
The Differential Roles for Neurodevelopmental and Neuroendocrine Genes in Shaping GnRH Neuron Physiology and Deficiency


Abstract
1. Introduction
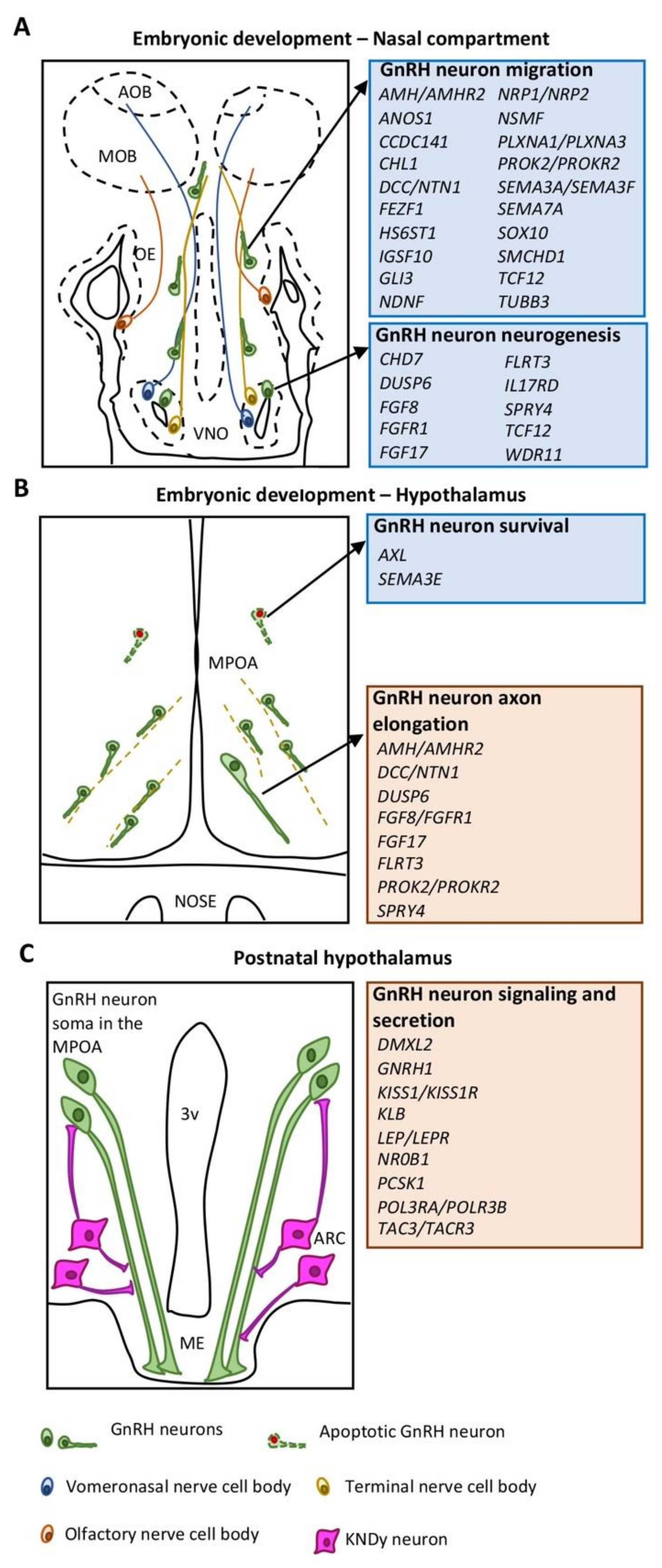
2. GnRH Neuron Development and Function
2.1. GnRH Neuron Development in the Nasal Compartment
2.2. GnRH Neuron Development in the Hypothalamus
3. Congenital Hypogonadotropic Hypogonadism (CHH)
4. Genetics of CHH
4.1. Neuroendocrine Genes
4.1.1. Gonadotropin Releasing Hormone 1 (GNRH1)
4.1.2. Kisspeptin (KISS1) and Kisspeptin Receptor (KISS1R)
4.1.3. Neurokinin B (TAC3) and Neurokinin B Receptor (TACR3)
4.1.4. Leptin (LEP) and Leptin Receptor (LEPR)
4.1.5. Proprotein Convertase Subtilisin/Kexin Type 1 (PCSK1)
4.1.6. β-klotho (KLB)
4.1.7. Nuclear Receptor Subfamily 0 Group B Member 1 (NR0B1)
4.1.8. Dmx-like 2 (DMXL2)
4.1.9. Patatin-like Phospholipase Domain Containing Protein (PNPLA6), OTU Deubiquitinase 4 (OTUD4), Ring Finger Protein 216 (RNF216), STIP1 Homology and U-Box Containing Protein 1 (STUB1)
4.1.10. Polymerase III, RNA Subunit A/B (POLR3A/POLR3B)
4.2. Neurodevelopmental Genes
4.2.1. Anosmin 1 (ANOS1)
4.2.2. Heparan Sulphate 6-O Sulfotransferase 1 (HS6ST1)
4.2.3. NMDA Receptor Synaptonuclear Signaling and Neuronal Migration Factor (NSMF)
4.2.4. AXL Tyrosine Kinase Receptor (AXL)
4.2.5. FEZ Family Zinc Finger 1 (FEZF1) and Coiled-Coil Domain Containing 141 (CCDC141)
4.2.6. Neuron-Derived Neurotrophic Factor (NDNF)
4.2.7. WD Repeat Domain 11 (WDR11)
4.2.8. Semaphorins and Receptors
- Semaphorin 3A and 3F (SEMA3A/3F), Neuropilin 1 and 2 (NRP1/2), Plexin A1 and A3 (PLXNA1/A3)
- 2.
- Semaphorin 3E (SEMA3E)
- 3.
- Semaphorin 7A (SEMA7A)
4.2.9. Immunoglobulin Superfamily Member 10 (IGSF10)
4.2.10. Chromodomain Helicase DNA Binding Protein 7 (CHD7)
4.2.11. Sex-Determining Region (SRY) Box 10 (SOX10)
4.2.12. Structural Maintenance of Chromosomes Flexible Hinge Domain Containing 1 (SMCHD1)
4.2.13. Transcription Factor 12 (TCF12)
4.2.14. Gli-Kruppel Family Member 3 (GLI3)
4.2.15. Tubulin Beta 3 Isotype III (TUBB3)
4.3. Neurodevelopmental and Neuroendocrine Genes
4.3.1. Fibroblast Growth Factor 8 (FGF8) and Fibroblast Growth Factor Receptor 1 (FGFR1)
4.3.2. FGF Signaling Genes
4.3.3. Anti-Mullerian Hormone (AMH) and Anti-Mullerian Hormone Receptor 2 (AMHR2)
4.3.4. Prokineticin 2 (PROK2) and Prokineticin Receptor 2 (PROKR2)
4.3.5. Deleted in Colorectal Cancer (DCC) and Netrin 1 (NTN1)
4.4. Additional Genes
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Kaprara, A.; Huhtaniemi, I.T. The hypothalamus-pituitary-gonad axis: Tales of mice and men. Metabolism 2018, 86, 3–17. [Google Scholar] [CrossRef]
- Boehm, U.; Bouloux, P.-M.; Dattani, M.T.; de Roux, N.; Dodé, C.; Dunkel, L.; Dwyer, A.; Giacobini, P.; Hardelin, J.-P.; Juul, A.; et al. Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism—pathogenesis, diagnosis and treatment. Nat. Rev. Endocrinol. 2015, 11, 547–564. [Google Scholar] [CrossRef]
- Casoni, F.; Malone, S.; Belle, M.; Luzzati, F.; Collier, F.; Allet, C.; Hrabovszky, E.; Rasika, S.; Prevot, V.; Chedotal, A.; et al. Development of the neurons controlling fertility in humans: New insights from 3D imaging and transparent fetal brains. Development 2016, 143, 3969–3981. [Google Scholar] [CrossRef] [PubMed]
- Wray, S. From Nose to Brain: Development of Gonadotrophin-Releasing Hormone -1 Neurones. J. Neuroendocr. 2010, 22, 743–753. [Google Scholar] [CrossRef]
- Rance, N.E.; Young, W.S.; McMullen, N.T. Topography of neurons expressing luteinizing hormone-releasing hormone gene transcripts in the human hypothalamus and basal forebrain. J. Comp. Neurol. 1994, 339, 573–586. [Google Scholar] [CrossRef]
- Maier, E.C.; Saxena, A.; Alsina, B.; Bronner, M.E.; Whitfield, T.T. Sensational placodes: Neurogenesis in the otic and olfactory systems. Dev. Biol. 2014, 389, 50–67. [Google Scholar] [CrossRef]
- Forni, P.E.; Wray, S. GnRH, anosmia and hypogonadotropic hypogonadism—Where are we? Front. Neuroendocr. 2014, 36, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Forni, P.E.; Taylor-Burds, C.; Melvin, V.S.; Williams, T.; Wray, S.; Williams, T. Neural Crest and Ectodermal Cells Intermix in the Nasal Placode to Give Rise to GnRH-1 Neurons, Sensory Neurons, and Olfactory Ensheathing Cells. J. Neurosci. 2011, 31, 6915–6927. [Google Scholar] [CrossRef]
- Cho, H.-J.; Shan, Y.; Whittington, N.C.; Wray, S. Nasal Placode Development, GnRH Neuronal Migration and Kallmann Syndrome. Front. Cell Dev. Biol. 2019, 7, 121. [Google Scholar] [CrossRef]
- Taroc, E.Z.M.; Katreddi, R.R.; Forni, P.E. Identifying Isl1 Genetic Lineage in the Developing Olfactory System and in GnRH-1 Neurons. Front. Physiol. 2020, 11, 601923. [Google Scholar] [CrossRef]
- Lund, C.; Yellapragada, V.; Vuoristo, S.; Balboa, D.; Trova, S.; Allet, C.; Eskici, N.; Pulli, K.; Giacobini, P.; Tuuri, T.; et al. Characterization of the human GnRH neuron developmental transcriptome using a GNRH1-TdTomato reporter line in human pluripotent stem cells. Dis. Model. Mech. 2020, 13, dmm040105. [Google Scholar] [CrossRef]
- Cariboni, A.; Maggi, R.; Parnavelas, J.G. From nose to fertility: The long migratory journey of gonadotropin-releasing hormone neurons. Trends Neurosci. 2007, 30, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Wierman, M.E.; Kiseljak-Vassiliades, K.; Tobet, S. Gonadotropin-releasing hormone (GnRH) neuron migration: Initiation, maintenance and cessation as critical steps to ensure normal reproductive function. Front. Neuroendocr. 2011, 32, 43–52. [Google Scholar] [CrossRef]
- Yoshida, K.; Tobet, S.A.; Crandall, J.E.; Jimenez, T.P.; Schwarting, G.A. The migration of luteinizing hormone-releasing hormone neurons in the developing rat is associated with a transient, caudal projection of the vomeronasal nerve. J. Neurosci. 1995, 15, 7769–7777. [Google Scholar] [CrossRef] [PubMed]
- Taroc, E.Z.; Prasad, A.; Lin, J.M.; Forni, P.E. The terminal nerve plays a prominent role in GnRH-1 neuronal migration independent from proper olfactory and vomeronasal connections to the olfactory bulbs. Biol. Open 2017, 6, 1552–1568. [Google Scholar] [CrossRef]
- Messina, A.; Giacobini, P. Semaphorin Signaling in the Development and Function of the Gonadotropin Hormone-Releasing Hormone System. Front. Endocrinol. 2013, 4, 133. [Google Scholar] [CrossRef]
- Lettieri, A.; Oleari, R.; Gimmelli, J.; André, V.; Cariboni, A. The role of semaphorin signaling in the etiology of hypogonadotropic hypogonadism. Minerva Endocrinol. 2016, 41, 266–278. [Google Scholar]
- Oleari, R.; Lettieri, A.; Paganoni, A.; Zanieri, L.; Cariboni, A. Semaphorin Signaling in GnRH Neurons: From Development to Disease. Neuroendocrinology 2018, 109, 193–199. [Google Scholar] [CrossRef]
- Prevot, V. Puberty in Mice and Rats. In Knobil and Neill’s Physiology of Reproduction; Elsevier: Amsterdam, The Netherlands, 2015; Volume 2, pp. 1395–1439. ISBN 9780123977694. [Google Scholar]
- Pierce, A.; Bliesner, B.; Xu, M.; Nielsen-Preiss, S.; Lemke, G.; Tobet, S.; Wierman, M.E. Axl and Tyro3 Modulate Female Reproduction by Influencing Gonadotropin-Releasing Hormone Neuron Survival and Migration. Mol. Endocrinol. 2008, 22, 2481–2495. [Google Scholar] [CrossRef]
- Muscatelli, F.; Abrous, D.N.; Massacrier, A.; Boccaccio, I.; Le Moal, M.; Cau, P.; Cremer, H. Disruption of the mouse Necdin gene results in hypothalamic and behavioral alterations reminiscent of the human Prader-Willi syndrome. Hum. Mol. Genet. 2000, 9, 3101–3110. [Google Scholar] [CrossRef]
- Cogliati, T.; Delgado-Romero, P.; Norwitz, E.R.; Guduric-Fuchs, J.; Kaiser, U.B.; Wray, S.; Kirsch, I.R. Pubertal Impairment in Nhlh2 Null Mice Is Associated with Hypothalamic and Pituitary Deficiencies. Mol. Endocrinol. 2007, 21, 3013–3027. [Google Scholar] [CrossRef] [PubMed]
- Cariboni, A.; André, V.; Chauvet, S.; Cassatella, D.; Davidson, K.; Caramello, A.; Fantin, A.; Bouloux, P.; Mann, F.; Ruhrberg, C. Dysfunctional SEMA3E signaling underlies gonadotropin-releasing hormone neuron deficiency in Kallmann syndrome. J. Clin. Investig. 2015, 125, 2413–2428. [Google Scholar] [CrossRef] [PubMed]
- Pitteloud, N.; Zhang, C.; Pignatelli, D.; Li, J.-D.; Raivio, T.; Cole, L.W.; Plummer, L.; Jacobson-Dickman, E.E.; Mellon, P.L.; Zhou, Q.-Y.; et al. Loss-of-function mutation in the prokineticin 2 gene causes Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc. Natl. Acad. Sci. USA 2007, 104, 17447–17452. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.C.; Tsai, P.-S. Expression of a Dominant Negative FGF Receptor in Developing GNRH1 Neurons Disrupts Axon Outgrowth and Targeting to the Median Eminence. Biol. Reprod. 2006, 74, 463–472. [Google Scholar] [CrossRef]
- Tsai, P.-S.; Moenter, S.M.; Postigo, H.R.; El Majdoubi, M.; Pak, T.R.; Gill, J.C.; Paruthiyil, S.; Werner, S.; Weiner, R.I. Targeted Expression of a Dominant-Negative Fibroblast Growth Factor (FGF) Receptor in Gonadotropin-Releasing Hormone (GnRH) Neurons Reduces FGF Responsiveness and the Size of GnRH Neuronal Population. Mol. Endocrinol. 2005, 19, 225–236. [Google Scholar] [CrossRef]
- Low, V.F.; Fiorini, Z.; Fisher, L.; Jasoni, C.L. Netrin-1 Stimulates Developing GnRH Neurons to Extend Neurites to the Median Eminence in a Calcium- Dependent Manner. PLoS ONE 2012, 7, e46999. [Google Scholar] [CrossRef]
- Parkash, J.; Cimino, I.; Ferraris, N.; Casoni, F.; Wray, S.; Cappy, H.; Prevot, V.; Giacobini, P. Suppression of β1-integrin in gonadotropin-releasing hormone cells disrupts migration and axonal extension resulting in severe reproductive alterations. J. Neurosci. 2012, 32, 16992–17002. [Google Scholar] [CrossRef]
- Herde, M.; Iremonger, K.; Constantin, S.; Herbison, A.E. GnRH Neurons Elaborate a Long-Range Projection with Shared Axonal and Dendritic Functions. J. Neurosci. 2013, 33, 12689–12697. [Google Scholar] [CrossRef]
- Moore, A.M.; Prescott, M.; Czieselsky, K.; Desroziers, E.; Yip, S.H.; Campbell, R.E.; Herbison, A.E. Synaptic Innervation of the GnRH Neuron Distal Dendron in Female Mice. Endocrinology 2018, 159, 3200–3208. [Google Scholar] [CrossRef]
- Wang, L.; Guo, W.; Shen, X.; Yeo, S.; Long, H.; Wang, Z.; Lyu, Q.; Herbison, A.E.; Kuang, Y. Different dendritic domains of the GnRH neuron underlie the pulse and surge modes of GnRH secretion in female mice. eLife 2020, 9, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Clarke, I.; Arbabi, L. New concepts of the central control of reproduction, integrating influence of stress, metabolic state, and season. Domest. Anim. Endocrinol. 2016, 56, S165–S179. [Google Scholar] [CrossRef]
- Herbison, A.E. Control of puberty onset and fertility by gonadotropin-releasing hormone neurons. Nat. Rev. Endocrinol. 2016, 12, 452–466. [Google Scholar] [CrossRef] [PubMed]
- Spergel, D.J. Neuropeptidergic modulation of GnRH neuronal activity and GnRH secretion controlling reproduction: Insights from recent mouse studies. Cell Tissue Res. 2018, 375, 179–191. [Google Scholar] [CrossRef]
- Cravo, R.; Margatho, L.; Osborne-Lawrence, S.; Donato, J.; Atkin, S.; Bookout, A.; Rovinsky, S.; Frazao, R.; Lee, C.; Gautron, L.; et al. Characterization of Kiss1 neurons using transgenic mouse models. Neuroscience 2011, 173, 37–56. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.M.; Coolen, L.M.; Porter, D.T.; Goodman, R.L.; Lehman, M.N. KNDy Cells Revisited. Endocrinology 2018, 159, 3219–3234. [Google Scholar] [CrossRef] [PubMed]
- Navarro, V.M.; Gottsch, M.L.; Chavkin, C.; Okamura, H.; Clifton, D.K.; Steiner, R.A. Regulation of Gonadotropin-Releasing Hormone Secretion by Kisspeptin/Dynorphin/Neurokinin B Neurons in the Arcuate Nucleus of the Mouse. J. Neurosci. 2009, 29, 11859–11866. [Google Scholar] [CrossRef]
- Marino, M.; Moriondo, V.; Vighi, E.; Pignatti, E.; Simoni, M. Central Hypogonadotropic Hypogonadism: Genetic Complexity of a Complex Disease. Int. J. Endocrinol. 2014, 2014, 649154. [Google Scholar] [CrossRef] [PubMed]
- Stamou, M.I.; Georgopoulos, N.A. Kallmann syndrome: Phenotype and genotype of hypogonadotropic hypogonadism. Metabolism 2017, 86, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Young, J.; Xu, C.; Papadakis, G.E.; Acierno, J.S.; Maione, L.; Hietamäki, J.; Raivio, T.; Pitteloud, N. Clinical Management of Congenital Hypogonadotropic Hypogonadism. Endocr. Rev. 2019, 40, 669–710. [Google Scholar] [CrossRef] [PubMed]
- Cangiano, B.; Swee, D.S.; Quinton, R.; Bonomi, M. Genetics of congenital hypogonadotropic hypogonadism: Peculiarities and phenotype of an oligogenic disease. Hum. Ganet. 2020, 140, 77–111. [Google Scholar] [CrossRef] [PubMed]
- Renault, C.H.; Aksglaede, L.; Wøjdemann, D.; Hansen, A.B.; Jensen, R.B.; Juul, A. Minipuberty of human infancy—A window of opportunity to evaluate hypogonadism and differences of sex development? Ann. Pediatr. Endocrinol. Metab. 2020, 25, 84–91. [Google Scholar] [CrossRef]
- Kuiri-Hänninen, T.; Sankilampi, U.; Dunkel, L. Activation of the Hypothalamic-Pituitary-Gonadal Axis in Infancy: Minipuberty. Horm. Res. Paediatr. 2014, 82, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Pitteloud, N. Congenital Hypogonadotropic Hypogonadism (Isolated GnRH Deficiency). In Contemporary Endocrinology; Springer: Berlin/Heidelberg, Germany, 2019; pp. 229–250. [Google Scholar] [CrossRef]
- Varimo, T.; Hero, M.; Laitinen, E.-M.; Sintonen, H.; Raivio, T. Health-related quality of life in male patients with congenital hypogonadotropic hypogonadism. Clin. Endocrinol. 2015, 83, 141–143. [Google Scholar] [CrossRef] [PubMed]
- Laitinen, E.-M.; Hero, M.; Vaaralahti, K.; Tommiska, J.; Raivio, T. Bone mineral density, body composition and bone turnover in patients with congenital hypogonadotropic hypogonadism. Int. J. Androl. 2012, 35, 534–540. [Google Scholar] [CrossRef]
- Brand, J.S.; Rovers, M.; Yeap, B.B.; Schneider, H.J.; Tuomainen, T.-P.; Haring, R.; Corona, G.; Onat, A.; Maggio, M.; Bouchard, C.; et al. Testosterone, Sex Hormone-Binding Globulin and the Metabolic Syndrome in Men: An Individual Participant Data Meta-Analysis of Observational Studies. PLoS ONE 2014, 9, e100409. [Google Scholar] [CrossRef] [PubMed]
- Raivio, T.; Falardeau, J.; Dwyer, A.; Quinton, R.; Hayes, F.J.; Hughes, V.A.; Cole, L.W.; Pearce, S.; Lee, H.; Boepple, P.; et al. Reversal of Idiopathic Hypogonadotropic Hypogonadism. N. Engl. J. Med. 2007, 357, 863. [Google Scholar] [CrossRef] [PubMed]
- Sidhoum, V.F.; Chan, Y.-M.; Lippincott, M.; Balasubramanian, R.; Quinton, R.; Plummer, L.; Dwyer, A.; Pitteloud, N.; Hayes, F.J.; Hall, J.; et al. Reversal and Relapse of Hypogonadotropic Hypogonadism: Resilience and Fragility of the Reproductive Neuroendocrine System. J. Clin. Endocrinol. Metab. 2014, 99, 861–870. [Google Scholar] [CrossRef]
- Dodé, C.; Hardelin, J.-P. Kallmann syndrome. Eur. J. Hum. Genet. 2009, 17, 139–146. [Google Scholar] [CrossRef]
- Kim, S.-H. Congenital Hypogonadotropic Hypogonadism and Kallmann Syndrome: Past, Present, and Future. Endocrinol. Metab. 2015, 30, 456–466. [Google Scholar] [CrossRef]
- Nachtigall, L.B.; Boepple, P.A.; Pralong, F.P.; Crowley, W.F. Adult-Onset Idiopathic Hypogonadotropic Hypogonadism—A Treatable Form of Male Infertility. N. Engl. J. Med. 1997, 336, 410–415. [Google Scholar] [CrossRef]
- Tajar, A.; Forti, G.; O’Neill, T.W.; Lee, D.M.; Silman, A.J.; Finn, J.D.; Bartfai, G.; Boonen, S.; Casanueva, F.F.; Giwercman, A.; et al. Characteristics of Secondary, Primary, and Compensated Hypogonadism in Aging Men: Evidence from the European Male Ageing Study. J. Clin. Endocrinol. Metab. 2010, 95, 1810–1818. [Google Scholar] [CrossRef]
- Cangiano, B.; Duminuco, P.; Vezzoli, V.; Guizzardi, F.; Chiodini, I.; Corona, G.; Maggi, M.; Persani, L.; Bonomi, M. Evidence for a Common Genetic Origin of Classic and Milder Adult-Onset Forms of Isolated Hypogonadotropic Hypogonadism. J. Clin. Med. 2019, 8, 126. [Google Scholar] [CrossRef] [PubMed]
- Pitteloud, N.; Quinton, R.; Pearce, S.; Raivio, T.; Acierno, J.; Dwyer, A.; Plummer, L.; Hughes, V.; Seminara, S.; Cheng, Y.-Z.; et al. Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. J. Clin. Investig. 2007, 117, 457–463. [Google Scholar] [CrossRef]
- Sykiotis, G.; Plummer, L.; Hughes, V.A.; Au, M.; Durrani, S.; Nayak-Young, S.; Dwyer, A.; Quinton, R.; Hall, J.; Gusella, J.F.; et al. Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc. Natl. Acad. Sci. USA 2010, 107, 15140–15144. [Google Scholar] [CrossRef]
- Cassatella, D.; Howard, S.; Acierno, J.; Xu, C.; Papadakis, G.E.; Santoni, F.; Dwyer, A.; Santini, S.; Sykiotis, G.; Chambion, C.; et al. Congenital hypogonadotropic hypogonadism and constitutional delay of growth and puberty have distinct genetic architectures. Eur. J. Endocrinol. 2018, 178, 377–388. [Google Scholar] [CrossRef]
- Maione, L.; Dwyer, A.; Francou, B.; Guiochon-Mantel, A.; Binart, N.; Bouligand, J.; Young, J. Genetics in Endocrinology: Genetic counseling for congenital hypogonadotropic hypogonadism and Kallmann syndrome: New challenges in the era of oligogenism and next-generation sequencing. Eur. J. Endocrinol. 2018, 178, R55–R80. [Google Scholar] [CrossRef]
- Balasubramanian, R.; Crowley, W.F. Isolated Gonadotropin-Releasing Hormone (GnRH) Deficiency. In GeneReviews®; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Constantin, S. Physiology of the Gonadotrophin-Releasing Hormone (GnRH) Neurone: Studies from Embryonic GnRH Neurones. J. Neuroendocr. 2011, 23, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Palevitch, O.; Kight, K.; Abraham, E.; Wray, S.; Zohar, Y.; Gothilf, Y. Ontogeny of the GnRH systems in zebrafish brain: In situ hybridization and promoter-reporter expression analyses in intact animals. Cell Tissue Res. 2006, 327, 313–322. [Google Scholar] [CrossRef]
- Maggi, R.; Pimpinelli, F.; Molteni, L.; Milani, M.; Martini, L.; Piva, F. Immortalized Luteinizing Hormone-Releasing Hormone Neurons Show a Different Migratory Activity in Vitro. Endocrinology 2000, 141, 2105–2112. [Google Scholar] [CrossRef][Green Version]
- Louden, E.D.; Poch, A.; Kim, H.-G.; Ben-Mahmoud, A.; Kim, S.-H.; Layman, L.C. Genetics of hypogonadotropic Hypogonadism—Human and mouse genes, inheritance, oligogenicity, and genetic counseling. Mol. Cell. Endocrinol. 2021, 534, 111334. [Google Scholar] [CrossRef] [PubMed]
- Mason, A.; Hayflick, J.; Zoeller, R.; Young, W.; Phillips, H.; Nikolics, K.; Seeburg, P. A deletion truncating the gonadotropin-releasing hormone gene is responsible for hypogonadism in the hpg mouse. Science 1986, 234, 1366–1371. [Google Scholar] [CrossRef]
- Bouligand, J.; Ghervan, C.; Tello, J.A.; Brailly-Tabard, S.; Salenave, S.; Chanson, P.; Lombès, M.; Millar, R.P.; Guiochon-Mantel, A.; Young, J. Isolated Familial Hypogonadotropic Hypogonadism and aGNRH1Mutation. N. Engl. J. Med. 2009, 360, 2742–2748. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.-M.; de Guillebon, A.; Lang-Muritano, M.; Plummer, L.; Cerrato, F.; Tsiaras, S.; Gaspert, A.; Lavoie, H.B.; Wu, C.-H.; Crowley, W.F.; et al. GNRH1mutations in patients with idiopathic hypogonadotropic hypogonadism. Proc. Natl. Acad. Sci. USA 2009, 106, 11703–11708. [Google Scholar] [CrossRef] [PubMed]
- Mengen, E.; Tunc, S.; Kotan, L.D.; Nalbantoglu, O.; Demir, K.; Gürbüz, F.; Turan, I.; Seker, G.; Yuksel, B.; Topaloglu, A.K. Complete Idiopathic Hypogonadotropic Hypogonadism due to Homozygous GNRH1 Mutations in the Mutational Hot Spots in the Region Encoding the Decapeptide. Horm. Res. Paediatr. 2015, 85, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Horikoshi, M.; Day, F.R.; Akiyama, M.; Hirata, M.; Kamatani, Y.; Matsuda, K.; Ishigaki, K.; Kanai, M.; Wright, H.; Toro, C.A.; et al. Elucidating the genetic architecture of reproductive ageing in the Japanese population. Nat. Commun. 2018, 9, 1977. [Google Scholar] [CrossRef] [PubMed]
- Messager, S.; Chatzidaki, E.E.; Ma, D.; Hendrick, A.; Zahn, D.; Dixon, J.; Thresher, R.R.; Malinge, I.; Lomet, D.; Carlton, M.B.L.; et al. Kisspeptin directly stimulates gonadotropin-releasing hormone release via G protein-coupled receptor 54. Proc. Natl. Acad. Sci. USA 2005, 102, 1761–1766. [Google Scholar] [CrossRef]
- Topaloglu, A.K.; Tello, J.A.; Kotan, L.D.; Ozbek, M.N.; Yilmaz, M.B.; Erdogan, S.; Gürbüz, F.; Temiz, F.; Millar, R.P.; Yuksel, B. InactivatingKISS1Mutation and Hypogonadotropic Hypogonadism. N. Engl. J. Med. 2012, 366, 629–635. [Google Scholar] [CrossRef]
- De Roux, N.; Genin, E.; Carel, J.-C.; Matsuda, F.; Chaussain, J.-L.; Milgrom, E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc. Natl. Acad. Sci. USA 2003, 100, 10972–10976. [Google Scholar] [CrossRef]
- Seminara, S.B.; Messager, S.; Chatzidaki, E.E.; Thresher, R.R.; Acierno, J.S.; Shagoury, J.K.; Bo-Abbas, Y.; Kuohung, W.; Schwinof, K.M.; Hendrick, A.; et al. TheGPR54Gene as a Regulator of Puberty. N. Engl. J. Med. 2003, 349, 1614–1627. [Google Scholar] [CrossRef] [PubMed]
- Colledge, W. Transgenic mouse models to study Gpr54/kisspeptin physiology. Peptides 2009, 30, 34–41. [Google Scholar] [CrossRef] [PubMed]
- d’Anglemont de Tassigny, X.D.; Fagg, L.A.; Dixon, J.P.C.; Day, K.; Leitch, H.; Hendrick, A.; Zahn, D.; Franceschini, I.; Caraty, A.; Carlton, M.B.L.; et al. Hypogonadotropic hypogonadism in mice lacking a functional Kiss1 gene. Proc. Natl. Acad. Sci. USA 2007, 104, 10714–10719. [Google Scholar] [CrossRef] [PubMed]
- Lapatto, R.; Pallais, J.C.; Zhang, D.; Chan, Y.-M.; Mahan, A.; Cerrato, F.; Le, W.W.; Hoffman, G.E.; Seminara, S.B. Kiss1−/− Mice Exhibit More Variable Hypogonadism than Gpr54−/− Mice. Endocrinology 2007, 148, 4927–4936. [Google Scholar] [CrossRef]
- Chan, Y.-M.; Broder-Fingert, S.; Wong, K.M.; Seminara, S.B. Kisspeptin/Gpr54-Independent Gonadotrophin-Releasing Hormone Activity inKiss1andGpr54Mutant Mice. J. Neuroendocr. 2009, 21, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Fujiyama, T.; Miyashita, S.; Tsuneoka, Y.; Kanemaru, K.; Kakizaki, M.; Kanno, S.; Ishikawa, Y.; Yamashita, M.; Owa, T.; Nagaoka, M.; et al. Forebrain Ptf1a Is Required for Sexual Differentiation of the Brain. Cell Rep. 2018, 24, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, D.E.; Oleari, R.; Gregory, L.; Le Quesne-Stabej, P.; Williams, H.; GOSgene; Torpiano, J.; Formosa, N.; Cachia, M.; Field, D.; et al. A recessive PRDM13 mutation results in congenital hypogonadotropic hypogonadism and cerebellar hypoplasia. medRxiv 2021. [Google Scholar] [CrossRef]
- Hrabovszky, E.; Takács, S.; Göcz, B.; Skrapits, K. New Perspectives for Anatomical and Molecular Studies of Kisspeptin Neurons in the Aging Human Brain. Neuroendocrinology 2019, 109, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.A.; Dhillo, W.S. Kisspeptin across the human lifespan: Evidence from animal studies and beyond. J. Endocrinol. 2016, 229, R83–R98. [Google Scholar] [CrossRef]
- Topaloglu, A.K.; Reimann, F.; Guclu, M.; Yalin, A.S.; Kotan, L.D.; Porter, K.M.; Serin, A.; Mungan, N.O.; Cook, J.; Imamoglu, S.; et al. TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for Neurokinin B in the central control of reproduction. Nat. Genet. 2008, 41, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Young, J.; Bouligand, J.; Francou, B.; Raffin-Sanson, M.-L.; Gaillez, S.; Jeanpierre, M.; Grynberg, M.; Kamenicky, P.; Chanson, P.; Brailly-Tabard, S.; et al. TAC3andTACR3Defects Cause Hypothalamic Congenital Hypogonadotropic Hypogonadism in Humans. J. Clin. Endocrinol. Metab. 2010, 95, 2287–2295. [Google Scholar] [CrossRef]
- Gianetti, E.; Tusset, C.; Noel, S.D.; Au, M.G.; Dwyer, A.; Hughes, V.A.; Abreu, A.P.; Carroll, J.; Trarbach, E.; Silveira, L.; et al. TAC3/TACR3 Mutations Reveal Preferential Activation of Gonadotropin-Releasing Hormone Release by Neurokinin B in Neonatal Life Followed by Reversal in Adulthood. J. Clin. Endocrinol. Metab. 2010, 95, 2857–2867. [Google Scholar] [CrossRef]
- True, C.; Alam, S.N.; Cox, K.; Chan, Y.-M.; Seminara, S.B. Neurokinin B Is Critical for Normal Timing of Sexual Maturation but Dispensable for Adult Reproductive Function in Female Mice. Endocrinology 2015, 156, 1386–1397. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.J.; Caligioni, C.S.; Chan, Y.-M.; Seminara, S.B. Uncovering Novel Reproductive Defects in Neurokinin B Receptor Null Mice: Closing the Gap Between Mice and Men. Endocrinology 2012, 153, 1498–1508. [Google Scholar] [CrossRef] [PubMed]
- Strobel, A.; Issad, T.; Camoin, L.; Ozata, M.; Strosberg, A.D. A leptin missense mutation associated with hypogonadism and morbid obesity. Nat. Genet. 1998, 18, 213–215. [Google Scholar] [CrossRef]
- Clément, K.; Vaisse, C.; Lahlou, N.; Cabrol, S.; Pelloux, V.; Cassuto, D.; Gourmelen, M.; Dina, C.; Chambaz, J.; Lacorte, J.-M.; et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature 1998, 392, 398–401. [Google Scholar] [CrossRef]
- Manfredi-Lozano, M.; Roa, J.; Tena-Sempere, M. Connecting metabolism and gonadal function: Novel central neuropeptide pathways involved in the metabolic control of puberty and fertility. Front. Neuroendocr. 2018, 48, 37–49. [Google Scholar] [CrossRef]
- Quennell, J.H.; Mulligan, A.C.; Tups, A.; Liu, X.; Phipps, S.J.; Kemp, C.J.; Herbison, A.; Grattan, D.; Anderson, G.M. Leptin Indirectly Regulates Gonadotropin-Releasing Hormone Neuronal Function. Endocrinology 2009, 150, 2805–2812. [Google Scholar] [CrossRef]
- Landry, D.; Cloutier, F.; Martin, L.J. Implications of leptin in neuroendocrine regulation of male reproduction. Reprod. Biol. 2013, 13, 1–14. [Google Scholar] [CrossRef]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Zhao, C.; Cai, X.; Montez, J.M.; Rohani, S.C.; Feinstein, P.; Mombaerts, P.; Friedman, J.M. Selective deletion of leptin receptor in neurons leads to obesity. J. Clin. Investig. 2001, 108, 1113–1121. [Google Scholar] [CrossRef]
- Stijnen, P.; Ramos-Molina, B.; O’Rahilly, S.; Creemers, J.W.M. PCSK1 Mutations and Human Endocrinopathies: From Obesity to Gastrointestinal Disorders. Endocr. Rev. 2016, 37, 347–371. [Google Scholar] [CrossRef] [PubMed]
- O’Rahilly, S.; Gray, H.; Humphreys, P.J.; Krook, A.; Polonsky, K.S.; White, A.; Gibson, S.; Taylor, K.; Carr, C. Brief Report: Impaired Processing of Prohormones Associated with Abnormalities of Glucose Homeostasis and Adrenal Function. N. Engl. J. Med. 1995, 333, 1386–1391. [Google Scholar] [CrossRef]
- Jackson, R.S.; Creemers, J.W.M.; Ohagi, S.; Raffin-Sanson, M.-L.; Sanders, L.; Montague, C.; Hutton, J.C.; O’Rahilly, S. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat. Genet. 1997, 16, 303–306. [Google Scholar] [CrossRef]
- Farooqi, S.; O’Rahilly, S. Genetics of Obesity in Humans. Endocr. Rev. 2006, 27, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, I.S.; Volders, K.; Stanhope, R.; Heuschkel, R.; White, A.; Lank, E.; Keogh, J.; O’Rahilly, S.; Creemers, J.W.M. Hyperphagia and Early-Onset Obesity due to a Novel Homozygous Missense Mutation in Prohormone Convertase 1/3. J. Clin. Endocrinol. Metab. 2007, 92, 3369–3373. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.D.; Malabunga, M.; Tonra, J.R.; DiRenzo, R.; Carrick, F.E.; Zheng, H.; Berthoud, H.-R.; McGuinness, O.P.; Shen, J.; Bohlen, P.; et al. Monoclonal antibody antagonists of hypothalamic FGFR1 cause potent but reversible hypophagia and weight loss in rodents and monkeys. Am. J. Physiol. Metab. 2007, 292, E964–E976. [Google Scholar] [CrossRef]
- Tacer, K.F.; Bookout, A.L.; Ding, X.; Kurosu, H.; John, G.B.; Wang, L.; Goetz, R.; Mohammadi, M.; Kuro-O, M.; Mangelsdorf, D.; et al. Research Resource: Comprehensive Expression Atlas of the Fibroblast Growth Factor System in Adult Mouse. Mol. Endocrinol. 2010, 24, 2050–2064. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Messina, A.; Somm, E.; Miraoui, H.; Kinnunen, T.K.; Acierno, J.; Niederländer, N.J.; Bouilly, J.; Dwyer, A.; Sidis, Y.; et al. KLB, encoding β-Klotho, is mutated in patients with congenital hypogonadotropic hypogonadism. EMBO Mol. Med. 2017, 9, 1379–1397. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, Z. Current understanding of klotho. Ageing Res. Rev. 2009, 8, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Kuro-O, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-M.; Gao, J.-Z.; He, B.; Long, W.-J.; Luo, X.-P.; Chen, L. A Novel NR0B1 Gene Mutation Causes Different Phenotypes in Two Male Patients with Congenital Adrenal Hypoplasia. Curr. Med. Sci. 2020, 40, 172–177. [Google Scholar] [CrossRef]
- Reutens, A.T.; Achermann, J.C.; Ito, M.; Ito, M.; Gu, W.-X.; Habiby, R.L.; Donohoue, P.A.; Pang, S.; Hindmarsh, P.C.; Jameson, J.L. Clinical and Functional Effects of Mutations in theDAX-1Gene in Patients with Adrenal Hypoplasia Congenita. J. Clin. Endocrinol. Metab. 1999, 84, 504–511. [Google Scholar] [CrossRef]
- Iyer, A.K.; McCabe, E.R. Molecular mechanisms of DAX1 action. Mol. Genet. Metab. 2004, 83, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Liu, R.; Zhang, H.; Yang, J.; Sun, S.; Zhang, M.; Liu, Y.; Lu, Y.; Wang, W.; Mu, Y.; et al. Seven Novel DAX1 Mutations with Loss of Function Identified in Chinese Patients with Congenital Adrenal Hypoplasia. J. Clin. Endocrinol. Metab. 2010, 95, E104–E111. [Google Scholar] [CrossRef]
- Achermann, J.; Meeks, J.J.; Jameson, J.L. Phenotypic spectrum of mutations in DAX-1 and SF-1. Mol. Cell. Endocrinol. 2001, 185, 17–25. [Google Scholar] [CrossRef]
- Muscatelli, F.; Strom, T.M.; Walker, A.P.; Zanaria, E.; Récan, D.; Meindl, A.; Bardoni, B.; Guioli, S.; Zehetner, G.; Rabl, W.; et al. Mutations in the DAX-1 gene give rise to both X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism. Nature 1994, 372, 672–676. [Google Scholar] [CrossRef]
- Seminara, S.B.; Achermann, J.C.; Genel, M.; Jameson, J.L.; Crowley, W.F. X-Linked Adrenal Hypoplasia Congenita: A Mutation in DAX1 Expands the Phenotypic Spectrum in Males and Females. J. Clin. Endocrinol. Metab. 1999, 84, 4501–4509. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tata, B.; Huijbregts, L.; Jacquier, S.; Csaba, Z.; Genin, E.; Meyer, V.; Leka, S.; Dupont, J.; Charles, P.; Chevenne, D.; et al. Haploinsufficiency of Dmxl2, Encoding a Synaptic Protein, Causes Infertility Associated with a Loss of GnRH Neurons in Mouse. PLoS Biol. 2014, 12, e1001952. [Google Scholar] [CrossRef] [PubMed]
- Tata, B.K.; Harbulot, C.; Csaba, Z.; Peineau, S.; Jacquier, S.; De Roux, N. Rabconnectin-3α is required for the morphological maturation of GnRH neurons and kisspeptin responsiveness. Sci. Rep. 2017, 7, 42463. [Google Scholar] [CrossRef]
- Winrow, C.J.; Hemming, M.L.; Allen, D.M.; Quistad, G.B.; Casida, J.E.; Barlow, C. Loss of neuropathy target esterase in mice links organophosphate exposure to hyperactivity. Nat. Genet. 2003, 33, 477–485. [Google Scholar] [CrossRef]
- Topaloglu, A.K.; Lomniczi, A.; Kretzschmar, D.; Dissen, G.A.; Kotan, L.D.; McArdle, C.A.; Koc, A.F.; Hamel, B.C.; Guclu, M.; Papatya, E.D.; et al. Loss-of-Function Mutations inPNPLA6Encoding Neuropathy Target Esterase Underlie Pubertal Failure and Neurological Deficits in Gordon Holmes Syndrome. J. Clin. Endocrinol. Metab. 2014, 99, E2067–E2075. [Google Scholar] [CrossRef]
- Margolin, D.H.; Kousi, M.; Chan, Y.-M.; Lim, T.T.; Schmahmann, J.D.; Hadjivassiliou, M.; Hall, J.; Adam, I.; Dwyer, A.; Plummer, L.; et al. Ataxia, Dementia, and Hypogonadotropism Caused by Disordered Ubiquitination. N. Engl. J. Med. 2013, 368, 1992–2003. [Google Scholar] [CrossRef]
- Shi, C.-H.; Schisler, J.; Rubel, C.E.; Tan, S.; Song, B.; McDonough, H.; Xu, L.; Portbury, A.L.; Mao, C.-Y.; True, C.; et al. Ataxia and hypogonadism caused by the loss of ubiquitin ligase activity of the U box protein CHIP. Hum. Mol. Genet. 2013, 23, 1013–1024. [Google Scholar] [CrossRef]
- Melnick, A.; Gao, Y.; Liu, J.; Ding, D.; Predom, A.; Kelly, C.; Hess, R.A.; Chen, C. RNF216 is essential for spermatogenesis and male fertility. Biol. Reprod. 2019, 100, 1132–1134. [Google Scholar] [CrossRef]
- Li, F.; Li, D.; Liu, H.; Cao, B.-B.; Jiang, F.; Chen, D.-N.; Li, J.-D. RNF216 Regulates the Migration of Immortalized GnRH Neurons by Suppressing Beclin1-Mediated Autophagy. Front. Endocrinol. 2019, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- Dumay-Odelot, H.; Durrieu-Gaillard, S.; Da Silva, D.; Roeder, R.G.; Teichmann, M. Cell growth- and differentiation-dependent regulation of RNA polymerase III transcription. Cell Cycle 2010, 9, 3711–3723. [Google Scholar] [CrossRef]
- Wolff, A.; Koch, M.J.; Benzinger, S.; Van Waes, H.; Wolf, N.I.; Boltshauser, E.; Luder, H.U. Rare dental peculiarities associated with the hypomyelinating leukoencephalopathy 4H syndrome/ADDH. Pediatr. Dent. 2010, 32, 386–392. [Google Scholar] [PubMed]
- Daoud, H.; Tétreault, M.; Gibson, W.; Guerrero, K.; Cohen, A.; Gburek-Augustat, J.; Synofzik, M.; Brais, B.; Stevens, C.A.; Sanchez-Carpintero, R.; et al. Mutations inPOLR3AandPOLR3Bare a major cause of hypomyelinating leukodystrophies with or without dental abnormalities and/or hypogonadotropic hypogonadism. J. Med. Genet. 2013, 50, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Choquet, K.; Yang, S.; Moir, R.D.; Forget, D.; Larivière, R.; Bouchard, A.; Poitras, C.; Sgarioto, N.; Dicaire, M.-J.; Noohi, F.; et al. Absence of neurological abnormalities in mice homozygous for the Polr3a G672E hypomyelinating leukodystrophy mutation. Mol. Brain 2017, 10, 13. [Google Scholar] [CrossRef]
- Choquet, K.; Pinard, M.; Yang, S.; Moir, R.D.; Poitras, C.; Dicaire, M.-J.; Sgarioto, N.; Larivière, R.; Kleinman, C.L.; Willis, I.M.; et al. The leukodystrophy mutation Polr3b R103H causes homozygote mouse embryonic lethality and impairs RNA polymerase III biogenesis. Mol. Brain 2019, 12, 59. [Google Scholar] [CrossRef]
- Franco, B.; Guioli, S.; Pragliola, A.; Incerti, B.; Bardoni, B.; Tonlorenzi, R.; Carrozzo, R.; Maestrini, E.; Pieretti, M.; Taillon-Miller, P.; et al. A gene deleted in Kallmann’s syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature 1991, 353, 529–536. [Google Scholar] [CrossRef]
- Schwanzel-Fukuda, M.; Bick, D.; Pfaff, D.W. Luteinizing hormone-releasing hormone (LHRH)-expressing cells do not migrate normally in an inherited hypogonadal (Kallmann) syndrome. Brain Res. Mol. Brain Res. 1989, 6, 311–326. [Google Scholar] [CrossRef]
- Bulow, H.E.; Berry, K.; Topper, L.H.; Peles, E.; Hobert, O. Heparan sulfate proteoglycan-dependent induction of axon branching and axon misrouting by the Kallmann syndrome gene kal-1. Proc. Natl. Acad. Sci. USA 2002, 99, 6346–6351. [Google Scholar] [CrossRef] [PubMed]
- Soussi-Yanicostas, N.; de Castro, F.; Julliard, A.K.; Perfettini, I.; Chedotal, A.; Petit, C. Anosmin-1, Defective in the X-Linked Form of Kallmann Syndrome, Promotes Axonal Branch Formation from Olfactory Bulb Output Neurons. Cell 2002, 109, 217–228. [Google Scholar] [CrossRef]
- Cariboni, A.; Pimpinelli, F.; Colamarino, S.; Zaninetti, R.; Piccolella, M.; Rumio, C.; Piva, F.; Rugarli, E.I.; Maggi, R. The product of X-linked Kallmann’s syndrome gene (KAL1) affects the migratory activity of gonadotropin-releasing hormone (GnRH)-producing neurons. Hum. Mol. Genet. 2004, 13, 2781–2791. [Google Scholar] [CrossRef]
- Azzarelli, R.; Oleari, R.; Lettieri, A.; Andre’, V.; Cariboni, A. In Vitro, Ex Vivo and In Vivo Techniques to Study Neuronal Migration in the Developing Cerebral Cortex. Brain Sci. 2017, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Costa-Barbosa, F.A.; Balasubramanian, R.; Keefe, K.W.; Shaw, N.; Al Tassan, N.; Plummer, L.; Dwyer, A.; Buck, C.L.; Choi, J.-H.; Seminara, S.B.; et al. Prioritizing Genetic Testing in Patients With Kallmann Syndrome Using Clinical Phenotypes. J. Clin. Endocrinol. Metab. 2013, 98, E943–E953. [Google Scholar] [CrossRef]
- Layman, L.C. Mutations in human gonadotropin genes and their physiologic significance in puberty and reproduction. Fertil. Steril. 1999, 71, 201–218. [Google Scholar] [CrossRef]
- Tukiainen, T.; GTEx Consortium; Villani, A.-C.; Yen, A.; Rivas, M.A.; Marshall, J.L.; Satija, R.; Aguirre, M.; Gauthier, L.; Fleharty, M.; et al. Landscape of X chromosome inactivation across human tissues. Nature 2017, 550, 244–248. [Google Scholar] [CrossRef]
- Dodé, C.; Levilliers, J.; Dupont, J.-M.; De Paepe, A.; Le Dû, N.; Soussi-Yanicostas, N.; Coimbra, R.; Delmaghani, S.; Compain-Nouaille, S.; Baverel, F.; et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat. Genet. 2003, 33, 463–465. [Google Scholar] [CrossRef]
- Tornberg, J.; Sykiotis, G.; Keefe, K.; Plummer, L.; Hoang, X.; Hall, J.; Quinton, R.; Seminara, S.B.; Hughes, V.; Van Vliet, G.; et al. Heparan sulfate 6-O-sulfotransferase 1, a gene involved in extracellular sugar modifications, is mutated in patients with idiopathic hypogonadotrophic hypogonadism. Proc. Natl. Acad. Sci. USA 2011, 108, 11524–11529. [Google Scholar] [CrossRef] [PubMed]
- Condomitti, G.; De Wit, J. Heparan Sulfate Proteoglycans as Emerging Players in Synaptic Specificity. Front. Mol. Neurosci. 2018, 11, 14. [Google Scholar] [CrossRef]
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan Sulfate Proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3, a004952. [Google Scholar] [CrossRef] [PubMed]
- Habuchi, H.; Nagai, N.; Sugaya, N.; Atsumi, F.; Stevens, R.L.; Kimata, K. Mice Deficient in Heparan Sulfate 6-O-Sulfotransferase-1 Exhibit Defective Heparan Sulfate Biosynthesis, Abnormal Placentation, and Late Embryonic Lethality. J. Biol. Chem. 2007, 282, 15578–15588. [Google Scholar] [CrossRef]
- Howard, S.; Oleari, R.; Poliandri, A.; Chantzara, V.; Fantin, A.; Ruiz-Babot, G.; Metherell, L.A.; Cabrera, C.P.; Barnes, M.R.; Wehkalampi, K.; et al. HS6ST1 Insufficiency Causes Self-Limited Delayed Puberty in Contrast with Other GnRH Deficiency Genes. J. Clin. Endocrinol. Metab. 2018, 103, 3420–3429. [Google Scholar] [CrossRef] [PubMed]
- Kramer, P.; Wray, S. Novel gene expressed in nasal region influences outgrowth of olfactory axons and migration of luteinizing hormone-releasing hormone (LHRH) neurons. Genes Dev. 2000, 14, 1824–1834. [Google Scholar] [CrossRef] [PubMed]
- Palevitch, O.; Abraham, E.; Borodovsky, N.; Levkowitz, G.; Zohar, Y.; Gothilf, Y. Nasal embryonic LHRH factor plays a role in the developmental migration and projection of gonadotropin-releasing hormone 3 neurons in zebrafish. Dev. Dyn. 2009, 238, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Quaynor, S.D.; Ko, E.K.; Chorich, L.P.; Sullivan, M.E.; Demir, D.; Waller, J.L.; Kim, H.-G.; Cameron, R.S.; Layman, L.C. NELF knockout is associated with impaired pubertal development and subfertility. Mol. Cell. Endocrinol. 2015, 407, 26–36. [Google Scholar] [CrossRef]
- Miura, K.; Acierno, J.; Seminara, S.B. Characterization of the human nasal embryonic LHRH factor gene, NELF, and a mutation screening among 65 patients with idiopathic hypogonadotropic hypogonadism (IHH). J. Hum. Genet. 2004, 49, 265–268. [Google Scholar] [CrossRef]
- Lemke, G. Biology of the TAM Receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a009076. [Google Scholar] [CrossRef] [PubMed]
- Salian-Mehta, S.; Xu, M.; Knox, A.J.; Plummer, L.; Slavov, D.; Taylor, M.; Bevers, S.; Hodges, R.S.; Crowley, W.F.; Wierman, M.E. Functional consequences of AXL sequence variants in hypogonadotropic hypogonadism. J. Clin. Endocrinol. Metab. 2014, 99, 1452–1460. [Google Scholar] [CrossRef] [PubMed]
- Eckler, M.J.; Chen, B. Fez family transcription factors: Controlling neurogenesis and cell fate in the developing mammalian nervous system. BioEssays 2014, 36, 788–797. [Google Scholar] [CrossRef] [PubMed]
- Kotan, L.D.; Hutchins, B.I.; Ozkan, Y.; Demirel, F.; Stoner, H.; Cheng, P.J.; Esen, I.; Gürbüz, F.; Bicakci, Y.K.; Mengen, E.; et al. Mutations in FEZF1 Cause Kallmann Syndrome. Am. J. Hum. Genet. 2014, 95, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Hutchins, B.I.; Kotan, L.D.; Taylor-Burds, C.; Ozkan, Y.; Cheng, P.J.; Gürbüz, F.; Tiong, J.D.R.; Mengen, E.; Yuksel, B.; Topaloglu, A.K.; et al. CCDC141 Mutation Identified in Anosmic Hypogonadotropic Hypogonadism (Kallmann Syndrome) Alters GnRH Neuronal Migration. Endocrinology 2016, 157, 1956–1966. [Google Scholar] [CrossRef]
- Turan, I.; Hutchins, B.I.; Hacihamdioglu, B.; Kotan, L.D.; Gurbuz, F.; Ulubay, A.; Mengen, E.; Yuksel, B.; Wray, S.; Topaloglu, A.K. CCDC141 Mutations in Idiopathic Hypogonadotropic Hypogonadism. J. Clin. Endocrinol. Metab. 2017, 102, 1816–1825. [Google Scholar] [CrossRef]
- Watanabe, Y.; Inoue, K.; Okuyama-Yamamoto, A.; Nakai, N.; Nakatani, J.; Nibu, K.-I.; Sato, N.; Iiboshi, Y.; Yusa, K.; Kondoh, G.; et al. Fezf1is required for penetration of the basal lamina by olfactory axons to promote olfactory development. J. Comp. Neurol. 2009, 515, 565–584. [Google Scholar] [CrossRef]
- Kuang, X.-L.; Zhao, X.-M.; Xu, H.-F.; Shi, Y.-Y.; Deng, J.-B.; Sun, G.-T. Spatio-temporal expression of a novel neuron-derived neurotrophic factor (NDNF) in mouse brains during development. BMC Neurosci. 2010, 11, 137. [Google Scholar] [CrossRef]
- Messina, A.; Pulli, K.; Santini, S.; Acierno, J.; Känsäkoski, J.; Cassatella, D.; Xu, C.; Casoni, F.; Malone, S.A.; Ternier, G.; et al. Neuron-Derived Neurotrophic Factor Is Mutated in Congenital Hypogonadotropic Hypogonadism. Am. J. Hum. Genet. 2019, 106, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Tamaoka, S.; Suzuki, E.; Hattori, A.; Ogata, T.; Fukami, M.; Katoh-Fukui, Y. NDNF variants are rare in patients with congenital hypogonadotropic hypogonadism. Hum. Genome Var. 2021, 8, 5. [Google Scholar] [CrossRef]
- Kim, H.-G.; Ahn, J.-W.; Kurth, I.; Ullmann, R.; Kim, H.-T.; Kulharya, A.; Ha, K.-S.; Itokawa, Y.; Meliciani, I.; Wenzel, W.; et al. WDR11, a WD Protein that Interacts with Transcription Factor EMX1, Is Mutated in Idiopathic Hypogonadotropic Hypogonadism and Kallmann Syndrome. Am. J. Hum. Genet. 2010, 87, 465–479. [Google Scholar] [CrossRef]
- Kim, Y.; Osborn, D.; Lee, J.; Araki, M.; Araki, K.; Mohun, T.; Känsäkoski, J.; Brandstack, N.; Kim, H.; Miralles, F.; et al. WDR11-mediated Hedgehog signalling defects underlie a new ciliopathy related to Kallmann syndrome. EMBO Rep. 2017, 19, 269–289. [Google Scholar] [CrossRef] [PubMed]
- McCormack, S.E.; Li, N.; Kim, Y.J.; Lee, J.Y.; Kim, S.-H.; Rapaport, R.; Levine, M. Digenic Inheritance of PROKR2 and WDR11 Mutations in Pituitary Stalk Interruption Syndrome. J. Clin. Endocrinol. Metab. 2017, 102, 2501–2507. [Google Scholar] [CrossRef]
- Cariboni, A.; Davidson, K.; Rakic, S.; Maggi, R.; Parnavelas, J.G.; Ruhrberg, C. Defective gonadotropin-releasing hormone neuron migration in mice lacking SEMA3A signalling through NRP1 and NRP2: Implications for the aetiology of hypogonadotropic hypogonadism. Hum. Mol. Genet. 2010, 20, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Alto, L.T.; Terman, J.R. Semaphorins and their Signaling Mechanisms. Methods Mol. Biol. 2016, 1493, 1–25. [Google Scholar] [CrossRef]
- Cariboni, A.; Hickok, J.; Rakic, S.; Andrews, W.; Maggi, R.; Tischkau, S.; Parnavelas, J.G. Neuropilins and Their Ligands Are Important in the Migration of Gonadotropin-Releasing Hormone Neurons. J. Neurosci. 2007, 27, 2387–2395. [Google Scholar] [CrossRef]
- Hanchate, N.K.; Giacobini, P.; Lhuillier, P.; Parkash, J.; Espy, C.; Fouveaut, C.; Leroy, C.; Baron, S.; Campagne, C.; Vanacker, C.; et al. SEMA3A, a Gene Involved in Axonal Pathfinding, Is Mutated in Patients with Kallmann Syndrome. PLoS Genet. 2012, 8, e1002896. [Google Scholar] [CrossRef] [PubMed]
- Oleari, R.; Caramello, A.; Campinoti, S.; Lettieri, A.; Ioannou, E.; Paganoni, A.; Fantin, A.; Cariboni, A.; Ruhrberg, C. PLXNA1 and PLXNA3 cooperate to pattern the nasal axons that guide gonadotropin-releasing hormone neurons. Development 2019, 146, dev176461. [Google Scholar] [CrossRef] [PubMed]
- Marcos, S.; Monnier, C.; Rovira, X.; Fouveaut, C.; Pitteloud, N.; Ango, F.; Dodé, C.; Hardelin, J.-P. Defective signaling through plexin-A1 compromises the development of the peripheral olfactory system and neuroendocrine reproductive axis in mice. Hum. Mol. Genet. 2017, 26, 2006–2017. [Google Scholar] [CrossRef]
- Young, J.; Metay, C.; Bouligand, J.; Tou, B.; Francou, B.; Maione, L.; Tosca, L.; Sarfati, J.; Brioude, F.; Esteva, B.; et al. SEMA3A deletion in a family with Kallmann syndrome validates the role of semaphorin 3A in human puberty and olfactory system development. Hum. Reprod. 2012, 27, 1460–1465. [Google Scholar] [CrossRef]
- Känsäkoski, J.; Fagerholm, R.; Laitinen, E.-M.; Vaaralahti, K.; Hackman, P.; Pitteloud, N.; Raivio, T.; Tommiska, J. Mutation screening of SEMA3A and SEMA7A in patients with congenital hypogonadotropic hypogonadism. Pediatr. Res. 2014, 75, 641–644. [Google Scholar] [CrossRef]
- Kotan, L.D.; Isik, E.; Turan, I.; Mengen, E.; Akkus, G.; Tastan, M.; Gurbuz, F.; Yuksel, B.; Topaloglu, A.K. Prevalence and associated phenotypes of PLXNA1 variants in normosmic and anosmic idiopathic hypogonadotropic hypogonadism. Clin. Genet. 2018, 95, 320–324. [Google Scholar] [CrossRef]
- Kotan, L.D.; Ternier, G.; Cakir, A.D.; Emeksiz, H.C.; Turan, I.; Delpouve, G.; Kardelen, A.D.; Ozcabi, B.; Isik, E.; Mengen, E.; et al. Loss-of-function variants in SEMA3F and PLXNA3 encoding semaphorin-3F and its receptor plexin-A3 respectively cause idiopathic hypogonadotropic hypogonadism. Genet. Med. 2021, 23, 1008–1016. [Google Scholar] [CrossRef]
- Takeuchi, H.; Inokuchi, K.; Aoki, M.; Suto, F.; Tsuboi, A.; Matsuda, I.; Suzuki, M.; Aiba, A.; Serizawa, S.; Yoshihara, Y.; et al. Sequential Arrival and Graded Secretion of Sema3F by Olfactory Neuron Axons Specify Map Topography at the Bulb. Cell 2010, 141, 1056–1067. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, T.; Niu, Y.; Wang, D.; Liu, K.; Wang, T.; Wang, S.; Xu, H.; Liu, J. Cell adhesion molecule L1 like plays a role in the pathogenesis of idiopathic hypogonadotropic hypogonadism. J. Endocrinol. Investig. 2021, 44, 1739–1751. [Google Scholar] [CrossRef]
- Heyden, A.; Angenstein, F.; Sallaz, M.; Seidenbecher, C.; Montag, D. Abnormal axonal guidance and brain anatomy in mouse mutants for the cell recognition molecules close homolog of L1 and NgCAM-related cell adhesion molecule. Neuroscience 2008, 155, 221–233. [Google Scholar] [CrossRef]
- Chauvet, S.; Cohen, S.; Yoshida, Y.; Fekrane, L.; Livet, J.; Gayet, O.; Segu, L.; Buhot, M.-C.; Jessell, T.M.; Henderson, C.; et al. Gating of Sema3E/PlexinD1 Signaling by Neuropilin-1 Switches Axonal Repulsion to Attraction during Brain Development. Neuron 2007, 56, 807–822. [Google Scholar] [CrossRef]
- Bellon, A.; Luchino, J.; Haigh, K.; Rougon, G.; Haigh, J.; Chauvet, S.; Mann, F. VEGFR2 (KDR/Flk1) Signaling Mediates Axon Growth in Response to Semaphorin 3E in the Developing Brain. Neuron 2010, 66, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Lalani, S.R.; Safiullah, A.M.; Molinari, L.M.; Fernbach, S.D.; Martin, D.M.; Belmont, J.W. SEMA3E mutation in a patient with CHARGE syndrome. J. Med. Genet. 2004, 41, e94. [Google Scholar] [CrossRef] [PubMed]
- Lettieri, A.; Oleari, R.; Paganoni, A.J.J.; Gervasini, C.; Massa, V.; Fantin, A.; Cariboni, A. Semaphorin Regulation by the Chromatin Remodeler CHD7: An Emerging Genetic Interaction Shaping Neural Cells and Neural Crest in Development and Cancer. Front. Cell Dev. Biol. 2021, 9, 638674. [Google Scholar] [CrossRef]
- Messina, A.; Ferraris, N.; Wray, S.; Cagnoni, G.; Donohue, D.; Casoni, F.; Kramer, P.; Derijck, A.A.; Adolfs, Y.; Fasolo, A.; et al. Dysregulation of Semaphorin7A/β1-integrin signaling leads to defective GnRH-1 cell migration, abnormal gonadal development and altered fertility. Hum. Mol. Genet. 2011, 20, 4759–4774. [Google Scholar] [CrossRef] [PubMed]
- Howard, S.; Guasti, L.; Ruiz-Babot, G.; Mancini, A.; David, A.; Storr, H.; Metherell, L.; Sternberg, M.; Cabrera, C.P.; Warren, H.R.; et al. IGSF 10 mutations dysregulate gonadotropin-releasing hormone neuronal migration resulting in delayed puberty. EMBO Mol. Med. 2016, 8, 626–642. [Google Scholar] [CrossRef]
- Sanlaville, D.; Etchevers, H.; Gonzales, M.; Martinovic, J.; Clément-Ziza, M.; Delezoide, A.-L.; Aubry, M.-C.; Pelet, A.; Chemouny, S.; Cruaud, C.; et al. Phenotypic spectrum of CHARGE syndrome in fetuses with CHD7 truncating mutations correlates with expression during human development. J. Med. Genet. 2005, 43, 211–317. [Google Scholar] [CrossRef] [PubMed]
- Layman, W.; McEwen, D.; Beyer, L.; Lalani, S.; Fernbach, S.; Oh, E.; Swaroop, A.; Hegg, C.; Raphael, Y.; Martens, J.; et al. Defects in neural stem cell proliferation and olfaction in Chd7 deficient mice indicate a mechanism for hyposmia in human CHARGE syndrome. Hum. Mol. Genet. 2009, 18, 1909–1923. [Google Scholar] [CrossRef]
- Bergman, J.E.H.; Bosman, E.A.; Van Ravenswaaij-Arts, C.M.; Steel, K.P. Study of smell and reproductive organs in a mouse model for CHARGE syndrome. Eur. J. Hum. Genet. 2009, 18, 171–177. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Layman, W.S.; Hurd, E.A.; Martin, D.M. Reproductive dysfunction and decreased GnRH neurogenesis in a mouse model of CHARGE syndrome. Hum. Mol. Genet. 2011, 20, 3138–3150. [Google Scholar] [CrossRef]
- Vissers, L.; Van Ravenswaaij, C.M.A.; Admiraal, R.; Hurst, J.A.; De Vries, B.B.A.; Janssen, I.M.; Van Der Vliet, W.A.; Huys, E.H.L.P.G.; De Jong, P.J.; Hamel, B.C.J.; et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat. Genet. 2004, 36, 955–957. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, L.; Guimiot, F.; Dodé, C.; Fallet-Bianco, C.; Millar, R.P.; Delezoide, A.-L.; Hardelin, J.-P. Defective migration of neuroendocrine GnRH cells in human arrhinencephalic conditions. J. Clin. Investig. 2010, 120, 3668–3672. [Google Scholar] [CrossRef]
- Kim, H.-G.; Kurth, I.; Lan, F.; Meliciani, I.; Wenzel, W.; Eom, S.H.; Kang, G.B.; Rosenberger, G.; Tekin, M.; Ozata, M.; et al. Mutations in CHD7, Encoding a Chromatin-Remodeling Protein, Cause Idiopathic Hypogonadotropic Hypogonadism and Kallmann Syndrome. Am. J. Hum. Genet. 2008, 83, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Jongmans, M.C.J.; Van Ravenswaaij-Arts, C.M.A.; Pitteloud, N.; Ogata, T.; Sato, N.; Claahsen-van der Grinten, H.L.; Van Der Donk, K.; Seminara, S.; Bergman, J.; Brunner, H.G.; et al. CHD7mutations in patients initially diagnosed with Kallmann syndrome—The clinical overlap with CHARGE syndrome. Clin. Genet. 2008, 75, 65–71. [Google Scholar] [CrossRef]
- Xu, C.; Cassatella, D.; Van Der Sloot, A.M.; Quinton, R.; Hauschild, M.; De Geyter, C.; Flueck, C.; Feller, K.; Bartholdi, D.; Nemeth, A.; et al. Evaluating CHARGE syndrome in congenital hypogonadotropic hypogonadism patients harboring CHD7 variants. Genet. Med. 2017, 20, 872–881. [Google Scholar] [CrossRef]
- Schulz, Y.; Wehner, P.; Opitz, L.; Salinas-Riester, G.; Bongers, E.M.H.F.; Van Ravenswaaij-Arts, C.M.A.; Wincent, J.; Schoumans, J.; Kohlhase, J.; Borchers, A.; et al. CHD7, the gene mutated in CHARGE syndrome, regulates genes involved in neural crest cell guidance. Hum. Ganet. 2014, 133, 997–1009. [Google Scholar] [CrossRef] [PubMed]
- Ufartes, R.; Schwenty-Lara, J.; Freese, L.; Neuhofer, C.M.; Möller, J.; Wehner, P.; Van Ravenswaaij-Arts, C.M.A.; Wong, M.T.Y.; Schanze, I.; Tzschach, A.; et al. Sema3a plays a role in the pathogenesis of CHARGE syndrome. Hum. Mol. Genet. 2018, 27, 1343–1352. [Google Scholar] [CrossRef]
- Barraud, P.; John, J.S.; Stolt, C.C.; Wegner, M.; Baker, C.V.H. Olfactory ensheathing glia are required for embryonic olfactory axon targeting and the migration of gonadotropin-releasing hormone neurons. Biol. Open 2013, 2, 750–759. [Google Scholar] [CrossRef]
- Pingault, V.; Bodereau, V.; Baral, V.; Marcos, S.; Watanabe, Y.; Chaoui, A.; Fouveaut, C.; Leroy, C.; Vérier-Mine, O.; Francannet, C.; et al. Loss-of-Function Mutations in SOX10 Cause Kallmann Syndrome with Deafness. Am. J. Hum. Genet. 2013, 92, 707–724. [Google Scholar] [CrossRef]
- Inoue, K.; Khajavi, M.; Ohyama, T.; Hirabayashi, S.-I.; Wilson, J.H.; Reggin, J.D.; Mancias, P.; Butler, I.J.; Wilkinson, M.F.; Wegner, M.; et al. Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat. Genet. 2004, 36, 361–369. [Google Scholar] [CrossRef]
- Rojas, R.A.; Kutateladze, A.A.; Plummer, L.; Stamou, M.; Keefe, D.L.K., Jr.; Salnikov, K.B.; Delaney, A.; Hall, J.E.; Sadreyev, R.; Ji, F.; et al. Phenotypic continuum between Waardenburg syndrome and idiopathic hypogonadotropic hypogonadism in humans with SOX10 variants. Genet. Med. 2021, 23, 629–636. [Google Scholar] [CrossRef]
- Blewitt, M.; Gendrel, A.-V.; Pang, Z.; Sparrow, D.B.; Whitelaw, N.; Craig, J.M.; Apedaile, A.; Hilton, D.J.; Dunwoodie, S.L.; Brockdorff, N.; et al. SmcHD1, containing a structural-maintenance-of-chromosomes hinge domain, has a critical role in X inactivation. Nat. Genet. 2008, 40, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Shaw, N.; Brand, H.; Kupchinsky, Z.A.; Bengani, H.; Plummer, L.; Jones, T.I.; Erdin, S.; Williamson, K.A.; Rainger, J.; Stortchevoi, A.; et al. SMCHD1 mutations associated with a rare muscular dystrophy can also cause isolated arhinia and Bosma arhinia microphthalmia syndrome. Nat. Genet. 2017, 49, 238–248. [Google Scholar] [CrossRef]
- Gordon, C.T.; Xue, S.; Yigit, G.; Filali, H.; Chen, K.; Rosin, N.; Yoshiura, K.-I.; Oufadem, M.; Beck, T.J.; McGowan, R.; et al. De novo mutations in SMCHD1 cause Bosma arhinia microphthalmia syndrome and abrogate nasal development. Nat. Genet. 2017, 49, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Delaney, A.; Volochayev, R.; Meader, B.; Lee, J.; Almpani, K.; Noukelak, G.Y.; Henkind, J.; Chalmers, L.; Law, J.R.; Williamson, K.A.; et al. Insight Into the Ontogeny of GnRH Neurons From Patients Born Without a Nose. J. Clin. Endocrinol. Metab. 2020, 105, 1538–1551. [Google Scholar] [CrossRef]
- Sharma, V.P.; Mulliken, J.B.; Fenwick, A.L.; Brockop, M.S.; McGowan, S.; Goos, J.A.C.; Hoogeboom, A.J.M.; Brady, A.F.; Jeelani, N.U.O.; Lynch, S.A.; et al. Mutations in TCF12, encoding a basic helix-loop-helix partner of TWIST1, are a frequent cause of coronal craniosynostosis. Nat. Genet. 2013, 45, 304–307. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.E.; Balasubramanian, R.; Kupchinsky, Z.A.; Keefe, D.L.; Plummer, L.; Khan, K.; Meczekalski, B.; Heath, K.E.; Lopez-Gonzalez, V.; Ballesta-Martinez, M.J.; et al. TCF12 haploinsufficiency causes autosomal dominant Kallmann syndrome and reveals network-level interactions between causal loci. Hum. Mol. Genet. 2020, 29, 2435–2450. [Google Scholar] [CrossRef]
- Uittenbogaard, M.; Chiaramello, A. Expression of the bHLH transcription factor Tcf12 (ME1) gene is linked to the expansion of precursor cell populations during neurogenesis. Gene Expr. Patterns 2002, 1, 115–121. [Google Scholar] [CrossRef]
- Blümel, R.; Zink, M.; Klopocki, E.; Liedtke, D. On the traces of tcf12: Investigation of the gene expression pattern during development and cranial suture patterning in zebrafish (Danio rerio). PLoS ONE 2019, 14, e0218286. [Google Scholar] [CrossRef]
- Quaynor, S.D.; Bosley, M.E.; Duckworth, C.G.; Porter, K.R.; Kim, S.-H.; Kim, H.-G.; Chorich, L.P.; Sullivan, M.E.; Choi, J.-H.; Cameron, R.S.; et al. Targeted next generation sequencing approach identifies eighteen new candidate genes in normosmic hypogonadotropic hypogonadism and Kallmann syndrome. Mol. Cell. Endocrinol. 2016, 437, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.J.; Sapp, J.; Turner, J.T.; Amor, D.; Aftimos, S.; Aleck, K.A.; Bocian, M.; Bodurtha, J.N.; Cox, G.F.; Curry, C.J.; et al. Molecular analysis expands the spectrum of phenotypes associated with GLI3 mutations. Hum. Mutat. 2010, 31, 1142–1154. [Google Scholar] [CrossRef] [PubMed]
- Taroc, E.Z.M.; Naik, A.S.; Lin, J.M.; Peterson, N.B.; Keefe, D.L.; Genis, E.; Fuchs, G.; Balasubramanian, R.; Forni, P.E. Gli3 Regulates Vomeronasal Neurogenesis, Olfactory Ensheathing Cell Formation, and GnRH-1 Neuronal Migration. J. Neurosci. 2019, 40, 311–326. [Google Scholar] [CrossRef]
- Tischfield, M.A.; Baris, H.N.; Wu, C.; Rudolph, G.; Van Maldergem, L.; He, W.; Chan, W.-M.; Andrews, C.; Demer, J.L.; Robertson, R.; et al. Human TUBB3 Mutations Perturb Microtubule Dynamics, Kinesin Interactions, and Axon Guidance. Cell 2010, 140, 74–87. [Google Scholar] [CrossRef]
- Chew, S.; Balasubramanian, R.; Chan, W.-M.; Kang, P.; Andrews, C.; Webb, B.D.; MacKinnon, S.E.; Oystreck, D.T.; Rankin, J.; Crawford, T.O.; et al. A novel syndrome caused by the E410K amino acid substitution in the neuronal β-tubulin isotype 3. Brain 2013, 136, 522–535. [Google Scholar] [CrossRef]
- Shao, Q.; Yang, T.; Huang, H.; Majumder, T.; Khot, B.A.; Khouzani, M.M.; Alarmanazi, F.; Gore, Y.K.; Liu, G. Disease-associated mutations in human TUBB3 disturb netrin repulsive signaling. PLoS ONE 2019, 14, e0218811. [Google Scholar] [CrossRef] [PubMed]
- Latremoliere, A.; Cheng, L.; DeLisle, M.; Wu, C.; Chew, S.; Hutchinson, E.; Sheridan, A.; Alexandre, C.; Latremoliere, F.; Sheu, S.-H.; et al. Neuronal-Specific TUBB3 Is Not Required for Normal Neuronal Function but Is Essential for Timely Axon Regeneration. Cell Rep. 2018, 24, 1865–1879.e9. [Google Scholar] [CrossRef]
- Balasubramanian, R.; Zhang, X. Mechanisms of FGF gradient formation during embryogenesis. Semin. Cell Dev. Biol. 2015, 53, 94–100. [Google Scholar] [CrossRef]
- Chan, W.K.; Price, D.; Pratt, T. Fgf8 morphogen gradients are differentially regulated by heparan sulphotransferases Hs2st and Hs6st1 in the developing brain. Biol. Open 2017, 6, 1933–1942. [Google Scholar] [CrossRef] [PubMed]
- Kawauchi, S.; Shou, J.; Santos, R.; Heébert, J.M.; McConnell, S.K.; Mason, I.; Calof, A.L. Fgf8 expression defines a morphogenetic center required for olfactory neurogenesis and nasal cavity development in the mouse. Development 2005, 132, 5211–5223. [Google Scholar] [CrossRef] [PubMed]
- Falardeau, J.; Chung, W.; Beenken, A.; Raivio, T.; Plummer, L.; Sidis, Y.; Jacobson-Dickman, E.E.; Eliseenkova, A.V.; Ma, J.; Dwyer, A.; et al. Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J. Clin. Investig. 2008, 118, 2822–2831. [Google Scholar] [CrossRef]
- Chung, W.C.J.; Moyle, S.S.; Tsai, P.-S. Fibroblast Growth Factor 8 Signaling through Fibroblast Growth Factor Receptor 1 Is Required for the Emergence of Gonadotropin-Releasing Hormone Neurons. Endocrinology 2008, 149, 4997–5003. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.C.; Moenter, S.M.; Tsai, P.-S. Developmental Regulation of Gonadotropin-Releasing Hormone Neurons by Fibroblast Growth Factor Signaling. Endocrinology 2004, 145, 3830–3839. [Google Scholar] [CrossRef]
- Forni, P.E.; Bharti, K.; Flannery, E.M.; Shimogori, T.; Wray, S. The Indirect Role of Fibroblast Growth Factor-8 in Defining Neurogenic Niches of the Olfactory/GnRH Systems. J. Neurosci. 2013, 33, 19620–19634. [Google Scholar] [CrossRef]
- Pitteloud, N.; Acierno, J.S.; Meysing, A.; Eliseenkova, A.V.; Ma, J.; Ibrahimi, O.A.; Metzger, D.L.; Hayes, F.J.; Dwyer, A.A.; Hughes, V.A.; et al. Mutations in fibroblast growth factor receptor 1 cause both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc. Natl. Acad. Sci. USA 2006, 103, 6281–6286. [Google Scholar] [CrossRef]
- Simonis, N.; Migeotte, I.; Lambert, N.; Perazzolo, C.; De Silva, D.C.; Dimitrov, B.; Heinrichs, C.; Janssens, S.; Kerr, B.; Mortier, G.; et al. FGFR1mutations cause Hartsfield syndrome, the unique association of holoprosencephaly and ectrodactyly. J. Med Genet. 2013, 50, 585–592. [Google Scholar] [CrossRef]
- Villanueva, C.; Jacobson-Dickman, E.; Xu, C.; Manouvrier, S.; Dwyer, A.A.; Sykiotis, G.P.; Beenken, A.; Liu, Y.; Tommiska, J.; Hu, Y.; et al. Congenital hypogonadotropic hypogonadism with split hand/foot malformation: A clinical entity with a high frequency of FGFR1 mutations. Genet. Med. 2014, 17, 651–659. [Google Scholar] [CrossRef]
- Miraoui, H.; Dwyer, A.; Sykiotis, G.; Plummer, L.; Chung, W.; Feng, B.; Beenken, A.; Clarke, J.; Pers, T.; Dworzynski, P.; et al. Mutations in FGF17, IL17RD, DUSP6, SPRY4, and FLRT3 Are Identified in Individuals with Congenital Hypogonadotropic Hypogonadism. Am. J. Hum. Genet. 2013, 92, 725–743. [Google Scholar] [CrossRef] [PubMed]
- Men, M.; Wang, X.; Wu, J.; Zeng, W.; Jiang, F.; Zheng, R.; Li, J.-D. Prevalence and associated phenotypes of DUSP6, IL17RD and SPRY4 variants in a large Chinese cohort with isolated hypogonadotropic hypogonadism. J. Med. Genet. 2020, 58, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Josso, N.; di Clemente, N.; Gouédard, L. Anti-Müllerian hormone and its receptors. Mol. Cell. Endocrinol. 2001, 179, 25–32. [Google Scholar] [CrossRef]
- Josso, N.; Racine, C.; di Clemente, N.; Rey, R.A.; Xavier, F. The role of anti-Müllerian hormone in gonadal development. Mol. Cell. Endocrinol. 1998, 145, 3–7. [Google Scholar] [CrossRef]
- Behringer, R.R.; Finegold, M.J.; Cate, R.L. Müllerian-inhibiting substance function during mammalian sexual development. Cell 1994, 79, 415–425. [Google Scholar] [CrossRef]
- Cimino, I.; Casoni, F.; Liu, X.; Messina, A.; Parkash, J.; Jamin, S.; Catteau-Jonard, S.; Collier, F.; Baroncini, M.; Dewailly, D.; et al. Novel role for anti-Müllerian hormone in the regulation of GnRH neuron excitability and hormone secretion. Nat. Commun. 2016, 7, 10055. [Google Scholar] [CrossRef] [PubMed]
- Malone, S.A.; Papadakis, G.E.; Messina, A.; Mimouni, N.E.H.; Trova, S.; Imbernon, M.; Allet, C.; Cimino, I.; Acierno, J.; Cassatella, D.; et al. Defective AMH signaling disrupts GnRH neuron development and function and contributes to hypogonadotropic hypogonadism. eLife 2019, 8, 1–36. [Google Scholar] [CrossRef]
- Cannarella, R.; Paganoni, A.; Cicolari, S.; Oleari, R.; Condorelli, R.; La Vignera, S.; Cariboni, A.; Calogero, A.; Magni, P. Anti-Müllerian Hormone, Growth Hormone, and Insulin-Like Growth Factor 1 Modulate the Migratory and Secretory Patterns of GnRH Neurons. Int. J. Mol. Sci. 2021, 22, 2445. [Google Scholar] [CrossRef]
- Prosser, H.M.; Bradley, A.; Caldwell, M.A. Olfactory bulb hypoplasia in Prokr2 null mice stems from defective neuronal progenitor migration and differentiation. Eur. J. Neurosci. 2007, 26, 3339–3344. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.-I.; Yamazaki, C.; Masumoto, K.-H.; Nagano, M.; Naito, M.; Soga, T.; Hiyama, H.; Matsumoto, M.; Takasaki, J.; Kamohara, M.; et al. Abnormal development of the olfactory bulb and reproductive system in mice lacking prokineticin receptor PKR2. Proc. Natl. Acad. Sci. USA 2006, 103, 4140–4145. [Google Scholar] [CrossRef]
- Dodé, C.; Teixeira, L.; Levilliers, J.; Fouveaut, C.; Bouchard, P.; Kottler, M.-L.; Lespinasse, J.; Lienhardt-Roussie, A.; Mathieu, M.; Moerman, A.; et al. Kallmann Syndrome: Mutations in the Genes Encoding Prokineticin-2 and Prokineticin Receptor-2. PLoS Genet. 2006, 2, e175. [Google Scholar] [CrossRef]
- Cole, L.W.; Sidis, Y.; Zhang, C.; Quinton, R.; Plummer, L.; Pignatelli, D.; Hughes, V.A.; Dwyer, A.; Raivio, T.; Hayes, F.J.; et al. Mutations inProkineticin 2andProkineticin receptor 2genes in Human Gonadotrophin-Releasing Hormone Deficiency: Molecular Genetics and Clinical Spectrum. J. Clin. Endocrinol. Metab. 2008, 93, 3551–3559. [Google Scholar] [CrossRef]
- Cox, K.H.; Oliveira, L.M.B.; Plummer, L.; Corbin, B.; Gardella, T.; Balasubramanian, R.; Crowley, W.F. Modeling mutant/wild-type interactions to ascertain pathogenicity of PROKR2 missense variants in patients with isolated GnRH deficiency. Hum. Mol. Genet. 2017, 27, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Schwarting, G.A.; Kostek, C.; Bless, E.P.; Ahmad, N.; Tobet, S.A. Deleted in Colorectal Cancer (DCC) Regulates the Migration of Luteinizing Hormone-Releasing Hormone Neurons to the Basal Forebrain. J. Neurosci. 2001, 21, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Schwarting, G.A.; Raitcheva, D.; Bless, E.P.; Ackerman, S.L.; Tobet, S. Netrin 1-mediated chemoattraction regulates the migratory pathway of LHRH neurons. Eur. J. Neurosci. 2004, 19, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Bouilly, J.; Messina, A.; Papadakis, G.E.; Cassatella, D.; Xu, C.; Acierno, J.; Tata, B.; Sykiotis, G.; Santini, S.; Sidis, Y.; et al. DCC/NTN1 complex mutations in patients with congenital hypogonadotropic hypogonadism impair GnRH neuron development. Hum. Mol. Genet. 2017, 27, 359–372. [Google Scholar] [CrossRef]
- Kotan, L.D.; Cooper, C.; Darcan, Ş.; Carr, I.M.; Özen, S.; Yan, Y.; Hamedani, M.K.; Gürbüz, F.; Mengen, E.; Turan, I.; et al. Idiopathic Hypogonadotropic Hypogonadism Caused by Inactivating Mutations in SRA1. J. Clin. Res. Pediatr. Endocrinol. 2016, 8, 125–134. [Google Scholar] [CrossRef]
- Bamshad, M.; Lin, R.C.; Law, D.J.; Watkins, W.S.; Krakowiak, P.A.; Moore, M.E.; Franceschini, P.; Lala, R.; Holmes, L.B.; Gebuhr, T.C.; et al. Mutations in human TBX3 alter limb, apocrine and genital development in ulnar-mammary syndrome. Nat. Genet. 1997, 16, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Galazzi, E.; Duminuco, P.; Moro, M.; Guizzardi, F.; Marazzi, N.; Sartorio, A.; Avignone, S.; Bonomi, M.; Persani, L.; Bonati, M.T. Hypogonadotropic hypogonadism and pituitary hypoplasia as recurrent features in Ulnar-Mammary syndrome. Endocr. Connect. 2018, 7, 1432–1441. [Google Scholar] [CrossRef]
- Eriksson, K.S.; Mignot, E. T-box 3 is expressed in the adult mouse hypothalamus and medulla. Brain Res. 2009, 1302, 233–239. [Google Scholar] [CrossRef]
- Huisman, C.; Cho, H.; Brock, O.; Lim, S.J.; Youn, S.M.; Park, Y.; Kim, S.; Lee, S.-K.; Delogu, A.; Lee, J.W. Single cell transcriptome analysis of developing arcuate nucleus neurons uncovers their key developmental regulators. Nat. Commun. 2019, 10, 3696. [Google Scholar] [CrossRef] [PubMed]
- Quarta, C.; Fisette, A.; Xu, Y.; Colldén, G.; Legutko, B.; Tseng, Y.-T.; Reim, A.; Wierer, M.; De Rosa, M.C.; Klaus, V.; et al. Functional identity of hypothalamic melanocortin neurons depends on Tbx3. Nat. Metab. 2019, 1, 222–235. [Google Scholar] [CrossRef]
- Zhu, Z.; Han, X.; Li, Y.; Han, C.; Deng, M.; Zhang, Y.; Shen, Q.; Cao, Y.; Li, Z.; Wang, X.; et al. Identification of ROBO1/2 and SCEL as candidate genes in Kallmann syndrome with emerging bioinformatic analysis. Endocrine 2019, 67, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Cariboni, A.; Andrews, W.D.; Memi, F.; Ypsilanti, A.R.; Zelina, P.; Chedotal, A.; Parnavelas, J.G. Slit2 and Robo3 modulate the migration of GnRH-secreting neurons. Development 2012, 139, 3326–3331. [Google Scholar] [CrossRef]
- Taroc, E.Z.; Lin, J.M.; Tulloch, A.J.; Jaworski, A.; Forni, P.E. GnRH-1 Neural Migration from the Nose to the Brain Is Independent From Slit2, Robo3 and NELL2 Signaling. Front. Cell. Neurosci. 2019, 13, 70. [Google Scholar] [CrossRef]
- Oleari, R.; André, V.; Lettieri, A.; Tahir, S.; Roth, L.; Paganoni, A.; Eberini, I.; Parravicini, C.; Scagliotti, V.; Cotellessa, L.; et al. A Novel SEMA3G Mutation in Two Siblings Affected by Syndromic GnRH Deficiency. Neuroendocrinology 2020, 111, 421–441. [Google Scholar] [CrossRef] [PubMed]
- Jee, Y.H.; Won, S.; Lui, J.C.; Jennings, M.; Whalen, P.; Yue, S.; Temnycky, A.G.; Barnes, K.M.; Cheetham, T.; Boden, M.G.; et al. DLG2 variants in patients with pubertal disorders. Genet. Med. 2020, 22, 1329–1337. [Google Scholar] [CrossRef]
- Barraud, S.; Delemer, B.; Poirsier-Violle, C.; Bouligand, J.; Mérol, J.-C.; Grange, F.; Higel-Chaufour, B.; Decoudier, B.; Zalzali, M.; Dwyer, A.; et al. Congenital Hypogonadotropic Hypogonadism with Anosmia and Gorlin Features Caused by a PTCH1 Mutation Reveals a New Candidate Gene for Kallmann Syndrome. Neuroendocrinology 2020, 111, 99–114. [Google Scholar] [CrossRef]
- Messina, A.; Langlet, F.; Chachlaki, K.; Roa, J.; Rasika, S.; Jouy, N.; Gallet, S.; Gaytan, F.; Parkash, J.; Tena-Sempere, M.; et al. A microRNA switch regulates the rise in hypothalamic GnRH production before puberty. Nat. Neurosci. 2016, 19, 835–844. [Google Scholar] [CrossRef]
- Ahmed, K.; Lapierre, M.P.; Gasser, E.; Denzler, R.; Yang, Y.; Ruelicke, T.; Kero, J.; Latreille, M.; Stoffel, M. Loss of microRNA-7a2 induces hypogonadotropic hypogonadism and infertility. J. Clin. Investig. 2017, 127, 1061–1074. [Google Scholar] [CrossRef] [PubMed]
- Iivonen, A.-P.; Känsäkoski, J.; Vaaralahti, K.; Raivio, T. Screening for mutations in selected miRNA genes in hypogonadotropic hypogonadism patients. Endocr. Connect. 2019, 8, 506–509. [Google Scholar] [CrossRef] [PubMed]
- Stamou, M.; Ng, S.-Y.; Brand, H.; Wang, H.; Plummer, L.; Best, L.; Havlicek, S.; Hibberd, M.; Khor, C.C.; Gusella, J.; et al. A Balanced Translocation in Kallmann Syndrome Implicates a Long Noncoding RNA, RMST, as a GnRH Neuronal Regulator. J. Clin. Endocrinol. Metab. 2019, 105, e231–e244. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Gene | Function | Role in GnRH System | Mouse CHH-Related Phenotype | Human Phenotype | MIM Number |
|---|---|---|---|---|---|
| AMH | D/E | Migration and axon elongation | NA | nCHH/KS | 261550 |
| AMHR2 | D/E | Migration and axon elongation | Abnormal nasal axon patterning; impaired GnRH neuron migration, reduced number of MPOA GnRH neuron; hypogonadism and subfertility | nCHH/KS | 261550 |
| ANOS1 | D | Migration | NA | KS | 308700 |
| AXL | D | Survival | Increased apoptotic GnRH neurons, reduced number of MPOA GnRH neuron; delayed puberty | nCHH/KS | NA |
| CCDC141 | D | Migration | Decreased GnRH neuron migration (nasal explants) | KS | NA |
| CHD7 | D | Neurogenesis | OB hypoplasia, defective olfactory neuron neurogenesis, impaired senses of smell; defective GnRH neuron neurogenesis, reduced number of MPOA GnRH neuron; hypogonadism and delayed puberty | nCHH/KS + CHARGE | 612370 |
| CHL1 | D | Migration and survival | Abnormal nasal axon patterning (hypothesized) | KS | NA |
| DCC | D/E | Migration and axon elongation | Abnormal nasal axon patterning; impaired GnRH neuron migration, reduced number of MPOA GnRH neuron | KS | NA |
| DMXL2 | E | Signaling and secretion | Reduced number of MPOA GnRH neuron; hypogonadism, delayed puberty, subfertility | nCHH + PEPNS | 616133 |
| DUSP6 | D/E | Neurogenesis and axon elongation (hypothesized) | NA | nCHH/KS | 615269 |
| FEZF1 | D | Migration | OB hypoplasia, abnormal nasal axon patterning; impaired GnRH neuron migration | KS | 616030 |
| FGF8 | D/E | Neurogenesis and axon elongation | Abnormal nasal axon patterning; absence of GnRH neurons | nCHH/KS + SOD | 612702 |
| FGF17 | D/E | Neurogenesis and axon elongation (hypothesized) | NA | CHH + DWS | 615270 |
| FGFR1 | D/E | Neurogenesis and axon elongation | Reduced number of nasal and MPOA GnRH neuron; delayed puberty, subfertility | nCHH/KS + HS and SHFM | 147950 |
| FLRT3 | D/E | Neurogenesis and axon elongation (hypothesized) | NA | KS | 615271 |
| GNRH1 | E | Signaling and secretion | Hypogonadism, infertility | nCHH | 614841 |
| GLI3 | D | Migration | Impaired OECs formation, abnormal nasal axon patterning; impaired GnRH neuron migration, reduced number of MPOA GnRH neuron | KS + GCPS | 175700 |
| HS6ST1 | D | Migration | Delayed puberty | nCHH/KS | 614880 |
| IGSF10 | D | Migration | NA | nCHH | NA |
| IL17RD | D | Neurogenesis (hypothesized) | NA | KS | 615267 |
| KISS1 | E | Signaling and secretion | Absent puberty, hypogonadism | nCHH | 614842 |
| KISS1R | E | Signaling and secretion | Absent puberty, hypogonadism | nCHH | 614837 |
| KLB | E | Signaling and secretion | Hypogonadism, delayed puberty, subfertility | nCHH | NA |
| LEP | E | Signaling and secretion | Hypogonadism, infertility | nCHH + obesity | 614962 |
| LEPR | E | Signaling and secretion | Infertility | nCHH + obesity | 614963 |
| NDNF | D | Migration | Abnormal nasal axon patterning; impaired GnRH neuron migration, reduced number of MPOA GnRH neuron | KS | 618841 |
| NR0B1 | E | Signaling and secretion | Hypogonadism, infertility | nCHH + CAH | 300200 |
| NRP1 | D | Migration | Abnormal nasal axon patterning; impaired GnRH neuron migration, reduced number of MPOA GnRH neuron | KS | NA |
| NRP2 | D | Migration | Abnormal nasal axon patterning; impaired GnRH neuron migration, reduced number of MPOA GnRH neuron; hypogonadism | KS | NA |
| NSMF | D | Migration | Abnormal nasal axon patterning; impaired GnRH neuron migration (nasal explants), reduced number of MPOA GnRH neuron; hypogonadism, delayed puberty, subfertility | nCHH/KS | 614838 |
| NTN1 | D/E | Migration and axon elongation | Abnormal nasal axon patterning; impaired GnRH neuron migration, reduced number of MPOA GnRH neuron | KS | 618264 |
| OTUD4 | D | Uncertain | NA | nCHH + GHS | NA |
| PCSK1 | E | Signaling and secretion | NA | nCHH + obesity | 600955 |
| PLXNA1 | D | Migration | Abnormal nasal axon patterning; impaired GnRH neuron migration, reduced number of MPOA GnRH neuron | KS | NA |
| PLXNA3 | D | Migration | Normal nasal axon patterning; normal GnRH neuron migration; normal gonadal size | nCHH/KS | NA |
| PNPLA6 | E | Uncertain | NA | nCHH + GHS | 215470 |
| POLR3A | E | Uncertain | NA | nCHH + 4H | 607694 |
| POLR3B | E | Uncertain | NA | nCHH + 4H | 614381 |
| PROK2 | D/E | Migration and axon elongation | OB hypoplasia; impaired GnRH neuron migration, reduced number of MPOA GnRH neuron; hypogonadism, absent puberty, infertility | nCHH/KS | 610628 |
| PROKR2 | D/E | Migration and axon elongation | OB hypoplasia, abnormal nasal axon patterning; reduced number of MPOA GnRH neuron; hypogonadism | nCHH/KS + SOD | 244200 |
| RNF216 | E | Uncertain | Hypogonadism, infertility | nCHH + GHS | 212840 |
| SMCHD1 | D | Migration | NA | KS + BAMS | 603457 |
| SEMA3A | D | Migration | Abnormal nasal axon patterning; impaired GnRH neuron migration, reduced number of MPOA GnRH neuron; hypogonadism | nCHH/KS | 614897 |
| SEMA3E | D | Survival | Increased apoptotic GnRH neurons, reduced number of MPOA GnRH neuron; hypogonadism | KS + CHARGE? | 214800 |
| SEMA3F | D | Migration | Normal nasal axon patterning; normal GnRH neuron migration | nCHH/KS | NA |
| SEMA7A | D | Migration | Impaired GnRH neuron migration, reduced number of MPOA GnRH neuron; hypogonadism; hypogonadism, subfertility | nCHH/KS | NA |
| SOX10 | D | Migration | Impaired OECs migration, abnormal nasal axon patterning; impaired GnRH neuron migration, reduced number of MPOA GnRH neuron | KS + WS and HD | 602229 |
| SPRY4 | D/E | Migration and axon elongation (hypothesized) | NA | nCHH/KS | 615266 |
| STUB1 | E | Uncertain | Hypogonadism | nCHH + GHS | NA |
| TAC3 | E | Signaling and secretion | Delayed puberty | nCHH | 614839 |
| TACR3 | E | Signaling and secretion | Hypogonadism | nCHH | 618840 |
| TCF12 | D | Neurogenesis and migration | NA | KS + C | 615314 |
| TUBB3 | D | Migration (hypothesized) | NA | KS + CFEOM | 600638 |
| WDR11 | D | Neurogenesis | OB hypoplasia, reduced number of hypothalamic GnRH neurons; hypogonadism, delayed puberty, infertility | nCHH/KS + CPHD | 614858 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oleari, R.; Massa, V.; Cariboni, A.; Lettieri, A. The Differential Roles for Neurodevelopmental and Neuroendocrine Genes in Shaping GnRH Neuron Physiology and Deficiency. Int. J. Mol. Sci. 2021, 22, 9425. https://doi.org/10.3390/ijms22179425
Oleari R, Massa V, Cariboni A, Lettieri A. The Differential Roles for Neurodevelopmental and Neuroendocrine Genes in Shaping GnRH Neuron Physiology and Deficiency. International Journal of Molecular Sciences. 2021; 22(17):9425. https://doi.org/10.3390/ijms22179425
Chicago/Turabian StyleOleari, Roberto, Valentina Massa, Anna Cariboni, and Antonella Lettieri. 2021. "The Differential Roles for Neurodevelopmental and Neuroendocrine Genes in Shaping GnRH Neuron Physiology and Deficiency" International Journal of Molecular Sciences 22, no. 17: 9425. https://doi.org/10.3390/ijms22179425
APA StyleOleari, R., Massa, V., Cariboni, A., & Lettieri, A. (2021). The Differential Roles for Neurodevelopmental and Neuroendocrine Genes in Shaping GnRH Neuron Physiology and Deficiency. International Journal of Molecular Sciences, 22(17), 9425. https://doi.org/10.3390/ijms22179425