
A Novel Cre/lox-Based Genetic Tool for Repeated, Targeted and Markerless Gene Integration in Yarrowia lipolytica


 and
and 
Abstract
:1. Introduction
2. Results
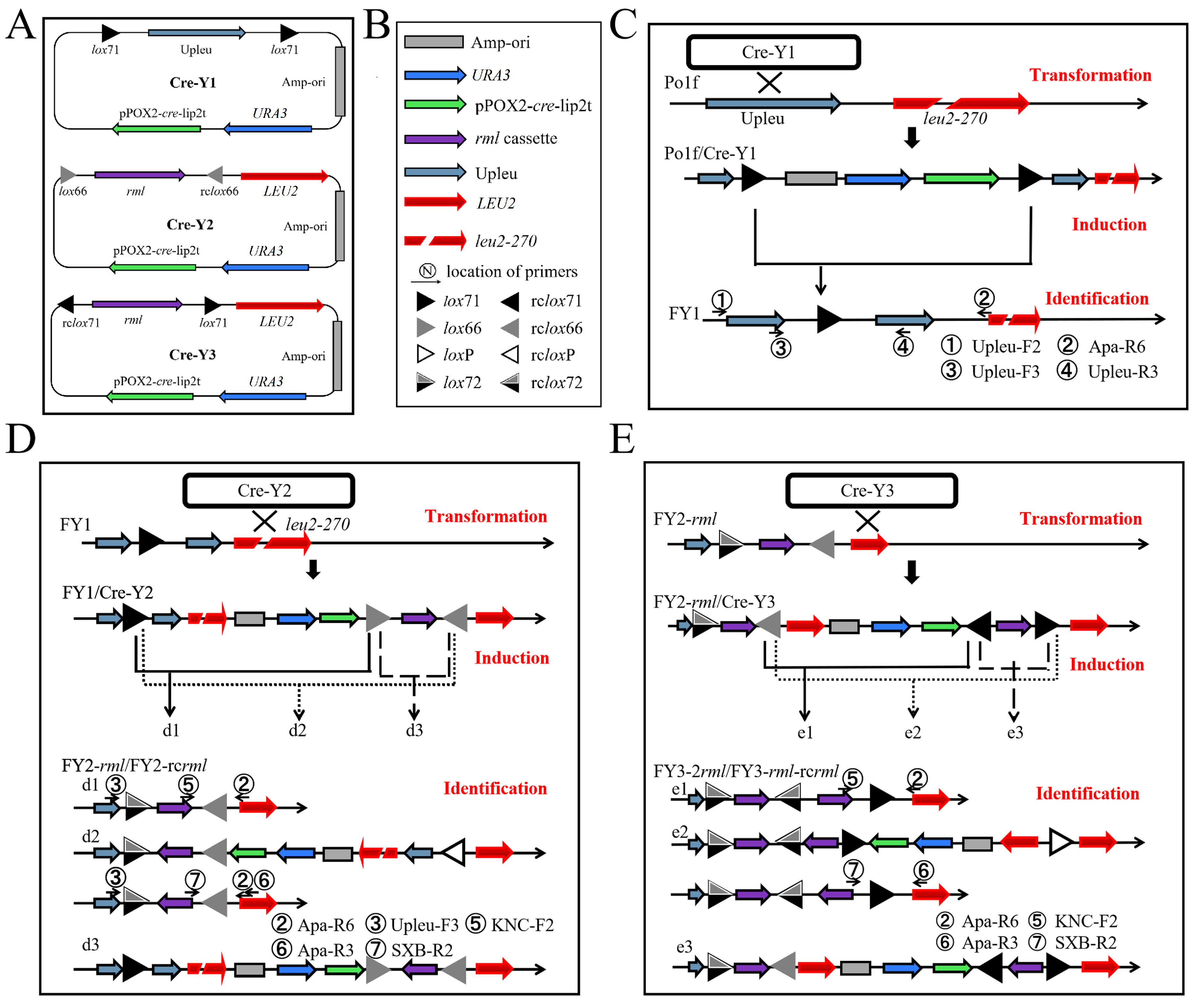
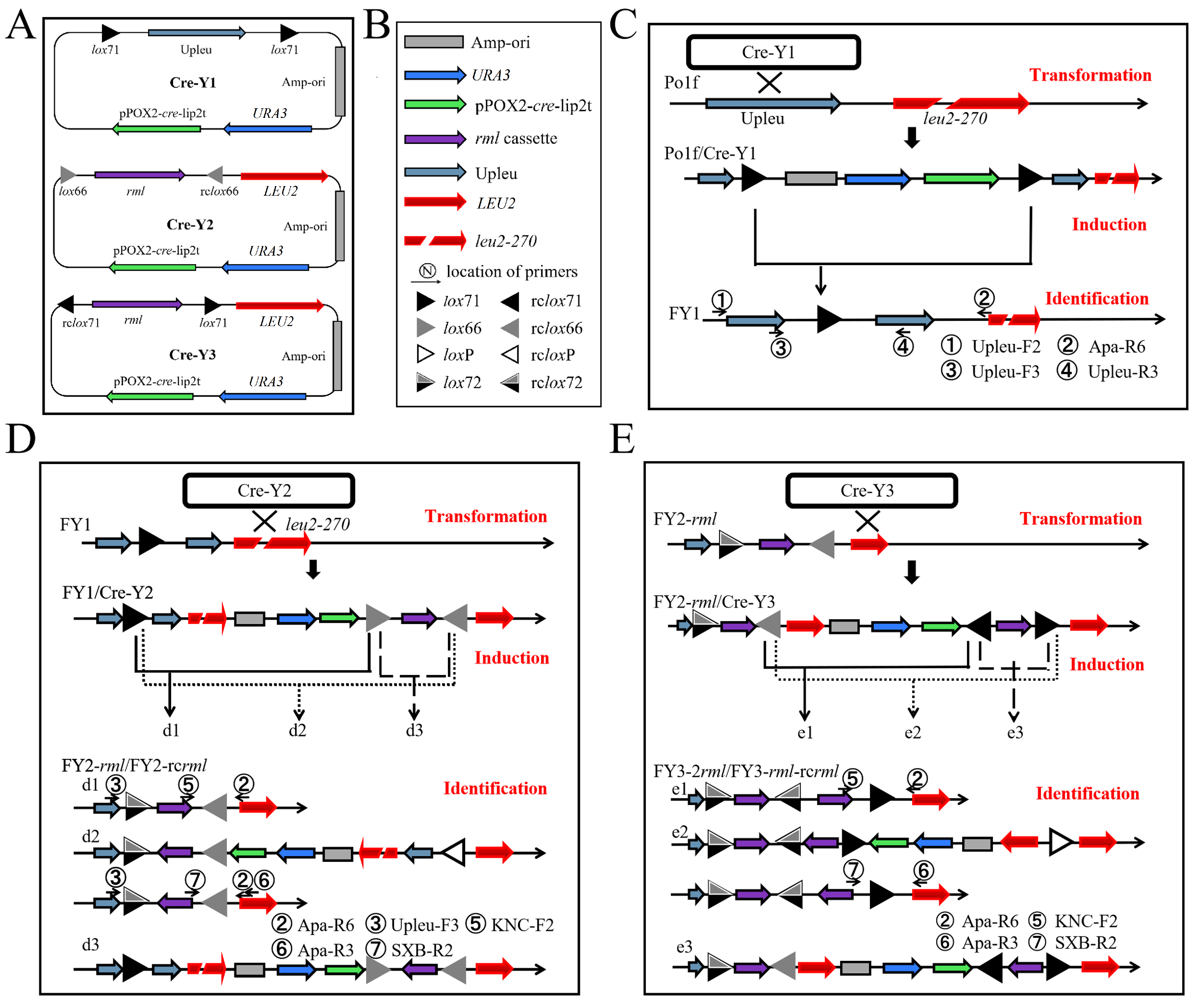
2.1. Genetic Tool Design
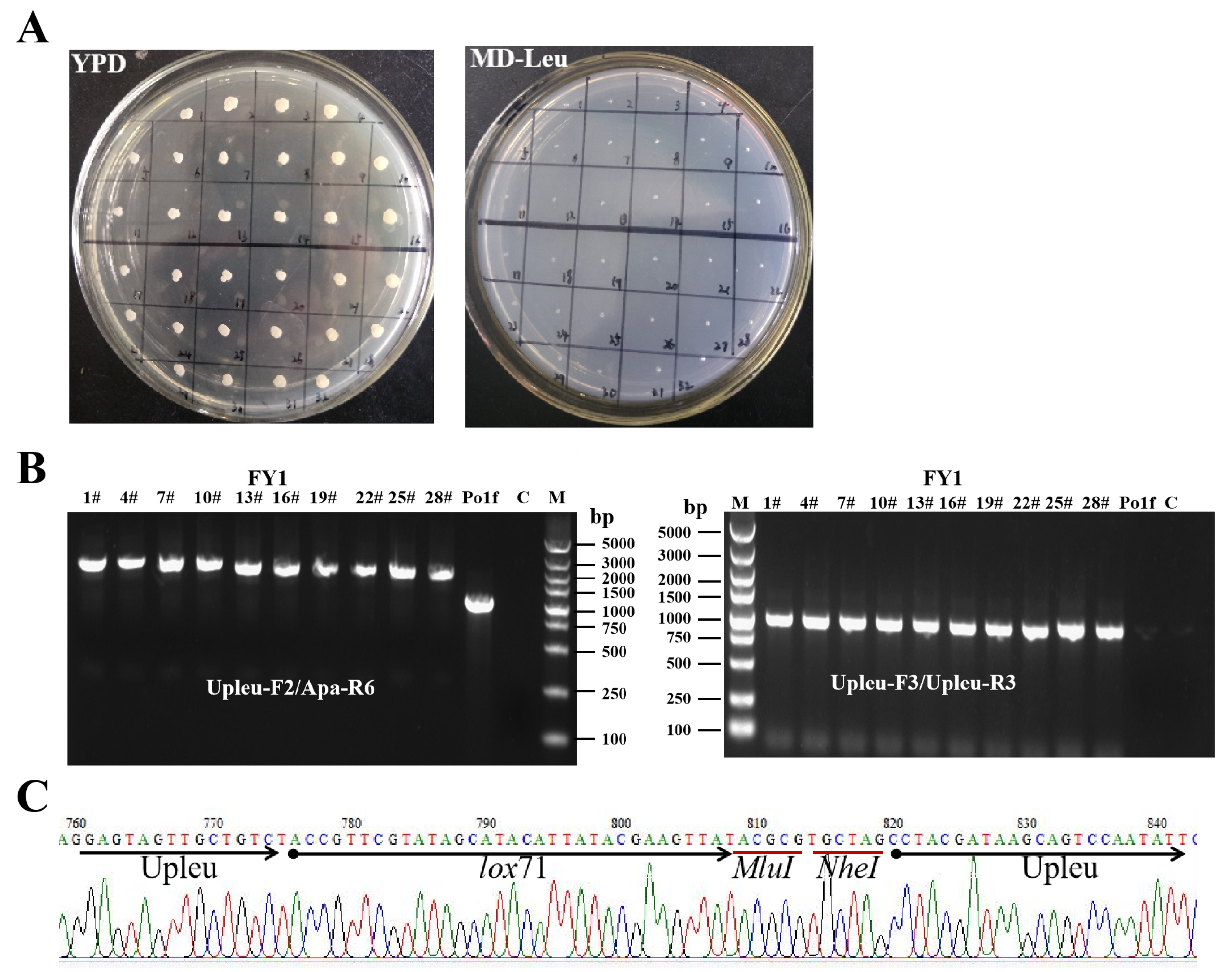
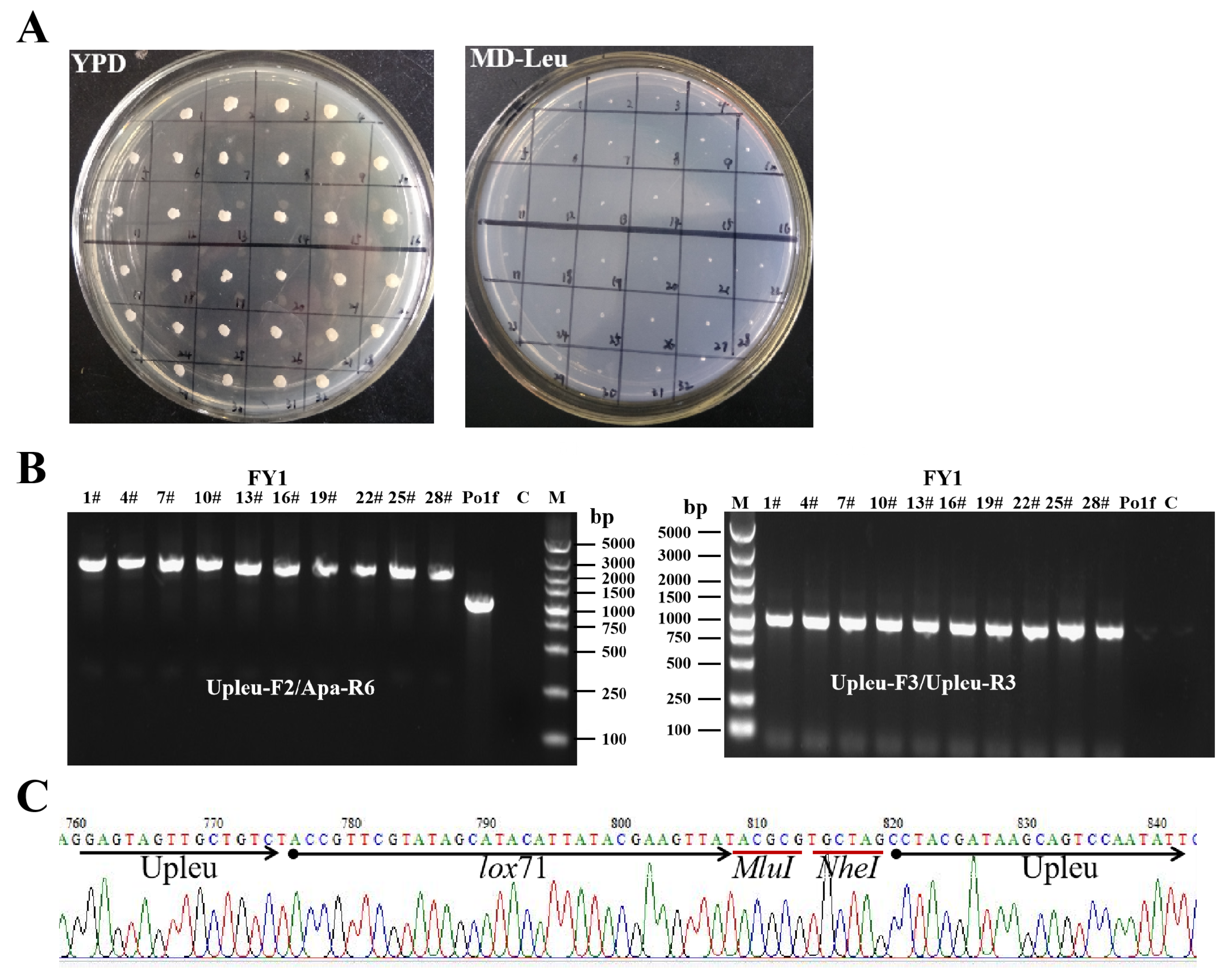
2.2. First-Round Integration to Construct the FY1 Strain
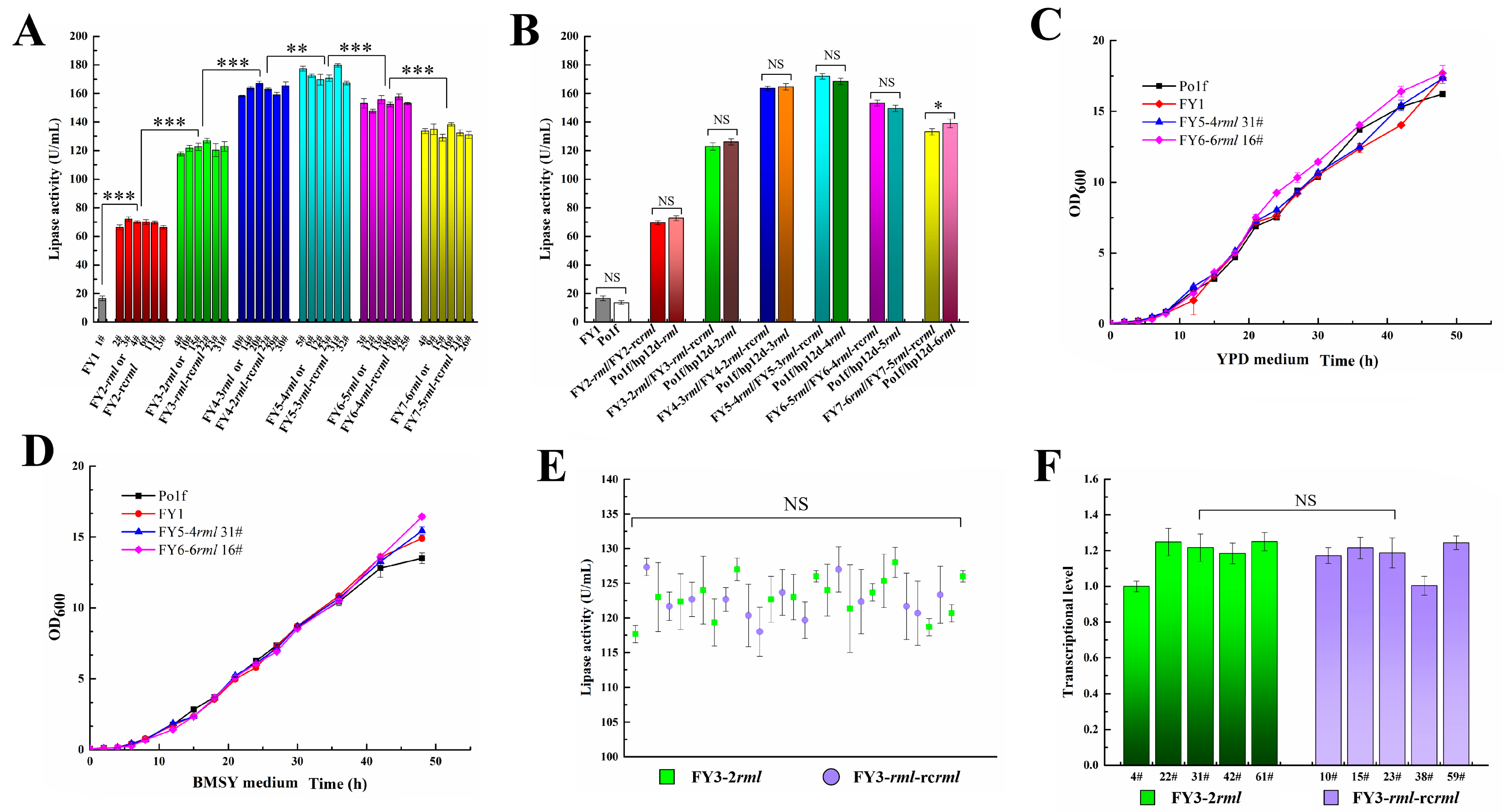
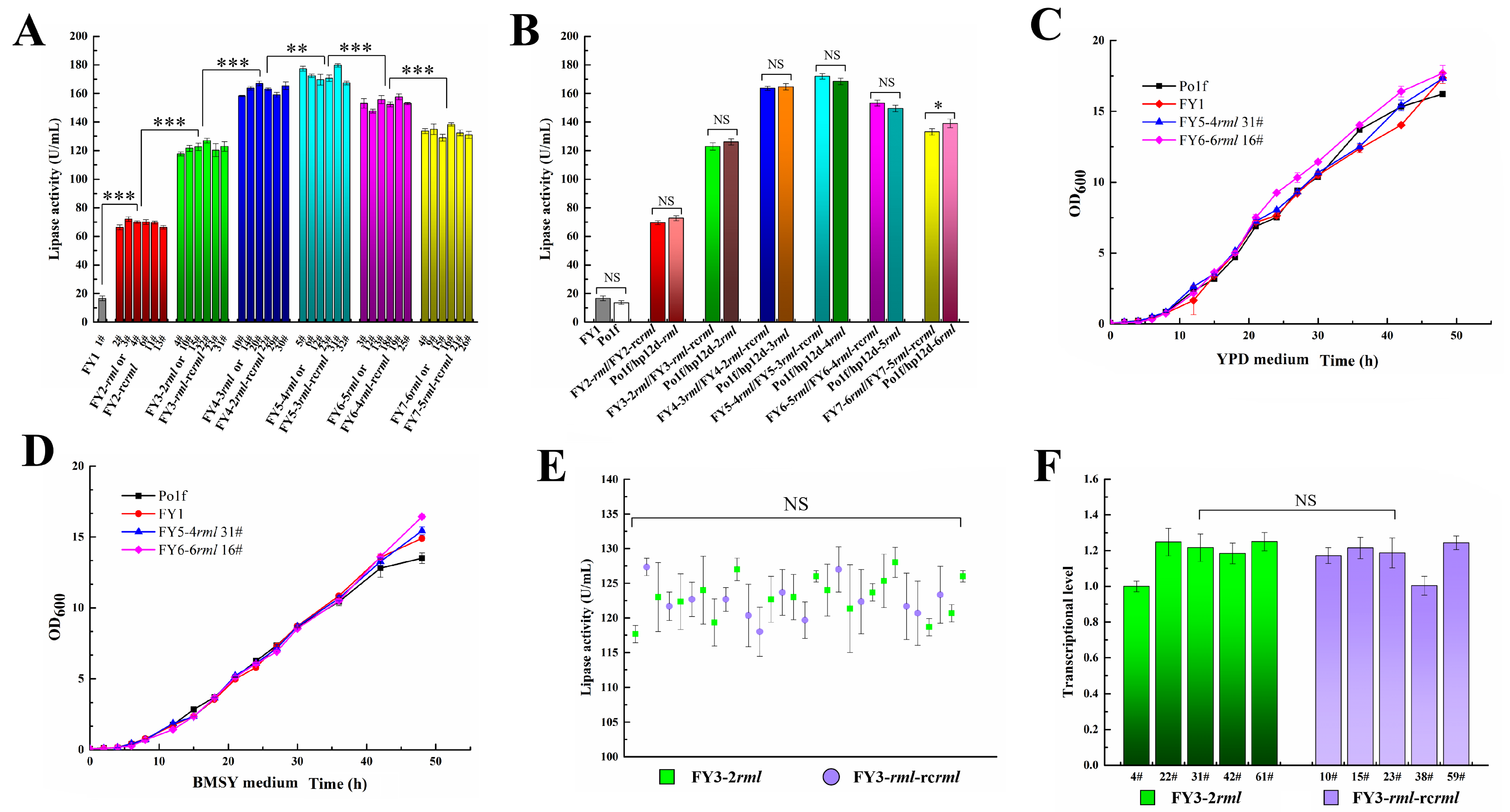
2.3. Second-Round Integration and Analysis
2.4. Induction Time Optimization in Second-Round Integration
2.5. Iterative Insertion of Cre-Y3 and Cre-Y2 Plasmids
2.6. Increasing Induction Time in Sixth and Seventh Rounds of Integration
2.7. Protein Expression Level and Growth Characteristics Evaluation
2.8. Comparison of Two-Genotype Recombinant Strains
2.9. Successfully Expanding to Six Different Genes and axp1-2 Locus
3. Discussion
4. Materials and Methods
4.1. Strains and Media
4.2. Plasmid Construction
4.3. Yeast Transformation, Screening and Induction
4.4. Shaking Flask Culture and Lipase Activity Determination
4.5. Determination of Gene Copy Number and Transcriptional Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blazeck, J.; Hill, A.; Liu, L.; Knight, R.; Miller, J.; Pan, A.; Otoupal, P.; Alper, H.S. Harnessing Yarrowia lipolytica lipogenesis to create a platform for lipid and biofuel production. Nat. Commun. 2014, 5, 3131. [Google Scholar] [CrossRef] [Green Version]
- Madzak, C. Yarrowia lipolytica: Recent achievements in heterologous protein expression and pathway engineering. Appl. Microbiol. Biotechnol. 2015, 99, 4559–4577. [Google Scholar] [CrossRef]
- Carly, F.; Vandermies, M.; Telek, S.; Steels, S.; Thomas, S.; Nicaud, J.M.; Fickers, P. Enhancing erythritol productivity in Yarrowia lipolytica using metabolic engineering. Metab. Eng. 2017, 42, 19–24. [Google Scholar] [CrossRef]
- Abdel-Mawgoud, A.M.; Markham, K.A.; Palmer, C.M.; Liu, N.; Stephanopoulos, G.; Alper, H.S. Metabolic engineering in the host Yarrowia lipolytica. Metab. Eng. 2018, 50, 192–208. [Google Scholar] [CrossRef]
- Czajka, J.J.; Nathenson, J.A.; Benites, V.T.; Baidoo, E.; Cheng, Q.; Wang, Y.; Tang, Y.J. Engineering the oleaginous yeast Yarrowia lipolytica to produce the aroma compound β-ionone. Microb. Cell Fact. 2018, 17, 136. [Google Scholar] [CrossRef]
- Madzak, C. Engineering Yarrowia lipolytica for use in biotechnological applications: A review of major achievements and recent innovations. Mol. Biotechnol. 2018, 60, 621–635. [Google Scholar] [CrossRef]
- Pignede, G.; Wang, H.J.; Fudalej, F.; Seman, M.; Gaillardin, C.; Nicaud, J.M. Autocloning and amplification of LIP2 in Yarrowia lipolytica. Appl. Environ. Microbiol. 2000, 66, 3283–3289. [Google Scholar] [CrossRef] [Green Version]
- Larroude, M.; Rossignol, T.; Nicaud, J.M.; Ledesma-Amaro, R. Synthetic biology tools for engineering Yarrowia lipolytica. Biotechnol. Adv. 2018, 36, 2150–2164. [Google Scholar] [CrossRef] [PubMed]
- Barth, G.; Gaillardin, C. Yarrowia lipolytica. In Nonconventional Yeasts in Biotechnology; Springer: Berlin/Heidelberg, Germany, 1996; pp. 313–388. [Google Scholar]
- Ledesma-Amaro, R.; Nicaud, J.M. Yarrowia lipolytica as a biotechnological chassis to produce usual and unusual fatty acids. Prog. Lipid Res. 2016, 61, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Larroude, M.; Celinska, E.; Back, A.; Thomas, S.; Nicaud, J.M.; Ledesma-Amaro, R. A synthetic biology approach to transform Yarrowia lipolytica into a competitive biotechnological producer of β-carotene. Biotechnol. Bioeng. 2018, 115, 464–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markham, K.A.; Palmer, C.M.; Chwatko, M.; Wagner, J.M.; Murray, C.; Vazquez, S.; Swaminathan, A.; Chakravarty, I.; Lynd, N.A.; Alper, H.S. Rewiring Yarrowia lipolytica toward triacetic acid lactone for materials generation. Proc. Natl. Acad. Sci. USA. 2018, 115, 2096–2101. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Qiao, Y.; Li, F.; Xu, Y.; Yan, Y.; Madzak, C.; Yan, J. Subcellular engineering of lipase dependent pathways directed towards lipid related organelles for highly effectively compartmentalized biosynthesis of triacylglycerol derived products in Yarrowia lipolytica. Metab. Eng. 2019, 55, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Gaj, T.; Gersbach, C.A.; Barbas, C.R. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends. Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Scott, D.A.; Kriz, A.J.; Chiu, A.C.; Hsu, P.D.; Dadon, D.B.; Cheng, A.W.; Trevino, A.E.; Konermann, S.; Chen, S.; et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat. Biotechnol. 2014, 32, 670–676. [Google Scholar] [CrossRef] [Green Version]
- Bae, S.J.; Park, B.G.; Kim, B.G.; Hahn, J.S. Multiplex gene disruption by targeted base editing of Yarrowia lipolytica genome using cytidine deaminase combined with the CRISPR/Cas9 system. Biotechnol. J. 2020, 15, e1900238. [Google Scholar] [CrossRef] [PubMed]
- Holkenbrink, C.; Dam, M.I.; Kildegaard, K.R.; Beder, J.; Dahlin, J.; Domenech, B.D.; Borodina, I. EasyCloneYALI: CRISPR/Cas9-based synthetic toolbox for engineering of the yeast Yarrowia lipolytica. Biotechnol. J. 2018, 13, e1700543. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, C.; Frogue, K.; Ramesh, A.; Misa, J.; Wheeldon, I. CRISPRi repression of nonhomologous end-joining for enhanced genome engineering via homologous recombination in Yarrowia lipolytica. Biotechnol. Bioeng. 2017, 114, 2896–2906. [Google Scholar] [CrossRef]
- Schwartz, C.; Shabbir-Hussain, M.; Frogue, K.; Blenner, M.; Wheeldon, I. Standardized markerless gene integration for pathway engineering in Yarrowia lipolytica. ACS Synth. Biol. 2017, 6, 402–409. [Google Scholar] [CrossRef]
- Gao, D.; Smith, S.; Spagnuolo, M.; Rodriguez, G.; Blenner, M. Dual CRISPR-Cas9 cleavage mediated gene excision and targeted integration in Yarrowia lipolytica. Biotechnol. J. 2018, 13, e1700590. [Google Scholar] [CrossRef]
- Kawano, F.; Okazaki, R.; Yazawa, M.; Sato, M. A photoactivatable Cre-loxP recombination system for optogenetic genome engineering. Nat. Chem. Biol. 2016, 12, 1059–1064. [Google Scholar] [CrossRef]
- Kuhn, R.; Torres, R.M. Cre/loxP recombination system and gene targeting. Methods Mol. Biol. 2002, 180, 175–204. [Google Scholar]
- Richardson, S.M.; Mitchell, L.A.; Stracquadanio, G.; Yang, K.; Dymond, J.S.; DiCarlo, J.E.; Lee, D.; Huang, C.L.; Chandrasegaran, S.; Cai, Y.; et al. Design of a synthetic yeast genome. Science 2017, 355, 1040–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Zhu, R.Y.; Mitchell, L.A.; Ma, L.; Liu, R.; Zhao, M.; Jia, B.; Xu, H.; Li, Y.X.; Yang, Z.M.; et al. In vitro DNA SCRaMbLE. Nat. Commun. 2018, 9, 1935. [Google Scholar] [CrossRef]
- Albert, H.; Dale, E.C.; Lee, E.; Ow, D.W. Site-specific integration of DNA into wild-type and mutant lox sites placed in the plant genome. Plant J. 1995, 7, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Leibig, M.; Krismer, B.; Kolb, M.; Friede, A.; Gotz, F.; Bertram, R. Marker removal in staphylococci via Cre recombinase and different lox sites. Appl. Environ. Microbiol. 2008, 74, 1316–1323. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Yu, H.J.; Hong, Q.; Li, S.P. Cre/lox system and PCR-based genome engineering in Bacillus subtilis. Appl. Environ. Microbiol. 2008, 74, 5556–5562. [Google Scholar] [CrossRef] [Green Version]
- Fickers, P.; Le Dall, M.T.; Gaillardin, C.; Thonart, P.; Nicaud, J.M. New disruption cassettes for rapid gene disruption and marker rescue in the yeast Yarrowia lipolytica. J. Microbiol. Methods 2003, 55, 727–737. [Google Scholar] [CrossRef]
- Hentges, P.; Van Driessche, B.; Tafforeau, L.; Vandenhaute, J.; Carr, A.M. Three novel antibiotic marker cassettes for gene disruption and marker switching in Schizosaccharomyces pombe. Yeast 2005, 22, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Hochrein, L.; Mitchell, L.A.; Schulz, K.; Messerschmidt, K.; Mueller-Roeber, B. L-SCRaMbLE as a tool for light-controlled Cre-mediated recombination in yeast. Nat. Commun. 2018, 9, 1931. [Google Scholar] [CrossRef]
- Gueldener, U.; Heinisch, J.; Koehler, G.J.; Voss, D.; Hegemann, J.H. A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res. 2002, 30, e23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, Y.; Edwards, H.; Zhou, J.; Xu, P. Combining 26s rDNA and the Cre-loxP system for iterative gene integration and efficient marker curation in Yarrowia lipolytica. ACS Synth. Biol. 2019, 8, 568–576. [Google Scholar] [CrossRef]
- Suzuki, N.; Nonaka, H.; Tsuge, Y.; Inui, M.; Yukawa, H. New multiple-deletion method for the Corynebacterium glutamicum genome, using a mutant lox sequence. Appl. Environ. Microbiol. 2005, 71, 8472–8480. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Jiao, L.; Qiao, Y.; Wang, Y.; Xu, L.; Yan, J.; Yan, Y. Overexpression of GRAS Rhizomucor miehei lipase in Yarrowia lipolytica via optimizing promoter, gene dosage and fermentation parameters. J. Biotechnol. 2019, 306, 16–23. [Google Scholar] [CrossRef]
- Eszterhas, S.K.; Bouhassira, E.E.; Martin, D.I.; Fiering, S. Transcriptional interference by independently regulated genes occurs in any relative arrangement of the genes and is influenced by chromosomal integration position. Mol. Cell. Biol. 2002, 22, 469–479. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.J.; Parikh, R.Y.; Weitz, J.S.; Kim, H.D. Suppression of expression between adjacent genes within heterologous modules in yeast. G3 (Bethesda) 2014, 4, 109–116. [Google Scholar] [CrossRef] [Green Version]
- Madzak, C.; Treton, B.; Blanchin-Roland, S. Strong hybrid promoters and integrative expression/secretion vectors for quasi-constitutive expression of heterologous proteins in the yeast Yarrowia lipolytica. J. Mol. Microbiol. Biotechnol. 2000, 2, 207–216. [Google Scholar] [PubMed]
- Madzak, C.; Gaillardin, C.; Beckerich, J.M. Heterologous protein expression and secretion in the non-conventional yeast Yarrowia lipolytica: A review. J. Biotechnol. 2004, 109, 63–81. [Google Scholar] [CrossRef]
- Chen, D.C.; Beckerich, J.M.; Gaillardin, C. One-step transformation of the dimorphic yeast Yarrowia lipolytica. Appl. Microbiol. Biotechnol. 1997, 48, 232–235. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Integration Round | Induction Time (h) | Positive Colonies/All Colonies | Proportion (%) | Selected Colonies | ||
|---|---|---|---|---|---|---|
| Genotype | Rate | rml Copy Number | ||||
| 1 | 12 | 32/32 | 100 | FY1 | - | 0 |
| 2 | 4 | 2/32 | 6.25 | FY2-rml, FY2-rcrml | 5/5 | 0.93 |
| 6 | 2/32 | 6.25 | ||||
| 8 | 14/32 | 43.75 | ||||
| 10 | 24/32 | 75 | ||||
| 12 | 32/32 | 100 | ||||
| 14 | 32/32 | 100 | ||||
| 3 | 12 | 32/32 | 100 | FY3-2rml, FY3-rml-rcrml | 17/13 | 1.99 |
| 4 | 12 | 31/32 | 96.88 | FY4-3rml, FY4-2rml-rcrml | 4/2 | 3.10 |
| 5 | 12 | 29/32 | 90.63 | FY5-4rml, FY5-3rml-rcrml | 3/3 | 4.08 |
| 6 | 12 | 17/32 | 53.12 | FY6-5rml, FY6-4rml-rcrml | 4/2 | 5.12 |
| 14 | 28/32 | 87.5 | ||||
| 16 | 32/32 | 100 | ||||
| 7 | 16 | 32/32 | 100 | FY7-6rml, FY7-5rml-rcrml | 3/3 | 5.94 |
| Gene | GenBank | Lenth (bp) | Integration Round | Positive Colonies/All Colonies | Proportion (%) |
|---|---|---|---|---|---|
| Integration locus: LEU2 gene of FY5-4rml; induction time: 16 h | |||||
| ire1 | XM_002142952.1 | 3663 | 1 | 32/32 | 100 |
| kar2 | U63136.1 | 2013 | 1 | 32/32 | 100 |
| 2 | 32/32 | 100 | |||
| 3 | 32/32 | 100 | |||
| pdi | XM_503481.1 | 1515 | 1 | 32/32 | 100 |
| 2 | 32/32 | 100 | |||
| 3 | 31/32 | 96.88 | |||
| 4 | 31/32 | 96.88 | |||
| sls1 | Z50154.1 | 1281 | 1 | 32/32 | 100 |
| 2 | 32/32 | 100 | |||
| hac1 | XM_500811.1 | 918 | 1 | 32/32 | 100 |
| vgb | AF292694.1 | 441 | 1 | 32/32 | 100 |
| 2 | 32/32 | 100 | |||
| Integration locus: axp1-2 of Po1h; induction time: 12 h | |||||
| rol | AF229435.1 | 1101 | 1 | 31/32 | 96.88 |
| 2 | 32/32 | 100 | |||
| 3 | 32/32 | 100 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Q.; Jiao, L.; Li, W.; Hu, Z.; Li, Y.; Zhang, H.; Yang, M.; Xu, L.; Yan, Y. A Novel Cre/lox-Based Genetic Tool for Repeated, Targeted and Markerless Gene Integration in Yarrowia lipolytica. Int. J. Mol. Sci. 2021, 22, 10739. https://doi.org/10.3390/ijms221910739
Zhou Q, Jiao L, Li W, Hu Z, Li Y, Zhang H, Yang M, Xu L, Yan Y. A Novel Cre/lox-Based Genetic Tool for Repeated, Targeted and Markerless Gene Integration in Yarrowia lipolytica. International Journal of Molecular Sciences. 2021; 22(19):10739. https://doi.org/10.3390/ijms221910739
Chicago/Turabian StyleZhou, Qinghua, Liangcheng Jiao, Wenjuan Li, Zhiming Hu, Yunchong Li, Houjin Zhang, Min Yang, Li Xu, and Yunjun Yan. 2021. "A Novel Cre/lox-Based Genetic Tool for Repeated, Targeted and Markerless Gene Integration in Yarrowia lipolytica" International Journal of Molecular Sciences 22, no. 19: 10739. https://doi.org/10.3390/ijms221910739
APA StyleZhou, Q., Jiao, L., Li, W., Hu, Z., Li, Y., Zhang, H., Yang, M., Xu, L., & Yan, Y. (2021). A Novel Cre/lox-Based Genetic Tool for Repeated, Targeted and Markerless Gene Integration in Yarrowia lipolytica. International Journal of Molecular Sciences, 22(19), 10739. https://doi.org/10.3390/ijms221910739