–5916. [Google Scholar] [CrossRef]
- Matarese, G.; Sanna, V.; Di Giacomo, A.; Lord, G.M.; Howard, J.K.; Bloom, S.R.; Lechler, R.I.; Fontana, S.; Zappacosta, S. Leptin potentiates experimental autoimmune encephalomyelitis in SJL female mice and confers susceptibility to males. Eur. J. Immunol. 2001, 31, 1324–1332. [Google Scholar] [CrossRef]
- Ouyang, S.; Hsuchou, H.; Kastin, A.J.; Mishra, P.K.; Wang, Y.; Pan, W. Leukocyte infiltration into spinal cord of EAE mice is attenuated by removal of endothelial leptin signaling. Brain Behav. Immun. 2014, 40, 61–73. [Google Scholar] [CrossRef][Green Version]
- Corem, N.; Anzi, S.; Gelb, S.; Ben-Zvi, A. Leptin receptor deficiency induces early, transient and hyperglycaemia-independent blood-brain barrier dysfunction. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Hung, W.; Wang, C.; Lin, S.; Cheng, S.; Liao, L.; Lu, L.; Chen, Y.; Huang, Y.; Lin, C.; Hsueh, C. Leptin protects brain from ischemia/reperfusion-induced infarction by stabilizing the blood–brain barrier to block brain infiltration by the blood-borne neutrophils. Eur. J. Neurosci. 2020, 52, 4890–4907. [Google Scholar] [CrossRef]
- Cheng, G.; Deng, Y.; Zhou, Z.; Yu, J.; Zhang, H.; Wang, X.; Li, X. Neuroprotective effect of leptin on a primate model of cerebral ischemia. Anim. Biotechnol. 2021, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.K.; Hsuchou, H.; Ouyang, S.; Kastin, A.J.; Wu, X.; Pan, W. Loss of astrocytic leptin signaling worsens experimental autoimmune encephalomyelitis. Brain Behav. Immun. 2013, 34, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Matoba, K.; Muramatsu, R.; Yamashita, T. Leptin sustains spontaneous remyelination in the adult central nervous system. Sci. Rep. 2017, 7, 40397. [Google Scholar] [CrossRef]
- Davis, C.; Mudd, J.; Hawkins, M. Neuroprotective effects of leptin in the context of obesity and metabolic disorders. Neurobiol. Dis. 2014, 72, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Xiaoying, L.; Li, T.; Yu, S.; Jiusheng, J.; Jilin, Z.; Jiayi, W.; DongXin, L.; Wengang, F.; Xinyue, Z.; Hao, Y.; et al. Resistin-Inhibited Neural Stem Cell-Derived Astrocyte Differentiation Contributes to Permeability Destruction of the Blood–Brain Barrier. Neurochem. Res. 2019, 44, 905–916. [Google Scholar] [CrossRef]
- Miao, J.; Benomar, Y.; Al Rifai, S.; Poizat, G.; Riffault, L.; Crépin, D.; Taouis, M. Resistin inhibits neuronal autophagy through Toll-like receptor 4. J. Endocrinol. 2018, 238, 77–89. [Google Scholar] [CrossRef]
- Sahebkar, A. Beyond anti-PCSK9 therapies: The potential role of resistin inhibitors. Nat. Rev. Cardiol. 2013, 11, 12. [Google Scholar] [CrossRef]
- Graham, K.L.; Zabel, B.A.; Loghavi, S.; Zuniga, L.A.; Ho, P.P.; Sobel, R.A.; Butcher, E.C. Chemokine-Like Receptor-1 Expression by Central Nervous System-Infiltrating Leukocytes and Involvement in a Model of Autoimmune Demyelinating Disease. J. Immunol. 2009, 183, 6717–6723. [Google Scholar] [CrossRef]
- Galli, U.; Colombo, G.; Travelli, C.; Tron, G.C.; Genazzani, A.A.; Grolla, A. Recent Advances in NAMPT Inhibitors: A Novel Immunotherapic Strategy. Front. Pharmacol. 2020, 11, 656. [Google Scholar] [CrossRef]
- Tan, Z.; Chen, L.; Ren, Y.; Jiang, X.; Gao, W. Neuroprotective effects of FK866 against traumatic brain injury: Involvement of p38/ERK pathway. Ann. Clin. Transl. Neurol. 2020, 7, 742–756. [Google Scholar] [CrossRef]
- Esposito, E.; Impellizzeri, D.; Mazzon, E.; Fakhfouri, G.; Rahimian, R.; Travelli, C.; Tron, G.C.; Genazzani, A.A.; Cuzzocrea, S. The NAMPT inhibitor FK866 reverts the damage in spinal cord injury. J. Neuroinflammation 2012, 9, 66. [Google Scholar] [CrossRef]
- Chen, C.-X.; Huang, J.; Tu, G.-Q.; Lu, J.-T.; Xie, X.; Zhao, B.; Wu, M.; Shi, Q.-J.; Fang, S.-H.; Wei, E.-Q.; et al. NAMPT inhibitor protects ischemic neuronal injury in rat brain via anti-neuroinflammation. Neuroscience 2017, 356, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Weng, Z.; Xia, Q.; Cao, C.; Leak, R.; Han, L.; Xiao, J.; Graham, S.H.; Cao, G. Intracerebroventricular Delivery of Recombinant NAMPT Deters Inflammation and Protects Against Cerebral Ischemia. Transl. Stroke Res. 2019, 10, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Bruzzone, S.; Fruscione, F.; Morando, S.; Ferrando, T.; Poggi, A.; Garuti, A.; D’Urso, A.; Selmo, M.; Benvenuto, F.; Cea, M.; et al. Catastrophic NAD+ Depletion in Activated T Lymphocytes through Nampt Inhibition Reduces Demyelination and Disability in EAE. PLoS ONE 2009, 4, e7897. [Google Scholar] [CrossRef] [PubMed]
- Lundt, S.; Zhang, N.; Li, J.-L.; Zhang, Z.; Zhang, L.; Wang, X.; Bao, R.; Cai, F.; Sun, W.; Ge, W.-P.; et al. Metabolomic and transcriptional profiling reveals bioenergetic stress and activation of cell death and inflammatory pathways in vivo after neuronal deletion of NAMPT. Br. J. Pharmacol. 2021, 41, 2116–2131. [Google Scholar] [CrossRef]
- Luo, H.; Xiang, Y.; Qu, X.; Liu, H.; Liu, C.; Li, G.; Han, L.; Qin, X. Apelin-13 Suppresses Neuroinflammation Against Cognitive Deficit in a Streptozotocin-Induced Rat Model of Alzheimer’s Disease Through Activation of BDNF-TrkB Signaling Pathway. Front Pharmacol. 2019, 10, 395. [Google Scholar] [CrossRef] [PubMed]
- Nasseri, B.; Zareian, P.; Alizade, H. Apelin attenuates streptozotocin-induced learning and memory impairment by modulating necroptosis signaling pathway. Int. Immunopharmacol. 2020, 84, 106546. [Google Scholar] [CrossRef] [PubMed]
- Saral, S.; Topçu, A.; Alkanat, M.; Mercantepe, T.; Akyıldız, K.; Yıldız, L.; Tümkaya, L.; Yazıcı, Z.A.; Yılmaz, A. Apelin-13 activates the hippocampal BDNF/TrkB signaling pathway and suppresses neuroinflammation in male rats with cisplatin-induced cognitive dysfunction. Behav. Brain Res. 2021, 408, 113290. [Google Scholar] [CrossRef]
- Xin, Q.; Cheng, B.; Pan, Y.; Liu, H.; Yang, C.; Chen, J.; Bai, B. Neuroprotective effects of apelin-13 on experimental ischemic stroke through suppression of inflammation. Peptides 2015, 63, 55–62. [Google Scholar] [CrossRef]
- Chen, D.; Lee, J.; Gu, X.; Wei, L.; Yu, S.P. Intranasal Delivery of Apelin-13 Is Neuroprotective and Promotes Angiogenesis After Ischemic Stroke in Mice. ASN Neuro 2015, 7, 1–15. [Google Scholar] [CrossRef]
- Duan, J.; Cui, Z.; Yang, C.; Guo, J.; Cao, M.; Xi, Y.; Weng, Y.; Yin, Y.; Wang, G.; Wei, B.; et al. Neuroprotective effect of Apelin 13 on ischemic stroke by activating AMPK/GSK-3beta/Nrf2 signaling. J. Neuroinflammation 2019, 16, 24. [Google Scholar] [CrossRef] [PubMed]
- Gold, R.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Tornatore, C.; Sweetser, M.T.; Yang, M.S.M.; Sheikh, S.I.; et al. Placebo-Controlled Phase 3 Study of Oral BG-12 for Relapsing Multiple Sclerosis. N. Engl. J. Med. 2012, 367, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Khalil, M.; Teunissen, C.E.; Otto, M.; Piehl, F.; Sormani, M.P.; Gattringer, T.; Barro, C.; Kappos, L.; Comabella, M.; Fazekas, F.; et al. Neurofilaments as biomarkers in neurological disorders. Nat. Rev. Neurol. 2018, 14, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, J.; Luo, S.; Zhan, Y.; Lu, Q. The roles of PPARgamma and its agonists in autoimmune diseases: A comprehensive review. J. Autoimmun. 2020, 113, 102510. [Google Scholar] [CrossRef]
- Piccio, L.; Stark, J.L.; Cross, A. Chronic calorie restriction attenuates experimental autoimmune encephalomyelitis. J. Leukoc. Biol. 2008, 84, 940–948. [Google Scholar] [CrossRef]
- Bai, M.; Wang, Y.; Han, R.; Xu, L.; Huang, M.; Zhao, J.; Lin, Y.; Song, S.; Chen, Y. Intermittent caloric restriction with a modified fasting-mimicking diet ameliorates autoimmunity and promotes recovery in a mouse model of multiple sclerosis. J. Nutr. Biochem. 2021, 87, 108493. [Google Scholar] [CrossRef]
- Choi, I.Y.; Piccio, L.; Childress, P.; Bollman, B.; Ghosh, A.; Brandhorst, S.; Suarez, J.; Michalsen, A.; Cross, A.; Morgan, T.E.; et al. A Diet Mimicking Fasting Promotes Regeneration and Reduces Autoimmunity and Multiple Sclerosis Symptoms. Cell Rep. 2016, 15, 2136–2146. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Adipokine | Primary Production Site (s) | Target Receptor (s) | Levels in Circulation | Role in Immunity | Changes in Circulating Levels in MS |
|---|---|---|---|---|---|
| Adiponectin | Adipocytes | AdipoR1 and 2 | 2.5–22 µg/mL [127] |
| ↑ early-onset RRMS females versus control [46] ↑ during remission [45] ↑ after IFN-β therapy [49] ↑ treatment-naïve patients, correlated with increased disability and progression [50] ↑ CSF, predicted worse prognosis and higher EDSS [52,53] newly diagnosed, treatment-naive patients ↓ after BMI adjustment [29] between CIS and other MS types [85] ↓ female MS versus control [48] ↓ RRMS females in relapse phase versus control [44] ↓ in patients with optic neuritis as first clinical episode [51] |
| Leptin | Adipocytes | LepR/OB-R | 5–50 ng/mL [127] |
| Levels: ↑ in MS patients versus control [62,64,70] ↑ in MS patients in relapse phase versus control [44,47,66,71] ↑ CSF in relapse RRMS versus control [66] ↑ in MS patients in remission phase versus control [51,72] between CIS and other MS types [85] in MS patients in relapse phase versus control [67] in MS patients in remission phase versus control [53,63,65,67,68,69,73] CSF in MS patients in remission phase versus control [53] ↓ in MS patients in relapse phase versus control [63,72] Correlations: ↑ plasma with EDSS in RRMS [75] ↑ plasma with EDSS in SPMS/PPMS [62,76] ↑ plasma with less T-reg cells [47,64,66] ↑ plasma correlated with TNF-α, IL-1β, CRP [64] no correlation with EDSS scores in RRMS patients [49,69,73,74] |
| Resistin | PBMC, adipocytes | CAP1, TLR4 | 38.78 ± 7.9 ng/mL [128] |
| ↑ plasma, correlated with pro-inflammatory cytokines and EDSS between CIS and other MS types [85] |
| Chemerin | Adipocytes, hepatocytes | CMKLR1, GPR1, CCRL2 | 62.1 ± 19.2 ng/mL [129] |
| between MS and control [97,98] |
| Visfatin | Adipocytes, macrophages, endothelial cells | InsR, TLR4 | 11.0 ± 2.0 ng/mL [130] |
| ↑ RRMS compared to PPMS and SPMS patients, positive correlation with TNF-α and IL-1β [64] ↑ newly diagnosed, treatment-naive patients <-> after BMI adjustment [29] |
| Apelin | Adipocytes | APLNR | 205 ± 108 pg/mL [130] |
| ↑ RRMS patients compared to controls, no correlations with EDSS and disease duration [118] ↓ RRMS females less than 1 year after onset, levels correlated positively with both EDSS and number of relapses [46] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rijnsburger, M.; Djuric, N.; Mulder, I.A.; de Vries, H.E. Adipokines as Immune Cell Modulators in Multiple Sclerosis. Int. J. Mol. Sci. 2021, 22, 10845. https://doi.org/10.3390/ijms221910845
Rijnsburger M, Djuric N, Mulder IA, de Vries HE. Adipokines as Immune Cell Modulators in Multiple Sclerosis. International Journal of Molecular Sciences. 2021; 22(19):10845. https://doi.org/10.3390/ijms221910845
Chicago/Turabian StyleRijnsburger, Merel, Niek Djuric, Inge A. Mulder, and Helga E. de Vries. 2021. "Adipokines as Immune Cell Modulators in Multiple Sclerosis" International Journal of Molecular Sciences 22, no. 19: 10845. https://doi.org/10.3390/ijms221910845
APA StyleRijnsburger, M., Djuric, N., Mulder, I. A., & de Vries, H. E. (2021). Adipokines as Immune Cell Modulators in Multiple Sclerosis. International Journal of Molecular Sciences, 22(19), 10845. https://doi.org/10.3390/ijms221910845