
Co-Exposure to Aristolochic Acids I and II Increases DNA Adduct Formation Responsible for Aristolochic Acid I-Mediated Carcinogenicity in Rats


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
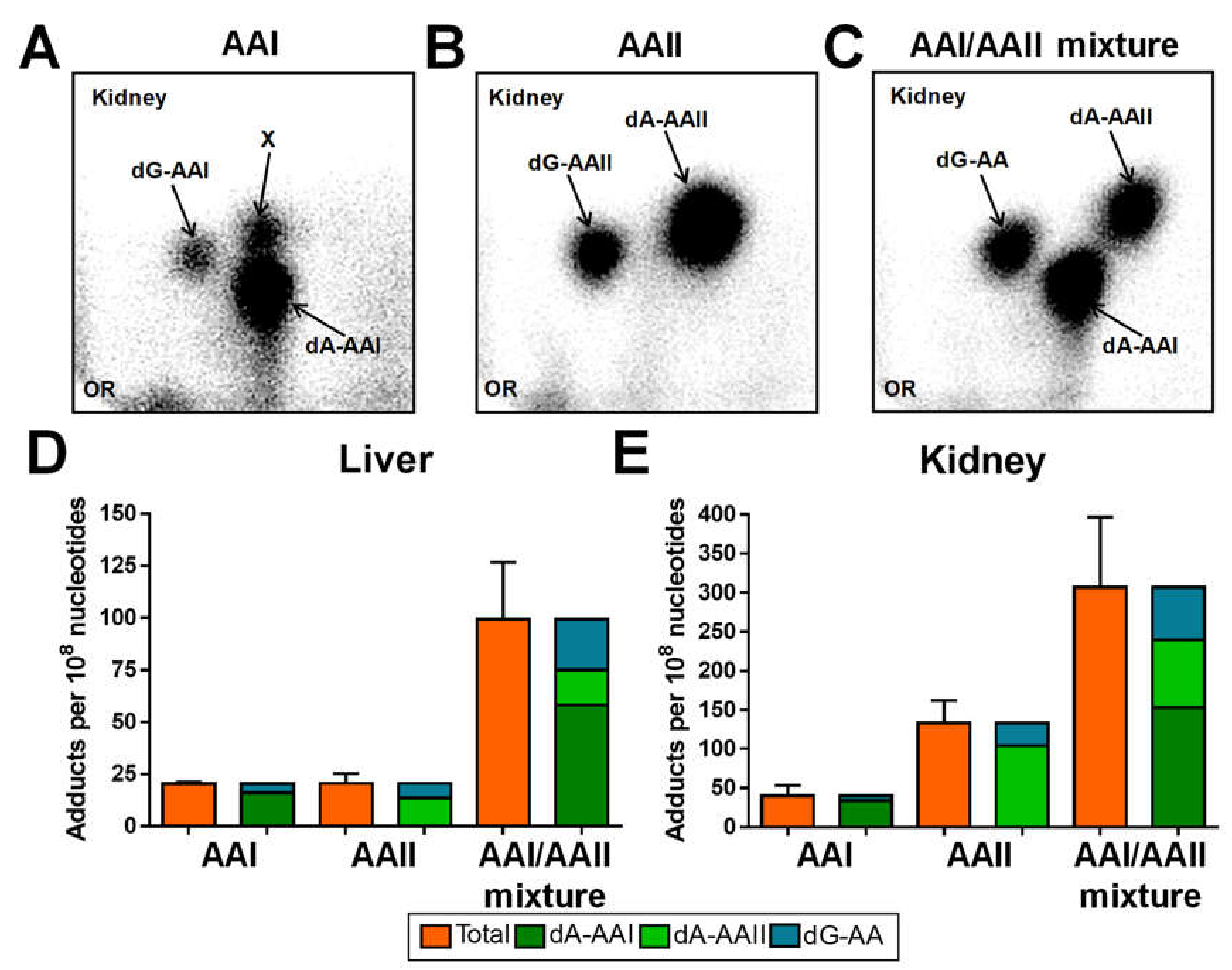
2.1. DNA Adduct Formation in Rats Treated with AAI, AAII or AAI/AAII Mixture
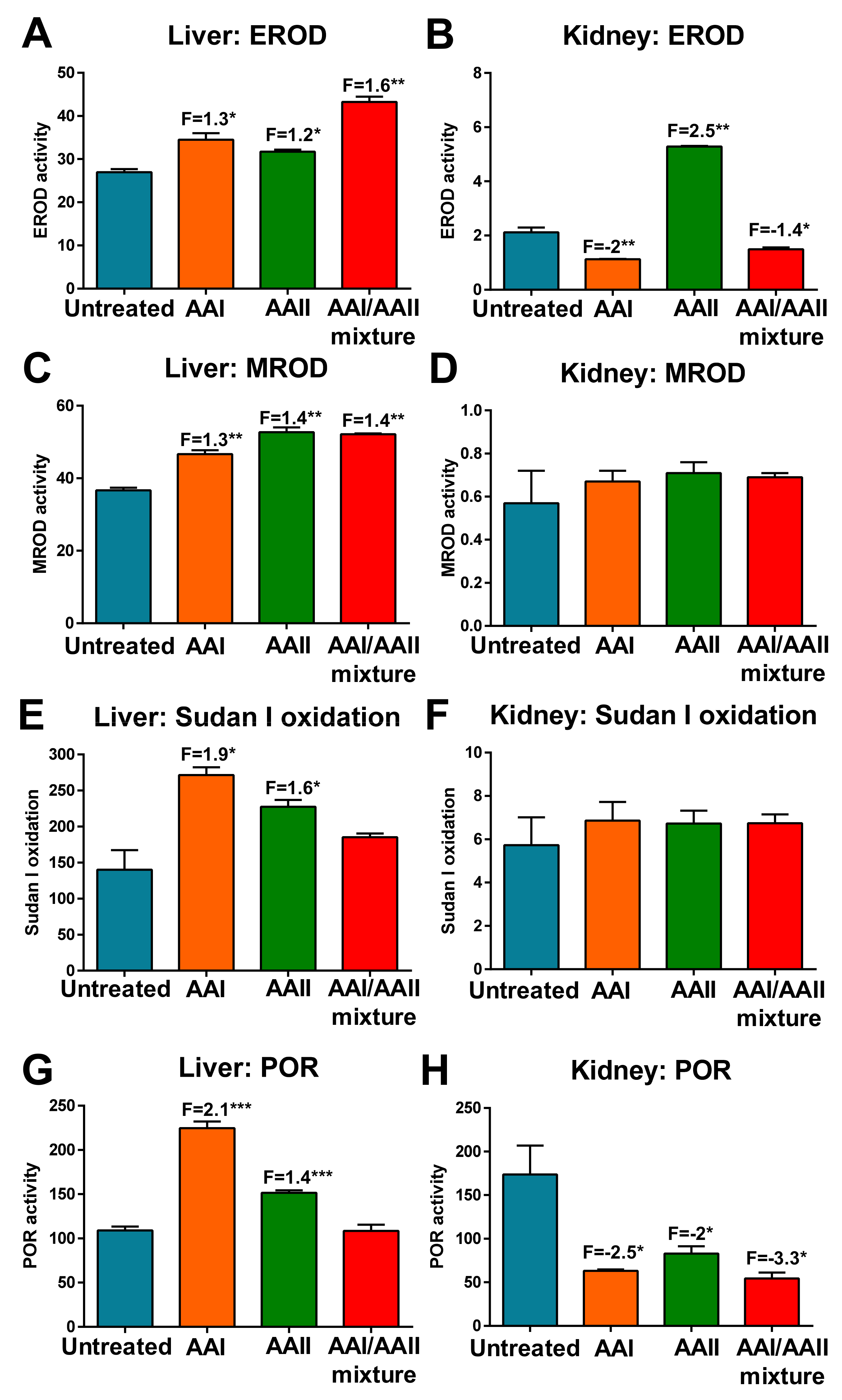
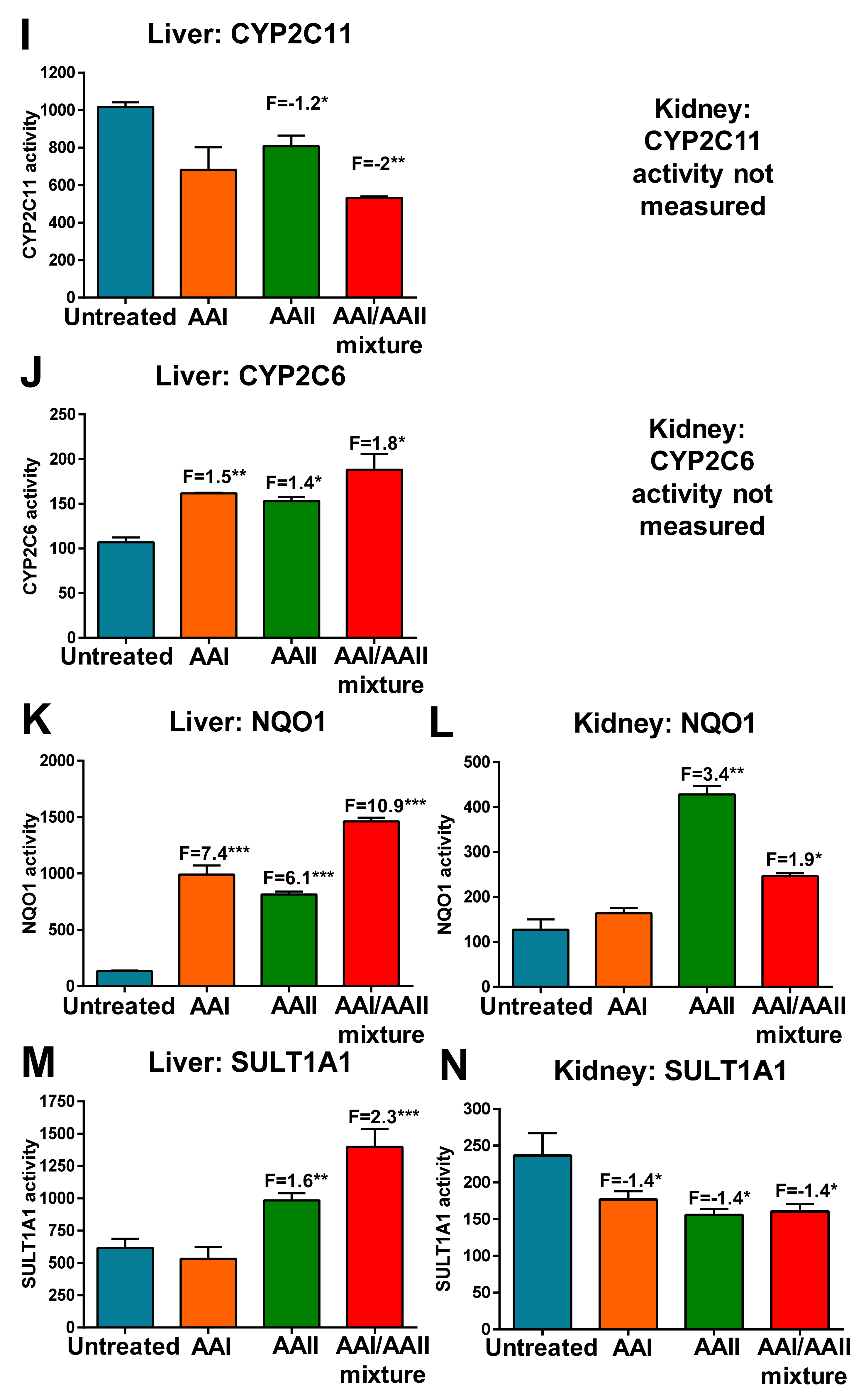
2.2. Activity Levels of Biotransformation Enzymes in the Liver and Kidneys of Rats Treated with AAI, AAII or AAI/AAII Mixture
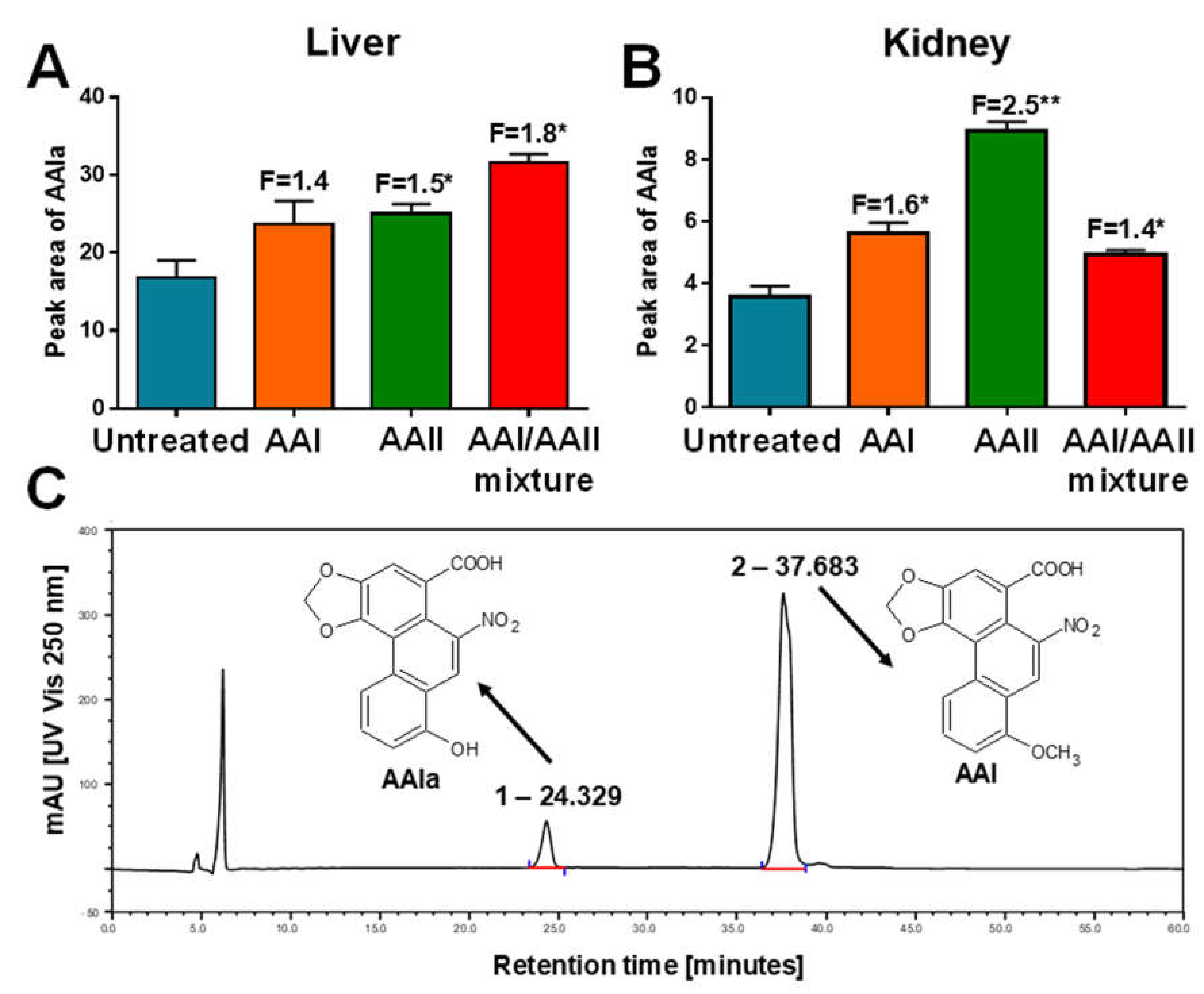
2.3. AAIa Formation in Hepatic and Renal Microsomes Isolated from Rats Treated with AAI, AAII or AAI/AAII Mixture
2.4. DNA Adduct Formation by AAI and AAII Ex Vivo Involving Incubation with Hepatic and Renal Microsomal and Cytosolic Fractions Isolated from Rats Treated with AAI, AAII or AAI/AAII Mixture
3. Discussion
4. Materials and Methods
4.1. Carcinogens
4.2. Animal Treatment and Sample Preparation
4.3. DNA Adduct Analysis by 32P-Postlabelling
4.4. Enzyme Activity Assays
4.5. Microsomal Incubation to Study AAI Demethylation
4.6. Cytosolic and Microsomal Formation of AAI- and AAII-Derived DNA Adducts
4.7. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heinrich, M.; Chan, J.; Wanke, S.; Neinhuis, C.; Simmonds, M.S.J. Local Uses of Aristolochia Species and Content of Nephrotoxic Aristolochic Acid 1 and 2-A Global Assessment Based on Bibliographic Sources. J. Ethnopharmacol. 2009, 125, 108–144. [Google Scholar] [CrossRef]
- Arlt, V.M.; Stiborova, M.; Schmeiser, H.H. Aristolochic Acid as a Probable Human Cancer Hazard in Herbal Remedies: A Review. Mutagenesis 2002, 17, 265–277. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Kucab, J.E.; Morganella, S.; Glodzik, D.; Alexandrov, L.B.; Arlt, V.M.; Weninger, A.; Hollstein, M.; Stratton, M.R.; Phillips, D.H. The Genome as a Record of Environmental Exposure. Mutagenesis 2015, 30, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Kucab, J.E.; Zou, X.; Morganella, S.; Joel, M.; Nanda, A.S.; Nagy, E.; Gomez, C.; Degasperi, A.; Harris, R.; Jackson, S.P.; et al. A Compendium of Mutational Signatures of Environmental Agents. Cell 2019, 177, 821–836.e16. [Google Scholar] [CrossRef]
- Matsumura, S.; Sato, H.; Otsubo, Y.; Tasaki, J.; Ikeda, N.; Morita, O. Genome-Wide Somatic Mutation Analysis via Hawk-SeqTM Reveals Mutation Profiles Associated with Chemical Mutagens. Arch. Toxicol. 2019, 93, 2689–2701. [Google Scholar] [CrossRef] [PubMed]
- Elhajouji, A.; Hove, T.T.; O’Connell, O.; Martus, H.; Dertinger, S.D. Pig-a Gene Mutation Assay Study Design: Critical Assessment of 3- versus 28-Day Repeat-Dose Treatment Schedules. Mutagenesis 2020, 35, 349–358. [Google Scholar] [CrossRef]
- Volkova, N.V.; Meier, B.; González-Huici, V.; Bertolini, S.; Gonzalez, S.; Vöhringer, H.; Abascal, F.; Martincorena, I.; Campbell, P.J.; Gartner, A.; et al. Mutational Signatures Are Jointly Shaped by DNA Damage and Repair. Nat. Commun. 2020, 11, 2169. [Google Scholar] [CrossRef]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans Pharmaceuticals. A Review of Human Carcinogens. IARC Monogr. Eval. Carcinog. Risks Hum. 2012, 100, 1–401. [Google Scholar]
- Gökmen, M.R.; Cosyns, J.-P.; Arlt, V.M.; Stiborová, M.; Phillips, D.H.; Schmeiser, H.H.; Simmonds, M.S.J.; Cook, H.T.; Vanherweghem, J.-L.; Nortier, J.L.; et al. The Epidemiology, Diagnosis, and Management of Aristolochic Acid Nephropathy: A Narrative Review. Ann. Intern. Med. 2013, 158, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Grollman, A.P. Aristolochic Acid Nephropathy: Harbinger of a Global Iatrogenic Disease. Environ. Mol. Mutagen. 2013, 54, 1–7. [Google Scholar] [CrossRef]
- Debelle, F.D.; Vanherweghem, J.-L.; Nortier, J.L. Aristolochic Acid Nephropathy: A Worldwide Problem. Kidney Int. 2008, 74, 158–169. [Google Scholar] [CrossRef]
- Arlt, V.M.; Stiborová, M.; vom Brocke, J.; Simões, M.L.; Lord, G.M.; Nortier, J.L.; Hollstein, M.; Phillips, D.H.; Schmeiser, H.H. Aristolochic Acid Mutagenesis: Molecular Clues to the Aetiology of Balkan Endemic Nephropathy-Associated Urothelial Cancer. Carcinogenesis 2007, 28, 2253–2261. [Google Scholar] [CrossRef] [PubMed]
- Grollman, A.P.; Shibutani, S.; Moriya, M.; Miller, F.; Wu, L.; Moll, U.; Suzuki, N.; Fernandes, A.; Rosenquist, T.; Medverec, Z.; et al. Aristolochic Acid and the Etiology of Endemic (Balkan) Nephropathy. Proc. Natl. Acad. Sci. USA 2007, 104, 12129–12134. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Frei, E.; Arlt, V.M.; Schmeiser, H.H. Metabolic Activation of Carcinogenic Aristolochic Acid, a Risk Factor for Balkan Endemic Nephropathy. Mutat. Res. 2008, 658, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Arlt, V.M.; Schmeiser, H.H. Balkan Endemic Nephropathy: An Update on Its Aetiology. Arch. Toxicol. 2016, 90, 2595–2615. [Google Scholar] [CrossRef] [PubMed]
- Schmeiser, H.H.; Kucab, J.E.; Arlt, V.M.; Phillips, D.H.; Hollstein, M.; Gluhovschi, G.; Gluhovschi, C.; Modilca, M.; Daminescu, L.; Petrica, L.; et al. Evidence of Exposure to Aristolochic Acid in Patients with Urothelial Cancer from a Balkan Endemic Nephropathy Region of Romania. Environ. Mol. Mutagen. 2012, 53, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Jelaković, B.; Dika, Ž.; Arlt, V.M.; Stiborova, M.; Pavlović, N.M.; Nikolić, J.; Colet, J.-M.; Vanherweghem, J.-L.; Nortier, J.L. Balkan Endemic Nephropathy and the Causative Role of Aristolochic Acid. Semin. Nephrol. 2019, 39, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Shibutani, S.; Dong, H.; Suzuki, N.; Ueda, S.; Miller, F.; Grollman, A.P. Selective Toxicity of Aristolochic Acids I and II. Drug Metab. Dispos. Biol. Fate Chem. 2007, 35, 1217–1222. [Google Scholar] [CrossRef]
- Stiborová, M.; Frei, E.; Schmeiser, H.H. Biotransformation Enzymes in Development of Renal Injury and Urothelial Cancer Caused by Aristolochic Acid. Kidney Int. 2008, 73, 1209–1211. [Google Scholar] [CrossRef]
- Stiborová, M.; Martínek, V.; Frei, E.; Arlt, V.M.; Schmeiser, H.H. Enzymes Metabolizing Aristolochic Acid and Their Contribution to the Development of Aristolochic Acid Nephropathy and Urothelial Cancer. Curr. Drug Metab. 2013, 14, 695–705. [Google Scholar] [CrossRef]
- Stiborová, M.; Frei, E.; Schmeiser, H.H.; Arlt, V.M.; Martínek, V. Mechanisms of Enzyme-Catalyzed Reduction of Two Carcinogenic Nitro-Aromatics, 3-Nitrobenzanthrone and Aristolochic Acid I: Experimental and Theoretical Approaches. Int. J. Mol. Sci. 2014, 15, 10271–10295. [Google Scholar] [CrossRef]
- Stiborová, M.; Frei, E.; Arlt, V.M.; Schmeiser, H.H. Knockout and Humanized Mice as Suitable Tools to Identify Enzymes Metabolizing the Human Carcinogen Aristolochic Acid. Xenobiotica Fate Foreign Compd. Biol. Syst. 2014, 44, 135–145. [Google Scholar] [CrossRef]
- Pfau, W.; Schmeiser, H.H.; Wiessler, M. Aristolochic Acid Binds Covalently to the Exocyclic Amino Group of Purine Nucleotides in DNA. Carcinogenesis 1990, 11, 313–319. [Google Scholar] [CrossRef]
- Pfau, W.; Schmeiser, H.H.; Wiessler, M. N6-Adenyl Arylation of DNA by Aristolochic Acid II and a Synthetic Model for the Putative Proximate Carcinogen. Chem. Res. Toxicol. 1991, 4, 581–586. [Google Scholar] [CrossRef]
- Stiborová, M.; Fernando, R.C.; Schmeiser, H.H.; Frei, E.; Pfau, W.; Wiessler, M. Characterization of DNA Adducts Formed by Aristolochic Acids in the Target Organ (Forestomach) of Rats by 32P-Postlabelling Analysis Using Different Chromatographic Procedures. Carcinogenesis 1994, 15, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Frei, E.; Breuer, A.; Wiessler, M.; Schmeiser, H.H. Evidence for Reductive Activation of Carcinogenic Aristolochic Acids by Prostaglandin H Synthase—32P-Postlabeling Analysis of DNA Adduct Formation. Mutat. Res. 2001, 493, 149–160. [Google Scholar] [CrossRef]
- Stiborová, M.; Frei, E.; Wiessler, M.; Schmeiser, H.H. Human Enzymes Involved in the Metabolic Activation of Carcinogenic Aristolochic Acids: Evidence for Reductive Activation by Cytochromes P450 1A1 and 1A2. Chem. Res. Toxicol. 2001, 14, 1128–1137. [Google Scholar] [CrossRef]
- Stiborová, M.; Hájek, M.; Frei, E.; Schmeiser, H.H. Carcinogenic and Nephrotoxic Alkaloids Aristolochic Acids upon Activation by NADPH: Cytochrome P450 Reductase Form Adducts Found in DNA of Patients with Chinese Herbs Nephropathy. Gen. Physiol. Biophys. 2001, 20, 375–392. [Google Scholar]
- Schmeiser, H.H.; Frei, E.; Wiessler, M.; Stiborova, M. Comparison of DNA Adduct Formation by Aristolochic Acids in Various in Vitro Activation Systems by 32P-Post-Labelling: Evidence for Reductive Activation by Peroxidases. Carcinogenesis 1997, 18, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Martinek, V.; Kubickova, B.; Arlt, V.M.; Frei, E.; Schmeiser, H.H.; Hudecek, J.; Stiborova, M. Comparison of Activation of Aristolochic Acid I and II with NADPH:Quinone Oxidoreductase, Sulphotransferases and N-Acetyltranferases. Neuro Endocrinol. Lett. 2011, 32 (Suppl. S1), 57–70. [Google Scholar] [PubMed]
- Schmeiser, H.H.; Bieler, C.A.; Wiessler, M.; van Ypersele de Strihou, C.; Cosyns, J.P. Detection of DNA Adducts Formed by Aristolochic Acid in Renal Tissue from Patients with Chinese Herbs Nephropathy. Cancer Res. 1996, 56, 2025–2028. [Google Scholar]
- Schmeiser, H.H.; Nortier, J.L.; Singh, R.; Gamboa da Costa, G.; Sennesael, J.; Cassuto-Viguier, E.; Ambrosetti, D.; Rorive, S.; Pozdzik, A.; Phillips, D.H.; et al. Exceptionally Long-Term Persistence of DNA Adducts Formed by Carcinogenic Aristolochic Acid I in Renal Tissue from Patients with Aristolochic Acid Nephropathy. Int. J. Cancer 2014, 135, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Bieler, C.A.; Stiborova, M.; Wiessler, M.; Cosyns, J.P.; van Ypersele de Strihou, C.; Schmeiser, H.H. 32P-Post-Labelling Analysis of DNA Adducts Formed by Aristolochic Acid in Tissues from Patients with Chinese Herbs Nephropathy. Carcinogenesis 1997, 18, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Nortier, J.L.; Martinez, M.C.; Schmeiser, H.H.; Arlt, V.M.; Bieler, C.A.; Petein, M.; Depierreux, M.F.; De Pauw, L.; Abramowicz, D.; Vereerstraeten, P.; et al. Urothelial Carcinoma Associated with the Use of a Chinese Herb (Aristolochia Fangchi). N. Engl. J. Med. 2000, 342, 1686–1692. [Google Scholar] [CrossRef] [PubMed]
- Arlt, V.M.; Pfohl-Leszkowicz, A.; Cosyns, J.; Schmeiser, H.H. Analyses of DNA Adducts Formed by Ochratoxin A and Aristolochic Acid in Patients with Chinese Herbs Nephropathy. Mutat. Res. 2001, 494, 143–150. [Google Scholar] [CrossRef]
- Arlt, V.M.; Ferluga, D.; Stiborova, M.; Pfohl-Leszkowicz, A.; Vukelic, M.; Ceovic, S.; Schmeiser, H.H.; Cosyns, J.-P. Is Aristolochic Acid a Risk Factor for Balkan Endemic Nephropathy-Associated Urothelial Cancer? Int. J. Cancer 2002, 101, 500–502. [Google Scholar] [CrossRef]
- Jelaković, B.; Karanović, S.; Vuković-Lela, I.; Miller, F.; Edwards, K.L.; Nikolić, J.; Tomić, K.; Slade, N.; Brdar, B.; Turesky, R.J.; et al. Aristolactam-DNA Adducts Are a Biomarker of Environmental Exposure to Aristolochic Acid. Kidney Int. 2012, 81, 559–567. [Google Scholar] [CrossRef]
- Lord, G.M.; Hollstein, M.; Arlt, V.M.; Roufosse, C.; Pusey, C.D.; Cook, T.; Schmeiser, H.H. DNA Adducts and P53 Mutations in a Patient with Aristolochic Acid-Associated Nephropathy. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2004, 43, e11–e17. [Google Scholar] [CrossRef]
- Chen, C.-H.; Dickman, K.G.; Moriya, M.; Zavadil, J.; Sidorenko, V.S.; Edwards, K.L.; Gnatenko, D.V.; Wu, L.; Turesky, R.J.; Wu, X.-R.; et al. Aristolochic Acid-Associated Urothelial Cancer in Taiwan. Proc. Natl. Acad. Sci. USA 2012, 109, 8241–8246. [Google Scholar] [CrossRef]
- Hoang, M.L.; Chen, C.-H.; Sidorenko, V.S.; He, J.; Dickman, K.G.; Yun, B.H.; Moriya, M.; Niknafs, N.; Douville, C.; Karchin, R.; et al. Mutational Signature of Aristolochic Acid Exposure as Revealed by Whole-Exome Sequencing. Sci. Transl. Med. 2013, 5, 197ra102. [Google Scholar] [CrossRef]
- Poon, S.L.; Pang, S.-T.; McPherson, J.R.; Yu, W.; Huang, K.K.; Guan, P.; Weng, W.-H.; Siew, E.Y.; Liu, Y.; Heng, H.L.; et al. Genome-Wide Mutational Signatures of Aristolochic Acid and Its Application as a Screening Tool. Sci. Transl. Med. 2013, 5, 197ra101. [Google Scholar] [CrossRef] [PubMed]
- Poon, S.L.; Huang, M.N.; Choo, Y.; McPherson, J.R.; Yu, W.; Heng, H.L.; Gan, A.; Myint, S.S.; Siew, E.Y.; Ler, L.D.; et al. Mutation Signatures Implicate Aristolochic Acid in Bladder Cancer Development. Genome Med. 2015, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Schmeiser, H.H.; Schoepe, K.B.; Wiessler, M. DNA Adduct Formation of Aristolochic Acid I and II in Vitro and in Vivo. Carcinogenesis 1988, 9, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Arlt, V.M.; Meinl, W.; Florian, S.; Nagy, E.; Barta, F.; Thomann, M.; Mrizova, I.; Krais, A.M.; Liu, M.; Richards, M.; et al. Impact of Genetic Modulation of SULT1A Enzymes on DNA Adduct Formation by Aristolochic Acids and 3-Nitrobenzanthrone. Arch. Toxicol. 2017, 91, 1957–1975. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Frei, E.; Sopko, B.; Wiessler, M.; Schmeiser, H.H. Carcinogenic Aristolochic Acids upon Activation by DT-Diaphorase Form Adducts Found in DNA of Patients with Chinese Herbs Nephropathy. Carcinogenesis 2002, 23, 617–625. [Google Scholar] [CrossRef]
- Kumar, V.; Poonam; Prasad, A.K.; Parmar, V.S. Naturally Occurring Aristolactams, Aristolochic Acids and Dioxoaporphines and Their Biological Activities. Nat. Prod. Rep. 2003, 20, 565–583. [Google Scholar] [CrossRef]
- Michl, J.; Kite, G.C.; Wanke, S.; Zierau, O.; Vollmer, G.; Neinhuis, C.; Simmonds, M.S.J.; Heinrich, M. LC-MS- and (1)H NMR-Based Metabolomic Analysis and in Vitro Toxicological Assessment of 43 Aristolochia Species. J. Nat. Prod. 2016, 79, 30–37. [Google Scholar] [CrossRef]
- Dedíková, A.; Bárta, F.; Martínek, V.; Kotalík, K.; Dušková, Š.; Mráz, J.; Arlt, V.M.; Stiborová, M.; Hodek, P. In Vivo Metabolism of Aristolochic Acid I and II in Rats Is Influenced by Their Coexposure. Chem. Res. Toxicol. 2020, 33, 2804–2818. [Google Scholar] [CrossRef]
- Martínek, V.; Bárta, F.; Hodek, P.; Frei, E.; Schmeiser, H.H.; Arlt, V.M.; Stiborová, M. Comparison of the Oxidation of Carcinogenic Aristolochic Acid I and II by Microsomal Cytochromes P450 in Vitro: Experimental and Theoretical Approaches. Monatshefte Chem. 2017, 148, 1971–1981. [Google Scholar] [CrossRef]
- Rendic, S.; Di Carlo, F.J. Human Cytochrome P450 Enzymes: A Status Report Summarizing Their Reactions, Substrates, Inducers, and Inhibitors. Drug Metab. Rev. 1997, 29, 413–580. [Google Scholar] [CrossRef]
- Stiborová, M.; Frei, E.; Hodek, P.; Wiessler, M.; Schmeiser, H.H. Human Hepatic and Renal Microsomes, Cytochromes P450 1A1/2, NADPH:Cytochrome P450 Reductase and Prostaglandin H Synthase Mediate the Formation of Aristolochic Acid-DNA Adducts Found in Patients with Urothelial Cancer. Int. J. Cancer 2005, 113, 189–197. [Google Scholar] [CrossRef]
- Stiborová, M.; Bárta, F.; Levová, K.; Hodek, P.; Schmeiser, H.H.; Arlt, V.M.; Martínek, V. A Mechanism of O-Demethylation of Aristolochic Acid I by Cytochromes P450 and Their Contributions to This Reaction in Human and Rat Livers: Experimental and Theoretical Approaches. Int. J. Mol. Sci. 2015, 16, 27561–27575. [Google Scholar] [CrossRef]
- Ban, T.H.; Min, J.-W.; Seo, C.; Kim, D.R.; Lee, Y.H.; Chung, B.H.; Jeong, K.-H.; Lee, J.W.; Kim, B.S.; Lee, S.-H.; et al. Update of Aristolochic Acid Nephropathy in Korea. Korean J. Intern. Med. 2018, 33, 961–969. [Google Scholar] [CrossRef]
- Chen, C.-J.; Yang, Y.-H.; Lin, M.-H.; Lee, C.-P.; Tsan, Y.-T.; Lai, M.-N.; Yang, H.-Y.; Ho, W.-C.; Chen, P.-C.; Health Data Analysis in Taiwan (hDATa) Research Group. Herbal Medicine Containing Aristolochic Acid and the Risk of Hepatocellular Carcinoma in Patients with Hepatitis B Virus Infection. Int. J. Cancer 2018, 143, 1578–1587. [Google Scholar] [CrossRef] [PubMed]
- Hoang, M.L.; Chen, C.-H.; Chen, P.-C.; Roberts, N.J.; Dickman, K.G.; Yun, B.H.; Turesky, R.J.; Pu, Y.-S.; Vogelstein, B.; Papadopoulos, N.; et al. Aristolochic Acid in the Etiology of Renal Cell Carcinoma. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 2016, 25, 1600–1608. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, T.; Wu, J.; Wen, J.; Tao, D.; Wan, T.; Zhu, W. Prognosis and Risk Factors of Patients with Upper Urinary Tract Urothelial Carcinoma and Postoperative Recurrence of Bladder Cancer in Central China. BMC Urol. 2019, 19, 24. [Google Scholar] [CrossRef]
- Zhong, W.; Zhang, L.; Ma, J.; Shao, S.; Lin, R.; Li, X.; Xiong, G.; Fang, D.; Zhou, L. Impact of Aristolochic Acid Exposure on Oncologic Outcomes of Upper Tract Urothelial Carcinoma after Radical Nephroureterectomy. OncoTargets Ther. 2017, 10, 5775–5782. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.L.; Ohyama, C.; Hatakeyama, S.; Wang, H.J.; Yoneyama, T.; Yang, W.C.; Chuang, Y.C.; Chen, Y.T.; Lee, W.C.; Cheng, Y.T.; et al. Unusual Presentation of Upper Urinary Tract Urothelial Carcinoma in Taiwan: Direct Comparison from Taiwan-Japan UTUC Collaboration Cohort. Int. J. Urol. Off. J. Jpn. Urol. Assoc. 2020, 27, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-H.; Dickman, K.G.; Huang, C.-Y.; Shun, C.-T.; Tai, H.-C.; Huang, K.-H.; Wang, S.-M.; Lee, Y.-J.; Grollman, A.P.; Pu, Y.-S. Recurrence Pattern and TP53 Mutation in Upper Urinary Tract Urothelial Carcinoma. Oncotarget 2016, 7, 45225–45236. [Google Scholar] [CrossRef] [PubMed]
- Dračínská, H.; Bárta, F.; Levová, K.; Hudecová, A.; Moserová, M.; Schmeiser, H.H.; Kopka, K.; Frei, E.; Arlt, V.M.; Stiborová, M. Induction of Cytochromes P450 1A1 and 1A2 Suppresses Formation of DNA Adducts by Carcinogenic Aristolochic Acid I in Rats in Vivo. Toxicology 2016, 344–346, 7–18. [Google Scholar] [CrossRef]
- Stiborová, M.; Levová, K.; Bárta, F.; Šulc, M.; Frei, E.; Arlt, V.M.; Schmeiser, H.H. The Influence of Dicoumarol on the Bioactivation of the Carcinogen Aristolochic Acid I in Rats. Mutagenesis 2014, 29, 189–200. [Google Scholar] [CrossRef]
- Stiborová, M.; Bárta, F.; Levová, K.; Hodek, P.; Frei, E.; Arlt, V.M.; Schmeiser, H.H. The Influence of Ochratoxin A on DNA Adduct Formation by the Carcinogen Aristolochic Acid in Rats. Arch. Toxicol. 2015, 89, 2141–2158. [Google Scholar] [CrossRef]
- Xing, G.; Qi, X.; Chen, M.; Wu, Y.; Yao, J.; Gong, L.; Nohmi, T.; Luan, Y.; Ren, J. Comparison of the Mutagenicity of Aristolochic Acid I and Aristolochic Acid II in the Gpt Delta Transgenic Mouse Kidney. Mutat. Res. 2012, 743, 52–58. [Google Scholar] [CrossRef]
- Bárta, F.; Levová, K.; Frei, E.; Schmeiser, H.H.; Arlt, V.M.; Stiborová, M. The Effect of Aristolochic Acid I on Expression of NAD(P)H:Quinone Oxidoreductase in Mice and Rats--a Comparative Study. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2014, 768, 1–7. [Google Scholar] [CrossRef]
- Chen, M.; Gong, L.; Qi, X.; Xing, G.; Luan, Y.; Wu, Y.; Xiao, Y.; Yao, J.; Li, Y.; Xue, X.; et al. Inhibition of Renal NQO1 Activity by Dicoumarol Suppresses Nitroreduction of Aristolochic Acid I and Attenuates Its Nephrotoxicity. Toxicol. Sci. Off. J. Soc. Toxicol. 2011, 122, 288–296. [Google Scholar] [CrossRef]
- Sborchia, M.; De Prez, E.G.; Antoine, M.-H.; Bienfait, L.; Indra, R.; Valbuena, G.; Phillips, D.H.; Nortier, J.L.; Stiborová, M.; Keun, H.C.; et al. The Impact of P53 on Aristolochic Acid I-Induced Nephrotoxicity and DNA Damage in Vivo and in Vitro. Arch. Toxicol. 2019, 93, 3345–3366. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Talalay, P. NAD(P)H:Quinone Acceptor Oxidoreductase 1 (NQO1), a Multifunctional Antioxidant Enzyme and Exceptionally Versatile Cytoprotector. Arch. Biochem. Biophys. 2010, 501, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, A.K. Regulation of Genes Encoding NAD(P)H:Quinone Oxidoreductases. Free Radic. Biol. Med. 2000, 29, 254–262. [Google Scholar] [CrossRef]
- Ma, Q.; Kinneer, K.; Bi, Y.; Chan, J.Y.; Kan, Y.W. Induction of Murine NAD(P)H:Quinone Oxidoreductase by 2,3,7,8-Tetrachlorodibenzo-p-Dioxin Requires the CNC (Cap “n” Collar) Basic Leucine Zipper Transcription Factor Nrf2 (Nuclear Factor Erythroid 2-Related Factor 2): Cross-Interaction between AhR (Aryl Hydrocarbon Receptor) and Nrf2 Signal Transduction. Biochem. J. 2004, 377, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Dračínská, H.; Martínek, V.; Svášková, D.; Hodek, P.; Milichovský, J.; Hejduková, Ž.; Brotánek, J.; Schmeiser, H.H.; Frei, E. Induced Expression of Cytochrome P450 1A and NAD(P)H:Quinone Oxidoreductase Determined at MRNA, Protein, and Enzyme Activity Levels in Rats Exposed to the Carcinogenic Azo Dye 1-Phenylazo-2-Naphthol (Sudan I). Chem. Res. Toxicol. 2013, 26, 290–299. [Google Scholar] [CrossRef]
- Talalay, P.; Prochaska, H. Mechanisms of Induction of Nad(p)h—Quinone Reductase. Chem. Scr. 1987, 27A, 61–66. [Google Scholar]
- Wang, L.; He, X.; Szklarz, G.D.; Bi, Y.; Rojanasakul, Y.; Ma, Q. The Aryl Hydrocarbon Receptor Interacts with Nuclear Factor Erythroid 2-Related Factor 2 to Mediate Induction of NAD(P)H:Quinoneoxidoreductase 1 by 2,3,7,8-Tetrachlorodibenzo-p-Dioxin. Arch. Biochem. Biophys. 2013, 537, 31–38. [Google Scholar] [CrossRef]
- Ross, D.; Kepa, J.K.; Winski, S.L.; Beall, H.D.; Anwar, A.; Siegel, D. NAD(P)H:Quinone Oxidoreductase 1 (NQO1): Chemoprotection, Bioactivation, Gene Regulation and Genetic Polymorphisms. Chem. Biol. Interact. 2000, 129, 77–97. [Google Scholar] [CrossRef]
- Ross, D. Quinone Reductases Multitasking in the Metabolic World. Drug Metab. Rev. 2004, 36, 639–654. [Google Scholar] [CrossRef]
- Hajirahimkhan, A.; Simmler, C.; Dong, H.; Lantvit, D.D.; Li, G.; Chen, S.-N.; Nikolić, D.; Pauli, G.F.; van Breemen, R.B.; Dietz, B.M.; et al. Induction of NAD(P)H:Quinone Oxidoreductase 1 (NQO1) by Glycyrrhiza Species Used for Women’s Health: Differential Effects of the Michael Acceptors Isoliquiritigenin and Licochalcone A. Chem. Res. Toxicol. 2015, 28, 2130–2141. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.-Y.; Wu, T.-S.; Chen, T.-W.; Liu, B.-H. Aristolochic Acid I Induced Oxidative DNA Damage Associated with Glutathione Depletion and ERK1/2 Activation in Human Cells. Toxicol. Vitro Int. J. Publ. Assoc. BIBRA 2011, 25, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Romanov, V.; Whyard, T.C.; Waltzer, W.C.; Grollman, A.P.; Rosenquist, T. Aristolochic Acid-Induced Apoptosis and G2 Cell Cycle Arrest Depends on ROS Generation and MAP Kinases Activation. Arch. Toxicol. 2015, 89, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.-H.; Lee, Y.-T.; Hsieh, H.-S.; Hwang, D.-F. Short-Term Toxicity of Aristolochic Acid, Aristolochic Acid-I and Aristolochic Acid-II in Rats. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2008, 46, 1157–1163. [Google Scholar] [CrossRef] [PubMed]
- Levová, K.; Moserová, M.; Kotrbová, V.; Sulc, M.; Henderson, C.J.; Wolf, C.R.; Phillips, D.H.; Frei, E.; Schmeiser, H.H.; Mares, J.; et al. Role of Cytochromes P450 1A1/2 in Detoxication and Activation of Carcinogenic Aristolochic Acid I: Studies with the Hepatic NADPH:Cytochrome P450 Reductase Null (HRN) Mouse Model. Toxicol. Sci. Off. J. Soc. Toxicol. 2011, 121, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Sistkova, J.; Hudecek, J.; Hodek, P.; Frei, E.; Schmeiser, H.H.; Stiborova, M. Human Cytochromes P450 1A1 and 1A2 Participate in Detoxication of Carcinogenic Aristolochic Acid. Neuro Endocrinol. Lett. 2008, 29, 733–737. [Google Scholar]
- Stiborova, M.; Mares, J.; Levova, K.; Pavlickova, J.; Barta, F.; Hodek, P.; Frei, E.; Schmeiser, H.H. Role of Cytochromes P450 in Metabolism of Carcinogenic Aristolochic Acid I: Evidence of Their Contribution to Aristolochic Acid I Detoxication and Activation in Rat Liver. Neuro Endocrinol. Lett. 2011, 32 (Suppl. S1), 121–130. [Google Scholar]
- Stiborová, M.; Levová, K.; Bárta, F.; Shi, Z.; Frei, E.; Schmeiser, H.H.; Nebert, D.W.; Phillips, D.H.; Arlt, V.M. Bioactivation versus Detoxication of the Urothelial Carcinogen Aristolochic Acid I by Human Cytochrome P450 1A1 and 1A2. Toxicol. Sci. Off. J. Soc. Toxicol. 2012, 125, 345–358. [Google Scholar] [CrossRef]
- Rosenquist, T.A.; Einolf, H.J.; Dickman, K.G.; Wang, L.; Smith, A.; Grollman, A.P. Cytochrome P450 1A2 Detoxicates Aristolochic Acid in the Mouse. Drug Metab. Dispos. Biol. Fate Chem. 2010, 38, 761–768. [Google Scholar] [CrossRef]
- Xiao, Y.; Ge, M.; Xue, X.; Wang, C.; Wang, H.; Wu, X.; Li, L.; Liu, L.; Qi, X.; Zhang, Y.; et al. Hepatic Cytochrome P450s Metabolize Aristolochic Acid and Reduce Its Kidney Toxicity. Kidney Int. 2008, 73, 1231–1239. [Google Scholar] [CrossRef]
- Xue, X.; Xiao, Y.; Zhu, H.; Wang, H.; Liu, Y.; Xie, T.; Ren, J. Induction of P450 1A by 3-Methylcholanthrene Protects Mice from Aristolochic Acid-I-Induced Acute Renal Injury. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2008, 23, 3074–3081. [Google Scholar] [CrossRef] [PubMed]
- Arlt, V.M.; Levová, K.; Bárta, F.; Shi, Z.; Evans, J.D.; Frei, E.; Schmeiser, H.H.; Nebert, D.W.; Phillips, D.H.; Stiborová, M. Role of P450 1A1 and P450 1A2 in Bioactivation versus Detoxication of the Renal Carcinogen Aristolochic Acid I: Studies in Cyp1a1-/-, Cyp1a2-/-, and Cyp1a1/1a2-/- Mice. Chem. Res. Toxicol. 2011, 24, 1710–1719. [Google Scholar] [CrossRef] [PubMed]
- Milichovský, J.; Bárta, F.; Schmeiser, H.H.; Arlt, V.M.; Frei, E.; Stiborová, M.; Martínek, V. Active Site Mutations as a Suitable Tool Contributing to Explain a Mechanism of Aristolochic Acid I Nitroreduction by Cytochromes P450 1A1, 1A2 and 1B1. Int. J. Mol. Sci. 2016, 17, 213. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, H.; Li, C.; Yi, Y.; Liu, J.; Zhao, Y.; Tian, J.; Zhang, Y.; Wei, X.; Gao, Y.; et al. Omeprazole Alleviates Aristolochia Manshuriensis Kom-Induced Acute Nephrotoxicity. PLoS ONE 2016, 11, e0164215. [Google Scholar] [CrossRef]
- Luan, Y.; Xing, G.; Ren, J.; Gu, J. Role of Hepatic Cytochrome P450 Enzymes in the Detoxication of Aristolochic Acid I; Effects on DNA Adduct, Mutation, and Tumor Formation. Genes Environ. Off. J. Jpn. Environ. Mutagen Soc. 2015, 37, 11. [Google Scholar] [CrossRef]
- Nedelcheva, V.; Gut, I. P450 in the Rat and Man: Methods of Investigation, Substrate Specificities and Relevance to Cancer. Xenobiotica Fate Foreign Compd. Biol. Syst. 1994, 24, 1151–1175. [Google Scholar] [CrossRef] [PubMed]
- Haduch, A.; Wójcikowski, J.; Daniel, W.A. Effect of Neuroleptics on Cytochrome P450 2C11 (CYP2C11) in Rat Liver. Pharmacol. Rep. 2011, 63, 1491–1499. [Google Scholar] [CrossRef]
- Večeřa, R.; Zachařová, A.; Orolin, J.; Strojil, J.; Skottová, N.; Anzenbacher, P. Fenofibrate-Induced Decrease of Expression of CYP2C11 and CYP2C6 in Rat. Biopharm. Drug Dispos. 2011, 32, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Zachařová, A.; Siller, M.; Spičáková, A.; Anzenbacherová, E.; Skottová, N.; Anzenbacher, P.; Večeřa, R. Rosuvastatin Suppresses the Liver Microsomal CYP2C11 and CYP2C6 Expression in Male Wistar Rats. Xenobiotica Fate Foreign Compd. Biol. Syst. 2012, 42, 731–736. [Google Scholar] [CrossRef]
- Goeptar, A.R.; Scheerens, H.; Vermeulen, N.P. Oxygen and Xenobiotic Reductase Activities of Cytochrome P450. Crit. Rev. Toxicol. 1995, 25, 25–65. [Google Scholar] [CrossRef] [PubMed]
- Hanukoglu, I. Electron Transfer Proteins of Cytochrome P450 Systems. Adv. Mol. Cell Biol. 1996, 14, 29–56. [Google Scholar] [CrossRef]
- Laursen, T.; Jensen, K.; Møller, B.L. Conformational Changes of the NADPH-Dependent Cytochrome P450 Reductase in the Course of Electron Transfer to Cytochromes P450. Biochim. Biophys. Acta 2011, 1814, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Pudney, C.R.; Khara, B.; Johannissen, L.O.; Scrutton, N.S. Coupled Motions Direct Electrons along Human Microsomal P450 Chains. PLoS Biol. 2011, 9, e1001222. [Google Scholar] [CrossRef]
- Jerabek, P.; Martinek, V.; Stiborova, M. Theoretical Investigation of Differences in Nitroreduction of Aristolochic Acid I by Cytochromes P450 1A1, 1A2 and 1B1. Neuro Endocrinol. Lett. 2012, 33 (Suppl. S3), 25–32. [Google Scholar]
- Levova, K.; Moserova, M.; Nebert, D.W.; Phillips, D.H.; Frei, E.; Schmeiser, H.H.; Arlt, V.M.; Stiborova, M. NAD(P)H:Quinone Oxidoreductase Expression in Cyp1a-Knockout and CYP1A-Humanized Mouse Lines and Its Effect on Bioactivation of the Carcinogen Aristolochic Acid I. Toxicol. Appl. Pharmacol. 2012, 265, 360–367. [Google Scholar] [CrossRef]
- Stiborová, M.; Stiborová-Rupertová, M.; Borek-Dohalská, L.; Wiessler, M.; Frei, E. Rat Microsomes Activating the Anticancer Drug Ellipticine to Species Covalently Binding to Deoxyguanosine in DNA Are a Suitable Model Mimicking Ellipticine Bioactivation in Humans. Chem. Res. Toxicol. 2003, 16, 38–47. [Google Scholar] [CrossRef]
- Stiborová, M.; Miksanová, M.; Sulc, M.; Rýdlová, H.; Schmeiser, H.H.; Frei, E. Identification of a Genotoxic Mechanism for the Carcinogenicity of the Environmental Pollutant and Suspected Human Carcinogen O-Anisidine. Int. J. Cancer 2005, 116, 667–678. [Google Scholar] [CrossRef]
- Schmeiser, H.H.; Stiborova, M.; Arlt, V.M. 32P-Postlabeling Analysis of DNA Adducts. Methods Mol. Biol. Clifton NJ 2013, 1044, 389–401. [Google Scholar] [CrossRef]
- Frame, L.T.; Ozawa, S.; Nowell, S.A.; Chou, H.C.; DeLongchamp, R.R.; Doerge, D.R.; Lang, N.P.; Kadlubar, F.F. A Simple Colorimetric Assay for Phenotyping the Major Human Thermostable Phenol Sulfotransferase (SULT1A1) Using Platelet Cytosols. Drug Metab. Dispos. Biol. Fate Chem. 2000, 28, 1063–1068. [Google Scholar] [PubMed]
- Krais, A.M.; Speksnijder, E.N.; Melis, J.P.M.; Singh, R.; Caldwell, A.; Gamboa da Costa, G.; Luijten, M.; Phillips, D.H.; Arlt, V.M. Metabolic Activation of 2-Amino-1-Methyl-6-Phenylimidazo [4,5-b]Pyridine and DNA Adduct Formation Depends on P53: Studies in Trp53(+/+),Trp53(+/−) and Trp53(−/−) Mice. Int. J. Cancer 2016, 138, 976–982. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.D.; Thompson, S.; Weaver, R.J.; Wolf, C.R.; Mayer, R.T. Cytochrome P450 Specificities of Alkoxyresorufin O-Dealkylation in Human and Rat Liver. Biochem. Pharmacol. 1994, 48, 923–936. [Google Scholar] [CrossRef]
- Stiborová, M.; Martínek, V.; Rýdlová, H.; Hodek, P.; Frei, E. Sudan I Is a Potential Carcinogen for Humans: Evidence for Its Metabolic Activation and Detoxication by Human Recombinant Cytochrome P450 1A1 and Liver Microsomes. Cancer Res. 2002, 62, 5678–5684. [Google Scholar] [PubMed]
- Arlt, V.M.; Stiborova, M.; Hewer, A.; Schmeiser, H.H.; Phillips, D.H. Human Enzymes Involved in the Metabolic Activation of the Environmental Contaminant 3-Nitrobenzanthrone: Evidence for Reductive Activation by Human NADPH:Cytochrome P450 Reductase. Cancer Res. 2003, 63, 2752–2761. [Google Scholar]
- Kobayashi, K.; Urashima, K.; Shimada, N.; Chiba, K. Substrate Specificity for Rat Cytochrome P450 (CYP) Isoforms: Screening with CDNA-Expressed Systems of the Rat. Biochem. Pharmacol. 2002, 63, 889–896. [Google Scholar] [CrossRef]
- Yamazaki, H.; Shimizu, M.; Nagashima, T.; Minoshima, M.; Murayama, N. Rat Cytochrome P450 2C11 in Liver Microsomes Involved in Oxidation of Anesthetic Agent Propofol and Deactivated by Prior Treatment with Propofol. Drug Metab. Dispos. Biol. Fate Chem. 2006, 34, 1803–1805. [Google Scholar] [CrossRef]
- Stiborová, M.; Frei, E.; Sopko, B.; Sopková, K.; Marková, V.; Lanková, M.; Kumstýrová, T.; Wiessler, M.; Schmeiser, H.H. Human Cytosolic Enzymes Involved in the Metabolic Activation of Carcinogenic Aristolochic Acid: Evidence for Reductive Activation by Human NAD(P)H:Quinone Oxidoreductase. Carcinogenesis 2003, 24, 1695–1703. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bárta, F.; Dedíková, A.; Bebová, M.; Dušková, Š.; Mráz, J.; Schmeiser, H.H.; Arlt, V.M.; Hodek, P.; Stiborová, M. Co-Exposure to Aristolochic Acids I and II Increases DNA Adduct Formation Responsible for Aristolochic Acid I-Mediated Carcinogenicity in Rats. Int. J. Mol. Sci. 2021, 22, 10479. https://doi.org/10.3390/ijms221910479
Bárta F, Dedíková A, Bebová M, Dušková Š, Mráz J, Schmeiser HH, Arlt VM, Hodek P, Stiborová M. Co-Exposure to Aristolochic Acids I and II Increases DNA Adduct Formation Responsible for Aristolochic Acid I-Mediated Carcinogenicity in Rats. International Journal of Molecular Sciences. 2021; 22(19):10479. https://doi.org/10.3390/ijms221910479
Chicago/Turabian StyleBárta, František, Alena Dedíková, Michaela Bebová, Šárka Dušková, Jaroslav Mráz, Heinz H. Schmeiser, Volker M. Arlt, Petr Hodek, and Marie Stiborová. 2021. "Co-Exposure to Aristolochic Acids I and II Increases DNA Adduct Formation Responsible for Aristolochic Acid I-Mediated Carcinogenicity in Rats" International Journal of Molecular Sciences 22, no. 19: 10479. https://doi.org/10.3390/ijms221910479
APA StyleBárta, F., Dedíková, A., Bebová, M., Dušková, Š., Mráz, J., Schmeiser, H. H., Arlt, V. M., Hodek, P., & Stiborová, M. (2021). Co-Exposure to Aristolochic Acids I and II Increases DNA Adduct Formation Responsible for Aristolochic Acid I-Mediated Carcinogenicity in Rats. International Journal of Molecular Sciences, 22(19), 10479. https://doi.org/10.3390/ijms221910479