
A Method to Monitor the NAD+ Metabolome—From Mechanistic to Clinical Applications

 ,
,  ,
,
Abstract
:
1. Introduction
2. Results and Discussion
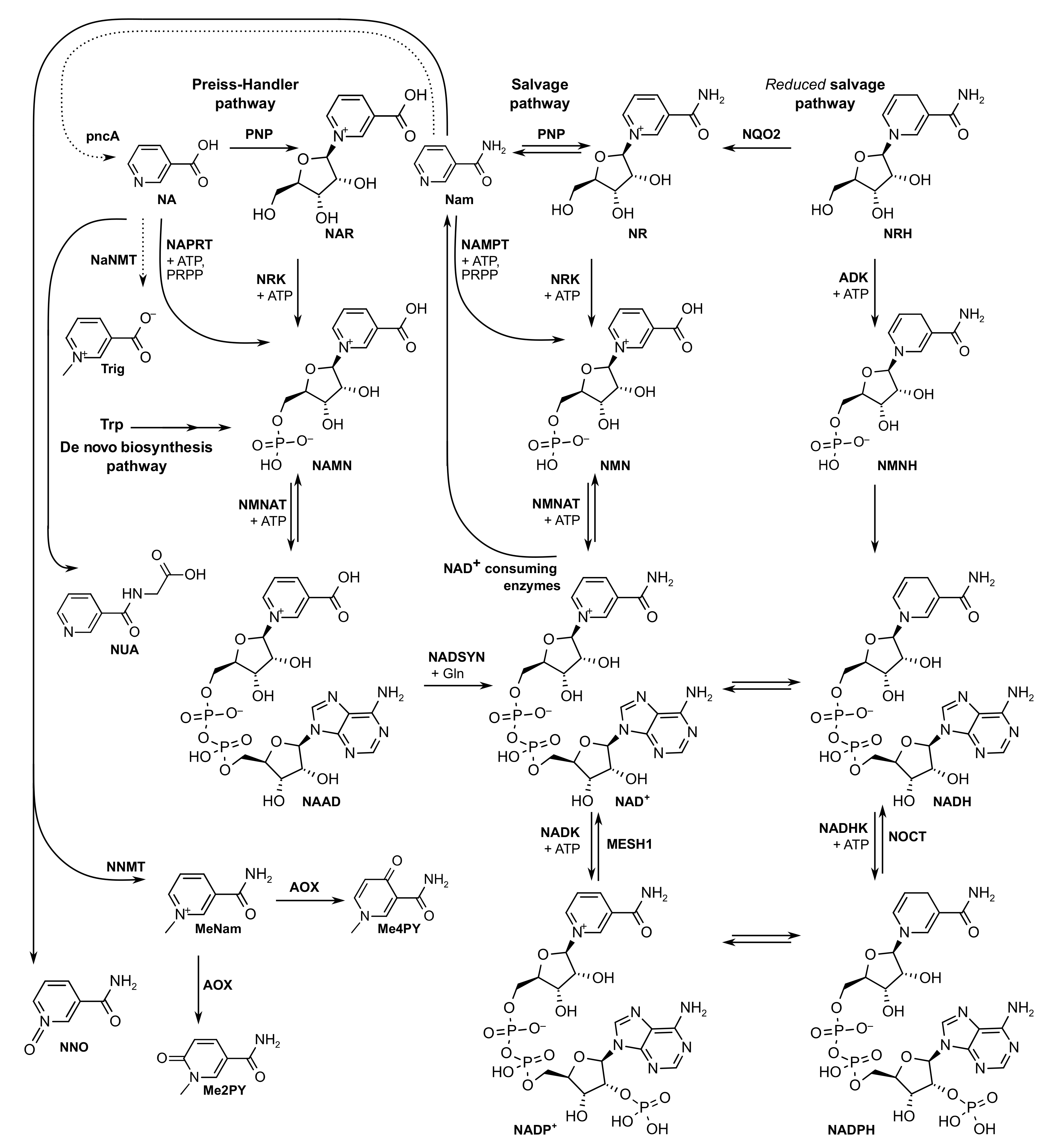
2.1. Setting up the NAD+ Metabolome LC-MS/MS Method
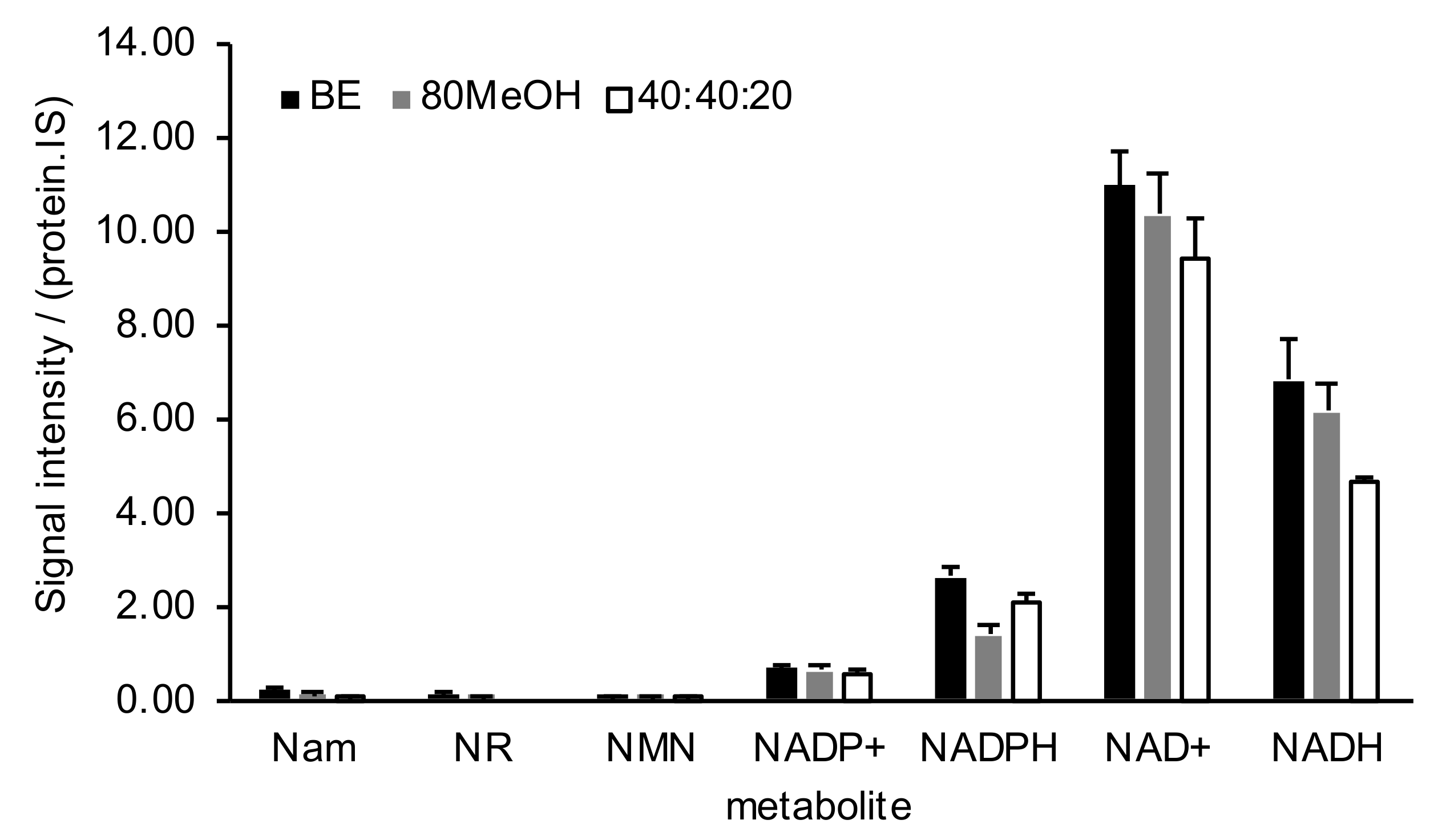
2.2. NAD+ Metabolome Sample Preparation
2.3. NAD+ Metabolome Is Sensitive to Precursor Administration
2.4. NAD+ Metabolome across Pre-Clinical Samples
2.5. NAD+ Metabolome in Clinical Samples
2.5.1. Selection of Blood Matrix
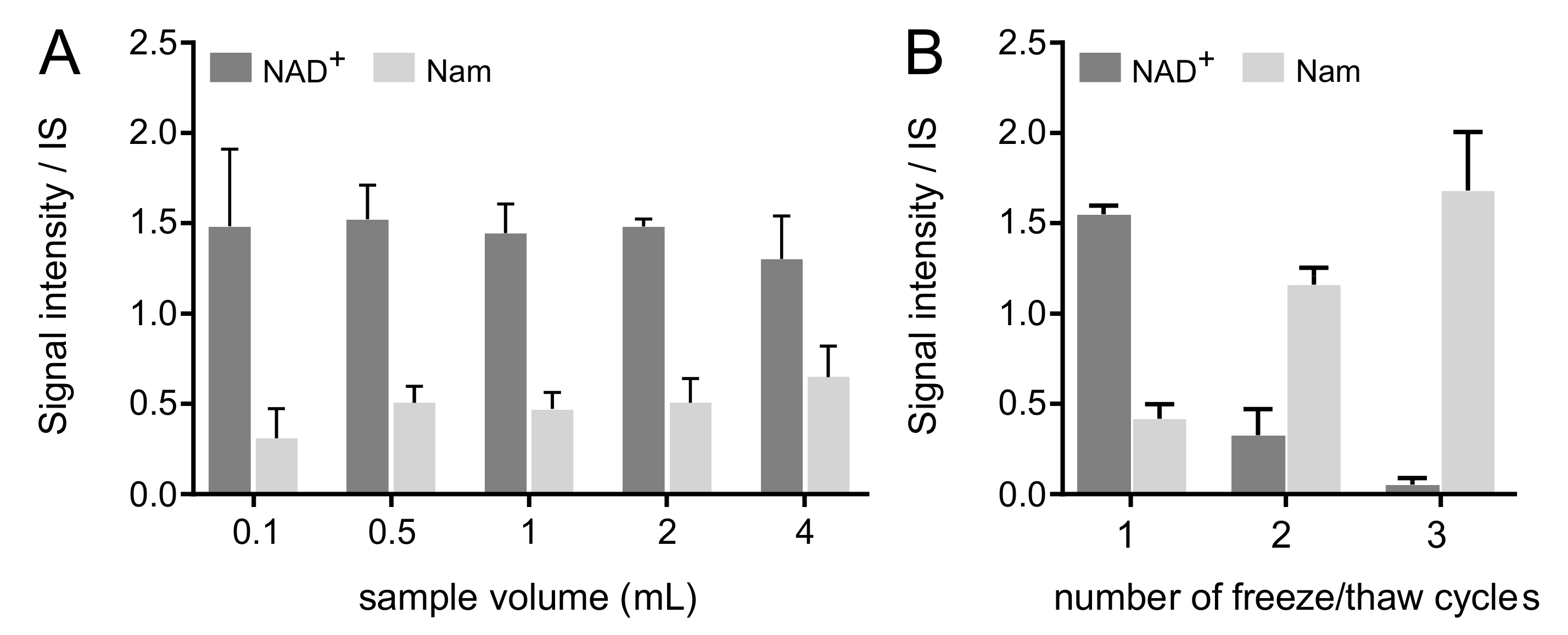
2.5.2. Effect of Aliquot Volume at Blood Draw
2.5.3. Effect of Freeze–Thaw Cycles
2.5.4. Setting up a Whole Blood Quantitative NAD+ Metabolome Method
3. Materials and Methods
3.1. Chemicals
3.2. Standard Compounds
3.3. Mammalian Cell Culture
3.4. Animal Tissues
3.5. Clinical samples
3.5.1. Human Blood (Whole Blood, Plasma, Serum)
3.5.2. Human Urine
3.6. Sample Extraction
3.6.1. Eighty Percent Cold Methanol (‘80MeOH’)
3.6.2. Volume Proportion 40:40:20 with 0.1M Formic Acid (’40:20:20′)
3.6.3. Biphasic Extraction (‘BE’)
3.7. Liquid Chromatography Mass Spectrometry
3.8. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fessel, J.P.; Oldham, W.M. Pyridine Dinucleotides from Molecules to Man. Antioxid. Redox Signal. 2018, 28, 180–212. [Google Scholar] [CrossRef]
- Xiao, W.; Wang, R.S.; Handy, D.E.; Loscalzo, J. NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism. Antioxid. Redox Signal. 2018, 28, 251–272. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Chiarugi, A.; Dölle, C.; Felici, R.; Ziegler, M. The NAD metabolome—A key determinant of cancer cell biology. Nat. Rev. Cancer 2012, 12, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.P.; Price, N.L.; Ling, A.J.Y.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S.I. Nicotinamide mononucleotide, a key NAD + intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gariani, K.; Menzies, K.J.; Ryu, D.; Wegner, C.J.; Wang, X.; Ropelle, E.R.; Moullan, N.; Zhang, H.; Perino, A.; Lemos, V.; et al. Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology 2016, 63, 1190–1204. [Google Scholar] [CrossRef]
- Poyan Mehr, A.; Tran, M.T.; Ralto, K.M.; Leaf, D.E.; Washco, V.; Messmer, J.; Lerner, A.; Kher, A.; Kim, S.H.; Khoury, C.C.; et al. De novo NAD+ biosynthetic impairment in acute kidney injury in humans. Nat. Med. 2018, 24, 1351–1359. [Google Scholar] [CrossRef]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef] [Green Version]
- Elvehjem, C.A.; Madden, R.J.; Strong, F.M.; Woolley, D.W. The isolation and identification of the anti-black tongue factor. J. Biol. Chem. 1938, 123, 137–149. [Google Scholar] [CrossRef]
- Preiss, J.; Handler, P. Biosynthesis of diphosphopyridine nucleotide. I. Identification of intermediates. J. Biol. Chem. 1958, 233, 488–492. [Google Scholar] [CrossRef]
- Bieganowski, P.; Brenner, C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a preiss-handler independent route to NAD+ in fungi and humans. Cell 2004, 117, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Giroud-Gerbetant, J.; Joffraud, M.; Giner, M.P.; Cercillieux, A.; Bartova, S.; Makarov, M.V.; Zapata-Pérez, R.; Sánchez-García, J.L.; Houtkooper, R.H.; Migaud, M.E.; et al. A reduced form of nicotinamide riboside defines a new path for NAD+ biosynthesis and acts as an orally bioavailable NAD+ precursor. Mol. Metab. 2019, 30, 192–202. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, N.; Zhang, G.; Sauve, A.A. NRH salvage and conversion to NAD+ requires NRH kinase activity by adenosine kinase. Nat. Metab. 2020, 2, 364–379. [Google Scholar] [CrossRef]
- Ying, W. NAD+/NADH and NADP+/NADPH in Cellular Functions and Cell Death: Regulation and Biological Consequences. Antioxid. Redox Signal. 2008, 10, 179–206. [Google Scholar] [CrossRef] [Green Version]
- Mrochek, J.E.; Jolley, R.L.; Young, D.S.; Turner, W.J. Metabolic response of humans to ingestion of nicotinic acid and nicotinamide. Clin. Chem. 1976, 22, 1821–1827. [Google Scholar] [CrossRef]
- Wiederkehr, A.; Demaurex, N. Illuminating redox biology using NADH- and NADPH-specific sensors. Nat. Methods 2017, 14, 671–672. [Google Scholar] [CrossRef]
- Moco, S.; Bino, R.J.; De Vos, R.C.H.; Vervoort, J. Metabolomics technologies and metabolite identification. TrAC Trends Anal. Chem. 2007, 26, 855–866. [Google Scholar] [CrossRef]
- Shulman, R.G.; Brown, T.R.; Ugurbil, K.; Ogawa, S.; Cohen, S.M.; Hollander, J.A. Den Cellular applications of 31P and 13C nuclear magnetic resonance. Science 1979, 205, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Shabalin, K.; Nerinovski, K.; Yakimov, A.; Kulikova, V.; Svetlova, M.; Solovjeva, L.; Khodorkovskiy, M.; Gambaryan, S.; Cunningham, R.; Migaud, M.E.; et al. NAD metabolome analysis in human cells using 1 H NMR spectroscopy. Int. J. Mol. Sci. 2018, 19, 3906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagana Gowda, G.A.; Raftery, D. Whole Blood Metabolomics by1H NMR Spectroscopy Provides a New Opportunity to Evaluate Coenzymes and Antioxidants. Anal. Chem. 2017, 89, 4620–4627. [Google Scholar] [CrossRef]
- Buescher, J.M.; Moco, S.; Sauer, U.; Zamboni, N. Ultrahigh Performance Liquid Chromatography-Tandem Mass Spectrometry Method for Fast and Robust Quantification of Anionic and Aromatic Metabolites. Anal. Chem. 2010, 82, 4403–4412. [Google Scholar] [CrossRef]
- Lu, W.; Wang, L.; Chen, L.; Hui, S.; Rabinowitz, J.D. Extraction and Quantitation of Nicotinamide Adenine Dinucleotide Redox Cofactors. Antioxid. Redox Signal. 2018, 28, 167–179. [Google Scholar] [CrossRef]
- Aronov, P.; Kawahara, Y.; Cox, J.; Christison, T.; Lopez, L. Determination of NADH and NADPH Using Ion Chromatography and High Resolution Accurate Mass Spectrometry. Thermo Sci. Tech. Note 2014, 149, 1–5. [Google Scholar]
- Taguchi, K.; Fukusaki, E.; Bamba, T. Determination of Niacin and Its Metabolites Using Supercritical Fluid Chromatography Coupled to Tandem Mass Spectrometry. Mass Spectrom. 2014, 3, A0029. [Google Scholar] [CrossRef] [Green Version]
- Bustamante, S.; Jayasena, T.; Richani, D.; Gilchrist, R.B.; Wu, L.E.; Sinclair, D.A.; Sachdev, P.S.; Braidy, N. Quantifying the cellular NAD+ metabolome using a tandem liquid chromatography mass spectrometry approach. Metabolomics 2018, 14, 1–15. [Google Scholar] [CrossRef]
- Yin, P.; Lehmann, R.; Xu, G. Effects of pre-analytical processes on blood samples used in metabolomics studies. Anal. Bioanal. Chem. 2015, 407, 4879–4892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Airhart, S.E.; Shireman, L.M.; Risler, L.J.; Anderson, G.D.; Gowda, G.A.N.; Raftery, D.; Tian, R.; Shen, D.D.; O’Brien, K.D. An open-label, non-randomized study of the pharmacokinetics of the nutritional supplement nicotinamide riboside (NR) and its effects on blood NAD+ levels in healthy volunteers. PLoS ONE 2017, 12, e0186459. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 2014, 42, D199–D205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LogD Predictor, ChemAxon, 2021. Available online: https://disco.chemaxon.com/calculators/demo/plugins/logd/ (accessed on 30 September 2021).
- Ma, S.; Subramanian, R.; Xu, Y.; Schrag, M.; Shou, M. Structural Characterization of Novel Adenine Dinucleotide Phosphate Conjugates of Imatinib in Incubations with Rat and Human Liver Microsomes. Drug Metab. Dispos. 2008, 36, 2414–2418. [Google Scholar] [CrossRef] [Green Version]
- Periat, A.; Kohler, I.; Thomas, A.; Nicoli, R.; Boccard, J.; Veuthey, J.L.; Schappler, J.; Guillarme, D. Systematic evaluation of matrix effects in hydrophilic interaction chromatography versus reversed phase liquid chromatography coupled to mass spectrometry. J. Chromatogr. A 2016, 1439, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, M.A.; Burant, C.F.; Kennedy, R.T. Reducing time and increasing sensitivity in sample preparation for adherent mammalian cell metabolomics. Anal. Chem. 2011, 83, 3406–3414. [Google Scholar] [CrossRef] [Green Version]
- Elia, I.; Broekaert, D.; Christen, S.; Boon, R.; Radaelli, E.; Orth, M.F.; Verfaillie, C.; Grünewald, T.G.P.; Fendt, S.-M. Proline metabolism supports metastasis formation and could be inhibited to selectively target metastasizing cancer cells. Nat. Commun. 2017, 8, 15267. [Google Scholar] [CrossRef]
- Ratajczak, J.; Joffraud, M.; Trammell, S.A.J.; Ras, R.; Canela, N.; Boutant, M.; Kulkarni, S.S.; Rodrigues, M.; Redpath, P.; Migaud, M.E.; et al. NRK1 controls nicotinamide mononucleotide and nicotinamide riboside metabolism in mammalian cells. Nat. Commun. 2016, 7, 13103. [Google Scholar] [CrossRef] [PubMed]
- Zamporlini, F.; Ruggieri, S.; Mazzola, F.; Amici, A.; Orsomando, G.; Raffaelli, N. Novel assay for simultaneous measurement of pyridine mononucleotides synthesizing activities allows dissection of the NAD + biosynthetic machinery in mammalian cells. FEBS J. 2014, 281, 5104–5119. [Google Scholar] [CrossRef]
- Sambeat, A.; Ratajczak, J.; Joffraud, M.; Sanchez-Garcia, J.L.; Giner, M.P.; Valsesia, A.; Giroud-Gerbetant, J.; Valera-Alberni, M.; Cercillieux, A.; Boutant, M.; et al. Endogenous nicotinamide riboside metabolism protects against diet-induced liver damage. Nat. Commun. 2019, 10, 4291. [Google Scholar] [CrossRef]
- Trammell, S.A.J.; Schmidt, M.S.; Weidemann, B.J.; Redpath, P.; Jaksch, F.; Dellinger, R.W.; Li, Z.; Abel, E.D.; Migaud, M.E.; Brenner, C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun. 2016, 7, 12948. [Google Scholar] [CrossRef]
- Dellinger, R.W.; Santos, S.R.; Morris, M.; Evans, M.; Alminana, D.; Guarente, L.; Marcotulli, E. Repeat dose NRPT (nicotinamide riboside and pterostilbene) increases NAD+ levels in humans safely and sustainably: A randomized, double-blind, placebo-controlled study. NPJ Aging Mech. Dis. 2017, 3, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, K.; Tanaka, K. Simple measurement of blood nadp and blood levels of nad and nadp in humans. Agric. Biol. Chem. 1986, 50, 2941–2942. [Google Scholar] [CrossRef]
- Stocchi, V.; Cucchiarini, L.; Magnani, M.; Chiarantini, L.; Palma, P.; Crescentini, G. Simultaneous extraction and reverse-phase high-performance liquid chromatographic determination of adenine and pyridine nucleotides in human red blood cells. Anal. Biochem. 1985, 146, 118–124. [Google Scholar] [CrossRef]
- Trammell, S.A.; Brenner, C. Targeted, LCMS-based Metabolomics for Quantitative Measurement of NAD+ Metabolites. Comput. Struct. Biotechnol. J. 2013, 4, e201301012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, C.T.; Tu, B.P.; Tang, Y. Eight Kinetically Stable but Thermodynamically Activated Molecules that Power Cell Metabolism. Chem. Rev. 2018, 118, 1460–1494. [Google Scholar] [CrossRef] [PubMed]
- Makarov, M.V.; Harris, N.W.; Rodrigues, M.; Migaud, M.E. Scalable syntheses of traceable ribosylated NAD+ precursors. Org. Biomol. Chem. 2019, 17, 8716–8720. [Google Scholar] [CrossRef]
- Christen, S.; Lorendeau, D.; Schmieder, R.; Broekaert, D.; Metzger, K.; Veys, K.; Elia, I.; Buescher, J.M.; Orth, M.F.; Davidson, S.M.; et al. Breast Cancer-Derived Lung Metastases Show Increased Pyruvate Carboxylase-Dependent Anaplerosis. Cell Rep. 2016, 17, 837–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | CAS nr | Rt | MF | MM | MIM | Q1 > Q3 | CE | S-Lens |
|---|---|---|---|---|---|---|---|---|
| Nam | 98-92-0 | 2.9 | C6H6N2O | 122.12 | 122.05 | 123 > 80, 53 | 26, 32 | 102 |
| Nam-d4 | 347841-88-7 | 2.9 | C6H22H4N2O | 126.15 | 126.07 | 127 > 84, 81 | 22, 18 | 99 |
| MeXPY (Me2PY /Me4PY) | 701-44-0, 769-49-3 | 3.2 | C7H8N2O2 | 152.15 | 152.06 | 153 > 136, 92 | 15, 23 | 62 |
| NA | 59-67-6 | 3.9 | C6H5NO2 | 123.11 | 123.03 | 124 > 78, 53 | 22, 31 | 102 |
| NNO | 1986-81-8 | 4.8 | C6H6N2O2 | 138.12 | 138.04 | 139 > 106, 78 | 21, 31 | 118 |
| NUA | 583-08-4 | 5.3 | C8H8N2O3 | 180.16 | 180.05 | 181 > 135, 80 | 16, 34 | 107 |
| NRH | 19132-12-8 | 5.4 | C11H16N2O5 | 256.26 | 256.11 | 257 > 150, 108 | 24, 31 | 99 |
| Trig | 535-83-1 | 9.2 | C7H7NO2 | 137.14 | 137.05 | 138 > 92, 94 | 21, 20 | 78 |
| Trig-d3 | - | 9.2 | C7H42H3NO2 | 140.15 | 140.07 | 141 > 97, 95 | 21, 23 | 73 |
| NAR | 17720-18-2 | 10.5 | C11H13NO6 | 255.22 | 255.07 | 256 > 124, 78 | 13, 40 | 146, 64 |
| NADH | 58-68-4 | 10.6 | C21H29N7O14P2 | 665.44 | 665.12 | 666 > 649, 302 | 20, 40 | 119 |
| 13C-NADH | 1259998-13-4 | 10.6 | 13C21H29N7O14P2 | 686.29 | 686.20 | 687 > 670, 311 | 20, 40 | 119 |
| NMNH | 4229-56-5 | 10.8 | C11H17N2O8P | 336.24 | 336.07 | 337 > 320, 108 | 13, 28 | 76 |
| NAAD | 6450-77-7 | 11.1 | C21H26N6O15P2 | 664.41 | 664.09 | 665 > 136, 119 | 40 | 144 |
| NAMN | 321-02-8 | 11.4 | C11H14NO9P | 335.20 | 335.04 | 336 > 124, 97 | 13, 22 | 92 |
| NAD+ | 53-84-9 | 11.5 | C21H27N7O14P2 | 663.43 | 663.11 | 664 > 524, 428 | 20 | 137 |
| 13C-NAD+ | 1259998-09-8 | 11.5 | 13C21H27N7O14P2 | 684.27 | 684.18 | 685 > 539, 438 | 20 | 137 |
| NADPH | 53-57-6 | 11.7 | C21H30N7O17P3 | 745.42 | 745.09 | 746 > 729, 428 | 20, 40 | 206 |
| 13C-NADPH | 1259998-18-9 | 11.7 | 13C21H30N7O17P3 | 766.27 | 766.16 | 767 > 438, 750 | 20 | 150 |
| NADP+ | 53-59-8 | 11.9 | C21H28N7O17P3 | 743.41 | 743.08 | 744 > 604, 508 | 20 | 150 |
| 13C-NADP+ | 1259998-15-6 | 11.9 | 13C21H28N7O17P3 | 764.25 | 764.15 | 765 > 518, 619 | 20 | 150 |
| NMN | 1094-61-7 | 12.0 | C11H15N2O8P | 334.22 | 334.06 | 335 > 123, 80 | 14, 46 | 85 |
| MeNam | 3106-60-3 | 16.0 | C7H9N2O | 137.16 | 137.07 | 137 > 79, 52 | 31, 37 | 89 |
| NR | 1341-23-7 | 17.1 | C11H15N2O5 | 255.25 | 255.10 | 255 > 123, 80 | 13, 38 | 64 |
| NR-d4 | - | 17.1 | C11H112H4N2O5 | 259.27 | 259.12 | 259 > 127, 84 | 16, 34 | 46, 49 |
| ID | Urine | Plasma | Serum | Whole Blood | Whole Blood | |||
|---|---|---|---|---|---|---|---|---|
| LOD | LOQ | r2 | C (Whole Blood) Min–Max ± SD | |||||
| Nam | X | - | - | X | 0.3 | 1.0 | 0.991 | 5.0 ± 0.3–10.0 ± 1.4 |
| MeXPY | X | X | X | X | - | - | - | - |
| NRH | X | X | X | X | - | - | - | - |
| Trig | X | X | X | X | - | - | - | - |
| NADH | - | - | - | X | 0.2 | 0.6 | 0.996 | 8.0 ± 1.0–16.0 ± 0.6 |
| NADPH | - | - | - | X | 0.3 | 1.0 | 0.991 | 7.0 ± 0.8–17.0 ± 1.0 |
| NAD+ | - | - | - | X | 0.2 | 0.7 | 0.995 | 10.0 ± 1.4–18.0 ± 0.1 |
| NADP+ | - | - | - | X | 0.3 | 1.1 | 0.989 | 13.0 ± 1.2–19.0 ± 5.9 |
| NMN | - | - | - | X | 0.5 | 1.5 | 0.978 | 2.0 ± 0.0–3.0 ± 0.2 |
| MeNam | - | - | - | X | - | - | - | - |
| NR | X | - | - | X | - | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giner, M.P.; Christen, S.; Bartova, S.; Makarov, M.V.; Migaud, M.E.; Canto, C.; Moco, S. A Method to Monitor the NAD+ Metabolome—From Mechanistic to Clinical Applications. Int. J. Mol. Sci. 2021, 22, 10598. https://doi.org/10.3390/ijms221910598
Giner MP, Christen S, Bartova S, Makarov MV, Migaud ME, Canto C, Moco S. A Method to Monitor the NAD+ Metabolome—From Mechanistic to Clinical Applications. International Journal of Molecular Sciences. 2021; 22(19):10598. https://doi.org/10.3390/ijms221910598
Chicago/Turabian StyleGiner, Maria Pilar, Stefan Christen, Simona Bartova, Mikhail V. Makarov, Marie E. Migaud, Carles Canto, and Sofia Moco. 2021. "A Method to Monitor the NAD+ Metabolome—From Mechanistic to Clinical Applications" International Journal of Molecular Sciences 22, no. 19: 10598. https://doi.org/10.3390/ijms221910598
APA StyleGiner, M. P., Christen, S., Bartova, S., Makarov, M. V., Migaud, M. E., Canto, C., & Moco, S. (2021). A Method to Monitor the NAD+ Metabolome—From Mechanistic to Clinical Applications. International Journal of Molecular Sciences, 22(19), 10598. https://doi.org/10.3390/ijms221910598