
IL-33/ST2 Axis Plays a Protective Effect in Streptococcus pyogenes Infection through Strengthening of the Innate Immunity

 ,
,
Abstract
:1. Introduction
2. Results
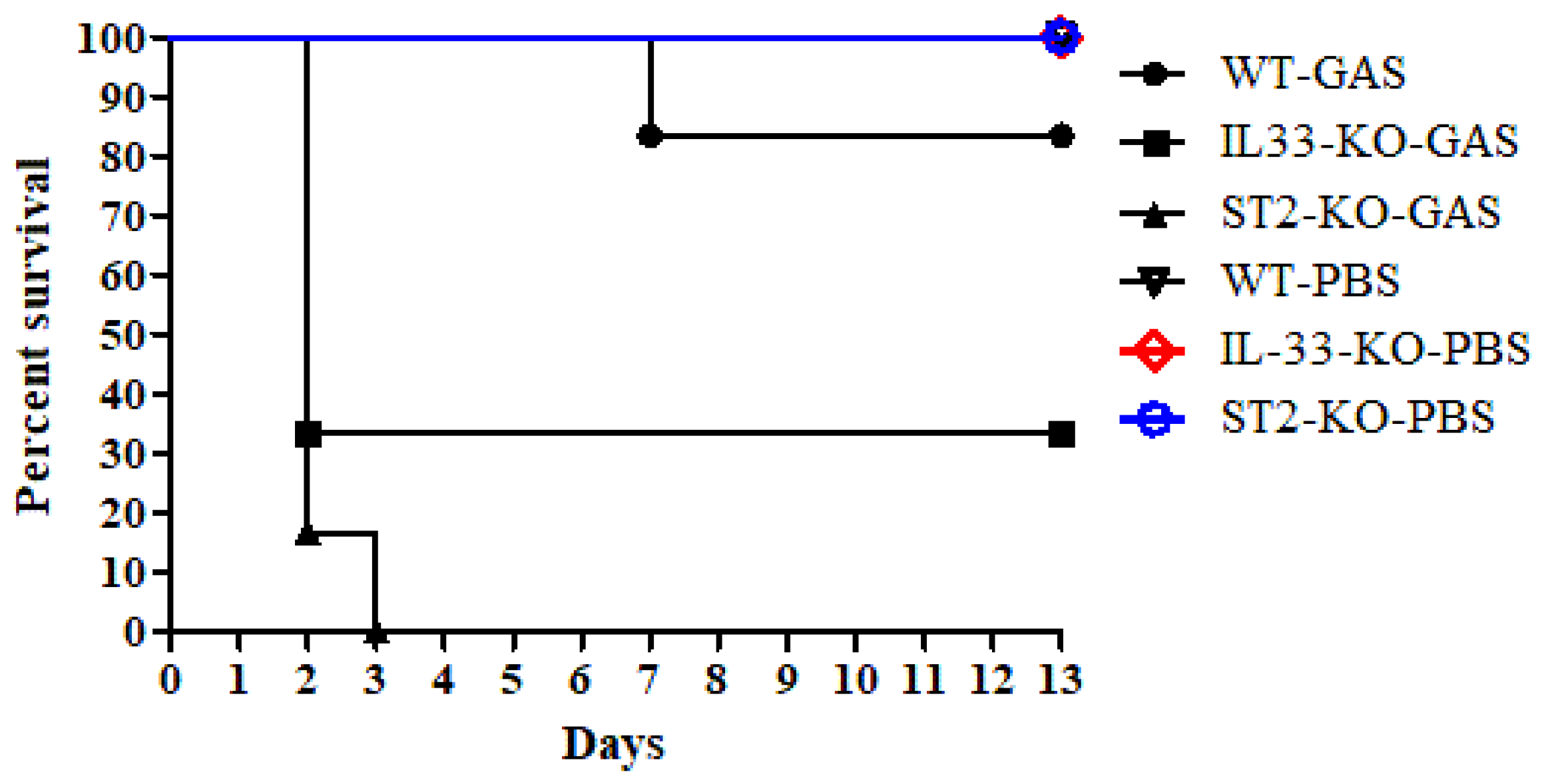
2.1. IL-33/ST2 Axis Played a Protective Effect on GAS Infection
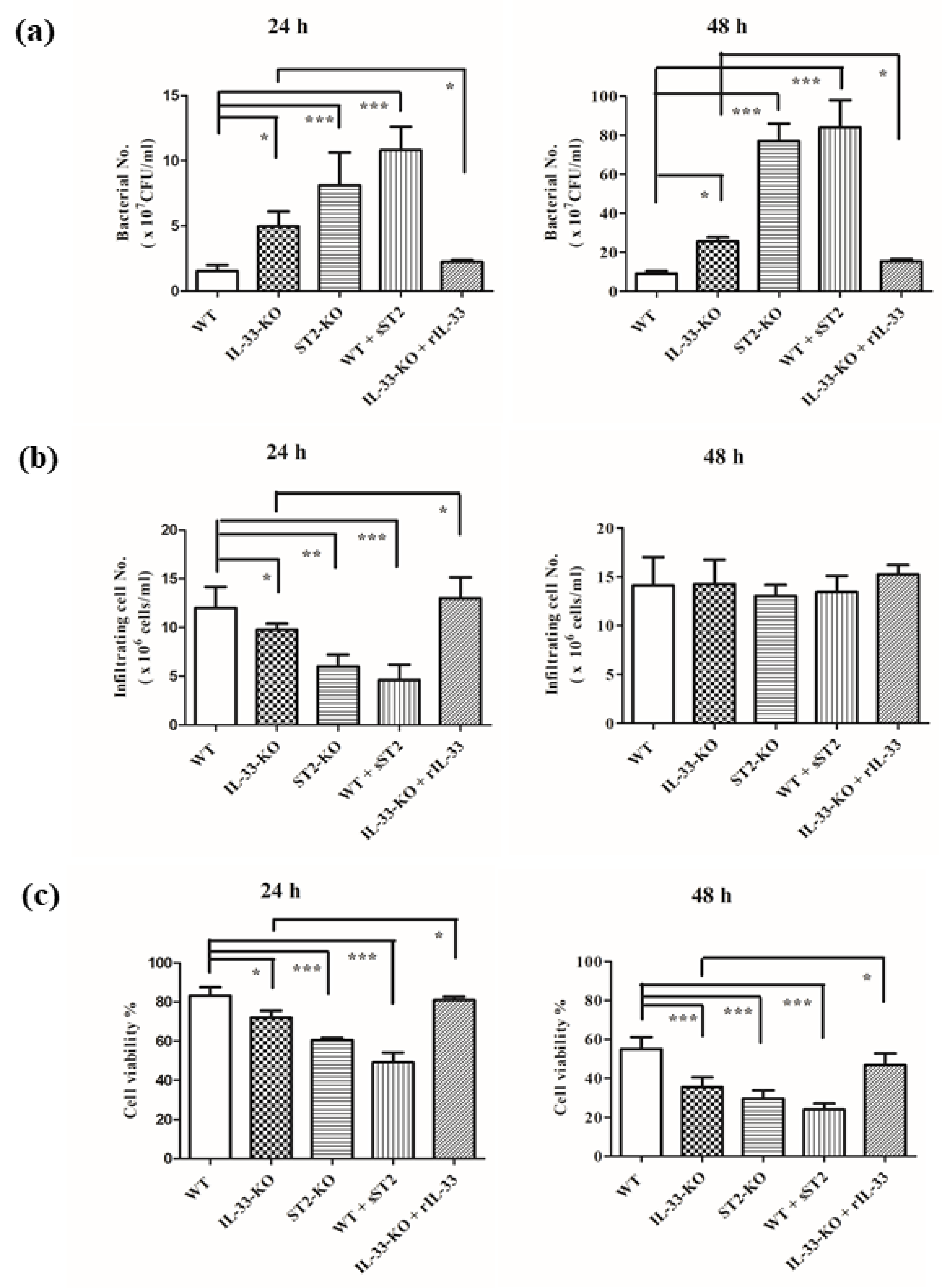
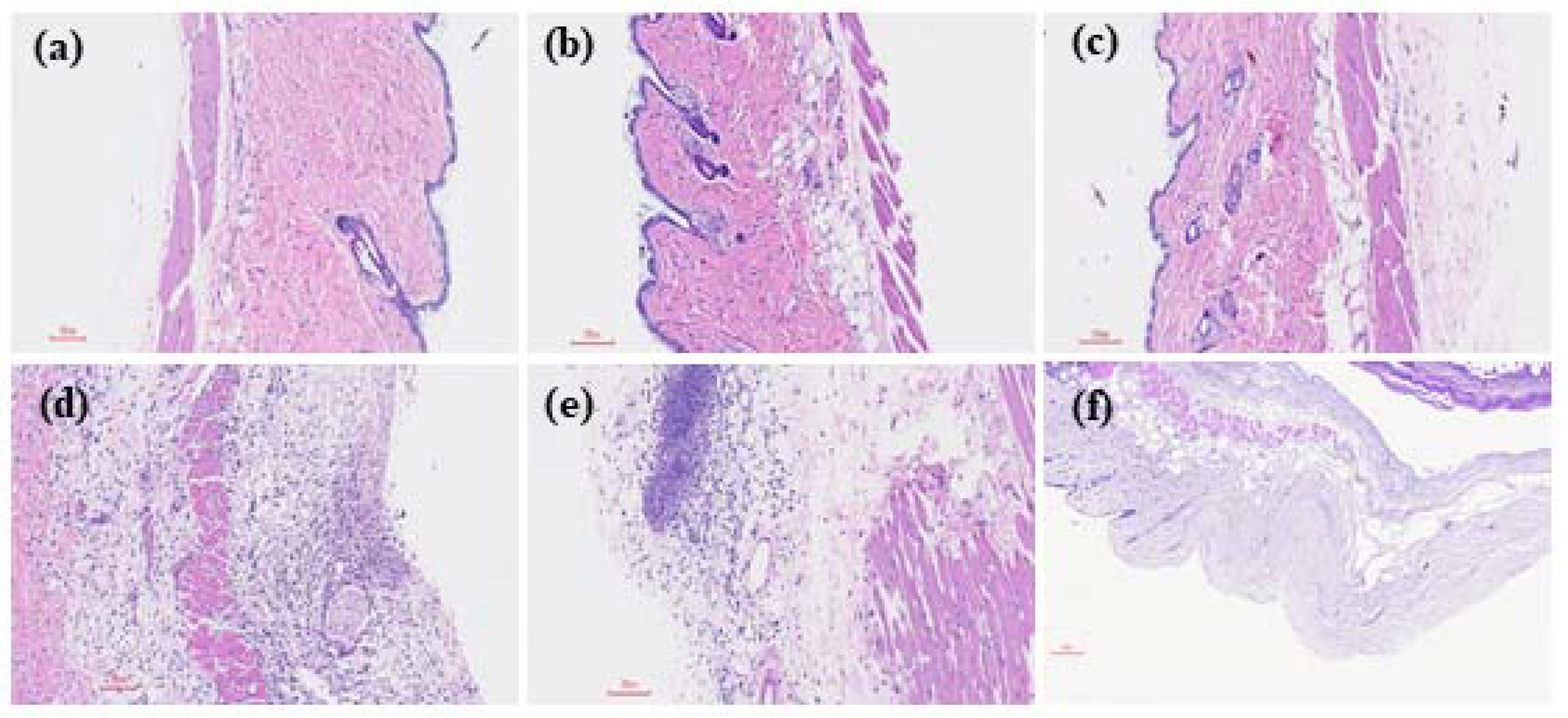
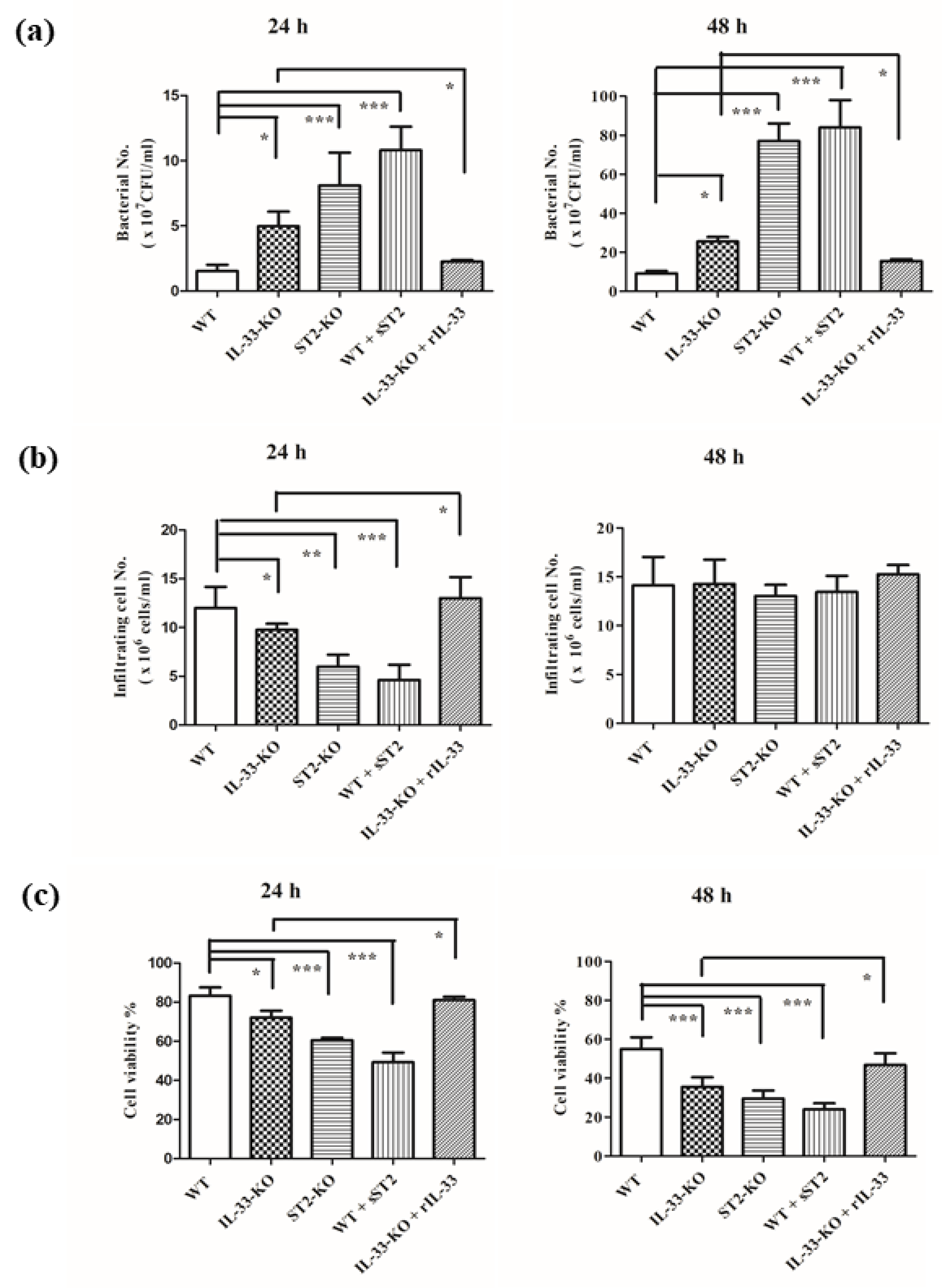
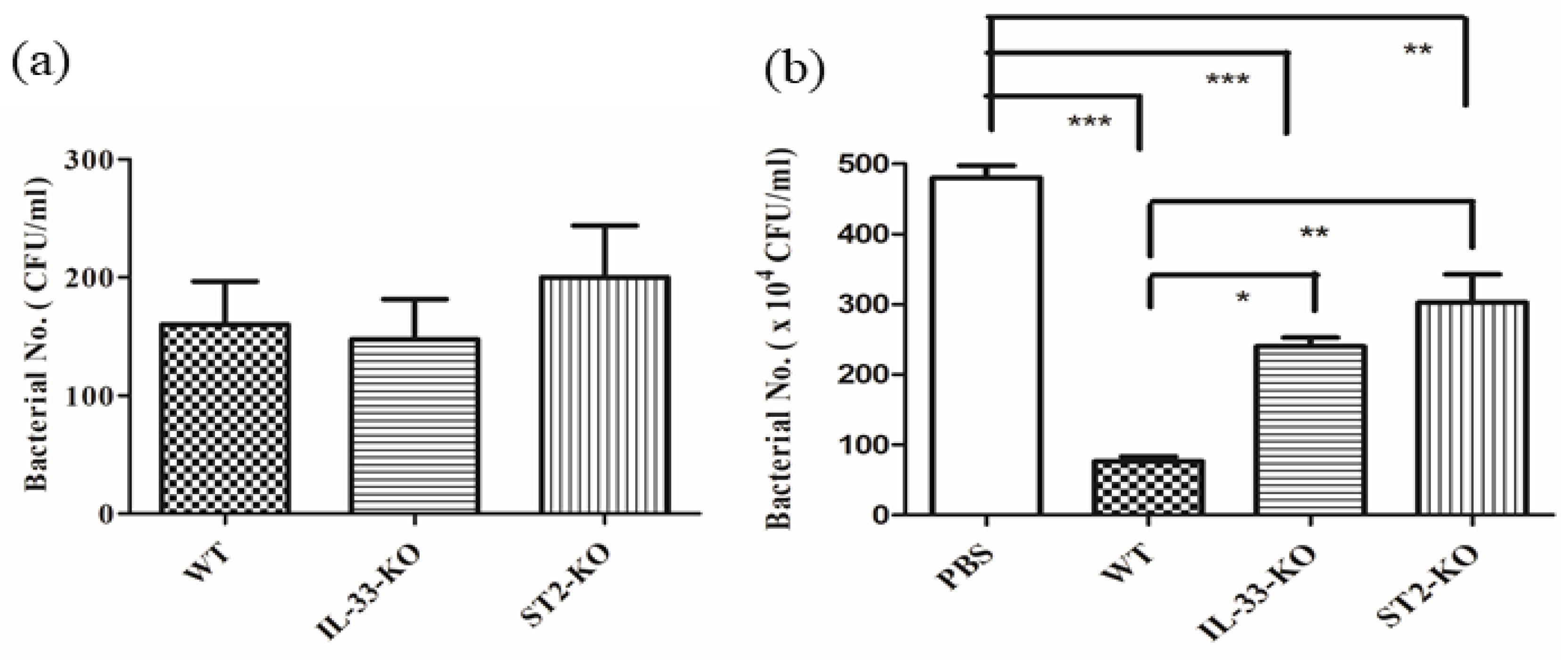
2.2. IL-33/ST2 Axis Affected Bacterial Burdens, Cell Infiltration and Cell Viability in Air Pouches of GAS-Infected Mice
2.3. Absence of IL-33/ST2 Axis Enhanced The Expression of Interleukin-6 and Interleukin-1β but Did Not Affect the Balance of TH1 and TH2 Responses
2.4. The Predominant Effector Cells in GAS-Infected Air Pouch Model Were Neutrophils
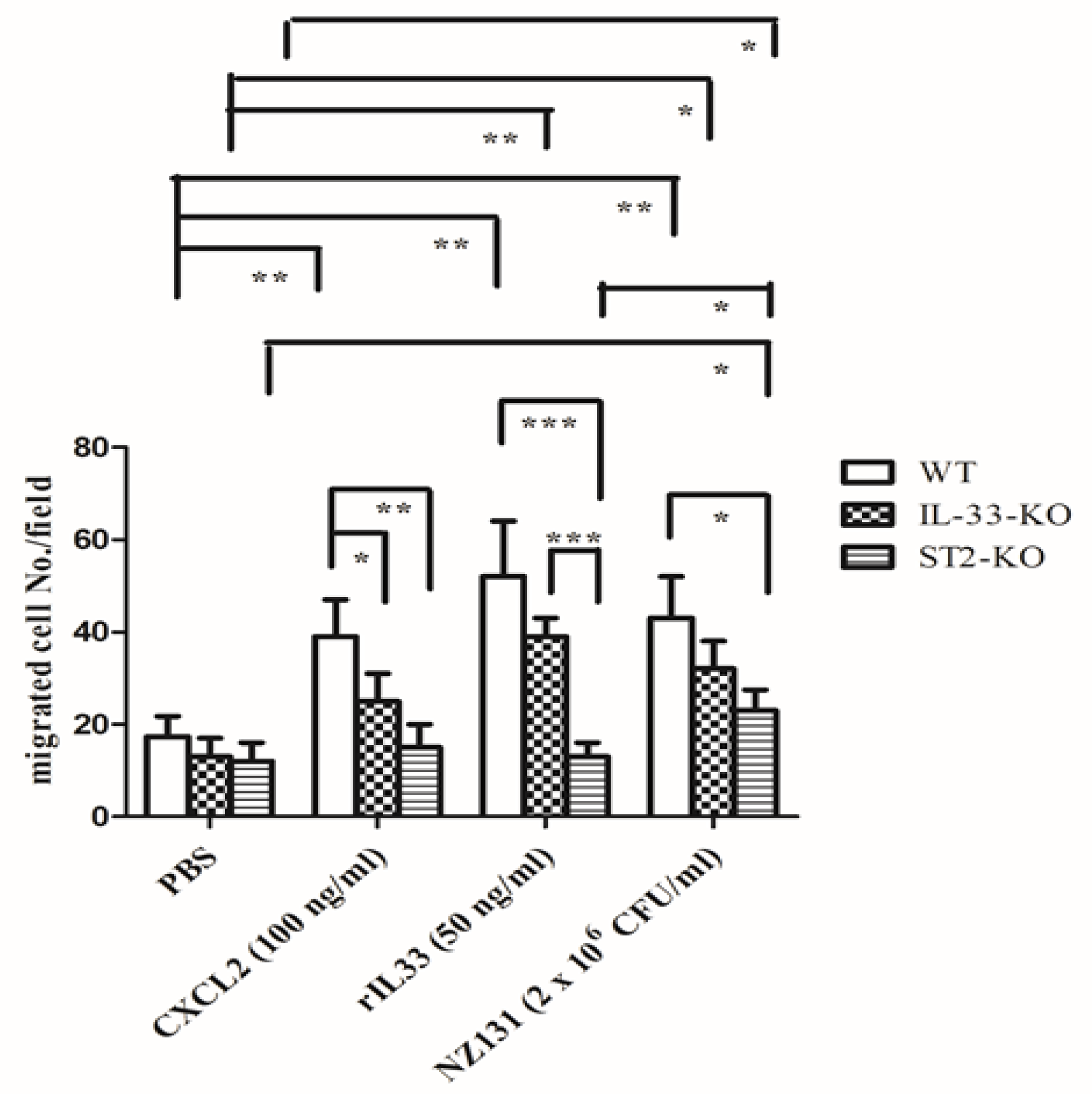
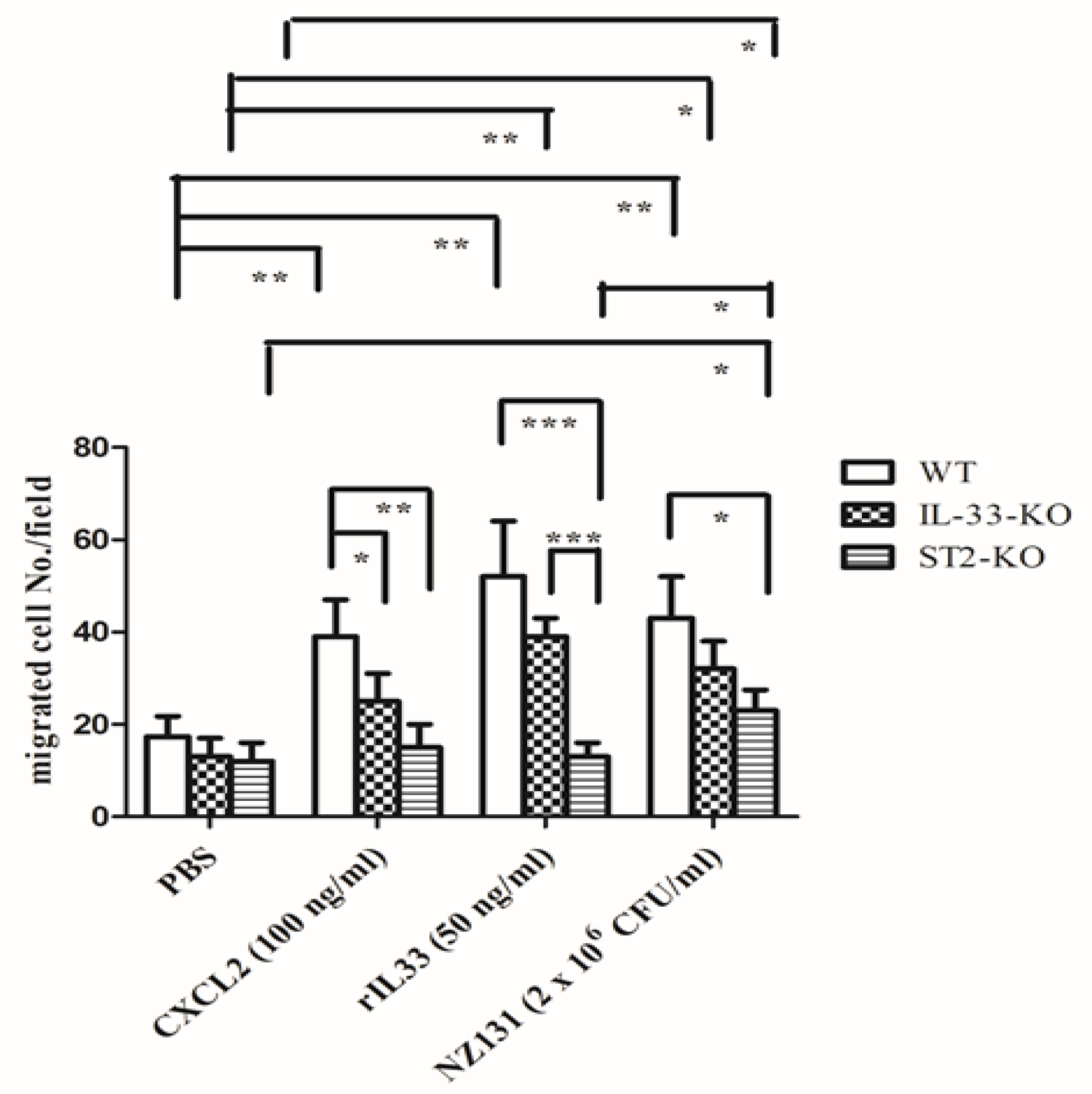
2.5. IL-33/ST2 Axis Affected Neutrophil Migration in the In Vitro Model
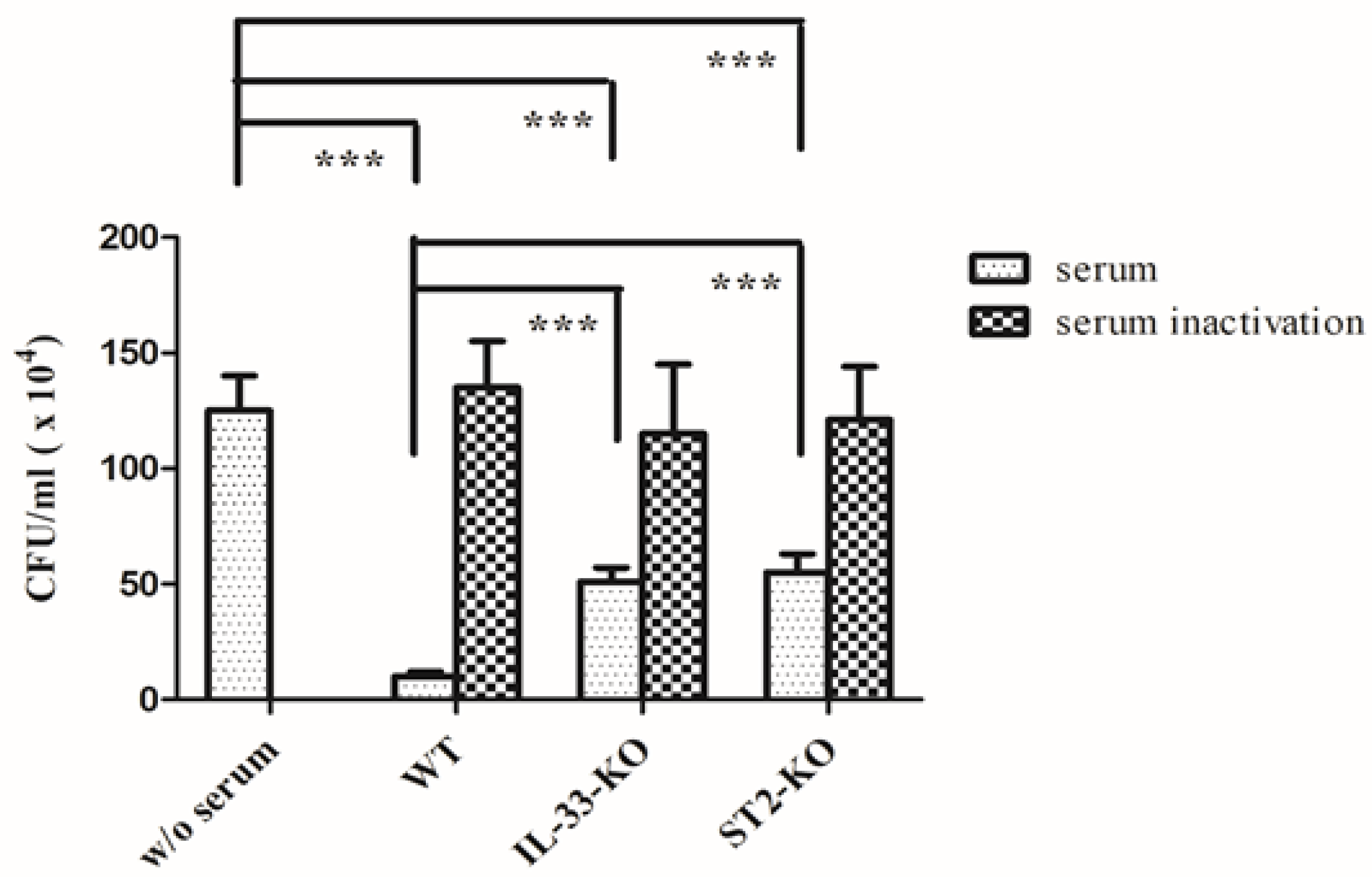
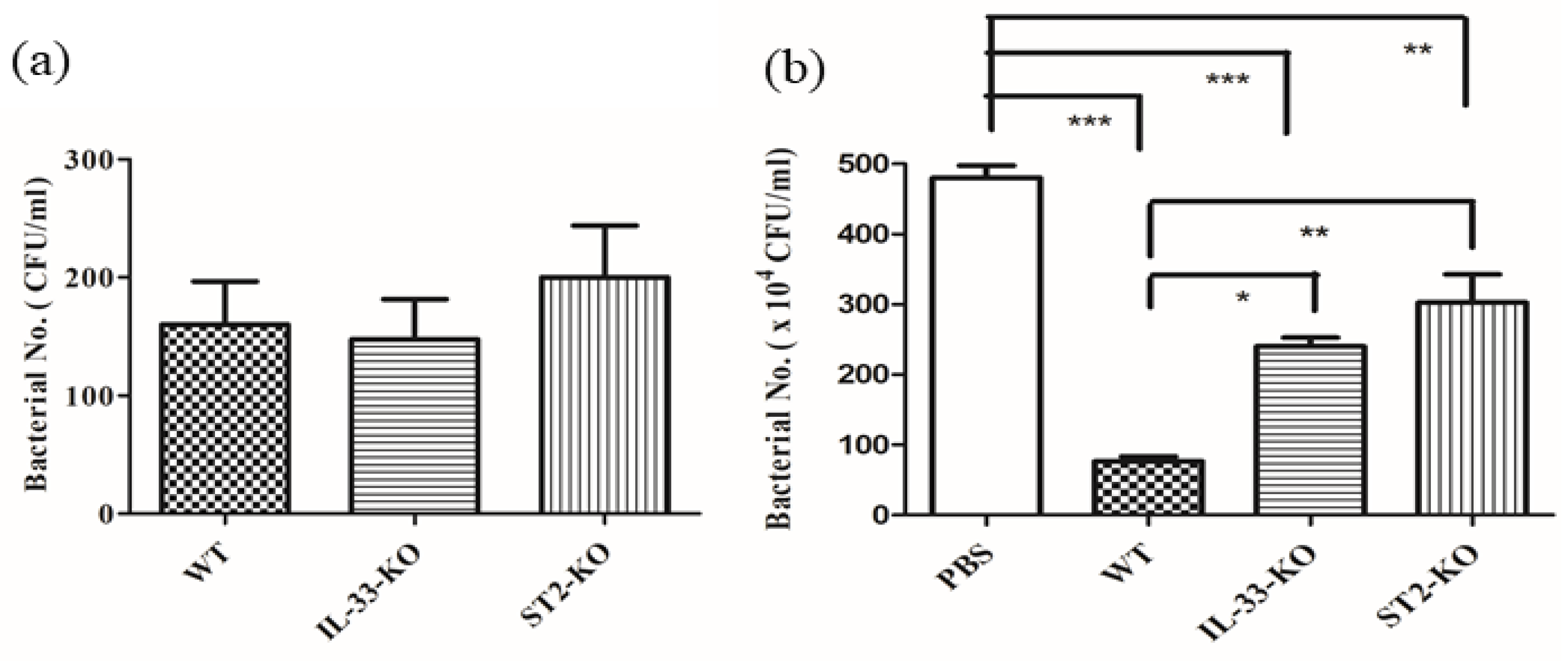
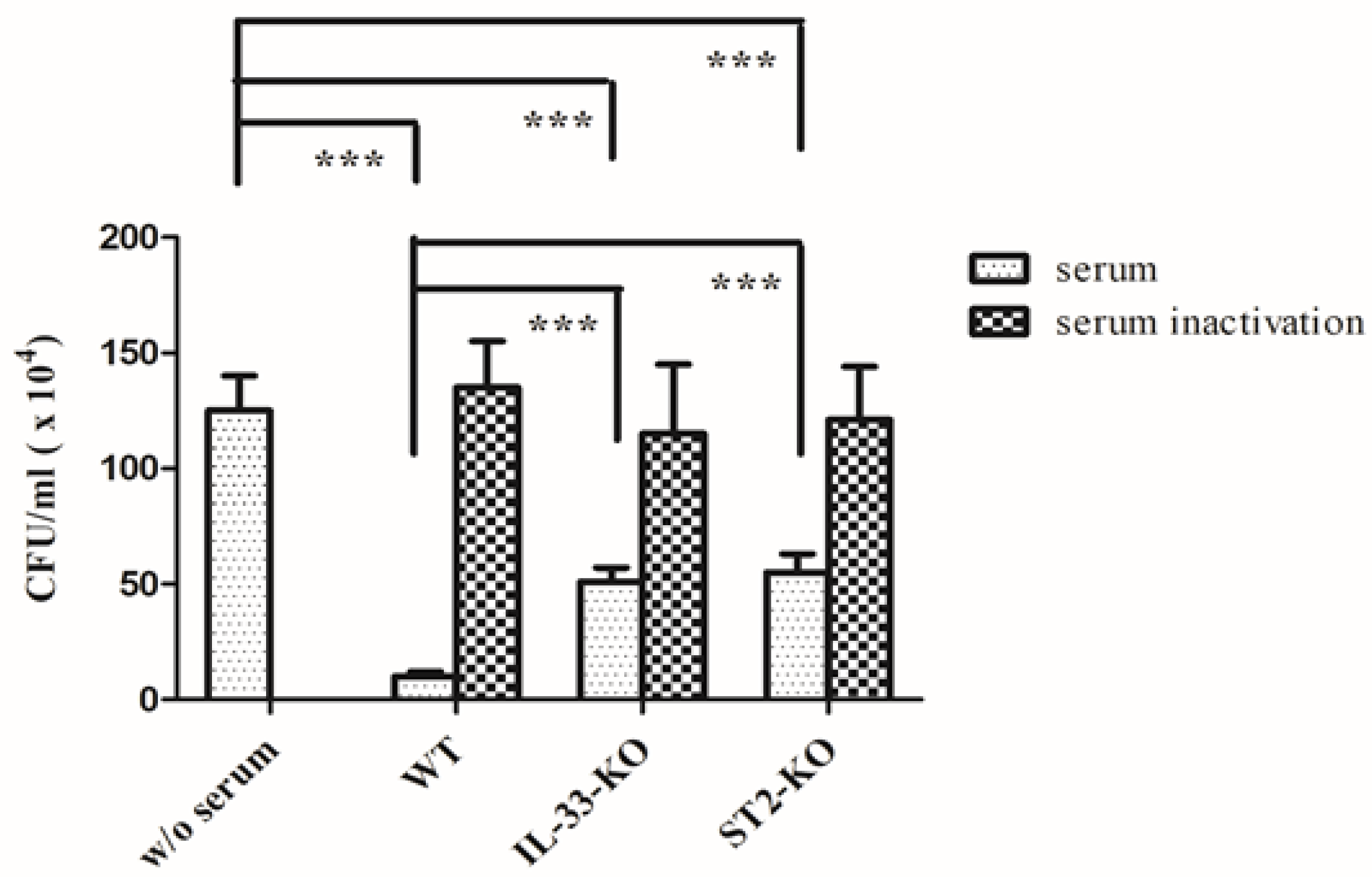
2.6. Absence of IL-33/ST2 Impaired the Bactericidal Activity of Sera and Neutrophils
3. Discussion
4. Materials and Methods
4.1. Bacterial Strain
4.2. Mice
4.3. Air Pouch Model of GAS Infection and Evaluation of Skin Lesion
4.4. Examination of the Inflammation and the Populations of Infiltrating Cells within the Air Pouches
4.5. Chemotaxis Assay of Mouse Neutrophils Isolated from Bone Marrow
4.6. Serum Bactericidal Assay
4.7. Phagocytosis and Bactericidal Assay of Neutrophils
4.8. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walker, M.J.; Barnett, T.C.; McArthur, J.D.; Cole, J.N.; Gillen, C.M.; Henningham, A.; Sriprakash, K.S.; Sanderson-Smith, M.L.; Nizet, V. Disease manifestations and pathogenic mechanisms of group A Streptococcus. Clin. Microbiol. Rev. 2014, 27, 264–301. [Google Scholar] [CrossRef] [Green Version]
- Norrby-Teglund, A.; Chatellier, S.; Low, D.E.; McGeer, A.; Green, K.; Kotb, M. Host variation in cytokine responses to superantigens determine the severity of invasive group A streptococcal infection. Eur. J. Immunol. 2000, 30, 3247–3255. [Google Scholar] [CrossRef]
- Påhlman, L.I.; Olin, A.I.; Darenberg, J.; Mörgelin, M.; Kotb, M.; Herwald, H.; Norrby-Teglund, A. Soluble M1 protein of Streptococcus pyogenes triggers potent T cell activation. Cell. Microbiol. 2008, 10, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Plainvert, C.; Doloy, A.; Loubinoux, J.; Lepoutre, A.; Collobert, G.; Touak, G.; Trieu-Cuot, P.; Bouvet, A.; Poyart, C.; CNR-Strep Network. Invasive group A streptococcal infections in adults, France (2006–2010). Clin. Microbiol. Infect. 2012, 18, 702–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liew, F.Y.; Pitman, N.I.; McInnes, I.B. Disease-associated functions of IL-33: The new kid in the IL-1 family. Nat. Rev. Immunol. 2010, 10, 103–110. [Google Scholar] [CrossRef]
- Milovanovic, M.; Volarevic, V.; Radosavljevic, G.; Jovanovic, I.; Pejnovic, N.; Arsenijevic, N.; Lukic, M.L. IL-33/ST2 axis in inflammation and immunopathology. Immunol. Res. 2012, 52, 89–99. [Google Scholar] [CrossRef]
- Rostan, O.; Arshad, M.I.; Piquet-Pellorce, C.; Robert-Gangneux, F.; Gangneux, J.P.; Samson, M. Crucial and diverse role of the interleukin-33/ST2 axis in infectious diseases. Infect. Immun. 2015, 83, 1738–1748. [Google Scholar] [CrossRef] [Green Version]
- Moussion, C.; Ortega, N.; Girard, J.P. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: A novel ‘alarmin’? PLoS ONE 2008, 3, e3331. [Google Scholar] [CrossRef] [Green Version]
- Sweet, M.J.; Leung, B.P.; Kang, D.; Sogaard, M.; Schulz, K.; Trajkovic, V.; Campbell, C.C.; Xu, D.; Liew, F.Y. A novel pathway regulating lipopolysaccharide-induced shock by ST2/T1 via inhibition of Toll-like receptor 4 expression. J. Immunol. 2001, 166, 6633–6639. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Buckley, J.M.; Redmond, H.P.; Wang, J.H. ST2 negatively regulates TLR2 signaling, but is not required for bacterial lipoprotein-induced tolerance. J. Immunol. 2010, 184, 5802–5808. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Mohs, A.; Thomas, M.; Klare, J.; Ross, R.; Schmitz, M.L.; Martin, M.U. The dual function cytokine IL-33 interacts with the transcription factor NF-κB to dampen NF-κB-stimulated gene transcription. J. Immunol. 2011, 187, 1609–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.S.; Park, J.A.; Kim, J.; Rho, S.S.; Park, H.; Kim, Y.M.; Kwon, Y.G. Nuclear IL-33 is a transcriptional regulator of NF-κB p65 and induces endothelial cell activation. Biochem. Biophys. Res. Commun. 2012, 421, 305–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cayrol, C.; Girard, J.P. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc. Natl. Acad. Sci. USA 2009, 106, 9021–9026. [Google Scholar] [CrossRef] [Green Version]
- Lüthi, A.U.; Cullen, S.P.; McNeela, E.A.; Duriez, P.J.; Afonina, I.S.; Sheridan, C.; Brumatti, G.; Taylor, R.C.; Kersse, K.; Vandenabeele, P.; et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity 2009, 31, 84–98. [Google Scholar] [CrossRef]
- Afferni, C.; Buccione, C.; Andreone, S.; Galdiero, M.R.; Varricchi, G.; Marone, G.; Mattei, F.; Schiavoni, G. The pleiotropic immunomodulatory functions of IL-33 and its implications in tumor immunity. Front. Immunol. 2018, 9, 2601. [Google Scholar] [CrossRef]
- Bergers, G.; Reikerstorfer, A.; Braselmann, S.; Graninger, P.; Busslinger, M. Alternative promoter usage of the Fos-responsive gene Fit-1 generates mRNA isoforms coding for either secreted or membrane-bound proteins related to the IL-1 receptor. EMBO J. 1994, 13, 1176–1188. [Google Scholar] [CrossRef]
- Alves-Filho, J.C.; Sônego, F.; Souto, F.O.; Freitas, A.; Verri, W.A., Jr.; Auxiliadora-Martins, M.; Basile-Filho, A.; McKenzie, A.N.; Xu, D.; Cunha, F.Q.; et al. Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat. Med. 2010, 16, 708–712. [Google Scholar] [CrossRef]
- Kuo, C.F.; Wu, J.J.; Lin, K.Y.; Tsai, P.J.; Lee, S.C.; Jin, Y.T.; Lei, H.Y.; Lin, Y.S. Role of streptococcal pyrogenic exotoxin B in the mouse model of group A streptococcal infection. Infect. Immun. 1998, 66, 3931–3935. [Google Scholar] [CrossRef] [Green Version]
- Hung, C.H.; Tsao, N.; Zeng, Y.F.; Lu, S.L.; Chuan, C.N.; Lin, Y.S.; Wu, J.J.; Kuo, C.F. Synergistic effects of streptolysin S and streptococcal pyrogenic exotoxin B on the mouse model of group A streptococcal infection. Med. Microbiol. Immunol. 2012, 201, 357–369. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Morrow, K.N.; Coopersmith, C.M.; Ford, M.L. IL-17, IL-27, and IL-33: A novel axis linked to immunological dysfunction during sepsis. Front. Immunol. 2019, 10, 1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Turnquist, H.R.; Hoffman, R.; Billiar, T.R. Role of the IL-33-ST2 axis in sepsis. Mil. Med. Res. 2017, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evrard, M.; Kwok, I.W.H.; Chong, S.Z.; Teng, K.W.W.; Becht, E.; Chen, J.; Sieow, J.L.; Penny, H.L.; Ching, G.C.; Devi, S.; et al. Developmental analysis of bone marrow neutrophils reveals populations specialized in expansion, trafficking, and effector functions. Immunity 2018, 48, 364–379.e8. [Google Scholar] [CrossRef] [Green Version]
- Dunay, I.R.; Damatta, R.A.; Fux, B.; Presti, R.; Greco, S.; Colonna, M.; Sibley, L.D. Gr1(+) inflammatory monocytes are required for mucosal resistance to the pathogen Toxoplasma gondii. Immunity 2008, 29, 306–317. [Google Scholar] [CrossRef] [Green Version]
- Barlow, J.L.; McKenzie, A.N.J. Type-2 innate lymphoid cells in human allergic disease. Curr. Opin. Allergy Clin. Immunol. 2014, 14, 397–403. [Google Scholar] [CrossRef] [Green Version]
- Steer, A.C.; Lamagni, T.; Curtis, N.; Carapetis, J.R. Invasive group a streptococcal disease: Epidemiology, pathogenesis and management. Drugs 2012, 72, 1213–1227. [Google Scholar] [CrossRef]
- Steer, A.C.; Batzloff, M.R.; Mulholland, K.; Carapetis, J.R. Group A streptococcal vaccines: Facts versus fantasy. Curr. Opin. Infect. Dis. 2009, 22, 544–552. [Google Scholar] [CrossRef]
- Batzloff, M.R.; Hayman, W.A.; Davies, M.R.; Zeng, M.; Pruksakorn, S.; Brandt, E.R.; Good, M.F. Protection against group A streptococcus by immunization with J8-diphtheria toxoid: Contribution of J8- and diphtheria toxoid-specific antibodies to protection. J. Infect. Dis. 2003, 187, 1598–1608. [Google Scholar] [CrossRef] [Green Version]
- Gorton, D.; Sikder, S.; Williams, N.L.; Chilton, L.; Rush, C.M.; Govan, B.L.; Cunningham, M.W.; Ketheesan, N. Repeat exposure to group A streptococcal M protein exacerbates cardiac damage in a rat model of rheumatic heart disease. Autoimmunity 2016, 25, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Zhu, F.X.; Zhao, X.J.; An, Y.Z. The immunoprotective activity of interleukin-33 in mouse model of cecal ligation and puncture-induced sepsis. Immunol. Lett. 2016, 169, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lv, R.; Zhao, J.; Lei, M.; Xiao, D.; Yu, Y.; Xie, J. IL-33 attenuates sepsis by inhibiting IL-17 receptor signaling through upregulation of SOCS3. Cell. Physiol. Biochem. 2017, 42, 1961–1972. [Google Scholar] [CrossRef] [PubMed]
- Bao, Q.; Lv, R.; Lei, M. IL-33 attenuates mortality by promoting IFN-gamma production in sepsis. Inflamm. Res. 2018, 67, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Staurengo-Ferrari, L.; Trevelin, S.C.; Fattori, V.; Nascimento, D.C.; de Lima, K.A.; Pelayo, J.S.; Figueiredo, F.; Casagrande, R.; Fukada, S.Y.; Teixeira, M.M.; et al. Interleukin-33 Receptor (ST2) deficiency improves the outcome of Staphylococcus aureus-induced septic arthritis. Front. Immunol. 2018, 9, 962. [Google Scholar] [CrossRef] [PubMed]
- Krishack, P.A.; Louviere, T.J.; Decker, T.S.; Kuzel, T.G.; Greenberg, J.A.; Camacho, D.F.; Hrusch, C.L.; Sperling, A.I.; Verhoef, P.A. Protection against Staphylococcus aureus bacteremia-induced mortality depends on ILC2s and eosinophils. JCI Insight 2019, 4, e124168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, K.M.; Ramanan, K.; Clay, M.E.; McHugh, K.J.; Rich, H.E.; Alcorn, J.F. Novel protective mechanism for interleukin-33 at the mucosal barrier during influenza-associated bacterial superinfection. Mucosal Immunol. 2018, 11, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, D.C.; Melo, P.H.; Piñeros, A.R.; Ferreira, R.G.; Colón, D.F.; Donate, P.B.; Castanheira, F.V.; Gozzi, A.; Czaikoski, P.G.; Niedbala, W.; et al. IL-33 contributes to sepsis-induced long-term immunosuppression by expanding the regulatory T cell population. Nat. Commun. 2017, 8, 14919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez-Moral, I.; Blok, D.C.; Bernink, J.H.; Garcia-Laorden, M.I.; Florquin, S.; Boon, L.; Van’t Veer, C.; Mack, M.; Saluzzo, S.; Knapp, S.; et al. Interleukin-33 improves local immunity during Gram-negative pneumonia by a combined effect on neutrophils and inflammatory monocytes. J. Pathol. 2021, 253, 374–383. [Google Scholar] [CrossRef]
- Petri, B.; Sanz, M.J. Neutrophil chemotaxis. Cell Tissue Res. 2018, 371, 425–436. [Google Scholar] [CrossRef]
- Sakai, N.; Van Sweringen, H.L.; Quillin, R.C.; Schuster, R.; Blanchard, J.; Burns, J.M.; Tevar, A.D.; Edwards, M.J.; Lentsch, A.B. Interleukin-33 is hepatoprotective during liver ischemia/reperfusion in mice. Hepatology 2012, 56, 1468–1478. [Google Scholar] [CrossRef] [Green Version]
- Carlsson, F.; Sandin, C.; Lindahl, G. Human fibrinogen bound to Streptococcus pyogenes M protein inhibits complement deposition via the classical pathway. Mol. Micribiol. 2005, 56, 28–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsao, N.; Tsai, W.H.; Lin, Y.S.; Chuang, W.J.; Wang, C.H.; Kuo, C.F. Streptococcal pyrogenic exotoxin B cleaves properdin and inhibits complement-mediated opsonophagocytosis. Biochem. Biophys. Res. Commun. 2006, 339, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.F.; Lin, Y.S.; Chuang, W.J.; Wu, J.J.; Tsao, N. Degradation of complement 3 by streptococcal pyrogenic exotoxin B inhibits complement activation and neutrophil opsonophagocytosis. Infect. Immun. 2008, 76, 1163–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honda-Ogawa, M.; Ogawa, T.; Terao, Y.; Sumitomo, T.; Nakata, M.; Ikebe, K.; Maeda, Y.; Kawabata, S. Cysteine proteinase from Streptococcus pyogenes enables evasion of innate immunity via degradation of complement factors. J. Biol. Chem. 2013, 288, 15854–15864. [Google Scholar] [CrossRef] [Green Version]
- Buckley, J.M.; Liu, J.H.; Li, C.H.; Blankson, S.; Wu, Q.D.; Jiang, Y.; Redmond, H.P.; Wang, J.H. Increased susceptibility of ST2-deficient mice to polymicrobial sepsis is associated with an impaired bactericidal function. J. Immunol. 2011, 187, 4293–4299. [Google Scholar] [CrossRef] [Green Version]
- Tsao, N.; Kuo, C.F.; Cheng, M.H.; Lin, W.C.; Lin, C.F.; Lin, Y.S. Streptolysin S induces mitochondrial damage and macrophage death through inhibiting degradation of glycogen synthase kinase-3β in Streptococcus pyogenes infection. Sci. Rep. 2019, 9, 5371. [Google Scholar] [CrossRef]
- Chen, W.Y.; Chang, Y.J.; Su, C.H.; Tsai, T.H.; Chen, S.D.; Hsing, C.H.; Yang, J.L. Upregulation of Interleukin-33 in obstructive renal injury. Biochem. Biophys. Res. Commun. 2016, 473, 1026–1032. [Google Scholar] [CrossRef]
- Kuo, C.F.; Tsao, N.; Hsieh, I.C.; Lin, Y.S.; Wu, J.J.; Hung, Y.T. Immunization with a streptococcal multiple-epitope recombinant protein protects mice against invasive group A streptococcal infection. PLoS ONE 2017, 12, e0174464. [Google Scholar] [CrossRef] [Green Version]
- Tsao, N.; Kuo, C.F.; Lei, H.Y.; Lu, S.L.; Huang, K.J. Inhibition of group A streptococcal infection by Melaleuca alternifolia (tea tree) oil concentrate in the murine model. J. Appl. Microbiol. 2010, 108, 936–944. [Google Scholar] [CrossRef]
- El Chemaly, A.; Nunes, P.; Jimaja, W.; Castelbou, C.; Demaurex, N. Hv1 proton channels differentially regulate the pH of neutrophil and macrophage phagosomes by sustaining the production of phagosomal ROS that inhibit the delivery of vacuolar ATPases. J. Leukoc. Biol. 2014, 95, 827–839. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | TNFα | IL-17A | IL-6 | IL-1β | IL-12 | IFN-γ | IL-10 (pg/mL) |
|---|---|---|---|---|---|---|---|
| WT-PBS | <5 a | <5 a | <5 a | <5 a | <5 a | <5 a | <5 a |
| WT + rIL-33 | 6 ± 1 | <5 a | <5 a | <5 a | <5 a | <5 a | 7 ± 2 |
| WT + sST2 | 6 ± 1 | <5 a | <5 a | <5 a | <5 a | <5 a | <5 a |
| WT-GAS | 891 ± 86 | <5 a | 4601 ± 875 | 724 ± 98 | 7 ± 2 | 7 ± 2 | 66 ± 19 |
| IL-33-KO-GAS | 498 ± 95 * | <5 a | 6150 ± 345 * | 1324 ± 297 * | 9 ± 4 | <5 a | 53 ± 8 |
| ST2-KO-GAS | 501 ± 101 | 6 ± 1 | 7325 ± 215 * | 1678 ± 112 * | 8 ± 3 | <5 a | 28 ± 4 |
| WT-GAS + sST2 | 489 ± 198 | 7 ± 2 | 7370 ± 550 * | 1860 ± 276 * | 9 ± 1 | <5 a | 21 ± 6 |
| IL-33-KO-GAS + rIL-33 | 798 ± 120 | <5 a | 4150 ± 621 | 813 ± 113 | 8 ± 2 | 6 ± 2 | 68 ± 13 |
| Treatment | CD3 | CD19 | NK1.1 | Gr1 | CD11b (%) |
|---|---|---|---|---|---|
| WT-GAS | 0.38 ± 0.01 | 3.93 ± 0.3 | 0.24 ± 0.03 | 90 ± 5.2 | 95 ± 1.0 |
| IL-33-KO-GAS | 0.42 ± 0.03 | 3.71 ± 2.4 | 0.70 ± 0.10 | 89 ± 5.1 | 93 ± 2.8 |
| ST2-KO-GAS | 0.20 ± 0.03 | 3.10 ± 0.3 | 0.15 ± 0.02 | 90 ± 3.5 | 93 ± 4.2 |
| WT-GAS + sST2 | 0.10 ± 0.05 | 2.94 ± 0.7 | 0.10 ± 0.10 | 88 ± 5.2 | 96 ± 2.1 |
| IL-33-KO-GAS + rIL-33 | 0.28 ± 0.05 | 3.53 ± 0.4 | 0.33 ± 0.10 | 93 ± 3.4 | 96 ± 2.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuo, C.-F.; Chen, W.-Y.; Yu, H.-H.; Tsai, Y.-H.; Chang, Y.-C.; Chang, C.-P.; Tsao, N. IL-33/ST2 Axis Plays a Protective Effect in Streptococcus pyogenes Infection through Strengthening of the Innate Immunity. Int. J. Mol. Sci. 2021, 22, 10566. https://doi.org/10.3390/ijms221910566
Kuo C-F, Chen W-Y, Yu H-H, Tsai Y-H, Chang Y-C, Chang C-P, Tsao N. IL-33/ST2 Axis Plays a Protective Effect in Streptococcus pyogenes Infection through Strengthening of the Innate Immunity. International Journal of Molecular Sciences. 2021; 22(19):10566. https://doi.org/10.3390/ijms221910566
Chicago/Turabian StyleKuo, Chih-Feng, Wei-Yu Chen, Hai-Han Yu, Yu-Hsuan Tsai, Ya-Chu Chang, Chih-Peng Chang, and Nina Tsao. 2021. "IL-33/ST2 Axis Plays a Protective Effect in Streptococcus pyogenes Infection through Strengthening of the Innate Immunity" International Journal of Molecular Sciences 22, no. 19: 10566. https://doi.org/10.3390/ijms221910566
APA StyleKuo, C.-F., Chen, W.-Y., Yu, H.-H., Tsai, Y.-H., Chang, Y.-C., Chang, C.-P., & Tsao, N. (2021). IL-33/ST2 Axis Plays a Protective Effect in Streptococcus pyogenes Infection through Strengthening of the Innate Immunity. International Journal of Molecular Sciences, 22(19), 10566. https://doi.org/10.3390/ijms221910566