
S1P Stimulates Erythropoietin Production in Mouse Renal Interstitial Fibroblasts by S1P1 and S1P3 Receptor Activation and HIF-2α Stabilization



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
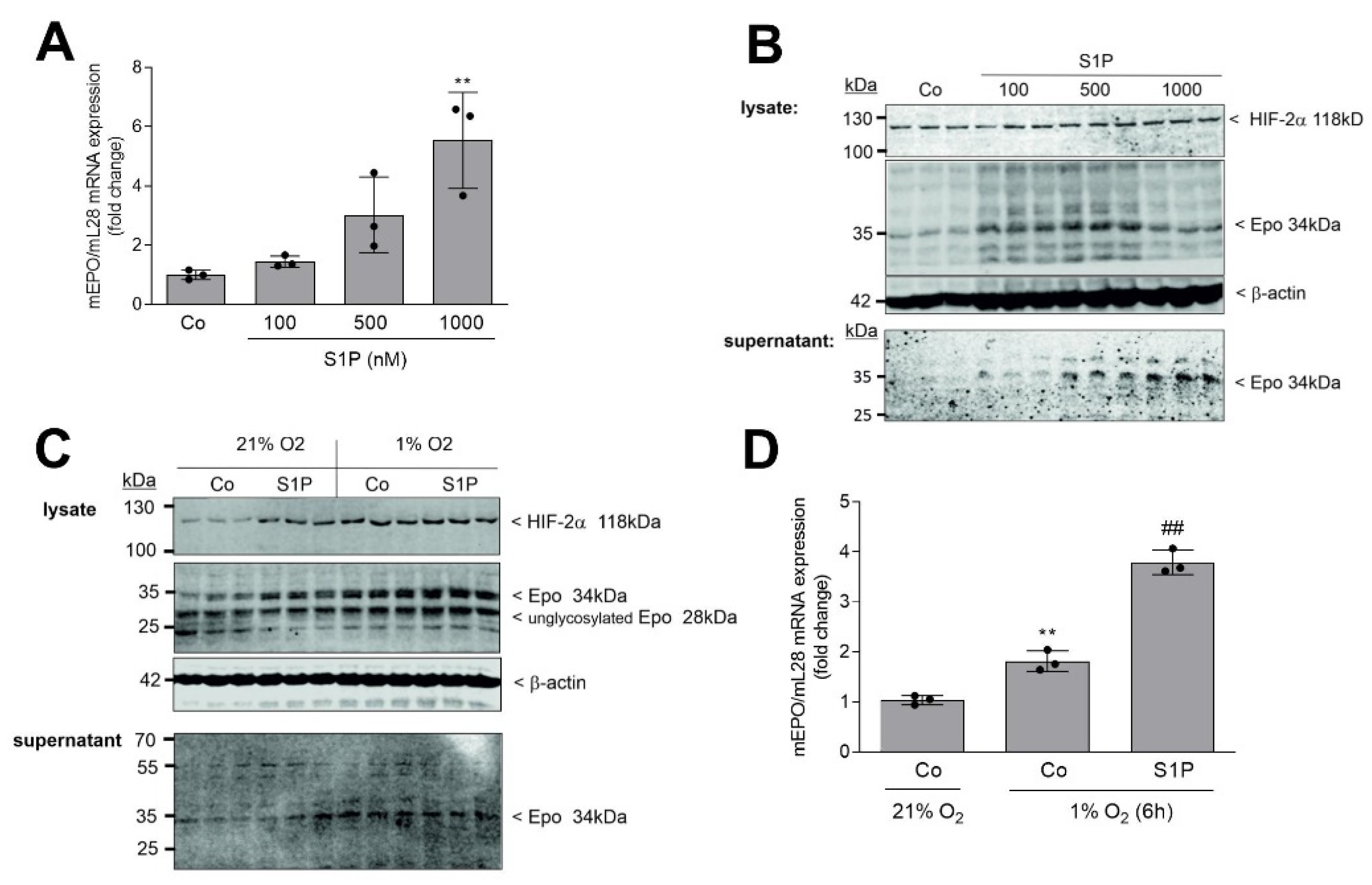
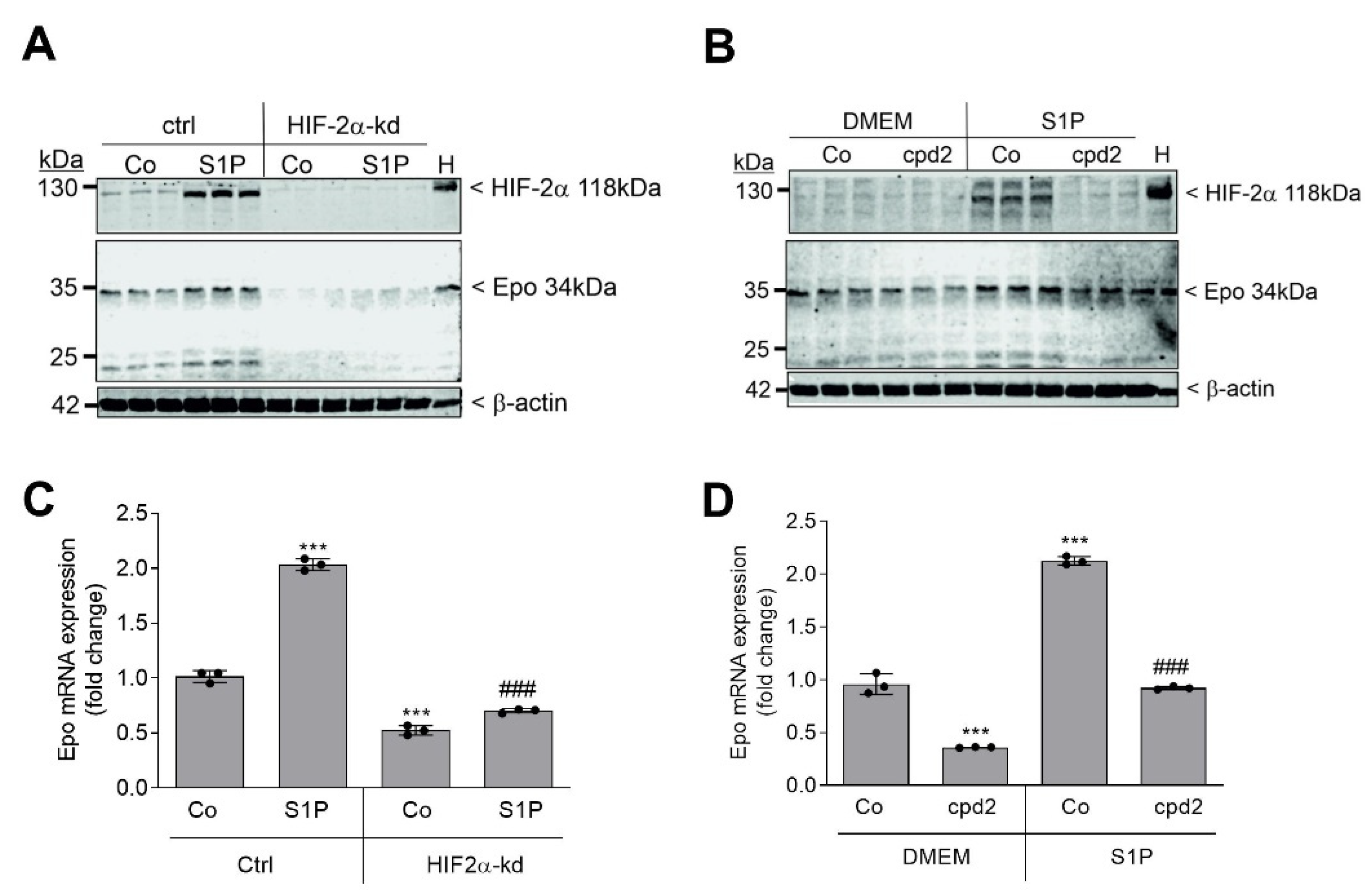
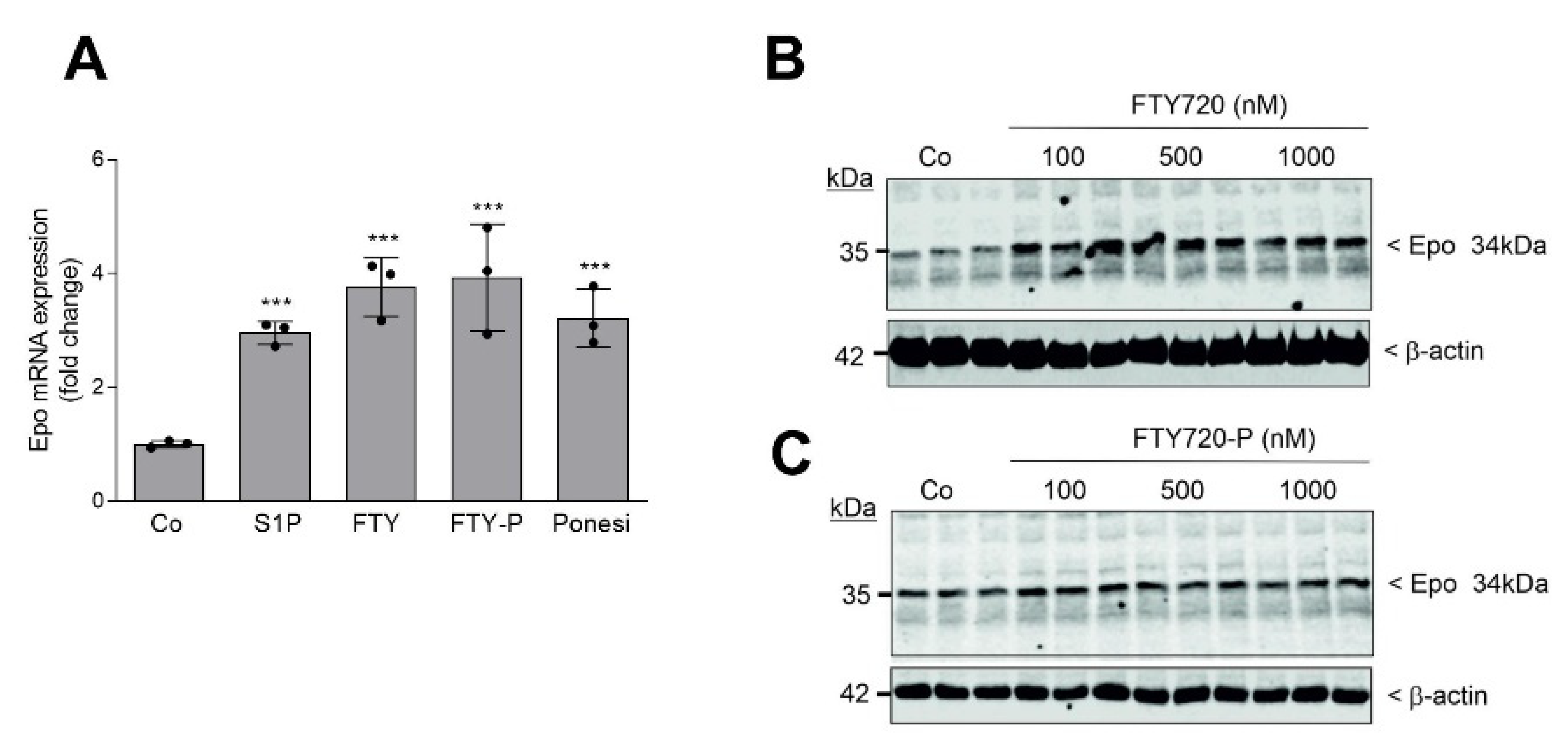
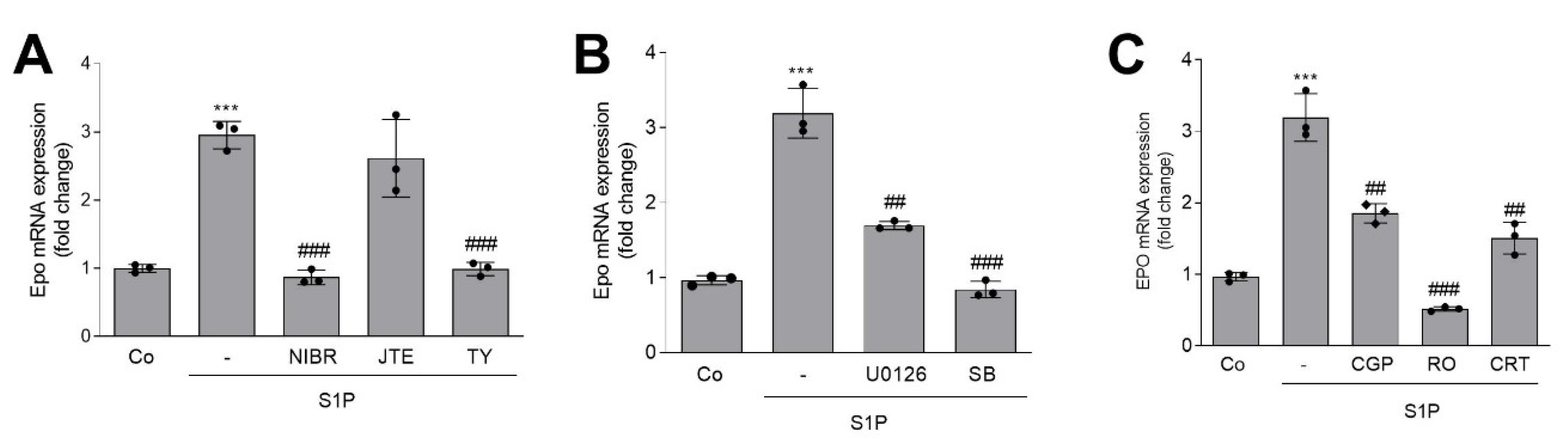
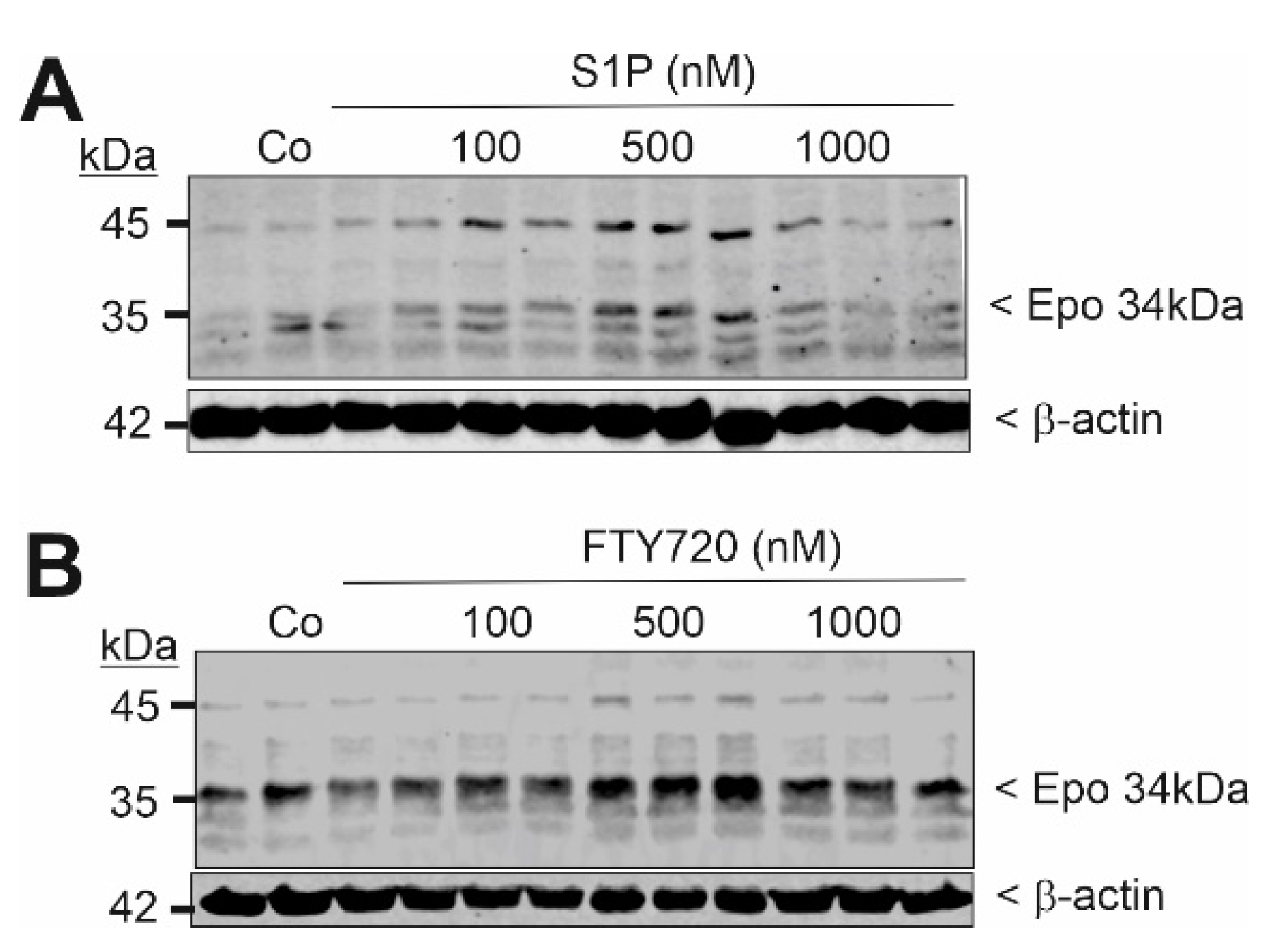
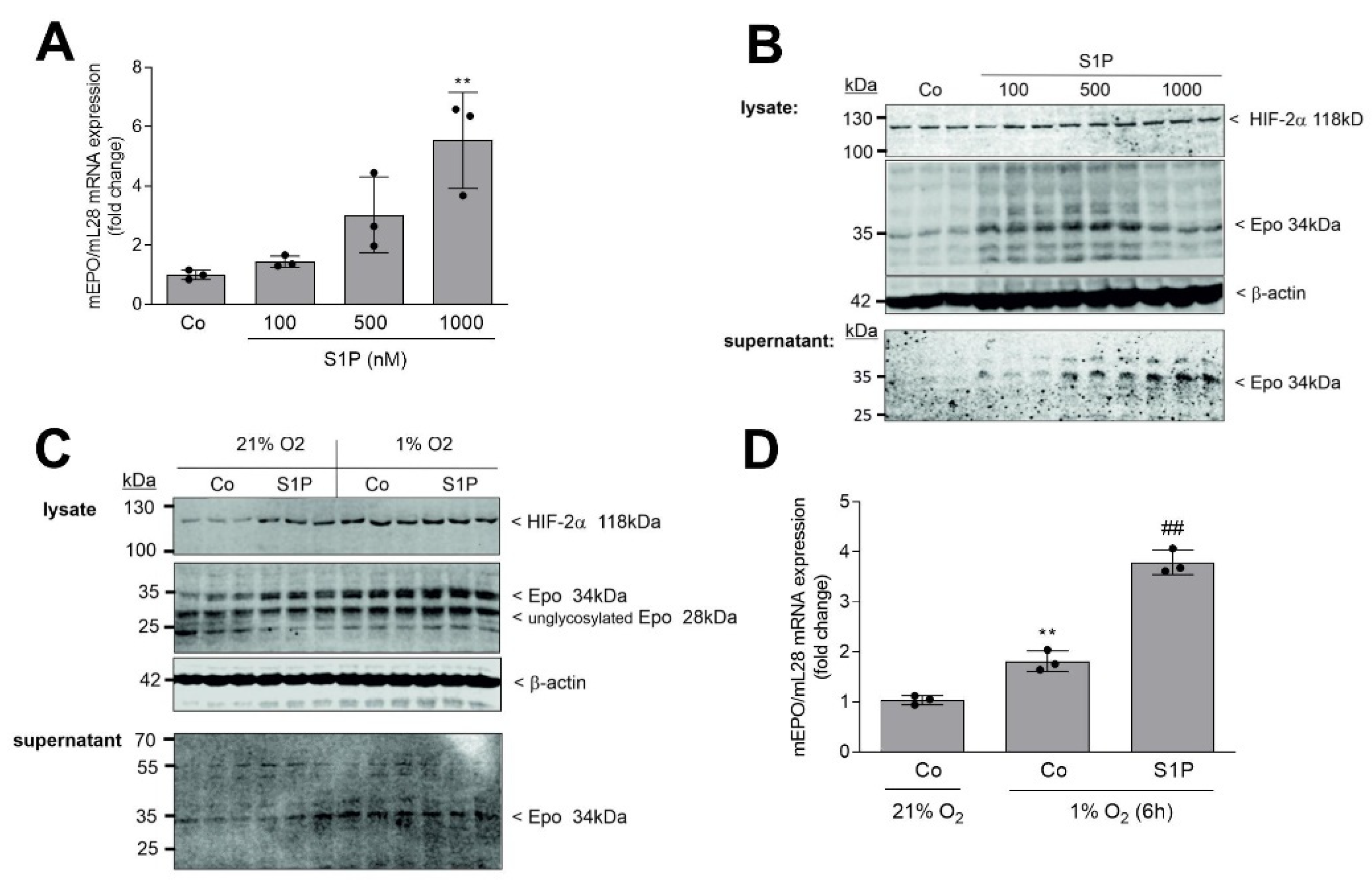
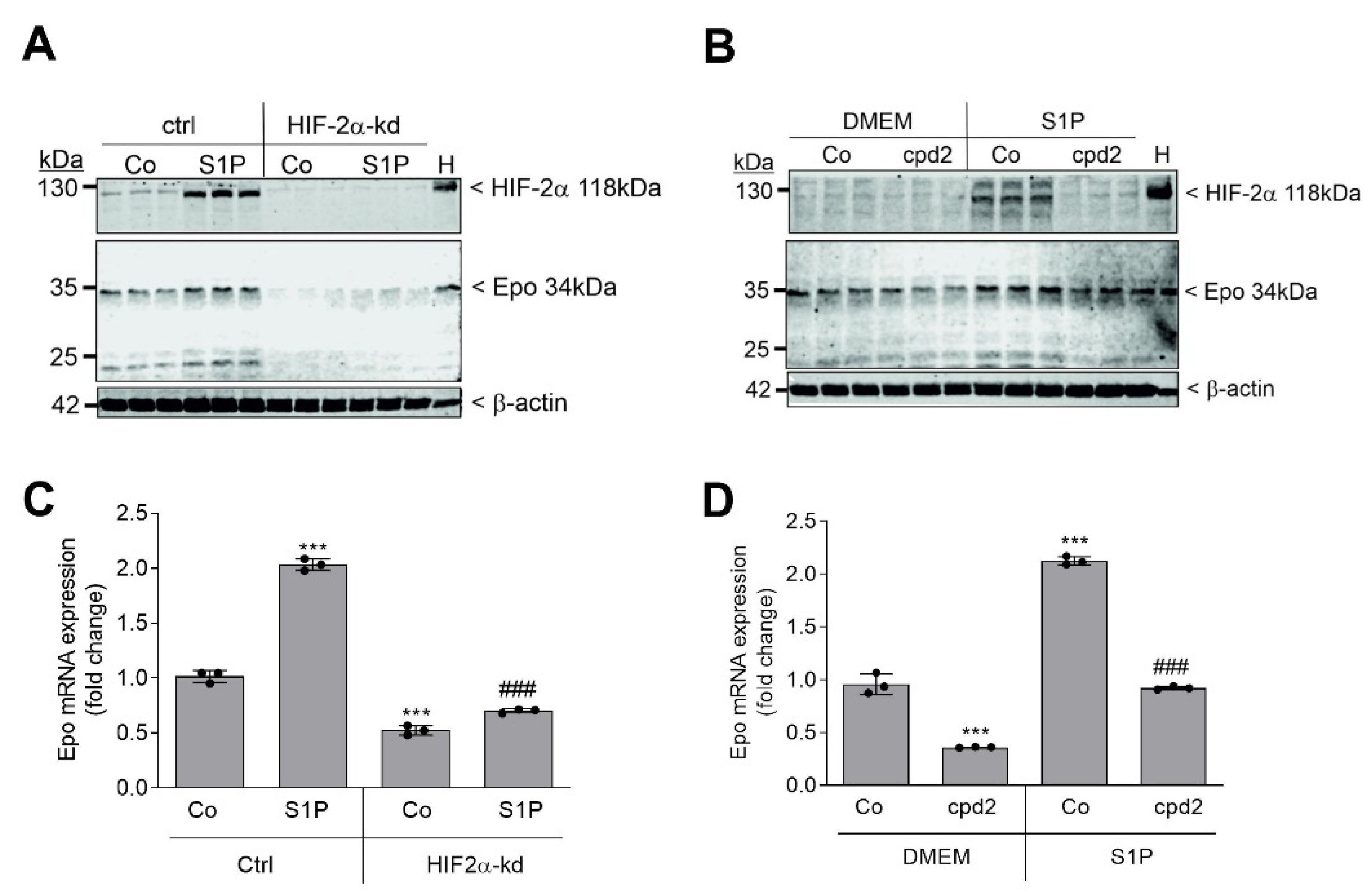
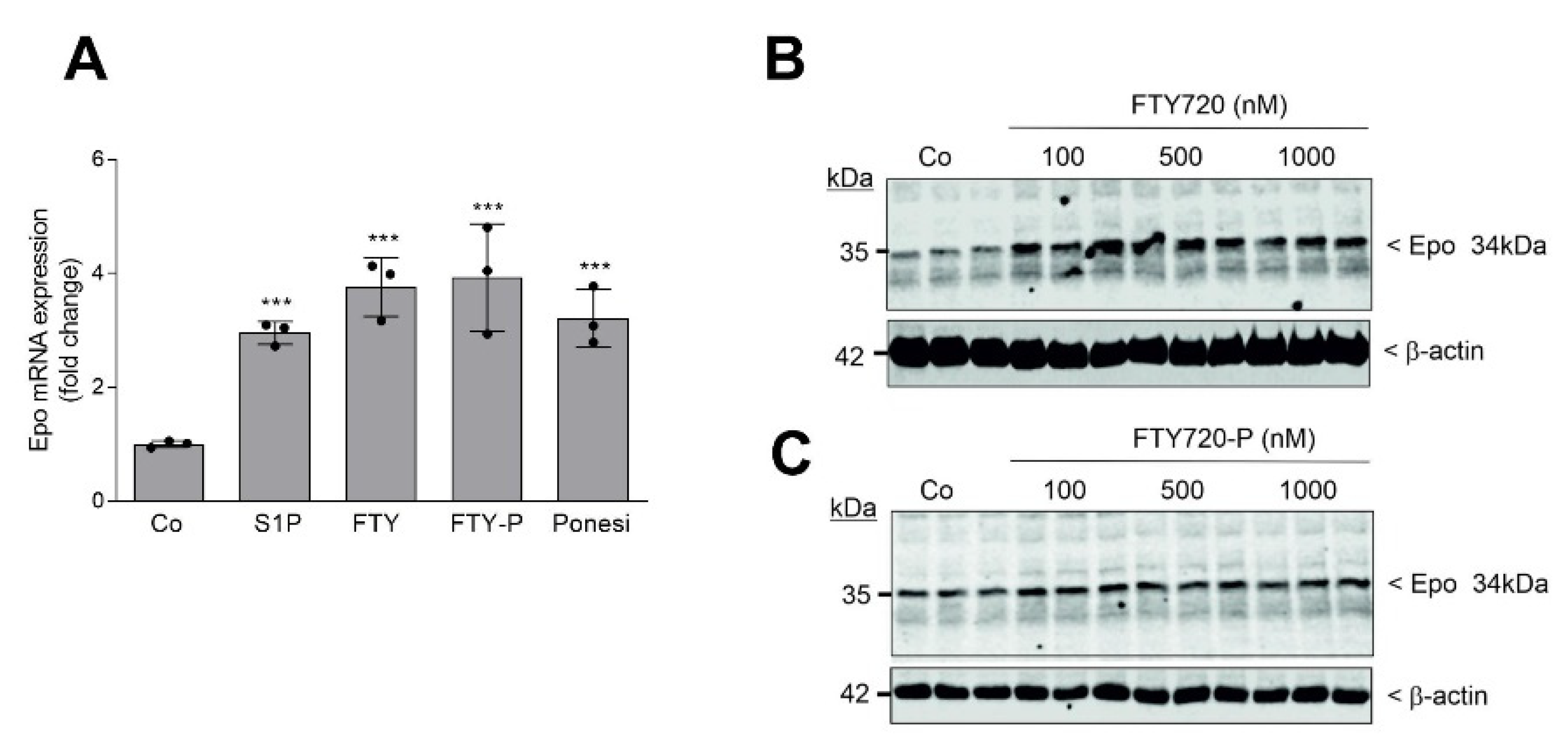
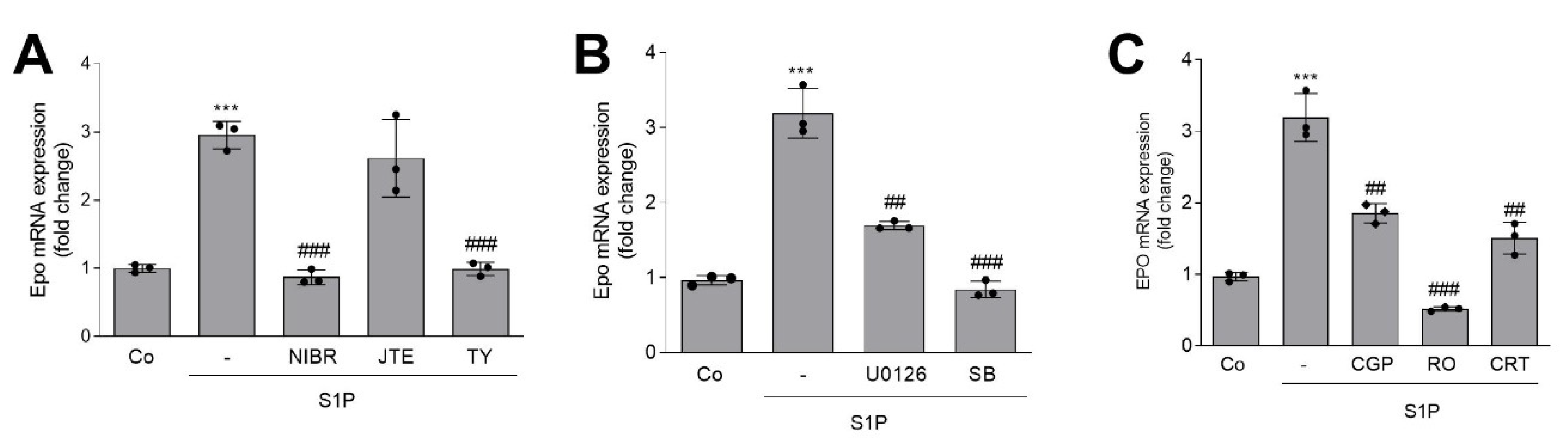
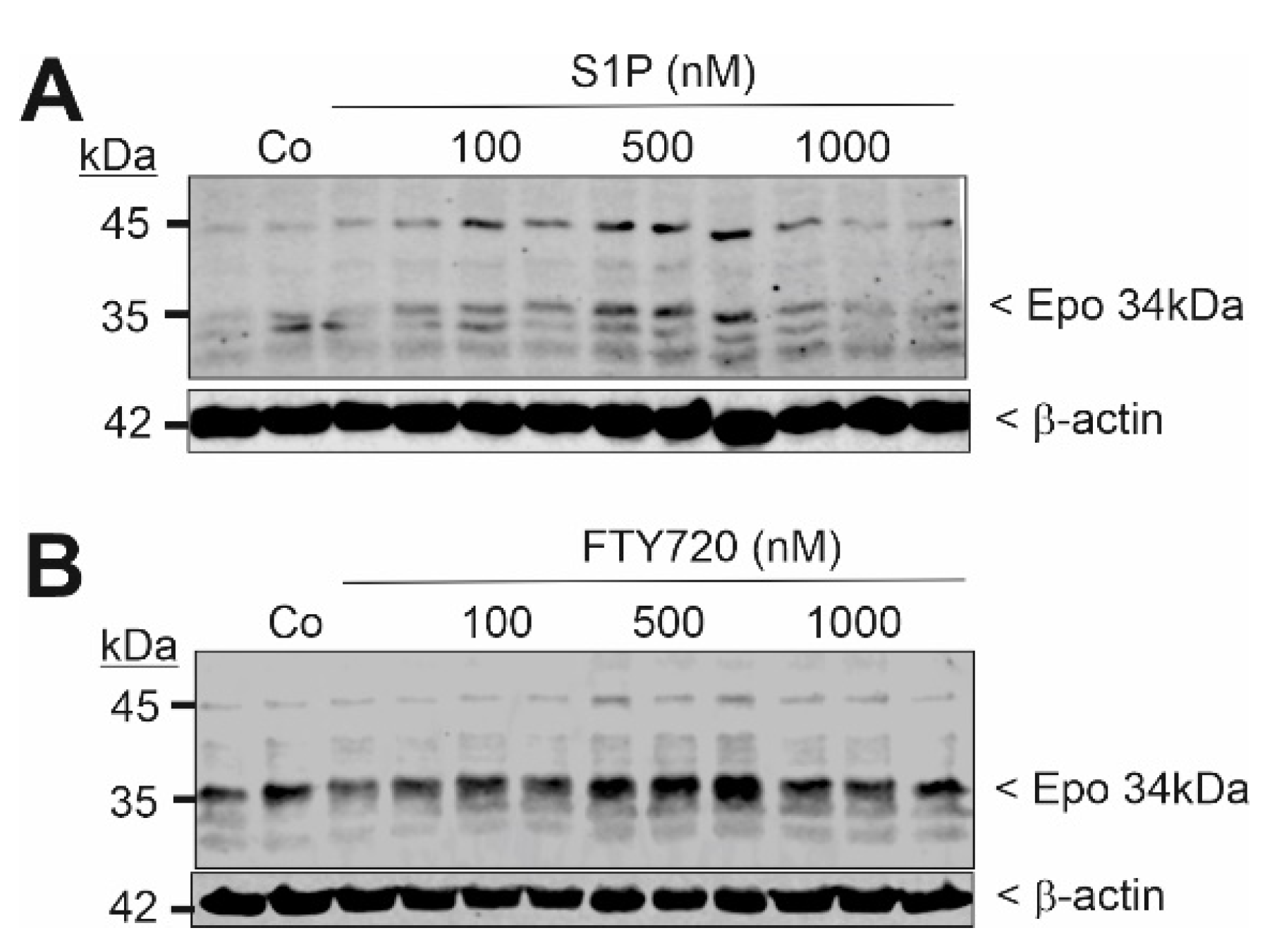
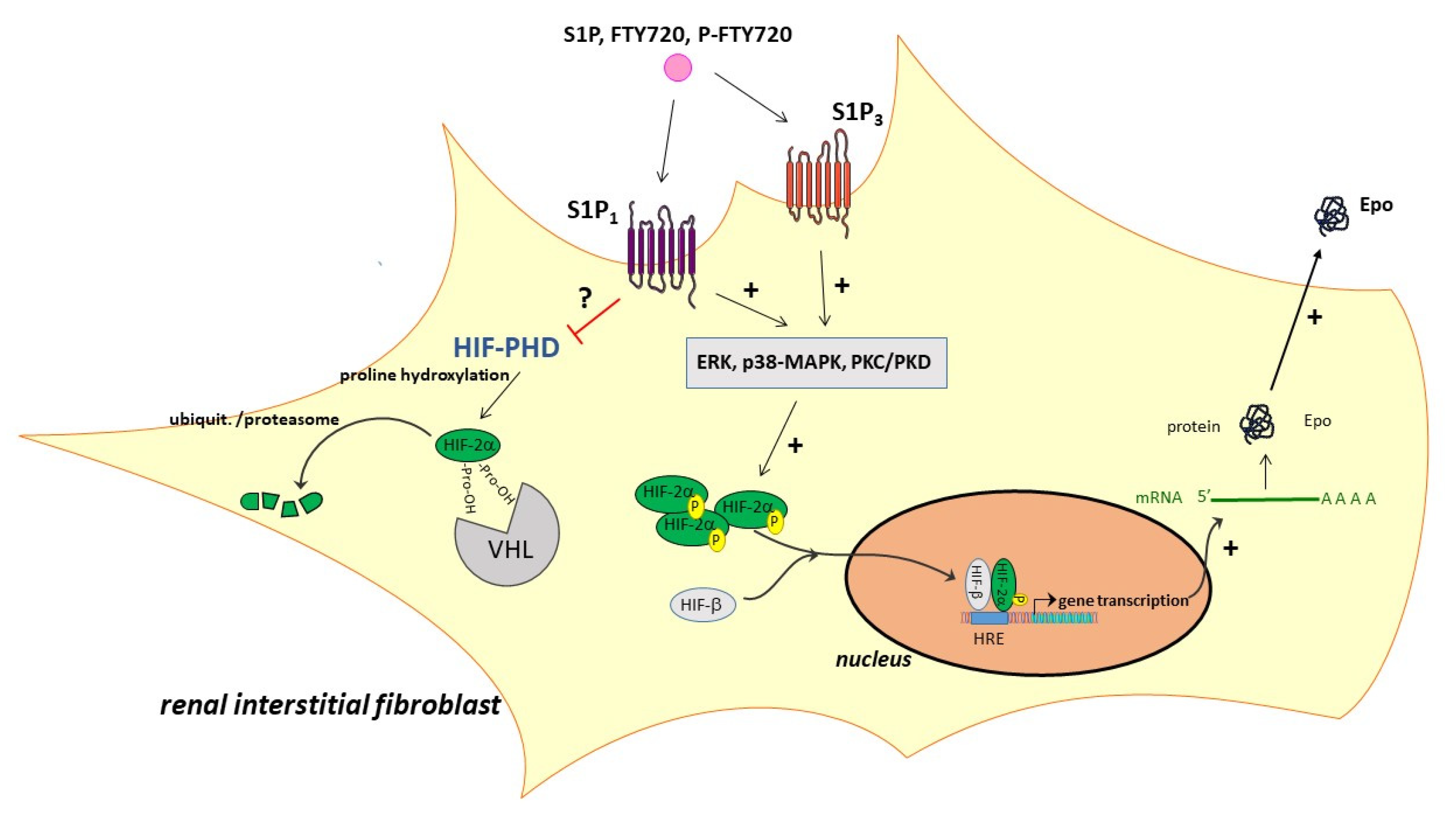
2. Results
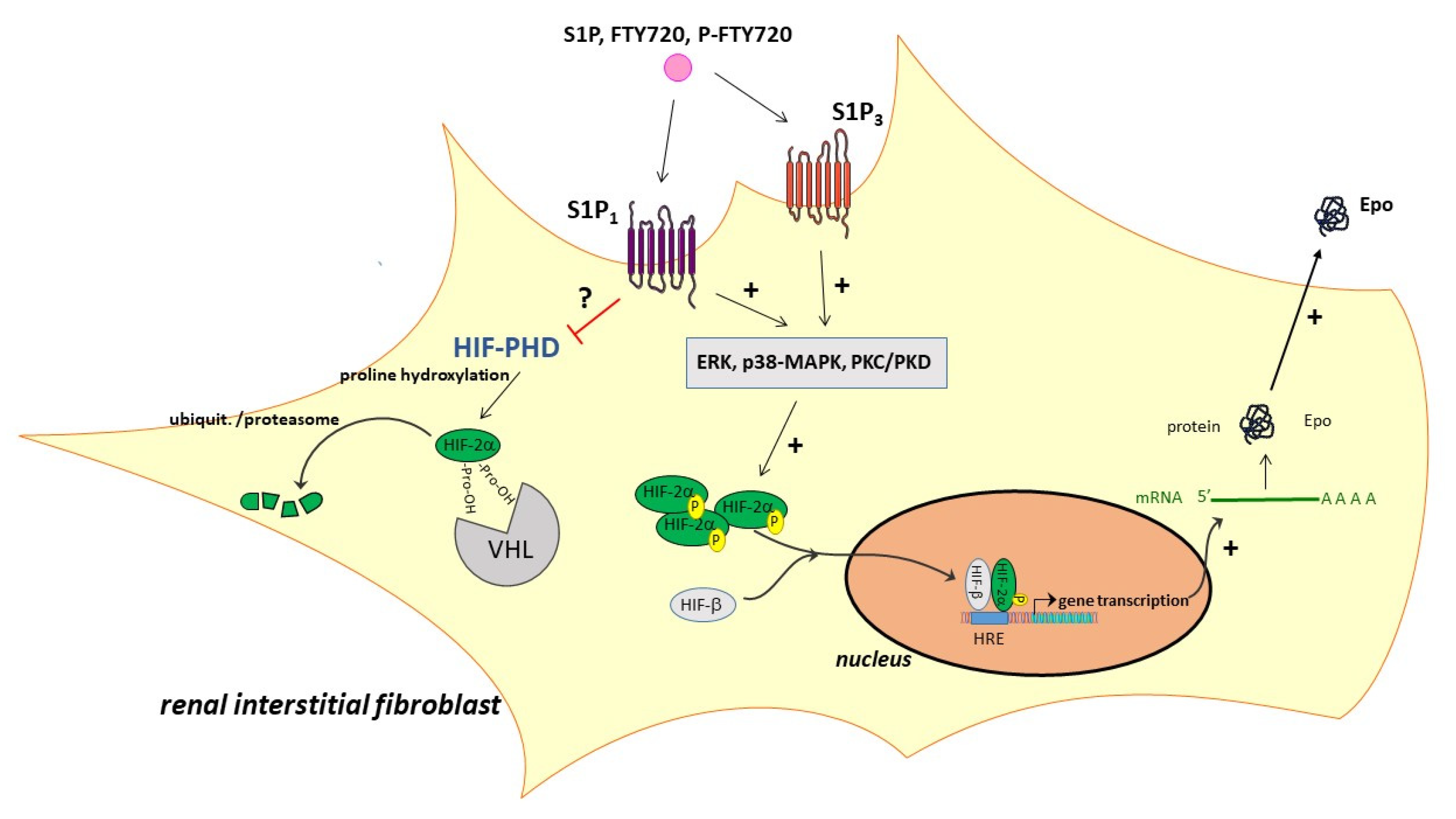
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Lines and Cell Culture Conditions
4.3. Cell Stimulation, Homogenization, and Western Blotting
4.4. RNA Extraction and Quantitative Real-Time PCR Analysis
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| βcR. | β-common receptor |
| BSA | bovine serum albumin |
| CKD | chronic kidney disease |
| DMEM | Dulbecco’s modified Eagle medium |
| Epo | erythropoietin |
| FBS | fetal bovine serum |
| GPCR | G protein-coupled receptor |
| HIF | hypoxia-inducible factor |
| HRE | hypoxia response element |
| kd | knockdown |
| PBS | phosphate-buffered saline |
| PI3K | phosphoinositide 3-kinase |
| PK | protein kinase |
| REPC | renal Epo producing cells |
| S1P | sphingosine 1-phosphate |
| SDS-PAGE | sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| shRNA | small hairpin RNA |
| SphK | sphingosine kinase |
| UUO | unilateral ureteral obstruction |
References
- Centers for Disease Control and Prevention. Chronic Kidney Disease in the United States, 2019; US Department of Health and Human Services, Centers for Disease Control and Prevention: Atlanta, GA, USA, 2019. [Google Scholar]
- Fogo, A.B. Mechanisms of progression of chronic kidney disease. Pediatr. Nephrol. 2007, 22, 2011–2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaissling, B.; Lehir, M.; Kriz, W. Renal epithelial injury and fibrosis. Biochim. Biophys. Acta 2013, 1832, 931–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, E.; Cogswell, M.; Egli, I.; Wojdyla, D.; de Benoist, B. Worldwide prevalence of anaemia, WHO Vitamin and Mineral Nutrition Information System, 1993–2005. Public Health Nutr. 2009, 12, 444–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eschbach, J.W.; Abdulhadi, M.H.; Browne, J.K.; Delano, B.G.; Downing, M.R.; Egrie, J.C.; Evans, R.W.; Friedman, E.A.; Graber, S.E.; Haley, N.R.; et al. Recombinant human erythropoietin in anemic patients with end-stage renal disease. Results of a phase III multicenter clinical trial. Ann. Intern. Med. 1989, 111, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Nangaku, M.; Eckardt, K.U. Pathogenesis of renal anemia. Semin. Nephrol. 2006, 26, 261–268. [Google Scholar] [CrossRef]
- Babitt, J.L.; Lin, H.Y. Mechanisms of anemia in CKD. J. Am. Soc. Nephrol. 2012, 23, 1631–1634. [Google Scholar] [CrossRef] [Green Version]
- Wenger, R.H.; Kurtz, A. Erythropoietin. Compr. Physiol. 2011, 1, 1759–1794. [Google Scholar]
- Calvillo, L.; Latini, R.; Kajstura, J.; Leri, A.; Anversa, P.; Ghezzi, P.; Salio, M.; Cerami, A.; Brines, M. Recombinant human erythropoietin protects the myocardium from ischemia-reperfusion injury and promotes beneficial remodeling. Proc. Natl. Acad. Sci. USA 2003, 100, 4802–4806. [Google Scholar] [CrossRef] [Green Version]
- Sharples, E.J.; Patel, N.; Brown, P.; Stewart, K.; Mota-Philipe, H.; Sheaff, M.; Kieswich, J.; Allen, D.; Harwood, S.; Raftery, M.; et al. Erythropoietin protects the kidney against the injury and dysfunction caused by ischemia-reperfusion. J. Am. Soc. Nephrol. 2004, 15, 2115–2124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharples, E.J.; Thiemermann, C.; Yaqoob, M.M. Novel applications of recombinant erythropoietin. Curr. Opin. Pharmacol. 2006, 6, 184–189. [Google Scholar] [CrossRef]
- Cuzzocrea, S.; Mazzon, E.; Di Paola, R.; Patel, N.S.; Genovese, T.; Muia, C.; De Sarro, A.; Thiemermann, C. Erythropoietin reduces the development of experimental inflammatory bowel disease. J. Pharmacol. Exp. Ther. 2004, 311, 1272–1280. [Google Scholar] [CrossRef]
- Calapai, G.; Marciano, M.C.; Corica, F.; Allegra, A.; Parisi, A.; Frisina, N.; Caputi, A.P.; Buemi, M. Erythropoietin protects against brain ischemic injury by inhibition of nitric oxide formation. Eur. J. Pharmacol. 2000, 401, 349–356. [Google Scholar] [CrossRef]
- Celik, M.; Gokmen, N.; Erbayraktar, S.; Akhisaroglu, M.; Konakc, S.; Ulukus, C.; Genc, S.; Genc, K.; Sagiroglu, E.; Cerami, A.; et al. Erythropoietin prevents motor neuron apoptosis and neurologic disability in experimental spinal cord ischemic injury. Proc. Natl. Acad. Sci. USA 2002, 99, 2258–2263. [Google Scholar] [CrossRef] [Green Version]
- Abdelrahman, M.; Sharples, E.J.; McDonald, M.C.; Collin, M.; Patel, N.S.; Yaqoob, M.M.; Thiemermann, C. Erythropoietin attenuates the tissue injury associated with hemorrhagic shock and myocardial ischemia. Shock 2004, 22, 63–69. [Google Scholar] [CrossRef]
- Vesey, D.A.; Cheung, C.; Pat, B.; Endre, Z.; Gobe, G.; Johnson, D.W. Erythropoietin protects against ischaemic acute renal injury. Nephrol. Dial. Transplant. 2004, 19, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Oba, S.; Suzuki, E.; Nishimatsu, H.; Kumano, S.; Hosoda, C.; Homma, Y.; Hirata, Y. Renoprotective effect of erythropoietin in ischemia/reperfusion injury: Possible roles of the Akt/endothelial nitric oxide synthase-dependent pathway. Int. J. Urol. 2012, 19, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Coldewey, S.M.; Khan, A.I.; Kapoor, A.; Collino, M.; Rogazzo, M.; Brines, M.; Cerami, A.; Hall, P.; Sheaff, M.; Kieswich, J.E.; et al. Erythropoietin attenuates acute kidney dysfunction in murine experimental sepsis by activation of the β-common receptor. Kidney Int. 2013, 84, 482–490. [Google Scholar] [CrossRef] [Green Version]
- Dang, J.Z.; Tu, Y.F.; Wang, J.; Yang, Y.J. Carbamylated Erythropoietin Alleviates Kidney Damage in Diabetic Rats by Suppressing Oxidative Stress. Curr. Med. Sci. 2021, 41, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Collino, M.; Thiemermann, C.; Cerami, A.; Brines, M. Flipping the molecular switch for innate protection and repair of tissues: Long-lasting effects of a non-erythropoietic small peptide engineered from erythropoietin. Pharmacol. Ther. 2015, 151, 32–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacombe, C.; Da Silva, J.L.; Bruneval, P.; Fournier, J.G.; Wendling, F.; Casadevall, N.; Camilleri, J.P.; Bariety, J.; Varet, B.; Tambourin, P. Peritubular cells are the site of erythropoietin synthesis in the murine hypoxic kidney. J. Clin. Investig. 1988, 81, 620–623. [Google Scholar] [CrossRef]
- Weidemann, A.; Johnson, R.S. Nonrenal regulation of EPO synthesis. Kidney Int. 2009, 75, 682–688. [Google Scholar] [CrossRef] [Green Version]
- Haase, V.H. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 2013, 27, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholz, H.; Schurek, H.J.; Eckardt, K.U.; Bauer, C. Role of erythropoietin in adaptation to hypoxia. Experientia 1990, 46, 1197–1201. [Google Scholar] [CrossRef] [PubMed]
- Jelkmann, W. Regulation of erythropoietin production. J. Physiol. 2011, 589 Pt 6, 1251–1258. [Google Scholar] [CrossRef]
- Fandrey, J.; Pagel, H.; Frede, S.; Wolff, M.; Jelkmann, W. Thyroid hormones enhance hypoxia-induced erythropoietin production in vitro. Exp. Hematol. 1994, 22, 272–277. [Google Scholar] [PubMed]
- Kambe, T.; Tada-Kambe, J.; Kuge, Y.; Yamaguchi-Iwai, Y.; Nagao, M.; Sasaki, R. Retinoic acid stimulates erythropoietin gene transcription in embryonal carcinoma cells through the direct repeat of a steroid/thyroid hormone receptor response element half-site in the hypoxia-response enhancer. Blood 2000, 96, 3265–3271. [Google Scholar] [CrossRef] [PubMed]
- Huwiler, A.; Pfeilschifter, J. Sphingolipid signaling in renal fibrosis. Matrix Biol. 2018, 68–69, 230–247. [Google Scholar] [CrossRef]
- Koch, A.; Pfeilschifter, J.; Huwiler, A. Sphingosine 1-phosphate in renal diseases. Cell. Physiol. Biochem. 2013, 31, 745–760. [Google Scholar] [CrossRef]
- Ueda, N. Sphingolipids in Genetic and Acquired Forms of Chronic Kidney Diseases. Curr. Med. Chem. 2017, 24, 1238–1275. [Google Scholar]
- Maceyka, M.; Milstien, S.; Spiegel, S. Sphingosine kinases, sphingosine-1-phosphate and sphingolipidomics. Prostaglandins Other Lipid Mediat. 2005, 77, 15–22. [Google Scholar] [CrossRef]
- Alemany, R.; van Koppen, C.J.; Danneberg, K.; Ter Braak, M.; Meyer Zu Heringdorf, D. Regulation and functional roles of sphingosine kinases. Naunyn-Schmiedebergs Arch. Pharmacol. 2007, 374, 413–428. [Google Scholar] [CrossRef] [PubMed]
- Schwalm, S.; Pfeilschifter, J.; Huwiler, A. Sphingosine-1-phosphate: A Janus-faced mediator of fibrotic diseases. Biochim. Biophys. Acta 2013, 1831, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Schwalm, S.; Pfeilschifter, J.; Huwiler, A. Targeting the sphingosine kinase/sphingosine 1-phosphate pathway to treat chronic inflammatory kidney diseases. Basic Clin. Pharmacol. Toxicol. 2014, 114, 44–49. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, S.; Milstien, S. Exogenous and intracellularly generated sphingosine 1-phosphate can regulate cellular processes by divergent pathways. Biochem. Soc. Trans. 2003, 31 Pt 6, 1216–1219. [Google Scholar] [CrossRef] [Green Version]
- Proia, R.L.; Hla, T. Emerging biology of sphingosine-1-phosphate: Its role in pathogenesis and therapy. J. Clin. Investig. 2015, 125, 1379–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, S.; Babelova, A.; Moreth, K.; Xin, C.; Eberhardt, W.; Doller, A.; Pavenstadt, H.; Schaefer, L.; Pfeilschifter, J.; Huwiler, A. Transforming growth factor-β2 upregulates sphingosine kinase-1 activity, which in turn attenuates the fibrotic response to TGF-β2 by impeding CTGF expression. Kidney Int. 2009, 76, 857–867. [Google Scholar] [CrossRef] [Green Version]
- Imeri, F.; Stepanovska Tanturovska, B.; Schwalm, S.; Saha, S.; Zeng-Brouwers, J.; Pavenstadt, H.; Pfeilschifter, J.; Schaefer, L.; Huwiler, A. Loss of sphingosine kinase 2 enhances Wilm’s tumor suppressor gene 1 and nephrin expression in podocytes and protects from streptozotocin-induced podocytopathy and albuminuria in mice. Matrix Biol. 2021, 98, 32–48. [Google Scholar] [CrossRef]
- Schwalm, S.; Beyer, S.; Frey, H.; Haceni, R.; Grammatikos, G.; Thomas, D.; Geisslinger, G.; Schaefer, L.; Huwiler, A.; Pfeilschifter, J. Sphingosine Kinase-2 Deficiency Ameliorates Kidney Fibrosis by Up-Regulating Smad7 in a Mouse Model of Unilateral Ureteral Obstruction. Am. J. Pathol. 2017, 187, 2413–2429. [Google Scholar] [CrossRef] [Green Version]
- Bajwa, A.; Huang, L.; Kurmaeva, E.; Ye, H.; Dondeti, K.R.; Chroscicki, P.; Foley, L.S.; Balogun, Z.A.; Alexander, K.J.; Park, H.; et al. Sphingosine Kinase 2 Deficiency Attenuates Kidney Fibrosis via IFN-γ. J. Am. Soc. Nephrol. 2017, 28, 1145–1161. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, M.; Thangada, S.; Dasgupta, O.; Khanna, K.M.; Yamase, H.T.; Kashgarian, M.; Hla, T.; Shapiro, L.H.; Ferrer, F.A. Cell-intrinsic sphingosine kinase 2 promotes macrophage polarization and renal inflammation in response to unilateral ureteral obstruction. PLoS ONE 2018, 13, e0194053. [Google Scholar] [CrossRef]
- Du, C.; Ren, Y.; Yao, F.; Duan, J.; Zhao, H.; Du, Y.; Xiao, X.; Duan, H.; Shi, Y. Sphingosine kinase 1 protects renal tubular epithelial cells from renal fibrosis via induction of autophagy. Int. J. Biochem. Cell Biol. 2017, 90, 17–28. [Google Scholar] [CrossRef]
- Schwalm, S.; Beyer, S.; Hafizi, R.; Trautmann, S.; Geisslinger, G.; Adams, D.R.; Pyne, S.; Pyne, N.; Schaefer, L.; Huwiler, A.; et al. Validation of highly selective sphingosine kinase 2 inhibitors SLM6031434 and HWG-35D as effective anti-fibrotic treatment options in a mouse model of tubulointerstitial fibrosis. Cell. Signal. 2021, 79, 109881. [Google Scholar] [CrossRef]
- Yun, J.K.; Kester, M. Regulatory role of sphingomyelin metabolites in hypoxia-induced vascular smooth muscle cell proliferation. Arch. Biochem. Biophys. 2002, 408, 78–86. [Google Scholar] [CrossRef]
- Schwalm, S.; Doll, F.; Romer, I.; Bubnova, S.; Pfeilschifter, J.; Huwiler, A. Sphingosine kinase-1 is a hypoxia-regulated gene that stimulates migration of human endothelial cells. Biochem. Biophys. Res. Commun. 2008, 368, 1020–1025. [Google Scholar] [CrossRef] [PubMed]
- Anelli, V.; Gault, C.R.; Cheng, A.B.; Obeid, L.M. Sphingosine kinase 1 is up-regulated during hypoxia in U87MG glioma cells. Role of hypoxia-inducible factors 1 and 2. J. Biol. Chem. 2008, 283, 3365–3375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ader, I.; Brizuela, L.; Bouquerel, P.; Malavaud, B.; Cuvillier, O. Sphingosine kinase 1: A new modulator of hypoxia inducible factor 1α during hypoxia in human cancer cells. Cancer Res. 2008, 68, 8635–8642. [Google Scholar] [CrossRef] [Green Version]
- Cuvillier, O.; Ader, I.; Bouquerel, P.; Brizuela, L.; Gstalder, C.; Malavaud, B. Hypoxia, therapeutic resistance, and sphingosine 1-phosphate. Adv. Cancer Res. 2013, 117, 117–141. [Google Scholar]
- Kalhori, V.; Kemppainen, K.; Asghar, M.Y.; Bergelin, N.; Jaakkola, P.; Tornquist, K. Sphingosine-1-Phosphate as a Regulator of Hypoxia-Induced Factor-1α in Thyroid Follicular Carcinoma Cells. PLoS ONE 2013, 8, e66189. [Google Scholar]
- Ader, I.; Gstalder, C.; Bouquerel, P.; Golzio, M.; Andrieu, G.; Zalvidea, S.; Richard, S.; Sabbadini, R.A.; Malavaud, B.; Cuvillier, O. Neutralizing S1P inhibits intratumoral hypoxia, induces vascular remodelling and sensitizes to chemotherapy in prostate cancer. Oncotarget 2015, 6, 13803–13821. [Google Scholar] [CrossRef] [Green Version]
- Bouquerel, P.; Gstalder, C.; Muller, D.; Laurent, J.; Brizuela, L.; Sabbadini, R.A.; Malavaud, B.; Pyronnet, S.; Martineau, Y.; Ader, I.; et al. Essential role for SphK1/S1P signaling to regulate hypoxia-inducible factor 2α expression and activity in cancer. Oncogenesis 2016, 5, e209. [Google Scholar] [CrossRef] [Green Version]
- Imeri, F.; Nolan, K.A.; Bapst, A.M.; Santambrogio, S.; Abreu-Rodriguez, I.; Spielmann, P.; Pfundstein, S.; Libertini, S.; Crowther, L.; Orlando, I.M.C.; et al. Generation of renal Epo-producing cell lines by conditional gene tagging reveals rapid HIF-2 driven Epo kinetics, cell autonomous feedback regulation, and a telocyte phenotype. Kidney Int. 2019, 95, 375–387. [Google Scholar] [CrossRef]
- Sato, K.; Hirano, I.; Sekine, H.; Miyauchi, K.; Nakai, T.; Kato, K.; Ito, S.; Yamamoto, M.; Suzuki, N. An immortalized cell line derived from renal erythropoietin-producing (REP) cells demonstrates their potential to transform into myofibroblasts. Sci. Rep. 2019, 9, 11254. [Google Scholar] [CrossRef]
- Darling, R.J.; Kuchibhotla, U.; Glaesner, W.; Micanovic, R.; Witcher, D.R.; Beals, J.M. Glycosylation of erythropoietin affects receptor binding kinetics: Role of electrostatic interactions. Biochemistry 2002, 41, 14524–14531. [Google Scholar] [CrossRef]
- Rosenberger, C.; Mandriota, S.; Jurgensen, J.S.; Wiesener, M.S.; Horstrup, J.H.; Frei, U.; Ratcliffe, P.J.; Maxwell, P.H.; Bachmann, S.; Eckardt, K.U. Expression of hypoxia-inducible factor-1α and -2α in hypoxic and ischemic rat kidneys. J. Am. Soc. Nephrol. 2002, 13, 1721–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, E.J.; Cho, Y.S.; Kim, M.S.; Park, J.W. Contribution of HIF-1α or HIF-2α to erythropoietin expression: In vivo evidence based on chromatin immunoprecipitation. Ann. Hematol. 2008, 87, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Paliege, A.; Rosenberger, C.; Bondke, A.; Sciesielski, L.; Shina, A.; Heyman, S.N.; Flippin, L.A.; Arend, M.; Klaus, S.J.; Bachmann, S. Hypoxia-inducible factor-2α-expressing interstitial fibroblasts are the only renal cells that express erythropoietin under hypoxia-inducible factor stabilization. Kidney Int. 2010, 77, 312–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheuermann, T.H.; Li, Q.; Ma, H.W.; Key, J.; Zhang, L.; Chen, R.; Garcia, J.A.; Naidoo, J.; Longgood, J.; Frantz, D.E.; et al. Allosteric inhibition of hypoxia inducible factor-2 with small molecules. Nat. Chem. Biol. 2013, 9, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Quancard, J.; Bollbuck, B.; Janser, P.; Angst, D.; Berst, F.; Buehlmayer, P.; Streiff, M.; Beerli, C.; Brinkmann, V.; Guerini, D.; et al. A potent and selective S1P(1) antagonist with efficacy in experimental autoimmune encephalomyelitis. Chem. Biol. 2012, 19, 1142–1151. [Google Scholar] [CrossRef] [Green Version]
- Ohmori, T.; Yatomi, Y.; Osada, M.; Kazama, F.; Takafuta, T.; Ikeda, H.; Ozaki, Y. Sphingosine 1-phosphate induces contraction of coronary artery smooth muscle cells via S1P2. Cardiovasc. Res. 2003, 58, 170–177. [Google Scholar] [CrossRef] [Green Version]
- Murakami, A.; Takasugi, H.; Ohnuma, S.; Koide, Y.; Sakurai, A.; Takeda, S.; Hasegawa, T.; Sasamori, J.; Konno, T.; Hayashi, K.; et al. Sphingosine 1-phosphate (S1P) regulates vascular contraction via S1P3 receptor: Investigation based on a new S1P3 receptor antagonist. Mol. Pharmacol. 2010, 77, 704–713. [Google Scholar] [CrossRef] [PubMed]
- Jelkmann, W.; Huwiler, A.; Fandrey, J.; Pfeilschifter, J. Inhibition of erythropoietin production by phorbol ester is associated with down-regulation of protein kinase C-α isoenzyme in hepatoma cells. Biochem. Biophys. Res. Commun. 1991, 179, 1441–1448. [Google Scholar] [CrossRef]
- Fandrey, J.; Huwiler, A.; Frede, S.; Pfeilschifter, J.; Jelkmann, W. Distinct signaling pathways mediate phorbol-ester-induced and cytokine-induced inhibition of erythropoietin gene expression. Eur. J. Biochem. 1994, 226, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Eckardt, K.U.; Ring, A.; Maier, M.; Gess, B.; Fabbro, D.; Kurtz, A. Hypoxia-induced accumulation of erythropoietin mRNA in isolated hepatocytes is inhibited by protein kinase C. Pflugers Arch. 1994, 426, 21–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtz, A.; Eckardt, K.U.; Pugh, C.; Corvol, P.; Fabbro, D.; Ratcliffe, P. Phorbol ester inhibits erythropoietin production in human hepatoma cells (Hep G2). Am. J. Physiol. 1992, 262 Pt 1, C1204–C1210. [Google Scholar] [CrossRef] [Green Version]
- Marte, B.M.; Meyer, T.; Stabel, S.; Standke, G.J.; Jaken, S.; Fabbro, D.; Hynes, N.E. Protein kinase C and mammary cell differentiation: Involvement of protein kinase C α in the induction of β-casein expression. Cell Growth Differ. 1994, 5, 239–247. [Google Scholar] [PubMed]
- Dieter, P.; Fitzke, E. RO 31-8220 and RO 31-7549 show improved selectivity for protein kinase C over staurosporine in macrophages. Biochem. Biophys. Res. Commun. 1991, 181, 396–401. [Google Scholar] [CrossRef]
- Harikumar, K.B.; Kunnumakkara, A.B.; Ochi, N.; Tong, Z.; Deorukhkar, A.; Sung, B.; Kelland, L.; Jamieson, S.; Sutherland, R.; Raynham, T.; et al. A novel small-molecule inhibitor of protein kinase D blocks pancreatic cancer growth in vitro and in vivo. Mol. Cancer Ther. 2010, 9, 1136–1146. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, K.; Roy, A. Evaluating the current status of protein kinase C (PKC)-protein kinase D (PKD) signalling axis as a novel therapeutic target in ovarian cancer. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188496. [Google Scholar] [CrossRef] [PubMed]
- Orlando, I.M.C.; Lafleur, V.N.; Storti, F.; Spielmann, P.; Crowther, L.; Santambrogio, S.; Schodel, J.; Hoogewijs, D.; Mole, D.R.; Wenger, R.H. Distal and proximal hypoxia response elements cooperate to regulate organ-specific erythropoietin gene expression. Haematologica 2020, 105, 2774–2784. [Google Scholar] [CrossRef] [Green Version]
- Luo, B.; Gan, W.; Liu, Z.; Shen, Z.; Wang, J.; Shi, R.; Liu, Y.; Liu, Y.; Jiang, M.; Zhang, Z.; et al. Erythropoeitin Signaling in Macrophages Promotes Dying Cell Clearance and Immune Tolerance. Immunity 2016, 44, 287–302. [Google Scholar] [CrossRef] [Green Version]
- Kapitsinou, P.P.; Liu, Q.; Unger, T.L.; Rha, J.; Davidoff, O.; Keith, B.; Epstein, J.A.; Moores, S.L.; Erickson-Miller, C.L.; Haase, V.H. Hepatic HIF-2 regulates erythropoietic responses to hypoxia in renal anemia. Blood 2010, 116, 3039–3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, D.E.; Berra, E.; Gothie, E.; Roux, D.; Pouyssegur, J. p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1α (HIF-1α) and enhance the transcriptional activity of HIF-1. J. Biol. Chem. 1999, 274, 32631–32637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sodhi, A.; Montaner, S.; Patel, V.; Zohar, M.; Bais, C.; Mesri, E.A.; Gutkind, J.S. The Kaposi’s sarcoma-associated herpes virus G protein-coupled receptor up-regulates vascular endothelial growth factor expression and secretion through mitogen-activated protein kinase and p38 pathways acting on hypoxia-inducible factor 1α. Cancer Res. 2000, 60, 4873–4880. [Google Scholar]
- Warfel, N.A.; Dolloff, N.G.; Dicker, D.T.; Malysz, J.; El-Deiry, W.S. CDK1 stabilizes HIF-1α via direct phosphorylation of Ser668 to promote tumor growth. Cell Cycle 2013, 12, 3689–3701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Yao, Y.; Lu, L.; Costa, M.; Dai, W. Plk3 functions as an essential component of the hypoxia regulatory pathway by direct phosphorylation of HIF-1α. J. Biol. Chem. 2010, 285, 38944–38950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flugel, D.; Gorlach, A.; Michiels, C.; Kietzmann, T. Glycogen synthase kinase 3 phosphorylates hypoxia-inducible factor 1α and mediates its destabilization in a VHL-independent manner. Mol. Cell. Biol. 2007, 27, 3253–3265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Na, Y.R.; Kim, S.Y.; Yang, E.G. Protein Kinase C Isoforms Differentially Regulate Hypoxia-Inducible Factor-1α Accumulation in Cancer Cells. J. Cell. Biochem. 2016, 117, 647–658. [Google Scholar] [CrossRef]
- Xia, J.; Ozaki, I.; Matsuhashi, S.; Kuwashiro, T.; Takahashi, H.; Anzai, K.; Mizuta, T. Mechanisms of PKC-Mediated Enhancement of HIF-1α Activity and its Inhibition by Vitamin K2 in Hepatocellular Carcinoma Cells. Int. J. Mol. Sci. 2019, 20, 1022. [Google Scholar] [CrossRef] [Green Version]
- Warnecke, C.; Zaborowska, Z.; Kurreck, J.; Erdmann, V.A.; Frei, U.; Wiesener, M.; Eckardt, K.U. Differentiating the functional role of hypoxia-inducible factor (HIF)-1α and HIF-2α (EPAS-1) by the use of RNA interference: Erythropoietin is a HIF-2α target gene in Hep3B and Kelly cells. FASEB J. 2004, 18, 1462–1464. [Google Scholar] [CrossRef]
- Gerl, K.; Nolan, K.A.; Karger, C.; Fuchs, M.; Wenger, R.H.; Stolt, C.C.; Willam, C.; Kurtz, A.; Kurt, B. Erythropoietin production by PDGFR-β(+) cells. Pflugers Arch. 2016, 468, 1479–1487. [Google Scholar] [CrossRef]
- Kierans, S.J.; Taylor, C.T. Regulation of glycolysis by the hypoxia-inducible factor (HIF): Implications for cellular physiology. J. Physiol. 2021, 599, 23–37. [Google Scholar] [CrossRef]
- Gstalder, C.; Ader, I.; Cuvillier, O. FTY720 (Fingolimod) Inhibits HIF1 and HIF2 Signaling, Promotes Vascular Remodeling, and Chemosensitizes in Renal Cell Carcinoma Animal Model. Mol. Cancer Ther. 2016, 15, 2465–2474. [Google Scholar] [CrossRef] [Green Version]
- Hait, N.C.; Maiti, A.; Xu, P.; Qi, Q.; Kawaguchi, T.; Okano, M.; Takabe, K.; Yan, L.; Luo, C. Regulation of hypoxia-inducible factor functions in the nucleus by sphingosine-1-phosphate. FASEB J. 2020, 34, 4293–4310. [Google Scholar] [CrossRef]
- Hait, N.C.; Allegood, J.; Maceyka, M.; Strub, G.M.; Harikumar, K.B.; Singh, S.K.; Luo, C.; Marmorstein, R.; Kordula, T.; Milstien, S.; et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 2009, 325, 1254–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberhard, M.; Ferlinz, K.; Alizzi, K.; Cacciato, P.M.; Faggio, C.; Foller, M.; Lang, F. FTY720-induced suicidal erythrocyte death. Cell. Physiol. Biochem. 2010, 26, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Poirier, B.; Briand, V.; Kadereit, D.; Schafer, M.; Wohlfart, P.; Philippo, M.C.; Caillaud, D.; Gouraud, L.; Grailhe, P.; Bidouard, J.P.; et al. A G protein-biased S1P1 agonist, SAR247799, protects endothelial cells without affecting lymphocyte numbers. Sci. Signal. 2020, 13, eaax8050. [Google Scholar] [CrossRef]
- Bergougnan, L.; Andersen, G.; Plum-Morschel, L.; Evaristi, M.F.; Poirier, B.; Tardat, A.; Ermer, M.; Herbrand, T.; Arrubla, J.; Coester, H.V.; et al. Endothelial-protective effects of a G-protein-biased sphingosine-1 phosphate receptor-1 agonist, SAR247799, in type-2 diabetes rats and a randomized placebo-controlled patient trial. Br. J. Clin. Pharmacol. 2021, 87, 2303–2320. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hafizi, R.; Imeri, F.; Wenger, R.H.; Huwiler, A. S1P Stimulates Erythropoietin Production in Mouse Renal Interstitial Fibroblasts by S1P1 and S1P3 Receptor Activation and HIF-2α Stabilization. Int. J. Mol. Sci. 2021, 22, 9467. https://doi.org/10.3390/ijms22179467
Hafizi R, Imeri F, Wenger RH, Huwiler A. S1P Stimulates Erythropoietin Production in Mouse Renal Interstitial Fibroblasts by S1P1 and S1P3 Receptor Activation and HIF-2α Stabilization. International Journal of Molecular Sciences. 2021; 22(17):9467. https://doi.org/10.3390/ijms22179467
Chicago/Turabian StyleHafizi, Redona, Faik Imeri, Roland H. Wenger, and Andrea Huwiler. 2021. "S1P Stimulates Erythropoietin Production in Mouse Renal Interstitial Fibroblasts by S1P1 and S1P3 Receptor Activation and HIF-2α Stabilization" International Journal of Molecular Sciences 22, no. 17: 9467. https://doi.org/10.3390/ijms22179467
APA StyleHafizi, R., Imeri, F., Wenger, R. H., & Huwiler, A. (2021). S1P Stimulates Erythropoietin Production in Mouse Renal Interstitial Fibroblasts by S1P1 and S1P3 Receptor Activation and HIF-2α Stabilization. International Journal of Molecular Sciences, 22(17), 9467. https://doi.org/10.3390/ijms22179467