
Translation Initiation Machinery as a Tumor Selective Target for Radiosensitization


Abstract
1. Introduction
2. Translation Initiation: eIF4F
2.1. eIF4E
2.2. eIF4G
2.3. eIF4A
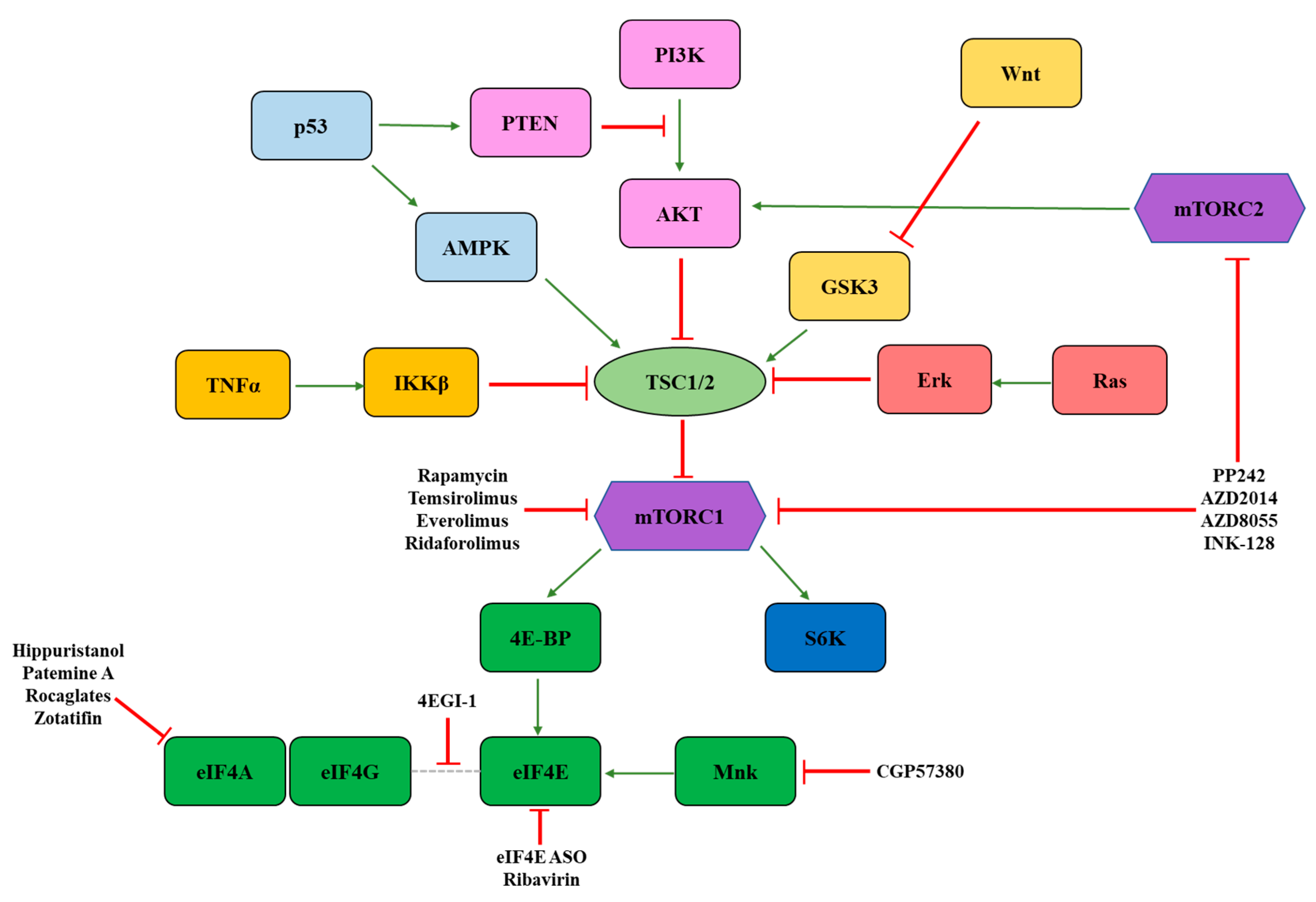
3. Signaling Molecules Regulating eIF4F Activity
3.1. mTOR
3.2. Mnk
4. Ribosome Biogenesis
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Berkey, F.J. Managing the adverse effects of radiation therapy. Am. Fam. Physician 2010, 82, 381–388. [Google Scholar] [PubMed]
- Liauw, S.L.; Connell, P.P.; Weichselbaum, R.R. New paradigms and future challenges in radiation oncology: An update of biological targets and technology. Sci. Transl. Med. 2013, 5, 173sr2. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; de la Pena, L.; Barker, C.; Camphausen, K.; Tofilon, P.J. Radiation-induced changes in gene expression involve recruitment of existing messenger RNAs to and away from polysomes. Cancer Res. 2006, 66, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Wahba, A.; Rath, B.H.; Bisht, K.; Camphausen, K.; Tofilon, P.J. Polysome Profiling Links Translational Control to the Radioresponse of Glioblastoma Stem-like Cells. Cancer Res. 2016, 76, 3078–3087. [Google Scholar] [CrossRef]
- Kumaraswamy, S.; Chinnaiyan, P.; Shankavaram, U.T.; Lu, X.; Camphausen, K.; Tofilon, P.J. Radiation-induced gene translation profiles reveal tumor type and cancer-specific components. Cancer Res. 2008, 68, 3819–3826. [Google Scholar] [CrossRef]
- Amundson, S.A.; Do, K.T.; Vinikoor, L.C.; Lee, R.A.; Koch-Paiz, C.A.; Ahn, J.; Reimers, M.; Chen, Y.; Scudiero, D.A.; Weinstein, J.N.; et al. Integrating global gene expression and radiation survival parameters across the 60 cell lines of the National Cancer Institute Anticancer Drug Screen. Cancer Res. 2008, 68, 415–424. [Google Scholar] [CrossRef]
- Szkanderova, S.; Port, M.; Stulik, J.; Hernychova, L.; Kasalova, I.; Van Beuningen, D.; Abend, M. Comparison of the abundance of 10 radiation-induced proteins with their differential gene expression in L929 cells. Int. J. Radiat. Biol. 2003, 79, 623–633. [Google Scholar] [CrossRef]
- Robichaud, N.; Sonenberg, N.; Ruggero, D.; Schneider, R.J. Translational Control in Cancer. Cold Spring Harb. Perspect. Biol. 2019, 11, a032896. [Google Scholar] [CrossRef]
- Merrick, W.C.; Pavitt, G.D. Protein Synthesis Initiation in Eukaryotic Cells. Cold Spring Harb. Perspect. Biol. 2018, 10, a033092. [Google Scholar] [CrossRef]
- Pisera, A.; Campo, A.; Campo, S. Structure and functions of the translation initiation factor eIF4E and its role in cancer development and treatment. J. Genet. Genom. 2018, 45, 13–24. [Google Scholar] [CrossRef]
- Das, S.; Das, B. eIF4G-an integrator of mRNA metabolism? FEMS Yeast Res. 2016, 16, fow087. [Google Scholar] [CrossRef]
- Heerma van Voss, M.R.; van Diest, P.J.; Raman, V. Targeting RNA helicases in cancer: The translation trap. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 510–520. [Google Scholar] [CrossRef]
- Lu, C.; Makala, L.; Wu, D.; Cai, Y. Targeting translation: eIF4E as an emerging anticancer drug target. Expert Rev. Mol. Med. 2016, 18, e2. [Google Scholar] [CrossRef]
- De Benedetti, A.; Harris, A.L. eIF4E expression in tumors: Its possible role in progression of malignancies. Int. J. Biochem. Cell Biol. 1999, 31, 59–72. [Google Scholar] [CrossRef]
- Pettersson, F.; Yau, C.; Dobocan, M.C.; Culjkovic-Kraljacic, B.; Retrouvey, H.; Puckett, R.; Flores, L.M.; Krop, I.E.; Rousseau, C.; Cocolakis, E.; et al. Ribavirin treatment effects on breast cancers overexpressing eIF4E, a biomarker with prognostic specificity for luminal B-type breast cancer. Clin. Cancer Res. 2011, 17, 2874–2884. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.N.; Hsieh, F.J.; Cheng, Y.M.; Lee, P.H.; Chang, K.J. Expression of eukaryotic initiation factor 4E in gastric adenocarcinoma and its association with clinical outcome. J. Surg. Oncol. 2004, 86, 22–27. [Google Scholar] [CrossRef]
- Hayman, T.J.; Williams, E.S.; Jamal, M.; Shankavaram, U.T.; Camphausen, K.; Tofilon, P.J. Translation initiation factor eIF4E is a target for tumor cell radiosensitization. Cancer Res. 2012, 72, 2362–2372. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.R.; Konicek, B.W.; Vincent, T.M.; Lynch, R.L.; Monteith, D.; Weir, S.N.; Schwier, P.; Capen, A.; Goode, R.L.; Dowless, M.S.; et al. Therapeutic suppression of translation initiation factor eIF4E expression reduces tumor growth without toxicity. J. Clin. Investig. 2007, 117, 2638–2648. [Google Scholar] [CrossRef]
- Jacobson, B.A.; Thumma, S.C.; Jay-Dixon, J.; Patel, M.R.; Dubear Kroening, K.; Kratzke, M.G.; Etchison, R.G.; Konicek, B.W.; Graff, J.R.; Kratzke, R.A. Targeting eukaryotic translation in mesothelioma cells with an eIF4E-specific antisense oligonucleotide. PLoS ONE 2013, 8, e81669. [Google Scholar] [CrossRef] [PubMed]
- Thumma, S.C.; Jacobson, B.A.; Patel, M.R.; Konicek, B.W.; Franklin, M.J.; Jay-Dixon, J.; Sadiq, A.; De, A.; Graff, J.R.; Kratzke, R.A. Antisense oligonucleotide targeting eukaryotic translation initiation factor 4E reduces growth and enhances chemosensitivity of non-small-cell lung cancer cells. Cancer Gene Ther. 2015, 22, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Kurzrock, R.; Oh, Y.; Wheler, J.; Naing, A.; Brail, L.; Callies, S.; Andre, V.; Kadam, S.K.; Nasir, A.; et al. A phase 1 dose escalation, pharmacokinetic, and pharmacodynamic evaluation of eIF-4E antisense oligonucleotide LY2275796 in patients with advanced cancer. Clin. Cancer Res. 2011, 17, 6582–6591. [Google Scholar] [CrossRef]
- Chinnaiyan, P.; Won, M.; Wen, P.Y.; Rojiani, A.M.; Werner-Wasik, M.; Shih, H.A.; Ashby, L.S.; Michael Yu, H.H.; Stieber, V.W.; Malone, S.C.; et al. A randomized phase II study of everolimus in combination with chemoradiation in newly diagnosed glioblastoma: Results of NRG Oncology RTOG 0913. Neuro. Oncol. 2018, 20, 666–673. [Google Scholar] [CrossRef]
- Kentsis, A.; Topisirovic, I.; Culjkovic, B.; Shao, L.; Borden, K.L. Ribavirin suppresses eIF4E-mediated oncogenic transformation by physical mimicry of the 7-methyl guanosine mRNA cap. Proc. Natl. Acad. Sci. USA 2004, 101, 18105–18110. [Google Scholar] [CrossRef] [PubMed]
- Kentsis, A.; Volpon, L.; Topisirovic, I.; Soll, C.E.; Culjkovic, B.; Shao, L.; Borden, K.L. Further evidence that ribavirin interacts with eIF4E. RNA 2005, 11, 1762–1766. [Google Scholar] [CrossRef] [PubMed]
- Borden, K.L.; Culjkovic-Kraljacic, B. Ribavirin as an anti-cancer therapy: Acute myeloid leukemia and beyond? Leuk. Lymphoma 2010, 51, 1805–1815. [Google Scholar] [CrossRef] [PubMed]
- Casaos, J.; Gorelick, N.L.; Huq, S.; Choi, J.; Xia, Y.; Serra, R.; Felder, R.; Lott, T.; Kast, R.E.; Suk, I.; et al. The Use of Ribavirin as an Anticancer Therapeutic: Will It Go Viral? Mol. Cancer Ther. 2019, 18, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Volpin, F.; Casaos, J.; Sesen, J.; Mangraviti, A.; Choi, J.; Gorelick, N.; Frikeche, J.; Lott, T.; Felder, R.; Scotland, S.J.; et al. Use of an anti-viral drug, Ribavirin, as an anti-glioblastoma therapeutic. Oncogene 2017, 36, 3037–3047. [Google Scholar] [CrossRef] [PubMed]
- Huq, S.; Casaos, J.; Serra, R.; Peters, M.; Xia, Y.; Ding, A.S.; Ehresman, J.; Kedda, J.N.; Morales, M.; Gorelick, N.L.; et al. Repurposing the FDA-Approved Antiviral Drug Ribavirin as Targeted Therapy for Nasopharyngeal Carcinoma. Mol. Cancer Ther. 2020, 19, 1797–1808. [Google Scholar] [CrossRef]
- Silvera, D.; Arju, R.; Darvishian, F.; Levine, P.H.; Zolfaghari, L.; Goldberg, J.; Hochman, T.; Formenti, S.C.; Schneider, R.J. Essential role for eIF4GI overexpression in the pathogenesis of inflammatory breast cancer. Nat. Cell Biol. 2009, 11, 903–908. [Google Scholar] [CrossRef]
- Braunstein, S.; Karpisheva, K.; Pola, C.; Goldberg, J.; Hochman, T.; Yee, H.; Cangiarella, J.; Arju, R.; Formenti, S.C.; Schneider, R.J. A hypoxia-controlled cap-dependent to cap-independent translation switch in breast cancer. Mol. Cell 2007, 28, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Comtesse, N.; Keller, A.; Diesinger, I.; Bauer, C.; Kayser, K.; Huwer, H.; Lenhof, H.P.; Meese, E. Frequent overexpression of the genes FXR1, CLAPM1 and EIF4G located on amplicon 3q26-27 in squamous cell carcinoma of the lung. Int. J. Cancer 2007, 120, 2538–2544. [Google Scholar] [CrossRef] [PubMed]
- Badura, M.; Braunstein, S.; Zavadil, J.; Schneider, R.J. DNA damage and eIF4G1 in breast cancer cells reprogram translation for survival and DNA repair mRNAs. Proc. Natl. Acad. Sci. USA 2012, 109, 18767–18772. [Google Scholar] [CrossRef] [PubMed]
- King, H.A.; Gerber, A.P. Translatome profiling: Methods for genome-scale analysis of mRNA translation. Brief. Funct. Genom. 2016, 15, 22–31. [Google Scholar] [CrossRef]
- Moerke, N.J.; Aktas, H.; Chen, H.; Cantel, S.; Reibarkh, M.Y.; Fahmy, A.; Gross, J.D.; Degterev, A.; Yuan, J.; Chorev, M.; et al. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell 2007, 128, 257–267. [Google Scholar] [CrossRef] [PubMed]
- De, A.; Jacobson, B.A.; Peterson, M.S.; Stelzner, M.E.; Jay-Dixon, J.; Kratzke, M.G.; Patel, M.R.; Bitterman, P.B.; Kratzke, R.A. Inhibition of oncogenic cap-dependent translation by 4EGI-1 reduces growth, enhances chemosensitivity and alters genome-wide translation in non-small cell lung cancer. Cancer Gene Ther. 2019, 26, 157–165. [Google Scholar] [CrossRef]
- Wang, W.; Li, J.; Wen, Q.; Luo, J.; Chu, S.; Chen, L.; Qing, Z.; Xie, G.; Xu, L.; Alnemah, M.M.; et al. 4EGI-1 induces apoptosis and enhances radiotherapy sensitivity in nasopharyngeal carcinoma cells via DR5 induction on 4E-BP1 dephosphorylation. Oncotarget 2016, 7, 21728–21741. [Google Scholar] [CrossRef]
- Modelska, A.; Turro, E.; Russell, R.; Beaton, J.; Sbarrato, T.; Spriggs, K.; Miller, J.; Graf, S.; Provenzano, E.; Blows, F.; et al. The malignant phenotype in breast cancer is driven by eIF4A1-mediated changes in the translational landscape. Cell Death Dis. 2015, 6, e1603. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Zhou, Y.; Chen, Y.; Ke, G.; Wen, H.; Wu, X. Decreased expression of EIF4A1 after preoperative brachytherapy predicts better tumor-specific survival in cervical cancer. Int. J. Gynecol. Cancer 2014, 24, 908–915. [Google Scholar] [CrossRef]
- Wolfe, A.L.; Singh, K.; Zhong, Y.; Drewe, P.; Rajasekhar, V.K.; Sanghvi, V.R.; Mavrakis, K.J.; Jiang, M.; Roderick, J.E.; Van der Meulen, J.; et al. RNA G-quadruplexes cause eIF4A-dependent oncogene translation in cancer. Nature 2014, 513, 65–70. [Google Scholar] [CrossRef]
- Bordeleau, M.E.; Mori, A.; Oberer, M.; Lindqvist, L.; Chard, L.S.; Higa, T.; Belsham, G.J.; Wagner, G.; Tanaka, J.; Pelletier, J. Functional characterization of IRESes by an inhibitor of the RNA helicase eIF4A. Nat. Chem. Biol. 2006, 2, 213–220. [Google Scholar] [CrossRef]
- Bordeleau, M.E.; Cencic, R.; Lindqvist, L.; Oberer, M.; Northcote, P.; Wagner, G.; Pelletier, J. RNA-mediated sequestration of the RNA helicase eIF4A by Pateamine A inhibits translation initiation. Chem. Biol. 2006, 13, 1287–1295. [Google Scholar] [CrossRef]
- Bordeleau, M.E.; Robert, F.; Gerard, B.; Lindqvist, L.; Chen, S.M.; Wendel, H.G.; Brem, B.; Greger, H.; Lowe, S.W.; Porco, J.A., Jr.; et al. Therapeutic suppression of translation initiation modulates chemosensitivity in a mouse lymphoma model. J. Clin. Investig. 2008, 118, 2651–2660. [Google Scholar] [CrossRef]
- Ernst, J.T.; Thompson, P.A.; Nilewski, C.; Sprengeler, P.A.; Sperry, S.; Packard, G.; Michels, T.; Xiang, A.; Tran, C.; Wegerski, C.J.; et al. Design of Development Candidate eFT226, a First in Class Inhibitor of Eukaryotic Initiation Factor 4A RNA Helicase. J. Med. Chem. 2020, 63, 5879–5955. [Google Scholar] [CrossRef]
- Thompson, P.A.; Eam, B.; Young, N.P.; Fish, S.; Chen, J.; Barrera, M.; Howard, H.; Sung, E.; Parra, A.; Staunton, J.; et al. Targeting Oncogene mRNA Translation in B-Cell Malignancies with eFT226, a Potent and Selective Inhibitor of eIF4A. Mol. Cancer Ther. 2021, 20, 26–36. [Google Scholar] [CrossRef]
- Webb, T.E.; Davies, M.; Maher, J.; Sarker, D. The eIF4A inhibitor silvestrol sensitizes T-47D ductal breast carcinoma cells to external-beam radiotherapy. Clin. Transl. Radiat. Oncol. 2020, 24, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Ju, X.; Zhou, Y.; Chen, Y.; Ke, G.; Wen, H.; Wu, X. Downregulation of eukaryotic initiation factor 4A1 improves radiosensitivity by delaying DNA double strand break repair in cervical cancer. Oncol. Lett. 2017, 14, 6976–6982. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, Y.; Zhou, X. Research progress of mTOR inhibitors. Eur. J. Med. Chem. 2020, 208, 112820. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Yonezawa, K.; Kozlowski, M.T.; Sugimoto, T.; Andrabi, K.; Weng, Q.P.; Kasuga, M.; Nishimoto, I.; Avruch, J. Regulation of eIF-4E BP1 phosphorylation by mTOR. J. Biol. Chem. 1997, 272, 26457–26463. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- Armengol, G.; Rojo, F.; Castellvi, J.; Iglesias, C.; Cuatrecasas, M.; Pons, B.; Baselga, J.; Ramon y Cajal, S. 4E-binding protein 1: A key molecular “funnel factor” in human cancer with clinical implications. Cancer Res. 2007, 67, 7551–7555. [Google Scholar] [CrossRef]
- Cai, S.L.; Tee, A.R.; Short, J.D.; Bergeron, J.M.; Kim, J.; Shen, J.; Guo, R.; Johnson, C.L.; Kiguchi, K.; Walker, C.L. Activity of TSC2 is inhibited by AKT-mediated phosphorylation and membrane partitioning. J. Cell Biol. 2006, 173, 279–289. [Google Scholar] [CrossRef]
- Ma, L.; Chen, Z.; Erdjument-Bromage, H.; Tempst, P.; Pandolfi, P.P. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 2005, 121, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.F.; Kuo, H.P.; Chen, C.T.; Hsu, J.M.; Chou, C.K.; Wei, Y.; Sun, H.L.; Li, L.Y.; Ping, B.; Huang, W.C.; et al. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 2007, 130, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hu, W.; de Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: Stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar] [CrossRef] [PubMed]
- Dumont, F.J.; Bischoff, P. Disrupting the mTOR signaling network as a potential strategy for the enhancement of cancer radiotherapy. Curr. Cancer Drug Targets 2012, 12, 899–924. [Google Scholar] [CrossRef]
- Wanigasooriya, K.; Tyler, R.; Barros-Silva, J.D.; Sinha, Y.; Ismail, T.; Beggs, A.D. Radiosensitising Cancer Using Phosphatidylinositol-3-Kinase (PI3K), Protein Kinase B (AKT) or Mammalian Target of Rapamycin (mTOR) Inhibitors. Cancers 2020, 12, 1278. [Google Scholar] [CrossRef]
- Holler, M.; Grottke, A.; Mueck, K.; Manes, J.; Jucker, M.; Rodemann, H.P.; Toulany, M. Dual Targeting of Akt and mTORC1 Impairs Repair of DNA Double-Strand Breaks and Increases Radiation Sensitivity of Human Tumor Cells. PLoS ONE 2016, 11, e0154745. [Google Scholar] [CrossRef]
- Eshleman, J.S.; Carlson, B.L.; Mladek, A.C.; Kastner, B.D.; Shide, K.L.; Sarkaria, J.N. Inhibition of the mammalian target of rapamycin sensitizes U87 xenografts to fractionated radiation therapy. Cancer Res. 2002, 62, 7291–7297. [Google Scholar]
- Weppler, S.A.; Krause, M.; Zyromska, A.; Lambin, P.; Baumann, M.; Wouters, B.G. Response of U87 glioma xenografts treated with concurrent rapamycin and fractionated radiotherapy: Possible role for thrombosis. Radiother. Oncol. 2007, 82, 96–104. [Google Scholar] [CrossRef]
- Benjamin, D.; Colombi, M.; Moroni, C.; Hall, M.N. Rapamycin passes the torch: A new generation of mTOR inhibitors. Nat. Rev. Drug Discov. 2011, 10, 868–880. [Google Scholar] [CrossRef] [PubMed]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 2009, 284, 8023–8032. [Google Scholar] [CrossRef] [PubMed]
- Hayman, T.J.; Kramp, T.; Kahn, J.; Jamal, M.; Camphausen, K.; Tofilon, P.J. Competitive but Not Allosteric mTOR Kinase Inhibition Enhances Tumor Cell Radiosensitivity. Transl. Oncol. 2013, 6, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Silvera, D.; Ernlund, A.; Arju, R.; Connolly, E.; Volta, V.; Wang, J.; Schneider, R.J. mTORC1 and -2 Coordinate Transcriptional and Translational Reprogramming in Resistance to DNA Damage and Replicative Stress in Breast Cancer Cells. Mol. Cell Biol. 2017, 37, e00577-16. [Google Scholar] [CrossRef] [PubMed]
- Kahn, J.; Hayman, T.J.; Jamal, M.; Rath, B.H.; Kramp, T.; Camphausen, K.; Tofilon, P.J. The mTORC1/mTORC2 inhibitor AZD2014 enhances the radiosensitivity of glioblastoma stem-like cells. Neuro. Oncol. 2014, 16, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.C.; Huang, H.B.; Hung, S.K.; Liao, H.F.; Lee, C.C.; Lin, H.Y.; Li, S.C.; Ho, H.C.; Hung, C.L.; Su, Y.C. AZD2014 Radiosensitizes Oral Squamous Cell Carcinoma by Inhibiting AKT/mTOR Axis and Inducing G1/G2/M Cell Cycle Arrest. PLoS ONE 2016, 11, e0151942. [Google Scholar] [CrossRef]
- Hayman, T.J.; Wahba, A.; Rath, B.H.; Bae, H.; Kramp, T.; Shankavaram, U.T.; Camphausen, K.; Tofilon, P.J. The ATP-competitive mTOR inhibitor INK128 enhances in vitro and in vivo radiosensitivity of pancreatic carcinoma cells. Clin. Cancer Res. 2014, 20, 110–119. [Google Scholar] [CrossRef]
- Miyahara, H.; Yadavilli, S.; Natsumeda, M.; Rubens, J.A.; Rodgers, L.; Kambhampati, M.; Taylor, I.C.; Kaur, H.; Asnaghi, L.; Eberhart, C.G.; et al. The dual mTOR kinase inhibitor TAK228 inhibits tumorigenicity and enhances radiosensitization in diffuse intrinsic pontine glioma. Cancer Lett 2017, 400, 110–116. [Google Scholar] [CrossRef]
- Waqar, S.N.; Robinson, C.; Bradley, J.; Goodgame, B.; Rooney, M.; Williams, K.; Gao, F.; Govindan, R. A phase I study of temsirolimus and thoracic radiation in non-small-cell lung cancer. Clin. Lung Cancer 2014, 15, 119–123. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Galanis, E.; Wu, W.; Dietz, A.B.; Kaufmann, T.J.; Gustafson, M.P.; Brown, P.D.; Uhm, J.H.; Rao, R.D.; Doyle, L.; et al. Combination of temsirolimus (CCI-779) with chemoradiation in newly diagnosed glioblastoma multiforme (GBM) (NCCTG trial N027D) is associated with increased infectious risks. Clin. Cancer Res. 2010, 16, 5573–5580. [Google Scholar] [CrossRef]
- Saba, N.F.; Force, S.; Staley, C.; Fernandez, F.; Willingham, F.; Pickens, A.; Cardona, K.; Chen, Z.; Goff, L.; Cardin, D.; et al. Phase IB Study of Induction Chemotherapy With XELOX, Followed by Radiation Therapy, Carboplatin, and Everolimus in Patients With Locally Advanced Esophageal Cancer. Am. J. Clin. Oncol. 2019, 42, 331–336. [Google Scholar] [CrossRef]
- Narayan, V.; Vapiwala, N.; Mick, R.; Subramanian, P.; Christodouleas, J.P.; Bekelman, J.E.; Deville, C.; Rajendran, R.; Haas, N.B. Phase 1 Trial of Everolimus and Radiation Therapy for Salvage Treatment of Biochemical Recurrence in Prostate Cancer Patients Following Prostatectomy. Int. J. Radiat. Oncol. Biol. Phys. 2017, 97, 355–361. [Google Scholar] [CrossRef]
- Naing, A.; Aghajanian, C.; Raymond, E.; Olmos, D.; Schwartz, G.; Oelmann, E.; Grinsted, L.; Burke, W.; Taylor, R.; Kaye, S.; et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of AZD8055 in advanced solid tumours and lymphoma. Br. J. Cancer 2012, 107, 1093–1099. [Google Scholar] [CrossRef]
- Asahina, H.; Nokihara, H.; Yamamoto, N.; Yamada, Y.; Tamura, Y.; Honda, K.; Seki, Y.; Tanabe, Y.; Shimada, H.; Shi, X.; et al. Safety and tolerability of AZD8055 in Japanese patients with advanced solid tumors; a dose-finding phase I study. Investig. New Drugs 2013, 31, 677–684. [Google Scholar] [CrossRef]
- Voss, M.H.; Gordon, M.S.; Mita, M.; Rini, B.; Makker, V.; Macarulla, T.; Smith, D.C.; Cervantes, A.; Puzanov, I.; Pili, R.; et al. Phase 1 study of mTORC1/2 inhibitor sapanisertib (TAK-228) in advanced solid tumours, with an expansion phase in renal, endometrial or bladder cancer. Br. J. Cancer 2020, 123, 1590–1598. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A., 3rd; Kurkjian, C.D.; Hart, L.; Pant, S.; Murphy, P.B.; Jones, S.F.; Neuwirth, R.; Patel, C.G.; Zohren, F.; Infante, J.R. TAK-228 (formerly MLN0128), an investigational dual TORC1/2 inhibitor plus paclitaxel, with/without trastuzumab, in patients with advanced solid malignancies. Cancer Chemother. Pharm. 2017, 80, 261–273. [Google Scholar] [CrossRef]
- Topisirovic, I.; Ruiz-Gutierrez, M.; Borden, K.L. Phosphorylation of the eukaryotic translation initiation factor eIF4E contributes to its transformation and mRNA transport activities. Cancer 2004, 64, 8639–8642. [Google Scholar] [CrossRef] [PubMed]
- Furic, L.; Rong, L.; Larsson, O.; Koumakpayi, I.H.; Yoshida, K.; Brueschke, A.; Petroulakis, E.; Robichaud, N.; Pollak, M.; Gaboury, L.A.; et al. eIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression. Proc. Natl. Acad. Sci. USA 2010, 107, 14134–14139. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Watanabe-Fukunaga, R.; Fukuyama, H.; Nagata, S.; Fukunaga, R. Mnk2 and Mnk1 are essential for constitutive and inducible phosphorylation of eukaryotic initiation factor 4E but not for cell growth or development. Cell Biol. 2004, 24, 6539–6549. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Yu, R.; Wang, X.; Proud, C.G.; Jiang, T. Progress in developing MNK inhibitors. Eur. Chem. 2021, 219, 113420. [Google Scholar] [CrossRef]
- Wang, W.; Wen, Q.; Luo, J.; Chu, S.; Chen, L.; Xu, L.; Zang, H.; Alnemah, M.M.; Li, J.; Zhou, J.; et al. Suppression Of beta-catenin Nuclear Translocation by CGP57380 Decelerates Poor Progression and Potentiates Radiation-Induced Apoptosis in Nasopharyngeal Carcinoma. Theranostics 2017, 7, 2134–2149. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Tian, L.; Feng, X.; Cheng, J.; Gong, Y.; Liu, X.; Zhang, Z.; Yang, X.; He, S.; Li, C.Y.; et al. eIF4E-phosphorylation-mediated Sox2 upregulation promotes pancreatic tumor cell repopulation after irradiation. Cancer Lett. 2016, 375, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, A.; Pagano, M.; Russo, G.; Russo, A. Ribosome Biogenesis and Cancer: Overview on Ribosomal Proteins. Int. J. Mol. Sci. 2021, 22, 5496. [Google Scholar] [CrossRef] [PubMed]
- Kressler, D.; Hurt, E.; Bassler, J. Driving ribosome assembly. Biochim. Biophys. Acta 2010, 1803, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Carotenuto, P.; Pecoraro, A.; Palma, G.; Russo, G.; Russo, A. Therapeutic Approaches Targeting Nucleolus in Cancer. Cells 2019, 8, 1090. [Google Scholar] [CrossRef] [PubMed]
- Burger, K.; Muhl, B.; Harasim, T.; Rohrmoser, M.; Malamoussi, A.; Orban, M.; Kellner, M.; Gruber-Eber, A.; Kremmer, E.; Holzel, M.; et al. Chemotherapeutic drugs inhibit ribosome biogenesis at various levels. J. Biol. Chem. 2010, 285, 12416–12425. [Google Scholar] [CrossRef]
- Ferreira, R.; Schneekloth, J.S., Jr.; Panov, K.I.; Hannan, K.M.; Hannan, R.D. Targeting the RNA Polymerase I Transcription for Cancer Therapy Comes of Age. Cells 2020, 9, 266. [Google Scholar] [CrossRef] [PubMed]
- Drygin, D.; Siddiqui-Jain, A.; O’Brien, S.; Schwaebe, M.; Lin, A.; Bliesath, J.; Ho, C.B.; Proffitt, C.; Trent, K.; Whitten, J.P.; et al. Anticancer activity of CX-3543: A direct inhibitor of rRNA biogenesis. Cancer Res. 2009, 69, 7653–7661. [Google Scholar] [CrossRef]
- Drygin, D.; Lin, A.; Bliesath, J.; Ho, C.B.; O’Brien, S.E.; Proffitt, C.; Omori, M.; Haddach, M.; Schwaebe, M.K.; Siddiqui-Jain, A.; et al. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011, 71, 1418–1430. [Google Scholar] [CrossRef]
- Peltonen, K.; Colis, L.; Liu, H.; Jaamaa, S.; Moore, H.M.; Enback, J.; Laakkonen, P.; Vaahtokari, A.; Jones, R.J.; af Hallstrom, T.M.; et al. Identification of novel p53 pathway activating small-molecule compounds reveals unexpected similarities with known therapeutic agents. PLoS ONE 2010, 5, e12996. [Google Scholar] [CrossRef]
- Kohler, A.; Hurt, E. Exporting RNA from the nucleus to the cytoplasm. Nat. Rev. Mol. Cell. Biol. 2007, 8, 761–773. [Google Scholar] [CrossRef]
- Azmi, A.S.; Uddin, M.H.; Mohammad, R.M. The nuclear export protein XPO1—From biology to targeted therapy. Nat. Rev. Clin. Oncol. 2021, 18, 152–169. [Google Scholar] [CrossRef]
- Wahba, A.; Rath, B.H.; O’Neill, J.W.; Camphausen, K.; Tofilon, P.J. The XPO1 Inhibitor Selinexor Inhibits Translation and Enhances the Radiosensitivity of Glioblastoma Cells Grown In Vitro and In Vivo. Mol. Cancer Ther. 2018, 17, 1717–1726. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Agent | Target | Clinical Usage | Radiotherapy Trial |
|---|---|---|---|
| LY2275796 (ASO) | eIF4E | Phase 1 complete; not as a sensitizer | no |
| Ribavirin | eIF4E | FDA approved; not as a sensitizer | no |
| Zotatifin (eFT226) | eIF4A | Phase 1 | no |
| Sirolimus (rapamycin) | mTORC1 | FDA approved; not as a sensitizer | no |
| Temsirolimus (CCI-779) | mTORC1 | FDA approved; not as a sensitizer | yes-ongoing |
| Everolimus (RAD-001) | mTORC1 | Phase II; as an RT sensitizer | yes-no benefit [22] |
| Ridaforolimus (AP23573) | mTORC1 | Phase III; drug only combinations | no |
| AZD8055 | mTORC1/2 | Phase 1 | no |
| Sapanisertib (Tak-228) | mTORC1/2 | Phase2/3; not as a sensitizer | no |
| BAY1143269 | MNK1 | Phase 1; not as a sensitizer | no |
| Tomivosertib (eFT508) | MNK1/2 | Phase 1/2; not as a sensitizer | no |
| ETC-1907206 | MNK1/2 | Phase 1; not as a sensitizer | no |
| Quarfloxin (CX-3543) | RNA pol I | Phase 1; not as a sensitizer | no |
| Pidnarulex (CX-5461) | RNA pol I | Phase 1; not as a sensitizer | no |
| Selinexor (KPT-330) | Ribosome Biogenesis | FDA approved; not as a sensitizer | yes-ongoing |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lehman, S.L.; Wilson, E.D.; Camphausen, K.; Tofilon, P.J. Translation Initiation Machinery as a Tumor Selective Target for Radiosensitization. Int. J. Mol. Sci. 2021, 22, 10664. https://doi.org/10.3390/ijms221910664
Lehman SL, Wilson ED, Camphausen K, Tofilon PJ. Translation Initiation Machinery as a Tumor Selective Target for Radiosensitization. International Journal of Molecular Sciences. 2021; 22(19):10664. https://doi.org/10.3390/ijms221910664
Chicago/Turabian StyleLehman, Stacey L., Evan D. Wilson, Kevin Camphausen, and Philip J. Tofilon. 2021. "Translation Initiation Machinery as a Tumor Selective Target for Radiosensitization" International Journal of Molecular Sciences 22, no. 19: 10664. https://doi.org/10.3390/ijms221910664
APA StyleLehman, S. L., Wilson, E. D., Camphausen, K., & Tofilon, P. J. (2021). Translation Initiation Machinery as a Tumor Selective Target for Radiosensitization. International Journal of Molecular Sciences, 22(19), 10664. https://doi.org/10.3390/ijms221910664