
The PPARα and PPARγ Epigenetic Landscape in Cancer and Immune and Metabolic Disorders


Abstract
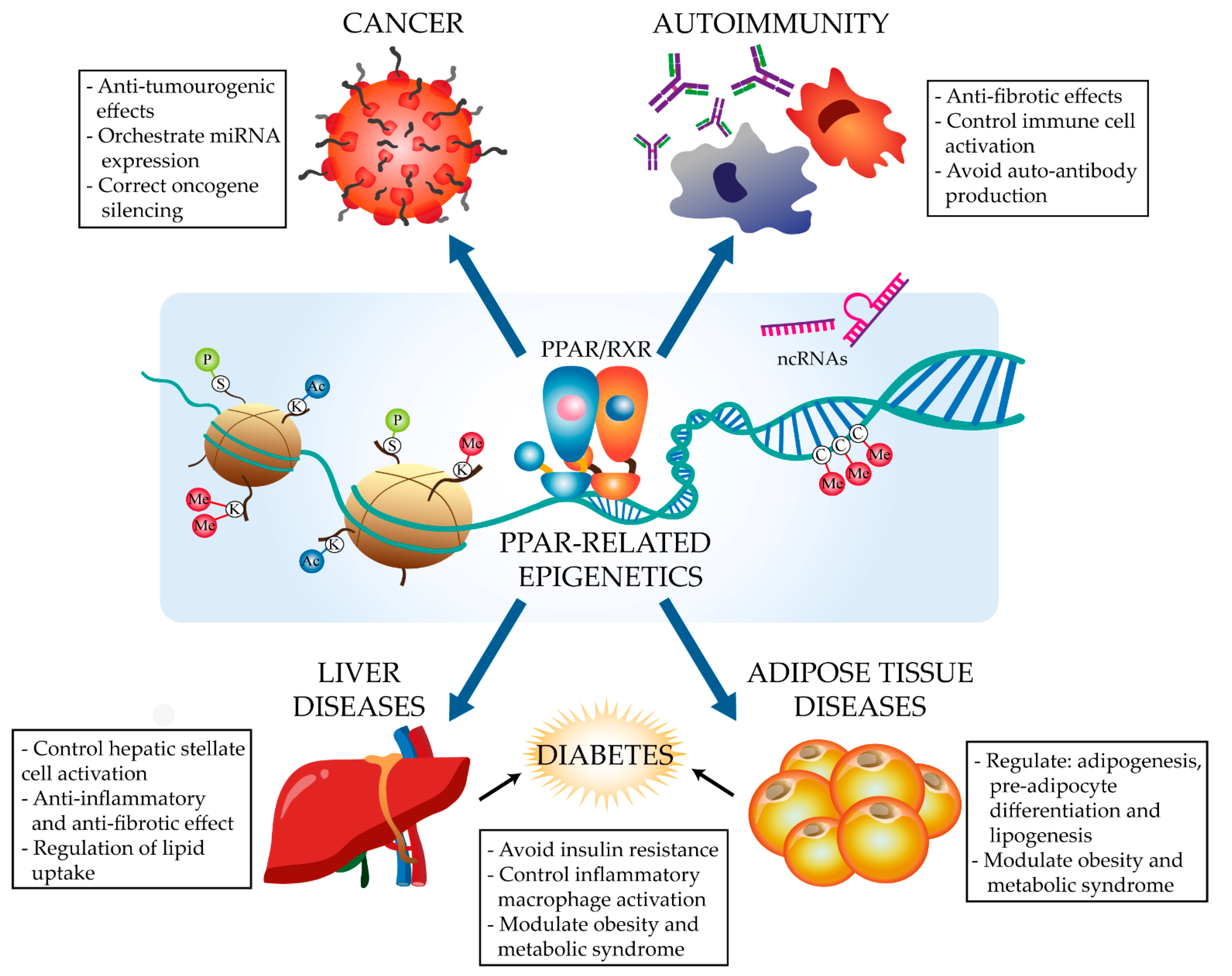
1. Introduction
1.1. Peroxisome Proliferator Activated Receptors
1.2. Epigenetics
1.2.1. Major Epigenetic Modifications
DNA Methylation
Histone Modification
Non-Coding RNAs
2. The PPARα and PPARγ Epigenetic Landscape in Disease
2.1. Cancer
2.1.1. Colorectal Cancer
2.1.2. Liver Cancer
2.1.3. Other Cancers

{kind=link}
| Condition | PPAR Isoform | Epigenetic Player | Effect | References |
|---|---|---|---|---|
| Colorectal cancer | PPARα | miR-506 | PPARα expression inhibition in a hydroxicamptothecin resistant colon cancer cell line. | [62] |
| DNMT1 | Absence of PPARα caused P21 and P27 methylation by DNMT1. | [68] | ||
| PPARγ | miR-27b, miR-130b and miR-138 | Potential downregulation of PPARγ. | [53] | |
| UHRF1 | Epigenetic PPARγ inactivation in human-derived CRC cell lines. | [64] | ||
| Promoter hypermethylation | Hypermethylation of Pparg promoter caused PPARγ suppression. | [53] | ||
| Hepatocellular carcinoma | PPARα | miR-9 | Putative biding sites to PPARα 3’ UTR. | [75] |
| PPARγ | miR-30, miR-29c and miR-338 | Antifibrotic miRNAs regulated by PPARγ during HCC-related liver fibrosis. | [71] | |
| miR-27a | PPARγ inhibition in hepatocarcinoma cells. | [72] | ||
| Thyroid cancer | PPARγ | miR-27a | no relation obsrved yet. | [77] |
| Lung cancer | PPARγ | Promoter methylation | Significantly loss of 5′-methylation. | [78] |
| Gingivo-buccal oral squamous cell carcinoma | PPARγ | DNMTs | DNA methyltransferase inhibitors could renew PPARγ transcription. | [79] |
| Prostate cancer | PPARα | miR-17/92 | Possible direct PPARα targetting and dowregulation. | [80] |
2.2. Immune Disorders
2.2.1. Asthma
2.2.2. Systemic Lupus Erythematosus
2.2.3. Systemic Sclerosis (Scleroderma)
| Condition | PPAR Isoform | Epigenetic Player | Effect | References |
|---|---|---|---|---|
| Asthma | PPARα | DNA methylation | Human white blood cells showed DNA methylation in several PPAR pathway. | [84] |
| PPARγ | miR-21 | The profibroti Smad-TGFβ1-miR-21c axis was supress upon PPARγ pioglitazone activation. | [87] | |
| miR-98 | This profibrotic miRNA was downregulated upon PPARγ rosiglitazone activation. | [80] | ||
| Not specified | set of lncRNAs | Modulation of PPAR signalling pathway in sputa from eosinophilic asthma patients. | [90] | |
| Systemic Lupus Erythematosus | PPARγ | H4K20me1 and HDAC9 | Decreased H3K9ac and H3K18ac in the Pparg promoter leading to pro-inflammatory T cell cytokines and B cell auto-antibodies. | [93,94] |
| PPARγ | Sirt1 | Reduced PPARγ expression due to H3 deacetylation, avoiding M2 monocytic transition. | [85] | |
| Systemic sclerosis | PPARγ | p300 | Ligand-activated PPARγ blocks histone acetylatransferase p300 avoiding Smad3 pathway activation and Col1a2 locus histone 4 hyperacetylation. | [99,100,101] |
2.3. Metabolism-Related Diseases
2.3.1. Liver Diseases
2.3.2. Adipose Tissue Diseases
| Condition | PPAR Isoform | Epigenetic Player | Effect | References |
|---|---|---|---|---|
| Adipose tissue diseases | PPARα | Lsd1 | Targets PPARα to control beige adipocyte numbers | [153] |
| Bta-miR-199a-3p, -154c, -320a and -432 | Control lipid metabolism through PPARα | [154] | ||
| miR-519d | Suppresses PPARα protein translation in obese patients | [155] |
| Condition | PPAR Isoform | Epigenetic Player | Effect | References |
|---|---|---|---|---|
| Adipose tissue diseases | PPARγ | U90926 | Inhibition of Pparg transcription activity | [180] |
| NEAT1 | Regulation of Pparg splicing | [178] | ||
| HOTAIR | Increased expression of PPARγ | [182] | ||
| miR-155, miR-221 and miR-122 | Decreased expression of PPARγ in human bone-marrow-derived stromal cells | [184] | ||
| miR-540 | Decreased expression of PPARγ in adipose tissue-derived stromal cells | [185] | ||
| miR-27a/b, miR-31, miR-130/b, miR301a, miR-302a and miR-548d5p | Negative regulation of PPARγ and adipogenesis | [186,187] | ||
| miR-103, miR-143, miR-200a, miR-335 and miR-375 | Upregulation of Pparg | [187,188] | ||
| p400/Brd8 complex | Incorporation of the histone variant H2A.Z, which facilitates the expression of PPARγ target genes | [156] | ||
| MLL3 and MLL4 | Complex with ASC-2. Migration to the Pparg locus and methylation of H3K4, promoting enhanced Pparg expression | [159] | ||
| EZH2 | H3K27 methylation in the Hdac9c promoter. Enhanced adipogenesis | [162] | ||
| SETD8 (KMT5A) | Enhanced H4K20me marks in PPARγ target genes. | [93] | ||
| JMJD2C | Downregulation of PPARγ transcriptional activation | [166] | ||
| JHDM2A (JMJD1A) | Decreased H3K9me2 marks and facilitated recruitment of PPARγ, RXRα and PGC1α | [167,168] | ||
| Cyclin D1 | Interaction with p300 and HDACs to inhibit Pparg expression | [172] | ||
| SIRT1 | Blocked PPARγ mechanism of action | [173,174] | ||
| LncRNA TUG1 and miR-294 | Control fatty acid accumulation through GLUT4/PPARγ/AKT axis | [183] |
2.3.3. Insulin Sensitivity and Resistance: Type 2 Diabetes
| Condition | PPAR Isoform | Epigenetic Player | Effect | References |
|---|---|---|---|---|
| Insulin sensitivity and resistance: Type 2 Diabetes | PPARγ | miR27-a | Target of Pparg transcripts, promoting insulin resistance. Induction of inflammatory ATM activation in obesity | [213,214] |
| HDAC3 | Decreased expression of PPARγ in E3 rat livers. Correlated with inflammation and insulin resistance | [196,197,198] | ||
| SIRT1 | Control of the PPARγ acetylation status and its activity | [175] | ||
| DNMT3b | Pparg promoter methylation. Increased inflammatory macrophage activation and insulin resistance | [209,210] | ||
| DNMT3a | Fgf21 hypermethylation in human adipocytes, insulin resistance | [211] | ||
| DNMT1 | Adiponectin promoter methylation in obese mice. Glucose intolerance | [212] |
3. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Mirza, A.Z.; AlThagafi, I.I.; Shamshad, H. Role of PPAR receptor in different diseases and their ligands: Physiological importance and clinical implications. Eur. J. Med. Chem. 2019, 166, 502–513. [Google Scholar] [CrossRef] [PubMed]
- Amber-Vitos, O.; Chaturvedi, N.; Nachliel, E.; Gutman, M.; Tsfadia, Y. The effect of regulating molecules on the structure of the PPAR-RXR complex. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2016, 1861, 1852–1863. [Google Scholar] [CrossRef] [PubMed]
- Feige, J.N.; Gelman, L.; Michalik, L.; Desvergne, B.; Wahli, W. From molecular action to physiological outputs: Peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Prog. Lipid Res. 2006, 45, 120–159. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.; Pan, S.; Guo, Y.; Xu, P.; Zhai, Y. PPARs as Nuclear Receptors for Nutrient and Energy Metabolism. Molecules 2019, 24, 2545. [Google Scholar] [CrossRef] [PubMed]
- Grygiel-Gorniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef]
- Hall, J.A.; Rusten, M.; AbuGhazaleh, R.D.; Wuertz, B.; Souksavong, V.; Escher, P.; Ondrey, F. Effects of PPAR-gamma agonists on oral cancer cell lines: Potential horizons for chemopreventives and adjunctive therapies. Head Neck 2020, 42, 2542–2554. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Grosdidier, A.; Michielin, O. Peroxisome proliferator-activated receptor structures: Ligand specificity, molecular switch and interactions with regulators. Biochim. Biophys. Acta 2007, 1771, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Font-Diaz, J.; Jimenez-Panizo, A.; Caelles, C.; Vivanco, M.D.; Perez, P.; Aranda, A.; Estébanez-Perpina, E.; Castrillo, A.; Ricote, M.; Valledor, A.F. Nuclear receptors: Lipid and hormone sensors with essential roles in the control of cancer development. Semin. Cancer Biol. 2021, 73, 58–75. [Google Scholar] [CrossRef]
- Fougerat, A.; Montagner, A.; Loiseau, N.; Guillou, H.; Wahli, W. Peroxisome Proliferator-Activated Receptors and Their Novel Ligands as Candidates for the Treatment of Non-Alcoholic Fatty Liver Disease. Cells 2020, 9, 1638. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, J.; Luo, S.; Zhan, Y.; Lu, Q. The roles of PPARgamma and its agonists in autoimmune diseases: A comprehensive review. J. Autoimmun. 2020, 113, 102510. [Google Scholar] [CrossRef]
- Nobs, S.P.; Natali, S.; Pohlmeier, L.; Okreglicka, K.; Schneider, C.; Kurrer, M.; Sallusto, F.; Kopf, M. PPARgamma in dendritic cells and T cells drives pathogenic type-2 effector responses in lung inflammation. J. Exp. Med. 2017, 214, 3015–3035. [Google Scholar] [CrossRef] [PubMed]
- Housley, W.J.; Adams, C.O.; Vang, A.G.; Brocke, S.; Nichols, F.C.; Lacombe, M.; Rajan, T.V.; Clark, R.B. Peroxisome Proliferator-Activated Receptor gamma Is Required for CD4+ T Cell-Mediated Lymphopenia-Associated Autoimmunity. J. Immunol. 2011, 187, 4161–4169. [Google Scholar] [CrossRef]
- Porcuna, J.; Menendez-Gutierrez, M.P.; Ricote, M. Molecular control of tissue-resident macrophage identity by nuclear receptors. Curr. Opin. Pharmacol. 2020, 53, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Tronick, E.; Hunter, R.G. Waddington, Dynamic Systems, and Epigenetics. Front. Behav. Neurosci. 2016, 10, 107. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Rajabi, H.; Tagde, A.; Alam, M.; Bouillez, A.; Pitroda, S.; Suzuki, Y.; Kufe, D. DNA methylation by DNMT1 and DNMT3b methyltransferases is driven by the MUC1-C oncoprotein in human carcinoma cells. Oncogene 2016, 35, 6439–6445. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.M.; Mahar, M.; Ewan, E.E.; Leahy, K.M.; Zhao, G.; Cavalli, V. Epigenetic regulator UHRF1 inactivates REST and growth suppressor gene expression via DNA methylation to promote axon regeneration. Proc. Natl. Acad. Sci. USA 2018, 115, E12417–E12426. [Google Scholar] [CrossRef]
- Ginno, P.A.; Gaidatzis, D.; Feldmann, A.; Hoerner, L.; Imanci, D.; Burger, L.; Zilbermann, F.; Peters, A.; Edenhofer, F.; Smallwood, S.A.; et al. A genome-scale map of DNA methylation turnover identifies site-specific dependencies of DNMT and TET activity. Nat. Commun. 2020, 11, 2680. [Google Scholar] [CrossRef]
- Giaimo, B.D.; Ferrante, F.; Herchenrother, A.; Hake, S.B.; Borggrefe, T. The histone variant H2A.Z in gene regulation. Epigenetics Chromatin 2019, 12, 37. [Google Scholar] [CrossRef]
- Miller, J.L.; Grant, P.A. The Role of DNA Methylation and Histone Modifications in Transcriptional Regulation in Humans. Subcell Biochem. 2013, 61, 289–317. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, D.; Avvakumov, N.; Cote, J. Histone phosphorylation: A chromatin modification involved in diverse nuclear events. Epigenetics 2012, 7, 1098–1108. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Blythe, S.A.; Cha, S.W.; Tadjuidje, E.; Heasman, J.; Klein, P.S. beta-Catenin Primes Organizer Gene Expression by Recruiting a Histone H3 Arginine 8 Methyltransferase, Prmt2. Dev. Cell 2010, 19, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Svobodova Kovarikova, A.; Legartova, S.; Krejci, J.; Bartova, E. H3K9me3 and H4K20me3 represent the epigenetic landscape for 53BP1 binding to DNA lesions. Aging (Albany NY) 2018, 10, 2585–2605. [Google Scholar] [CrossRef]
- Hervouet, E.; Peixoto, P.; Delage-Mourroux, R.; Boyer-Guittaut, M.; Cartron, P.-F. Specific or not specific recruitment of DNMTs for DNA methylation, an epigenetic dilemma. Clin. Epigenet. 2018, 10, 17. [Google Scholar] [CrossRef]
- Di Croce, L.; Raker, V.; Corsaro, M.; Fazi, F.; Fanelli, M.; Faretta, M.; Fuks, F.; Lo Coco, F.; Kouzarides, T.; Nervi, C.; et al. Methyltransferase Recruitment and DNA Hyermethylation of Target Promoters by an Oncogenic Transcription Factor. Science 2002, 295, 1079–1082. [Google Scholar] [CrossRef]
- Ferrari, K.J.; Scelfo, A.; Jammula, S.; Cuomo, A.; Barozzi, I.; Stutzer, A.; Fischle, W.; Bonaldi, T.; Pasini, D. Polycomb-Dependent H3K27me1 and H3K27me2 Regulate Active Transcription and Enhancer Fidelity. Mol. Cell 2014, 53, 49–62. [Google Scholar] [CrossRef]
- Kaikkonen, M.U.; Spann, N.J.; Heinz, S.; Romanoski, C.E.; Allison, K.A.; Stender, J.D.; Chun, H.B.; Tough, D.F.; Prinjha, R.K.; Benner, C.; et al. Remodeling of the Enhancer Landscape during Macrophage Activation Is Coupled to Enhancer Transcription. Mol. Cell 2013, 51, 310–325. [Google Scholar] [CrossRef]
- Lara-Astiaso, D.; Weiner, A.; Lorenzo-Vivas, E.; Zaretsky, I.; Jaitin, D.A.; David, E.; Keren-Shaul, H.; Mildner, A.; Winter, D.; Jung, S.; et al. Immunogenetics. Chromatin state dynamics during blood formation. Science 2014, 345, 943–949. [Google Scholar] [CrossRef]
- Zhang, P.; Wu, W.; Chen, Q.; Chen, M. Non-Coding RNAs and their Integrated Networks. J. Integr. Bioinform. 2019, 16. [Google Scholar] [CrossRef]
- Frias-Lasserre, D.; Villagra, C.A. The Importance of ncRNAs as Epigenetic Mechanisms in Phenotypic Variation and Organic Evolution. Front. Microbiol. 2017, 8, 2483. [Google Scholar] [CrossRef]
- Ratti, M.; Lampis, A.; Ghidini, M.; Salati, M.; Mirchev, M.B.; Valeri, N.; Hahne, J.C. MicroRNAs (miRNAs) and Long Non-Coding RNAs (lncRNAs) as New Tools for Cancer Therapy: First Steps from Bench to Bedside. Target. Oncol. 2020, 15, 261–278. [Google Scholar] [CrossRef]
- Zhang, W.; Qu, J.; Liu, G.-H.; Belmonte, J.C.I. The ageing epigenome and its rejuvenation. Nat. Rev. Mol. Cell Biol. 2020, 21, 137–150. [Google Scholar] [CrossRef]
- Zhao, Y.; Zheng, D.; Cvekl, A. Profiling of chromatin accessibility and identification of general cis-regulatory mechanisms that control two ocular lens differentiation pathways. Epigenet. Chromatin 2019, 12, 27. [Google Scholar] [CrossRef]
- Chen, S.; Yang, J.; Wei, Y.; Wei, X. Epigenetic regulation of macrophages: From homeostasis maintenance to host defense. Cell. Mol. Immunol. 2020, 17, 36–49. [Google Scholar] [CrossRef]
- Elnady, H.G.; Sherif, L.S.; Kholoussi, N.M.; Ali Azzam, M.; Foda, A.R.; Helwa, I.; Sabry, R.N.; Eissa, E.; Fahmy, R.F. Aberrant Expression of Immune-related MicroRNAs in Pediatric Patients with Asthma. Int. J. Mol. Cell Med. 2020, 9, 246–255. [Google Scholar]
- Neganova, M.E.; Klochkov, S.G.; Aleksandrova, Y.R.; Aliev, G. Histone modifications in epigenetic regulation of cancer: Perspectives and achieved progress. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2020. [Google Scholar] [CrossRef]
- Islam, A.; Mohammad, E.; Khan, M.A. Aberration of the modulatory functions of intronic microRNA hsa-miR-933 on its host gene ATF2 results in type II diabetes mellitus and neurodegenerative disease development. Hum. Genom. 2020, 14, 34. [Google Scholar] [CrossRef]
- Yang, Y.; Wicks, J.; Haitchi, H.M.; Powell, R.M.; Manuyakorn, W.; Howarth, P.H.; Holgate, S.T.; Davies, D.E. Regulation of A Disintegrin and Metalloprotease-33Expression by Transforming Growth Factor-β. Am. J. Respir. Cell Mol. Biol. 2012, 46, 633–640. [Google Scholar] [CrossRef]
- Harb, H.; Raedler, D.; Ballenberger, N.; Bock, A.; Kesper, D.A.; Renz, H.; Schaub, B. Childhood allergic asthma is associated with increased IL-13 and FOXP3 histone acetylation. J. Allergy Clin. Immunol. 2015, 136, 200–202. [Google Scholar] [CrossRef]
- Le, T.N.; Williams, S.R.; Alaimo, J.T.; Elsea, S.H. Genotype and phenotype correlation in 103 individuals with 2q37 deletion syndrome reveals incomplete penetrance and supports HDAC4 as the primary genetic contributor. Am. J. Med Genet. Part A 2019, 179, 782–791. [Google Scholar] [CrossRef]
- Wu, Y.; Cui, W.; Zhang, D.; Wu, W.; Yang, Z. The shortening of leukocyte telomere length relates to DNA hypermethylation of LINE-1 in type 2 diabetes mellitus. Oncotarget 2017, 8, 73964–73973. [Google Scholar] [CrossRef]
- Sun, Z.H.; Liu, Y.H.; Liu, J.D.; Xu, D.D.; Li, X.F.; Meng, X.M.; Ma, T.T.; Huang, C.; Li, J. MeCP2 Regulates PTCH1 Expression Through DNA Methylation in Rheumatoid Arthritis. Inflammation 2017, 40, 1497–1508. [Google Scholar] [CrossRef]
- Raveche, E.S.; Salerno, E.; Scaglione, B.J.; Manohar, V.; Abbasi, F.; Lin, Y.C.; Fredrickson, T.; Landgraf, P.; Ramachandra, S.; Huppi, K.; et al. Abnormal microRNA-16 locus with synteny to human 13q14 linked to CLL in NZB mice. Blood 2007, 109, 5079–5086. [Google Scholar] [CrossRef]
- Zernecke, A.; Bidzhekov, K.; Noels, H.; Shagdarsuren, E.; Gan, L.; Denecke, B.; Hristov, M.; Koppel, T.; Jahantigh, M.N.; Lutgens, E.; et al. Delivery of MicroRNA-126 by Apoptotic Bodies Induces CXCL12-Dependent Vascular Protection. Sci. Signal. 2009, 2, ra81. [Google Scholar] [CrossRef]
- Rana, Z.; Diermeier, S.; Hanif, M.; Rosengren, R.J. Understanding Failure and Improving Treatment Using HDAC Inhibitors for Prostate Cancer. Biomedicines 2020, 8, 22. [Google Scholar] [CrossRef]
- Dovey, O.M.; Foster, C.T.; Conte, N.; Edwards, S.A.; Edwards, J.M.; Singh, R.; Vassiliou, G.; Bradley, A.; Cowley, S.M. Histone deacetylase 1 and 2 are essential for normal T-cell development and genomic stability in mice. Blood 2013, 121, 1335–1344. [Google Scholar] [CrossRef]
- Tian, J.; Hu, L.; Li, X.; Geng, J.; Dai, M.; Bai, X. MicroRNA-130b promotes lung cancer progression via PPARγ/VEGF-A/BCL-2-mediated suppression of apoptosis. J. Exp. Clin. Cancer Res. 2016, 35, 105. [Google Scholar] [CrossRef]
- Koga, H.; Sakisaka, S.; Harada, M.; Takagi, T.; Hanada, S.; Taniguchi, E.; Kawaguchi, T.; Sasatomi, K.; Kimura, R.; Hashimoto, O.; et al. Involvement of p21(WAF1/Cip1), p27(Kip1), and p18(INK4c) in troglitazone-induced cell-cycle arrest in human hepatoma cell lines. Hepatology 2001, 33, 1087–1097. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Prz. Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef]
- Park, J.-I.; Kwak, J.-Y. The Role of Peroxisome Proliferator-Activated Receptors in Colorectal Cancer. PPAR Res. 2012, 2012, 876418. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.; Baldassarre, A.; Masotti, A.; Calura, E.; Martini, P.; Vari, R.; Scazzocchio, B.; Gessani, S.; Del Corno, M. Integrated Transcriptome Analysis of Human Visceral Adipocytes Unravels Dysregulated microRNA-Long Non-coding RNA-mRNA Networks in Obesity and Colorectal Cancer. Front. Oncol. 2020, 10, 1089. [Google Scholar] [CrossRef]
- Motawi, T.K.; Shaker, O.G.; Ismail, M.F.; Sayed, N.H. Peroxisome Proliferator-Activated Receptor Gamma in Obesity and Colorectal Cancer: The Role of Epigenetics. Sci. Rep. 2017, 7, 10714. [Google Scholar] [CrossRef]
- Baffa, R.; Fassan, M.; Volinia, S.; O’Hara, B.; Liu, C.G.; Palazzo, J.P.; Gardiman, M.; Rugge, M.; Gomella, L.G.; Croce, C.M.; et al. MicroRNA expression profiling of human metastatic cancers identifies cancer gene targets. J. Pathol. 2009, 219, 214–221. [Google Scholar] [CrossRef]
- Colangelo, T.; Fucci, A.; Votino, C.; Sabatino, L.; Pancione, M.; Laudanna, C.; Binaschi, M.; Bigioni, M.; Maggi, C.A.; Parente, D.; et al. MicroRNA-130b Promotes Tumor Development and Is Associated with Poor Prognosis in Colorectal Cancer. Neoplasia 2013, 15, 1086–1099. [Google Scholar] [CrossRef]
- Karbiener, M.; Fischer, C.; Nowitsch, S.; Opriessnig, P.; Papak, C.; Ailhaud, G.; Dani, C.; Amri, E.Z.; Scheideler, M. microRNA miR-27b impairs human adipocyte differentiation and targets PPARγ. Biochem. Biophys. Res. Commun. 2009, 390, 247–251. [Google Scholar] [CrossRef]
- Lin, Q.; Gao, Z.; Alarcon, R.M.; Ye, J.; Yun, Z. A role of miR-27 in the regulation of adipogenesis. FEBS J. 2009, 276, 2348–2358. [Google Scholar] [CrossRef]
- Yi, R.; Li, Y.; Wang, F.; Gu, J.; Isaji, T.; Li, J.; Qi, R.; Zhu, X.; Zhao, Y. Transforming growth factor (TGF) β1 acted through miR-130b to increase integrin α5 to promote migration of colorectal cancer cells. Tumor Biol. 2016, 37, 10763–10773. [Google Scholar] [CrossRef]
- Matsuyama, R.; Okuzaki, D.; Okada, M.; Oneyama, C. Micro RNA -27b suppresses tumor progression by regulating ARFGEF 1 and focal adhesion signaling. Cancer Sci. 2016, 107, 28–35. [Google Scholar] [CrossRef]
- Qin, Y.Z.; Xie, X.C.; Liu, H.Z.; Lai, H.; Qiu, H.; Ge, L.Y. Screening and preliminary validation of miRNAs with the regulation of hTERT in colorectal cancer. Oncol. Rep. 2015, 33, 2728–2736. [Google Scholar] [CrossRef]
- Zhao, L.; Yu, H.; Yi, S.; Peng, X.; Su, P.; Xiao, Z.; Liu, R.; Tang, A.; Li, X.; Liu, F.; et al. The tumor suppressor miR-138-5p targets PD-L1 in colorectal cancer. Oncotarget 2016, 7, 45370–45384. [Google Scholar] [CrossRef]
- Tong, J.L.; Zhang, C.P.; Nie, F.; Xu, X.T.; Zhu, M.M.; Xiao, S.D.; Ran, Z.H. MicroRNA 506 regulates expression of PPAR alpha in hydroxycamptothecin-resistant human colon cancer cells. FEBS Lett. 2011, 585, 3560–3568. [Google Scholar] [CrossRef]
- Liu, J.; Li, H.; Sun, L.; Shen, S.; Zhou, Q.; Yuan, Y.; Xing, C. Epigenetic Alternations of MicroRNAs and DNA Methylation Contribute to Liver Metastasis of Colorectal Cancer. Dig. Dis. Sci. 2019, 64, 1523–1534. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, L.; Fucci, A.; Pancione, M.; Carafa, V.; Nebbioso, A.; Pistore, C.; Babbio, F.; Votino, C.; Laudanna, C.; Ceccarelli, M.; et al. UHRF1 coordinates peroxisome proliferator activated receptor gamma (PPARG) epigenetic silencing and mediates colorectal cancer progression. Oncogene 2012, 31, 5061–5072. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.B.; Lee, E.J.; Jung, E.H.; Chun, H.K.; Chang, D.K.; Song, S.Y.; Park, J.; Kim, D.H. Aberrant Methylation of APC, MGMT, RASSF2A, and Wif-1 Genes in Plasma as a Biomarker for Early Detection of Colorectal Cancer. Clin. Cancer Res. 2009, 15, 6185–6191. [Google Scholar] [CrossRef]
- Yang, Y.; Chu, F.H.; Xu, W.R.; Sun, J.Q.; Sun, X.; Ma, X.M.; Yu, M.W.; Yang, G.W.; Wang, X.M. Identification of regulatory role of DNA methylation in colon cancer gene expression via systematic bioinformatics analysis. Medicine 2017, 96, e8487. [Google Scholar] [CrossRef] [PubMed]
- Pancione, M.; Sabatino, L.; Fucci, A.; Carafa, V.; Nebbioso, A.; Forte, N.; Febbraro, A.; Parente, D.; Ambrosino, C.; Normanno, N.; et al. Epigenetic Silencing of Peroxisome Proliferator-Activated Receptor γ Is a Biomarker for Colorectal Cancer Progression and Adverse Patients’ Outcome. PLoS ONE 2010, 5, e14229. [Google Scholar] [CrossRef]
- Luo, Y.; Xie, C.; Brocker, C.N.; Fan, J.; Wu, X.; Feng, L.; Wang, Q.; Zhao, J.; Lu, D.; Tandon, M.; et al. Intestinal PPARα Protects Against Colon Carcinogenesis via Regulation of Methyltransferases DNMT1 and PRMT6. Gastroenterology 2019, 157, 744–759.e4. [Google Scholar] [CrossRef] [PubMed]
- Yaghoubizadeh, M.; Pishkar, L.; Basati, G. Aberrant Expression of Peroxisome Proliferator-Activated Receptors in Colorectal Cancer and Their Association with Cancer Progression and Prognosis. Gastrointest. Tumors 2020, 7, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Nagano, T.; Shah, Y.; Cheung, C.; Ito, S.; Gonzalez, F.J. The PPARα-Humanized Mouse: A Model to Investigate Species Differences in Liver Toxicity Mediated by PPARα. Toxicol. Sci. 2008, 101, 132–139. [Google Scholar] [CrossRef]
- Winkler, I.; Bitter, C.; Winkler, S.; Weichenhan, D.; Thavamani, A.; Hengstler, J.G.; Borkham-Kamphorst, E.; Kohlbacher, O.; Plass, C.; Geffers, R.; et al. Identification of Pparγ-modulated miRNA hubs that target the fibrotic tumor microenvironment. Proc. Natl. Acad. Sci. USA 2020, 117, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, J.; Fei, B.-Y.; Shao, D.; Pan, Y.; Mo, Z.-H.; Sun, B.-Z.; Zhang, D.; Zheng, X.; Zhang, M.; et al. MiR-27a Promotes Hepatocellular Carcinoma Cell Proliferation Through Suppression of its Target Gene Peroxisome Proliferator-activated Receptor γ. Chin. Med. J. 2015, 128, 941–947. [Google Scholar] [CrossRef] [PubMed]
- Tombolan, L.; Zampini, M.; Casara, S.; Boldrin, E.; Zin, A.; Bisogno, G.; Rosolen, A.; De Pitta, C.; Lanfranchi, G. MicroRNA-27a Contributes to Rhabdomyosarcoma Cell Proliferation by Suppressing RARA and RXRA. PLoS ONE 2015, 10, e0125171. [Google Scholar] [CrossRef]
- Oda, Y.; Nakajima, M.; Tsuneyama, K.; Takamiya, M.; Aoki, Y.; Fukami, T.; Yokoi, T. Retinoid X receptor α in human liver is regulated by miR-34a. Biochem. Pharmacol. 2014, 90, 179–187. [Google Scholar] [CrossRef]
- Cui, M.; Xiao, Z.; Wang, Y.; Zheng, M.; Song, T.; Cai, X.; Sun, B.; Ye, L.; Zhang, X. Long Noncoding RNA HULC Modulates Abnormal Lipid Metabolism in Hepatoma Cells through an miR-9–Mediated RXRA Signaling Pathway. Cancer Res. 2015, 75, 846–857. [Google Scholar] [CrossRef]
- Catalano, M.G.; Poli, R.; Pugliese, M.; Fortunati, N.; Boccuzzi, G. Emerging molecular therapies of advanced thyroid cancer. Mol. Asp. Med. 2010, 31, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Toraih, E.A.; Fawzy, M.S.; Abushouk, A.I.; Shaheen, S.; Hobani, Y.H.; Alruwetei, A.M.; Mansouri, O.A.; Kandil, E.; Badran, D.I. Prognostic value of the miRNA-27a and PPAR/RXRα signaling axis in patients with thyroid carcinoma. Epigenomics 2020, 12, 1825–1843. [Google Scholar] [CrossRef]
- Herrera, C.L.; Kim, D.Y.; Kumar, S.R.; Bryan, J.N. Peroxisome proliferator activated receptor γ protein expression is asymmetrically distributed in primary lung tumor and metastatic to lung osteosarcoma samples and does not correlate with gene methylation. BMC Veter-Res. 2015, 11, 230. [Google Scholar] [CrossRef]
- Das, D.; Ghosh, S.; Maitra, A.; Biswas, N.K.; Panda, C.K.; Roy, B.; Sarin, R.; Majumder, P.P. Epigenomic dysregulation-mediated alterations of key biological pathways and tumor immune evasion are hallmarks of gingivo-buccal oral cancer. Clin. Epigenet. 2019, 11, 178. [Google Scholar] [CrossRef]
- Wang, W.-L.W.; Welsh, J.; Tenniswood, M. 1,25-Dihydroxyvitamin D3 modulates lipid metabolism in prostate cancer cells through miRNA mediated regulation of PPARA. J. Steroid Biochem. Mol. Biol. 2013, 136, 247–251. [Google Scholar] [CrossRef]
- Mohan, M.; Okeoma, C.M.; Sestak, K. Dietary Gluten and Neurodegeneration: A Case for Preclinical Studies. Int. J. Mol. Sci. 2020, 21, 5407. [Google Scholar] [CrossRef] [PubMed]
- Aprahamian, T.; Bonegio, R.G.; Weitzner, Z.; Gharakhanian, R.; Rifkin, I.R. Peroxisome proliferator-activated receptor gamma agonists in the prevention and treatment of murine systemic lupus erythematosus. Immunology 2014, 142, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Luo, G.; Li, X.; Chen, J.; Wu, J.; Peng, Y. PPARγ inhibits HMGB1 expression through upregulation of miR-142-3p in vitro and in vivo. Cell. Signal. 2016, 28, 158–164. [Google Scholar] [CrossRef]
- Jeong, A.; Imboden, M.; Ghantous, A.; Novoloaca, A.; Carsin, A.-E.; Kogevinas, M.; Schindler, C.; Lovison, G.; Herceg, Z.; Cuenin, C.; et al. DNA Methylation in Inflammatory Pathways Modifies the Association between BMI and Adult-Onset Non-Atopic Asthma. Int. J. Environ. Res. Public Health 2019, 16, 600. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Luo, S.; Zhan, Y.; Wang, J.; Zhao, R.; Li, Y.; Zeng, J.; Lu, Q. Increased Expression of PPAR-γ Modulates Monocytes Into a M2-Like Phenotype in SLE Patients: An Implicative Protective Mechanism and Potential Therapeutic Strategy of Systemic Lupus Erythematosus. Front. Immunol. 2021, 11, 579372. [Google Scholar] [CrossRef]
- Banno, A.; Reddy, A.; Lakshmi, S.; Reddy, A.R.C. PPARs: Key Regulators of Airway Inflammation and Potential Therapeutic Targets in Asthma. Nucl. Recept. Res. 2018, 5, 101306. [Google Scholar] [CrossRef]
- Wongtrakool, C.; Ko, J.; Jang, A.J.; Grooms, K.; Chang, S.; Sylber, C.; Kosmider, B.; Bahmed, K.; Blackburn, M.R.; Sutliff, R.L.; et al. MicroRNA-98 reduces nerve growth factor expression in nicotine-induced airway remodeling. J. Biol. Chem. 2020, 295, 18051–18064. [Google Scholar] [CrossRef]
- Liu, L.; Pan, Y.; Zhai, C.; Zhu, Y.; Ke, R.; Shi, W.; Wang, J.; Yan, X.; Su, X.; Song, Y.; et al. Activation of peroxisome proliferation–activated receptor-γ inhibits transforming growth factor-β1-induced airway smooth muscle cell proliferation by suppressing Smad–miR-21 signaling. J. Cell. Physiol. 2018, 234, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Wang, X.; Sun, Q.; Papakonstantinou, E.; S’Ng, C.; Tamm, M.; Stolz, D.; Roth, M. IgE Downregulates PTEN through MicroRNA-21-5p and Stimulates Airway Smooth Muscle Cell Remodeling. Int. J. Mol. Sci. 2019, 20, 875. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-J.; Mao, D.; Gao, W.; Hu, H. Peripheral whole blood lncRNA expression analysis in patients with eosinophilic asthma. Medicine 2018, 97, e9817. [Google Scholar] [CrossRef]
- Moulton, V.R.; Suárez-Fueyo, A.; Meidan, E.; Li, H.; Mizui, M.; Tsokos, G.C. Pathogenesis of Human Systemic Lupus Erythematosus: A Cellular Perspective. Trends Mol. Med. 2017, 23, 615–635. [Google Scholar] [CrossRef]
- Rőszer, T.; Menendez-Gutierrez, M.P.; Lefterova, M.I.; Alameda, D.; Nunez, V.; Lazar, M.A.; Fischer, T.; Ricote, M. Autoimmune kidney disease and impaired engulfment of apoptotic cells in mice with macrophage peroxisome proliferator-activated receptor gamma or retinoid X receptor alpha deficiency. J. Immunol. 2011, 186, 621–631. [Google Scholar] [CrossRef]
- Wakabayashi, K.-I.; Okamura, M.; Tsutsumi, S.; Nishikawa, N.S.; Tanaka, T.; Sakakibara, I.; Kitakami, J.-I.; Ihara, S.; Hashimoto, Y.; Hamakubo, T.; et al. The Peroxisome Proliferator-Activated Receptor γ/Retinoid X Receptor α Heterodimer Targets the Histone Modification Enzyme PR-Set7/Setd8 Gene and Regulates Adipogenesis through a Positive Feedback Loop. Mol. Cell. Biol. 2009, 29, 3544–3555. [Google Scholar] [CrossRef]
- Yan, K.; Cao, Q.; Reilly, C.; Young, N.; Garcia, B.A.; Mishra, N. Histone Deacetylase 9 Deficiency Protects against Effector T Cell-mediated Systemic Autoimmunity. J. Biol. Chem. 2011, 286, 28833–28843. [Google Scholar] [CrossRef]
- Hu, N.; Qiu, X.; Luo, Y.; Yuan, J.; Li, Y.; Lei, W.; Zhang, G.; Zhou, Y.; Su, Y.; Lu, Q. Abnormal histone modification patterns in lupus CD4+ T cells. J. Rheumatol. 2008, 35, 804–810. [Google Scholar]
- Xu, X.; Ramanujam, M.; Visvanathan, S.; Assassi, S.; Liu, Z.; Li, L. Transcriptional insights into pathogenesis of cutaneous systemic sclerosis using pathway driven meta-analysis assisted by machine learning methods. PLoS ONE 2020, 15, e0242863. [Google Scholar] [CrossRef]
- Wei, J.; Ghosh, A.K.; Sargent, J.L.; Komura, K.; Wu, M.; Huang, Q.-Q.; Jain, M.; Whitfield, M.L.; Feghali-Bostwick, C.; Varga, J. PPARγ Downregulation by TGFß in Fibroblast and Impaired Expression and Function in Systemic Sclerosis: A Novel Mechanism for Progressive Fibrogenesis. PLoS ONE 2010, 5, e13778. [Google Scholar] [CrossRef]
- Kohno, S.; Endo, H.; Hashimoto, A.; Hayashi, I.; Murakami, Y.; Kitasato, H.; Kojima, F.; Kawai, S.; Kondo, H. Inhibition of skin sclerosis by 15deoxy Δ12,14-prostaglandin J2 and retrovirally transfected prostaglandin D synthase in a mouse model of bleomycin-induced scleroderma. Biomed. Pharmacother. 2006, 60, 18–25. [Google Scholar] [CrossRef]
- Nuwormegbe, S.A.; Sohn, J.H.; Kim, S.W. A PPAR-Gamma Agonist Rosiglitazone Suppresses Fibrotic Response in Human Pterygium Fibroblasts by Modulating the p38 MAPK Pathway. Investig. Opthalmol. Vis. Sci. 2017, 58, 5217–5226. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Bhattacharyya, S.; Wei, J.; Kim, S.; Barak, Y.; Mori, Y.; Varga, J. Peroxisome proliferator-activated receptor-γ abrogates Smad-dependent collagen stimulation by targeting the p300 transcriptional coactivator. FASEB J. 2009, 23, 2968–2977. [Google Scholar] [CrossRef]
- Chen, J.; Xia, Y.; Lin, X.; Feng, X.-H.; Wang, Y. Smad3 signaling activates bone marrow-derived fibroblasts in renal fibrosis. Lab. Investig. 2014, 94, 545–556. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Bhattacharyya, S.; Lakos, G.; Chen, S.-J.; Mori, Y.; Varga, J. Disruption of transforming growth factor ? Signaling and profibrotic responses in normal skin fibroblasts by peroxisome proliferator-activated receptor? Arthritis Rheum. 2004, 50, 1305–1318. [Google Scholar] [CrossRef]
- Connolly, M.K. Systemic sclerosis (scleroderma): Remaining challenges. Ann. Transl. Med. 2021, 9, 438. [Google Scholar] [CrossRef]
- Ding, W.; Pu, W.; Wang, L.; Jiang, S.; Zhou, X.; Tu, W.; Yu, L.; Zhang, J.; Guo, S.; Liu, Q.; et al. Genome-Wide DNA Methylation Analysis in Systemic Sclerosis Reveals Hypomethylation of IFN-Associated Genes in CD4+ and CD8+ T Cells. J. Investig. Dermatol. 2018, 138, 1069–1077. [Google Scholar] [CrossRef]
- Hemmatazad, H.; Rodrigues, H.M.; Maurer, B.; Brentano, F.; Pileckyte, M.; Distler, J.H.W.; Gay, R.E.; Michel, B.A.; Gay, S.; Huber, L.C.; et al. Histone deacetylase 7, a potential target for the antifibrotic treatment of systemic sclerosis. Arthritis Rheum. 2009, 60, 1519–1529. [Google Scholar] [CrossRef]
- Huber, L.C.; Distler, J.H.W.; Moritz, F.; Hemmatazad, H.; Hauser, T.; Michel, B.A.; Gay, R.E.; Matucci-Cerinic, M.; Gay, S.; Distler, O.; et al. Trichostatin A prevents the accumulation of extracellular matrix in a mouse model of bleomycin-induced skin fibrosis. Arthritis Rheum. 2007, 56, 2755–2764. [Google Scholar] [CrossRef]
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef]
- Sherling, D.H.; Perumareddi, P.; Hennekens, C.H. Metabolic Syndrome. J. Cardiovasc. Pharmacol. Ther. 2017, 22, 365–367. [Google Scholar] [CrossRef]
- McCracken, E.; Monaghan, M.; Sreenivasan, S. Pathophysiology of the metabolic syndrome. Clin. Dermatol. 2018, 36, 14–20. [Google Scholar] [CrossRef]
- Hong, F.; Xu, P.; Zhai, Y. The Opportunities and Challenges of Peroxisome Proliferator-Activated Receptors Ligands in Clinical Drug Discovery and Development. Int. J. Mol. Sci. 2018, 19, 2189. [Google Scholar] [CrossRef]
- Bansal, G.; Thanikachalam, P.V.; Maurya, R.K.; Chawla, P.; Ramamurthy, S. An overview on medicinal perspective of thiazolidine-2,4-dione: A remarkable scaffold in the treatment of type 2 diabetes. J. Adv. Res. 2020, 23, 163–205. [Google Scholar] [CrossRef] [PubMed]
- Hevener, A.L.; Olefsky, J.M.; Reichart, D.; Nguyen, M.T.; Bandyopadyhay, G.; Leung, H.Y.; Watt, M.J.; Benner, C.; Febbraio, M.A.; Nguyen, A.K.; et al. Macrophage PPAR gamma is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. J. Clin. Investig. 2007, 117, 1658–1669. [Google Scholar] [CrossRef]
- Brunt, E.M.; Wong, V.W.-S.; Nobili, V.; Day, C.P.; Sookoian, S.; Maher, J.J.; Bugianesi, E.; Sirlin, C.B.; Neuschwander-Tetri, B.A.; Rinella, M.E. Nonalcoholic fatty liver disease. Nat. Rev. Dis. Prim. 2015, 1, 15080. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Hajri, T.; Zaiou, M.; Fungwe, T.; Ouguerram, K.; Besong, S. Epigenetic Regulation of Peroxisome Proliferator-Activated Receptor Gamma Mediates High-Fat Diet-Induced Non-Alcoholic Fatty Liver Disease. Cells 2021, 10, 1355. [Google Scholar] [CrossRef]
- Moran-Salvador, E.; Mann, J. Epigenetics and Liver Fibrosis. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 125–134. [Google Scholar] [CrossRef]
- Claveria-Cabello, A.; Colyn, L.; Arechederra, M.; Urman, J.M.; Berasain, C.; Avila, M.A.; Fernandez-Barrena, M.G. Epigenetics in Liver Fibrosis: Could HDACs Be a Therapeutic Target? Cells 2020, 9, 2321. [Google Scholar] [CrossRef]
- Sodum, N.; Kumar, G.; Bojja, S.L.; Kumar, N.; Rao, C.M. Epigenetics in NAFLD/NASH: Targets and therapy. Pharmacol. Res. 2021, 167, 105484. [Google Scholar] [CrossRef]
- Rodrigues, P.M.; Rodrigues, C.; Castro, R.E. Modulation of liver steatosis by miR-21/PPARα. Cell Death Discov. 2018, 4, 9. [Google Scholar] [CrossRef]
- Moody, L.; Xu, G.B.; Chen, H.; Pan, Y.-X. Epigenetic regulation of carnitine palmitoyltransferase 1 (Cpt1a) by high fat diet. Biochim. Biophys. Acta (BBA)-Bioenerg. 2018, 1862, 141–152. [Google Scholar] [CrossRef]
- Wang, L.; Chen, L.; Tan, Y.; Wei, J.; Chang, Y.; Jin, T.; Zhu, H. Betaine supplement alleviates hepatic triglyceride accumulation of apolipoprotein E deficient mice via reducing methylation of peroxisomal proliferator-activated receptor alpha promoter. Lipids Health Dis. 2013, 12, 34. [Google Scholar] [CrossRef]
- Ehara, T.; Kamei, Y.; Yuan, X.; Takahashi, M.; Kanai, S.; Tamura, E.; Tsujimoto, K.; Tamiya, T.; Nakagawa, Y.; Shimano, H.; et al. Ligand-Activated PPARα-Dependent DNA Demethylation Regulates the Fatty Acid β-Oxidation Genes in the Postnatal Liver. Diabetes 2014, 64, 775–784. [Google Scholar] [CrossRef]
- Byun, S.; Seok, S.; Kim, Y.-C.; Zhang, Y.; Yau, P.; Iwamori, N.; Xu, H.E.; Ma, J.; Kemper, B.; Kemper, J.K. Fasting-induced FGF21 signaling activates hepatic autophagy and lipid degradation via JMJD3 histone demethylase. Nat. Commun. 2020, 11, 807. [Google Scholar] [CrossRef]
- Huang, L.; Liu, J.; Zhang, X.-O.; Sibley, K.; Najjar, S.M.; Lee, M.M.; Wu, Q. Inhibition of protein arginine methyltransferase 5 enhances hepatic mitochondrial biogenesis. J. Biol. Chem. 2018, 293, 10884–10894. [Google Scholar] [CrossRef]
- Mann, J.; Chu, D.C.; Maxwell, A.; Oakley, F.; Zhu, N.; Tsukamoto, H.; Mann, D.A. MeCP2 Controls an Epigenetic Pathway That Promotes Myofibroblast Transdifferentiation and Fibrosis. Gastroenterology 2010, 138, 705–714.e4. [Google Scholar] [CrossRef]
- Liu, J.; Tang, T.; Wang, G.-D.; Liu, B. LncRNA-H19 promotes hepatic lipogenesis by directly regulating miR-130a/PPARγ axis in non-alcoholic fatty liver disease. Biosci. Rep. 2019, 39, BSR20181722. [Google Scholar] [CrossRef]
- Hardy, T.; Zeybel, M.; Day, C.P.; Dipper, C.; Masson, S.; McPherson, S.; Henderson, E.; Tiniakos, D.; White, S.; French, J.; et al. Plasma DNA methylation: A potential biomarker for stratification of liver fibrosis in non-alcoholic fatty liver disease. Gut 2016, 66, 1321–1328. [Google Scholar] [CrossRef]
- Hwang, J.W.; So, Y.; Bae, G.; Kim, S.; Kim, Y.K. Protein arginine methyltransferase 6 suppresses adipogenic differentiation by repressing peroxisome proliferator-activated receptor γ activity. Int. J. Mol. Med. 2019, 43, 2462–2470. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, S.; Zhao, Y.; Lin, C.; Zhong, F.; Jin, L.; He, F.; Wang, H. Histone H3K9 demethylase JMJD1A modulates hepatic stellate cells activation and liver fibrosis by epigenetically regulating peroxisome proliferator-activated receptor γ. FASEB J. 2015, 29, 1830–1841. [Google Scholar] [CrossRef]
- Kim, J.-H.; Jung, D.Y.; Nagappan, A.; Jung, M.H. Histone H3K9 demethylase JMJD2B induces hepatic steatosis through upregulation of PPARγ2. Sci. Rep. 2018, 8, 13734. [Google Scholar] [CrossRef]
- Pang, H.; Ling, D.; Cheng, Y.; Akbar, R.; Jin, L.; Ren, J.; Wu, H.; Chen, B.; Zhou, Y.; Zhu, H.; et al. Gestational high-fat diet impaired demethylation of Ppar α and induced obesity of offspring. J. Cell. Mol. Med. 2021, 25, 5404–5416. [Google Scholar] [CrossRef] [PubMed]
- Kawahori, K.; Kondo, Y.; Yuan, X.; Kawasaki, Y.; Hanzawa, N.; Tsujimoto, K.; Wada, F.; Kohda, T.; Ishigami, A.; Yamada, T.; et al. Ascorbic acid during the suckling period is required for proper DNA demethylation in the liver. Sci. Rep. 2020, 10, 21228. [Google Scholar] [CrossRef]
- Kharitonenkov, A.; Shiyanova, T.L.; Koester, A.; Ford, A.M.; Micanovic, R.; Galbreath, E.; Sandusky, G.E.; Hammond, L.J.; Moyers, J.S.; Owens, R.A.; et al. FGF-21 as a novel metabolic regulator. J. Clin. Investig. 2005, 115, 1627–1635. [Google Scholar] [CrossRef]
- Jimenez, V.; Jambrina, C.; Casana, E.; Sacristan, V.; Muñoz, S.; Darriba, S.; Rodó, J.; Mallol, C.; Garcia, M.; León, X.; et al. FGF21 gene therapy as treatment for obesity and insulin resistance. EMBO Mol. Med. 2018, 10, e8791. [Google Scholar] [CrossRef]
- Longo, R.; Peri, C.; Cricrì, D.; Coppi, L.; Caruso, D.; Mitro, N.; De Fabiani, E.; Crestani, M. Ketogenic Diet: A New Light Shining on Old but Gold Biochemistry. Nutrients 2019, 11, 2497. [Google Scholar] [CrossRef]
- Seok, S.; Kim, Y.-C.; Byun, S.; Choi, S.; Xiao, Z.; Iwamori, N.; Zhang, Y.; Wang, C.; Ma, J.; Ge, K.; et al. Fasting-induced JMJD3 histone demethylase epigenetically activates mitochondrial fatty acid β-oxidation. J. Clin. Investig. 2018, 128, 3144–3159. [Google Scholar] [CrossRef]
- Yuan, X.; Tsujimoto, K.; Hashimoto, K.; Kawahori, K.; Hanzawa, N.; Hamaguchi, M.; Seki, T.; Nawa, M.; Ehara, T.; Kitamura, Y.; et al. Epigenetic modulation of Fgf21 in the perinatal mouse liver ameliorates diet-induced obesity in adulthood. Nat. Commun. 2018, 9, 636. [Google Scholar] [CrossRef]
- Hazra, S.; Miyahara, T.; Rippe, R.A.; Tsukamoto, H. PPAR Gamma and Hepatic Stellate Cells. Comp. Hepatol. 2004, 3 (Suppl. 1), S7. [Google Scholar] [CrossRef]
- Perugorria, M.J.; Wilson, C.L.; Zeybel, M.; Walsh, M.; Amin, S.; Robinson, S.; White, S.A.; Burt, A.D.; Oakley, F.; Tsukamoto, H.; et al. Histone methyltransferase ASH1 orchestrates fibrogenic gene transcription during myofibroblast transdifferentiation. Hepatology 2012, 56, 1129–1139. [Google Scholar] [CrossRef]
- Byrd, K.N.; Shearn, A. ASH1, a Drosophila trithorax group protein, is required for methylation of lysine 4 residues on histone H3. Proc. Natl. Acad. Sci. USA 2003, 100, 11535–11540. [Google Scholar] [CrossRef]
- Zong, Y.; Yan, J.; Jin, L.; Xu, B.; He, Z.; Zhang, R.; Hu, C.; Jia, W. Relationship between circulating miR-132 and non-alcoholic fatty liver disease in a Chinese population. Hereditas 2020, 157, 22. [Google Scholar] [CrossRef] [PubMed]
- Lau-Corona, D.; Bae, W.K.; Hennighausen, L.; Waxman, D.J. Sex-biased genetic programs in liver metabolism and liver fibrosis are controlled by EZH1 and EZH2. PLoS Genet. 2020, 16, e1008796. [Google Scholar] [CrossRef]
- Moran-Salvador, E.; Garcia-Macia, M.; Sivaharan, A.; Sabater, L.; Zaki, M.Y.; Oakley, F.; Knox, A.; Page, A.; Luli, S.; Mann, J.; et al. Fibrogenic Activity of MECP2 Is Regulated by Phosphorylation in Hepatic Stellate Cells. Gastroenterology 2019, 157, 1398–1412.e9. [Google Scholar] [CrossRef]
- Jing, F.; Geng, Y.; Xu, X.-Y.; Xu, H.-Y.; Shi, J.-S.; Xu, Z.-H. MicroRNA29a Reverts the Activated Hepatic Stellate Cells in the Regression of Hepatic Fibrosis through Regulation of ATPase H+ Transporting V1 Subunit C1. Int. J. Mol. Sci. 2019, 20, 796. [Google Scholar] [CrossRef] [PubMed]
- Xuan, J.; Guo, S.-L.; Huang, A.; Xu, H.-B.; Shao, M.; Yang, Y.; Wen, W. MiR-29a and miR-652 Attenuate Liver Fibrosis by Inhibiting the Differentiation of CD4+ T Cells. Cell Struct. Funct. 2017, 42, 95–103. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Itami, S.; Kuroda, M.; Yoshizato, K.; Kawada, N.; Murakami, Y. MiR-29a Assists in Preventing the Activation of Human Stellate Cells and Promotes Recovery From Liver Fibrosis in Mice. Mol. Ther. 2016, 24, 1848–1859. [Google Scholar] [CrossRef]
- Galic, S.; Oakhill, J.; Steinberg, G.R. Adipose tissue as an endocrine organ. Mol. Cell. Endocrinol. 2010, 316, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Khandekar, M.J.; Cohen, P.; Spiegelman, B.M. Molecular mechanisms of cancer development in obesity. Nat. Rev. Cancer 2011, 11, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Heydari, H.; Ghiasi, R.; Ghaderpour, S.; Keyhanmanesh, R. The Mechanisms Involved in Obesity-Induced Male Infertility. Curr. Diabetes Rev. 2021, 17, 259–267. [Google Scholar] [CrossRef]
- Siersbaek, R.; Nielsen, R.; Mandrup, S. PPARγ in adipocyte differentiation and metabolism—Novel insights from genome-wide studies. FEBS Lett. 2010, 584, 3242–3249. [Google Scholar] [CrossRef]
- Lefterova, M.I.; Zhang, Y.; Steger, D.J.; Schupp, M.; Schug, J.; Cristancho, A.; Feng, D.; Zhuo, D.; Stoeckert, J.C.J.; Liu, X.S.; et al. PPAR and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev. 2008, 22, 2941–2952. [Google Scholar] [CrossRef] [PubMed]
- Fujiki, K.; Kano, F.; Shiota, K.; Murata, M. Expression of the peroxisome proliferator activated receptor γ gene is repressed by DNA methylation in visceral adipose tissue of mouse models of diabetes. BMC Biol. 2009, 7, 38. [Google Scholar] [CrossRef]
- Duteil, D.; Tosic, M.; Willmann, D.; Georgiadi, A.; Kanouni, T.; Schüle, R. Lsd1 prevents age-programed loss of beige adipocytes. Proc. Natl. Acad. Sci. USA 2017, 114, 5265–5270. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhang, B.; Lan, X.; Zhang, C.; Lei, C.; Chen, H. Comparative Transcriptome Analysis Reveals Significant Differences in MicroRNA Expression and Their Target Genes between Adipose and Muscular Tissues in Cattle. PLoS ONE 2014, 9, e102142. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, R.; Nardelli, C.; Pilone, V.; Buonomo, T.; Liguori, R.; Castanò, I.; Buono, P.; Masone, S.; Persico, G.; Forestieri, P.; et al. miR-519d Overexpression Is Associated With Human Obesity. Obesity 2010, 18, 2170–2176. [Google Scholar] [CrossRef] [PubMed]
- Couture, J.-P.; Nolet, G.; Beaulieu, E.; Blouin, R.; Gévry, N. The p400/Brd8 Chromatin Remodeling Complex Promotes Adipogenesis by Incorporating Histone Variant H2A.Z at PPARγ Target Genes. Endocrinology 2012, 153, 5796–5808. [Google Scholar] [CrossRef]
- Mikkelsen, T.S.; Xu, Z.; Zhang, X.; Wang, L.; Gimble, J.M.; Lander, E.S.; Rosen, E.D. Comparative Epigenomic Analysis of Murine and Human Adipogenesis. Cell 2010, 143, 156–169. [Google Scholar] [CrossRef]
- Park, U.; Hwang, J.; Youn, H.; Kim, E.; Um, S. Piperine inhibits adipocyte differentiation via dynamic regulation of histone modifications. Phytother. Res. 2019, 33, 2429–2439. [Google Scholar] [CrossRef]
- Lee, J.-E.; Wang, C.; Xu, S.; Cho, Y.-W.; Wang, L.; Feng, X.; Baldridge, A.; Sartorelli, V.; Zhuang, L.; Peng, W.; et al. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. Elife 2013, 2, e01503. [Google Scholar] [CrossRef]
- Cho, Y.-W.; Hong, S.; Jin, Q.; Wang, L.; Lee, J.-E.; Gavrilova, O.; Ge, K. Histone Methylation Regulator PTIP Is Required for PPARγ and C/EBPα Expression and Adipogenesis. Cell Metab. 2009, 10, 27–39. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, H.H.; Ye, B.J.; Lee-Kwon, W.; Choi, S.Y.; Kwon, H.M. TonEBP suppresses adipogenesis and insulin sensitivity by blocking epigenetic transition of PPARγ2. Sci. Rep. 2015, 5, 10937. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-H.; Chung, C.-C.; Liu, Y.-C.; Yeh, S.-P.; Hsu, J.L.; Hung, M.-C.; Su, H.-L.; Li, L.-Y. Enhancer of Zeste Homolog 2 and Histone Deacetylase 9c Regulate Age-Dependent Mesenchymal Stem Cell Differentiation into Osteoblasts and Adipocytes. Stem Cells 2016, 34, 2183–2193. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-H.; Yeh, F.-L.; Yeh, S.-P.; Ma, H.-T.; Hung, S.-C.; Hung, M.-C.; Li, L.-Y. Myocyte Enhancer Factor-2 Interacting Transcriptional Repressor (MITR) Is a Switch That Promotes Osteogenesis and Inhibits Adipogenesis of Mesenchymal Stem Cells by Inactivating Peroxisome Proliferator-activated Receptor γ-2. J. Biol. Chem. 2011, 286, 10671–10680. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Jang, Y.; Park, Y.-K.; Lee, J.-E.; Jain, S.; Froimchuk, E.; Broun, A.; Liu, C.; Gavrilova, O.; Ge, K. Depletion of Nsd2-mediated histone H3K36 methylation impairs adipose tissue development and function. Nat. Commun. 2018, 9, 1796. [Google Scholar] [CrossRef] [PubMed]
- Nic-Can, G.I.; Rodas-Junco, B.A.; Carrillo-Cocom, L.M.; Zepeda-Pedreguera, A.; Peñaloza-Cuevas, R.; Aguilar-Ayala, F.J.; Rojas-Herrera, R.A. Epigenetic Regulation of Adipogenic Differentiation by Histone Lysine Demethylation. Int. J. Mol. Sci. 2019, 20, 3918. [Google Scholar] [CrossRef] [PubMed]
- Lizcano, F.; Romero, C.; Vargas, D. Regulation of adipogenesis by nucelar receptor PPARγ is modulated by the histone demethylase JMJD2C. Genet. Mol. Biol. 2010, 34, 19–24. [Google Scholar] [CrossRef]
- Tateishi, K.; Okada, Y.; Kallin, E.M.; Zhang, Y. Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature 2009, 458, 757–761. [Google Scholar] [CrossRef]
- Inagaki, T.; Tachibana, M.; Magoori, K.; Kudo, H.; Tanaka, T.; Okamura, M.; Naito, M.; Kodama, T.; Shinkai, Y.; Sakai, J. Obesity and metabolic syndrome in histone demethylase JHDM2a-deficient mice. Genes Cells 2009, 14, 991–1001. [Google Scholar] [CrossRef]
- Lee, J.-E.; Ge, K. Transcriptional and epigenetic regulation of PPARγ expression during adipogenesis. Cell Biosci. 2014, 4, 29. [Google Scholar] [CrossRef]
- Steger, D.J.; Grant, G.; Schupp, M.; Tomaru, T.; Lefterova, M.I.; Schug, J.; Manduchi, E.; Stoeckert, C.J.; Lazar, M.A. Propagation of adipogenic signals through an epigenomic transition state. Genes Dev. 2010, 24, 1035–1044. [Google Scholar] [CrossRef]
- Erener, S.; Hesse, M.; Kostadinova, R.; Hottiger, M.O. Poly(ADP-Ribose)Polymerase-1 (PARP1) Controls Adipogenic Gene Expression and Adipocyte Function. Mol. Endocrinol. 2012, 26, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Rao, M.; Bouras, T.; Wang, C.; Wu, K.; Zhang, X.; Li, Z.; Yao, T.-P.; Pestell, R.G. Cyclin D1 Inhibits Peroxisome Proliferator-activated Receptor γ-mediated Adipogenesis through Histone Deacetylase Recruitment. J. Biol. Chem. 2005, 280, 16934–16941. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Zhang, F.; Yan, T.; Liu, Z.; Wang, C.; Ge, X.; Zhai, Q. C/EBPα regulates SIRT1 expression during adipogenesis. Cell Res. 2010, 20, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Picard, F.; Kurtev, M.; Chung, N.; Topark-Ngarm, A.; Senawong, T.; Machado De Oliveira, R.; Leid, M.; McBurney, M.W.; Guarente, L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature 2004, 429, 771–776. [Google Scholar] [CrossRef]
- Mayoral, R.; Osborn, O.; McNelis, J.; Johnson, A.M.; Oh, D.Y.; Izquierdo, C.L.; Chung, H.; Li, P.; Traves, P.G.; Bandyopadhyay, G.; et al. Adipocyte SIRT1 knockout promotes PPARγ activity, adipogenesis and insulin sensitivity in chronic-HFD and obesity. Mol. Metab. 2015, 4, 378–391. [Google Scholar] [CrossRef]
- Ferrari, A.; Fiorino, E.; Longo, R.; Barilla, S.; Mitro, N.; Cermenati, G.; Giudici, M.; Caruso, D.; Mai, A.; Guerrini, U.; et al. Attenuation of diet-induced obesity and induction of white fat browning with a chemical inhibitor of histone deacetylases. Int. J. Obes. 2016, 41, 289–298. [Google Scholar] [CrossRef]
- Aguilar, E.C.; Silva, J.; Navia-Pelaez, J.M.; Leonel, A.J.; Lopes, L.G.; Menezes-Garcia, Z.; Ferreira, A.; Capettini, L.; Teixeira, L.G.; Lemos, V.S.; et al. Sodium butyrate modulates adipocyte expansion, adipogenesis, and insulin receptor signaling by upregulation of PPAR-γ in obese Apo E knockout mice. Nutrition 2017, 47, 75–82. [Google Scholar] [CrossRef]
- Yoon, G.-E.; Jung, J.K.; Lee, Y.-H.; Jang, B.-C.; Kim, J.I. Histone deacetylase inhibitor CG200745 ameliorates high-fat diet-induced hypertension via inhibition of angiotensin II production. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2019, 393, 491–500. [Google Scholar] [CrossRef]
- Bian, F.; Ma, X.; Villivalam, S.D.; You, D.; Choy, L.R.; Paladugu, A.; Fung, S.; Kang, S. TET2 facilitates PPARγ agonist–mediated gene regulation and insulin sensitization in adipocytes. Metabolism 2018, 89, 39–47. [Google Scholar] [CrossRef]
- Chen, J.; Liu, Y.; Lu, S.; Yin, L.; Zong, C.; Cui, S.; Qin, D.; Yang, Y.; Guan, Q.; Li, X.; et al. The role and possible mechanism of lncRNA U90926 in modulating 3T3-L1 preadipocyte differentiation. Int. J. Obes. 2016, 41, 299–308. [Google Scholar] [CrossRef]
- Cooper, D.R.; Carter, G.; Li, P.; Patel, R.; Watson, J.E.; Patel, N.A. Long Non-Coding RNA NEAT1 Associates with SRp40 to Temporally Regulate PPARγ2 Splicing during Adipogenesis in 3T3-L1 Cells. Genes 2014, 5, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Divoux, A.; Karastergiou, K.; Xie, H.; Guo, W.; Perera, R.J.; Fried, S.K.; Smith, S.R. Identification of a novel lncRNA in gluteal adipose tissue and evidence for its positive effect on preadipocyte differentiation. Obesity 2014, 22, 1781–1785. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gu, M.; Ma, Y.; Peng, Y. LncRNA TUG1 reduces inflammation and enhances insulin sensitivity in white adipose tissue by regulating miR-204/SIRT1 axis in obesity mice. Mol. Cell. Biochem. 2020, 475, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Skårn, M.; Namløs, H.M.; Noordhuis, P.; Wang, M.-Y.; Meza-Zepeda, L.A.; Myklebost, O. Adipocyte Differentiation of Human Bone Marrow-Derived Stromal Cells Is Modulated by MicroRNA-155, MicroRNA-221, and MicroRNA-222. Stem Cells Dev. 2012, 21, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, Y.; Zhang, S.; Ye, L.; Cui, J.; Sun, Q.; Li, K.; Wu, H.; Liu, L. MiR-540 as a Novel Adipogenic Inhibitor Impairs Adipogenesis Via Suppression of PPARγ. J. Cell. Biochem. 2015, 116, 969–976. [Google Scholar] [CrossRef]
- Li, H.; Xue, M.; Xu, J.; Qin, X. MiR-301a is involved in adipocyte dysfunction during obesity-related inflammation via suppression of PPARgamma. Pharmazie 2016, 71, 84–88. [Google Scholar]
- Huang, Q.; Ma, C.; Chen, L.; Luo, D.; Chen, R.; Liang, F. Mechanistic Insights into the Interaction between Transcription Factors and Epigenetic Modifications and the Contribution to the Development of Obesity. Front. Endocrinol. 2018, 9, 370. [Google Scholar] [CrossRef]
- McGregor, R.A.; Choi, M.S. microRNAs in the Regulation of Adipogenesis and Obesity. Curr. Mol. Med. 2011, 11, 304–316. [Google Scholar] [CrossRef]
- Xie, H.; Lim, B.; Lodish, H.F. MicroRNAs Induced During Adipogenesis that Accelerate Fat Cell Development Are Downregulated in Obesity. Diabetes 2009, 58, 1050–1057. [Google Scholar] [CrossRef]
- Kolb, H.; Martin, S. Environmental/lifestyle factors in the pathogenesis and prevention of type 2 diabetes. BMC Med. 2017, 15, 131. [Google Scholar] [CrossRef]
- Ojo, O. Dietary Intake and Type 2 Diabetes. Nutrients 2019, 11, 2177. [Google Scholar] [CrossRef] [PubMed]
- Henning, R.J. Type-2 diabetes mellitus and cardiovascular disease. Future Cardiol. 2018, 14, 491–509. [Google Scholar] [CrossRef]
- Ogurtsova, K.; da Rocha Fernandes, J.D.; Huang, Y.; Linnenkamp, U.; Guariguata, L.; Cho, N.; Cavan, D.; Shaw, J.; Makaroff, L. IDF Diabetes Atlas: Global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res. Clin. Pract. 2017, 128, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, V.; Naidoo, M.; Ghai, M. Cell- and tissue-specific epigenetic changes associated with chronic inflammation in insulin resistance and type 2 diabetes mellitus. Scand. J. Immunol. 2018, 88, e12723. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Tsai, L.; Zhou, Y.; Evertts, A.G.; Xu, S.; Griffin, M.; Issner, R.; Whitton, H.J.; Garcia, B.A.; Epstein, C.B.; et al. Identification of nuclear hormone receptor pathways causing insulin resistance by transcriptional and epigenomic analysis. Nat. Cell. Biol. 2015, 17, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Ye, X.; Guo, W.; Lu, H.; Gao, Z. Inhibition of HDAC3 promotes ligand-independent PPARγ activation by protein acetylation. J. Mol. Endocrinol. 2014, 53, 191–200. [Google Scholar] [CrossRef]
- Li, D.; Wang, X.; Ren, W.; Ren, J.; Lan, X.; Wang, F.; Zhang, F.; Han, Y.; Song, T.; Holmdahl, R.; et al. High expression of liver histone deacetylase 3 contributes to high-fat-diet-induced metabolic syndrome by suppressing the PPAR-γ and LXR-α-pathways in E3 rats. Mol. Cell. Endocrinol. 2011, 344, 69–80. [Google Scholar] [CrossRef]
- Sathishkumar, C.; Prabu, P.; Balakumar, M.; Lenin, R.; Prabhu, D.; Anjana, R.M.; Mohan, V.; Balasubramanyam, M. Augmentation of histone deacetylase 3 (HDAC3) epigenetic signature at the interface of proinflammation and insulin resistance in patients with type 2 diabetes. Clin. Epigenet. 2016, 8, 125. [Google Scholar] [CrossRef]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef]
- Makkar, R.; Behl, T.; Arora, S. Role of HDAC inhibitors in diabetes mellitus. Curr. Res. Transl. Med. 2020, 68, 45–50. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, J.; Yuan, Y.; Chen, J.; Cheng, S.; Wang, H.; Xu, Y. Sodium butyrate mitigates type 2 diabetes by inhibiting PERK-CHOP pathway of endoplasmic reticulum stress. Environ. Toxicol. Pharmacol. 2018, 64, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Rafehi, H.; Kaspi, A.; Ziemann, M.; Okabe, J.; Karagiannis, T.C.; El-Osta, A. Systems approach to the pharmacological actions of HDAC inhibitors reveals EP300 activities and convergent mechanisms of regulation in diabetes. Epigenetics 2017, 12, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Fujisaka, S. The role of adipose tissue M1/M2 macrophages in type 2 diabetes mellitus. Diabetol. Int. 2020, 12, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zhao, J.; Meng, H.; Zhang, X. Adipose Tissue-Resident Immune Cells in Obesity and Type 2 Diabetes. Front. Immunol. 2019, 10, 1173. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; de Winther, M.P.; Bossche, J. Epigenetic mechanisms of macrophage activation in type 2 diabetes. Immunobiology 2017, 222, 937–943. [Google Scholar] [CrossRef]
- Bouhlel, M.A.; Derudas, B.; Rigamonti, E.; Dievart, R.; Brozek, J.; Haulon, S.; Zawadzki, C.; Jude, B.; Torpier, G.; Marx, N.; et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007, 6, 137–143. [Google Scholar] [CrossRef]
- Bouhlel, M.A.; Brozek, J.; Derudas, B.; Zawadzki, C.; Jude, B.; Staels, B.; Chinetti-Gbaguidi, G. Unlike PPARgamma, PPARalpha or PPARbeta/delta activation does not promote human monocyte differentiation toward alternative macrophages. Biochem. Biophys. Res. Commun. 2009, 386, 459–462. [Google Scholar] [CrossRef]
- Charo, I.F. Macrophage Polarization and Insulin Resistance: PPARγ in Control. Cell Metab. 2007, 6, 96–98. [Google Scholar] [CrossRef]
- Wang, X.; Cao, Q.; Yu, L.; Shi, H.; Xue, B.; Shi, H. Epigenetic regulation of macrophage polarization and inflammation by DNA methylation in obesity. JCI Insight 2016, 1, e87748. [Google Scholar] [CrossRef]
- Yang, X.; Wang, X.; Liu, D.; Yu, L.; Xue, B.; Shi, H. Epigenetic Regulation of Macrophage Polarization by DNA Methyltransferase 3b. Mol. Endocrinol. 2014, 28, 565–574. [Google Scholar] [CrossRef]
- You, D.; Nilsson, E.; Tenen, D.E.; Lyubetskaya, A.; Lo, J.C.; Jiang, R.; Deng, J.; Dawes, B.A.; Vaag, A.; Ling, C.; et al. Dnmt3a is an epigenetic mediator of adipose insulin resistance. Elife 2017, 6, e30766. [Google Scholar] [CrossRef]
- Kim, A.Y.; Park, Y.J.; Pan, X.; Shin, K.C.; Kwak, S.-H.; Bassas, A.F.; Sallam, R.M.; Park, K.S.; Alfadda, A.; Xu, A.; et al. Obesity-induced DNA hypermethylation of the adiponectin gene mediates insulin resistance. Nat. Commun. 2015, 6, 7585. [Google Scholar] [CrossRef]
- Chen, T.; Zhang, Y.; Liu, Y.; Zhu, D.; Yu, J.; Li, G.; Sun, Z.; Wang, W.; Jiang, H.; Hong, Z. MiR-27a promotes insulin resistance and mediates glucose metabolism by targeting PPAR-γ-mediated PI3K/AKT signaling. Aging (Albany NY) 2019, 11, 7510–7524. [Google Scholar] [CrossRef]
- Yao, F.; Yu, Y.; Feng, L.; Li, J.; Zhang, M.; Lan, X.; Yan, X.; Liu, Y.; Guan, F.; Zhang, M.; et al. Adipogenic miR-27a in adipose tissue upregulates macrophage activation via inhibiting PPARγ of insulin resistance induced by high-fat diet-associated obesity. Exp. Cell Res. 2017, 355, 105–112. [Google Scholar] [CrossRef]
- Liang, H.; Ward, W.F. PGC-1α: A key regulator of energy metabolism. Adv. Physiol. Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef]
- Jeninga, E.H.; Schoonjans, K.; Auwerx, J. Reversible acetylation of PGC-1: Connecting energy sensors and effectors to guarantee metabolic flexibility. Oncogene 2010, 29, 4617–4624. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.; Lerin, C.; Gerhart-Hines, Z.; Puigserver, P. Metabolic adaptations through the PGC-1α and SIRT1 pathways. FEBS Lett. 2007, 582, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Gerhart-Hines, Z.; Rodgers, J.; Bare, O.; Lerin, C.; Kim, S.-H.; Mostoslavsky, R.; Alt, F.W.; Wu, Z.; Puigserver, P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1α. EMBO J. 2007, 26, 1913–1923. [Google Scholar] [CrossRef] [PubMed]
- Sharabi, K.; Lin, H.; Tavares, C.D.; Dominy, J.E.; Camporez, J.P.; Perry, R.J.; Schilling, R.; Rines, A.K.; Lee, J.; Hickey, M.; et al. Selective Chemical Inhibition of PGC-1α Gluconeogenic Activity Ameliorates Type 2 Diabetes. Cell 2017, 169, 148–160.e15. [Google Scholar] [CrossRef] [PubMed]

| Condition | PPAR Isoform | Epigenetic Player | Effect | References |
|---|---|---|---|---|
| NASH | PPARα | miR-21 | Diminished PPARα expression and activation of HSCs in obesogenic models | [119] |
| TET1 and TET2 | Downregulated enzymes under high fat diet conditions, promoting Ppara hypermethylation | [121] | ||
| Ascorbic acid | Cofactor of TET enzymes. Its lack promotes PPARα target genes hypermethylation | [122] | ||
| JMJD3 | Phosphorylated upon fasting-induced FGF21 signaling. Direct interaction with PPARα for the upregulation of autophagy-related genes | [123] | ||
| PRMT5 | Downregulation of Ppara expression | [124] | ||
| PPARγ | miR-132 | miR-132 downregulation induces the expression of MeCP2 in HSCs | [125] | |
| miR-29a | Expressed upon Rosiglitazone-mediated PPARγ activation. Repression of profibrotic genes | [126] | ||
| MeCP2 | H3K9 and H3K27 methylation and HP1α repressor recruitment in Pparg locus of HSCs. MeCP2 also induces the expression of EZH2 and ASH1 in HSCs. | [125] | ||
| Pparg promoter CpG methylation | Downregulation of PPARγ. Potential non-invasive fibrosis marker in cell-free DNA in plasma. | [127] | ||
| PRMT6 | Repression of PPARγ activity | [128] | ||
| JMJD1A and JMJD2B | Upregulation of Pparg and increased lipid uptake | [129,130] | ||
| LncRNA-H19 | Control of hepatic lipogenesis through mi-130A/PPARγ axis | [126] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porcuna, J.; Mínguez-Martínez, J.; Ricote, M. The PPARα and PPARγ Epigenetic Landscape in Cancer and Immune and Metabolic Disorders. Int. J. Mol. Sci. 2021, 22, 10573. https://doi.org/10.3390/ijms221910573
Porcuna J, Mínguez-Martínez J, Ricote M. The PPARα and PPARγ Epigenetic Landscape in Cancer and Immune and Metabolic Disorders. International Journal of Molecular Sciences. 2021; 22(19):10573. https://doi.org/10.3390/ijms221910573
Chicago/Turabian StylePorcuna, Jesús, Jorge Mínguez-Martínez, and Mercedes Ricote. 2021. "The PPARα and PPARγ Epigenetic Landscape in Cancer and Immune and Metabolic Disorders" International Journal of Molecular Sciences 22, no. 19: 10573. https://doi.org/10.3390/ijms221910573
APA StylePorcuna, J., Mínguez-Martínez, J., & Ricote, M. (2021). The PPARα and PPARγ Epigenetic Landscape in Cancer and Immune and Metabolic Disorders. International Journal of Molecular Sciences, 22(19), 10573. https://doi.org/10.3390/ijms221910573